
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and solution equilibrium studies on half-sandwich organorhodium complexes of (N,N) donor bidentate ligands†
János P.
Mészáros
a,
Orsolya
Dömötör
a,
Carmen M.
Hackl
b,
Alexander
Roller
b,
Bernhard K.
Keppler
bc,
Wolfgang
Kandioller
 bc and
Éva A.
Enyedy
*a
bc and
Éva A.
Enyedy
*a
aDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: enyedy@chem.u-szeged.hu
bInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Str. 42, A-1090 Vienna, Austria
cResearch Cluster Translational Cancer Therapy Research, University of Vienna, Waehringer Str. 42, A-1090 Vienna, Austria
First published on 1st June 2018
Abstract
Complex formation equilibrium processes of [Rh(η5-C5Me5)(H2O)3]2+ with N,N′-dimethylethylenediamine (dmen), N,N,N′,N′-tetramethylethylenediamine (tmeda), 2-picolylamine (pin) and 1,10-phenanthroline (phen) were studied in aqueous solution by 1H NMR spectroscopy, UV-vis spectrophotometry and pH-potentiometry. Formation and deprotonation of [Rh(η5-C5Me5)(L)(H2O)]2+ complexes and exchange process of the aqua to chlorido ligand were characterized in addition to single-crystal X-ray diffraction analysis of [Rh(η5-C5Me5)(L)(Cl)]+ complexes (L = dmen, tmeda and pin). Formation of complexes with significantly high stability was found except tmeda due to the sterical hindrance between the methyl groups of the chelating ligand and the arenyl ring resulting in an increased methyl group-ring plane torsion angle. [Rh(η5-C5Me5)(L)(H2O)]2+ complexes of dmen, pin, phen predominate at pH 7.4 without decomposition even in the micromolar concentration range. The complexes were characterized by relatively high chloride affinity and a strong correlation was obtained between the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K′ (H2O/Cl−) and pKa of [Rh(η5-C5Me5)(L)(H2O)]2+ constants for a series of (O,O), (O,N) and (N,N)-chelated complexes. For this set of 12 complexes a relationship between logK′ (H2O/Cl−) values and certain crystallographic parameters was found using multiple linear regression approach. DNA binding of these complexes was also monitored and compared by ultrafiltration and fluorimetry.
K′ (H2O/Cl−) and pKa of [Rh(η5-C5Me5)(L)(H2O)]2+ constants for a series of (O,O), (O,N) and (N,N)-chelated complexes. For this set of 12 complexes a relationship between logK′ (H2O/Cl−) values and certain crystallographic parameters was found using multiple linear regression approach. DNA binding of these complexes was also monitored and compared by ultrafiltration and fluorimetry.
Introduction
The tremendous success of Pt(II) anticancer drugs, which currently are the best selling and most widely used antitumor compounds, has stimulated the exploration of other effective metal-based compounds. In this context Ru-based antineoplastic metal complexes with low side effects have been developed, e.g. trans-[tetrachloridobis(1H-indazole)ruthenate(III)] (KP1339/IT-139), which is currently under development against numerous human tumour types.1,2 Unfortunately, another clinically developed compound, trans-[tetrachlorido(DMSO)(imidazole)ruthenate(III)] (NAMI-A),3 failed to be successful under clinical studies. Ru(III) complexes are considered as prodrugs that are activated by reduction that provides the impetus for the development of various Ru(II) anticancer compounds. Ru is often stabilized in the +2 oxidation state by the coordination of η6-arene type ligands.4 Besides the numerous half-sandwich Ru(II) organometallics of the type [Ru(η6-arene)(X,Y)(Z)], in which (X,Y) is a chelating ligand and Z is leaving co-ligand, analogous complexes of the heavier congener Os(II) are also extensively being investigated.5,6 In addition a large number of the isoelectronic Rh(III) and Ir(III) η5-bound arenyl complexes were also developed showing promising in vitro anticancer activity.7 Notably, the half-sandwich organometallic compounds have attracted increasing attention not just as potential therapeutic agents, but this type of compounds offers a broad scope for the design of water-soluble catalysts for transfer hydrogenation reactions as well. In general, the type of the metal ion, the arene ring, the chelating bidentate ligand and the leaving group have a strong impact on the biological or the catalytic activity. Some structure–activity relationships have already been established8–11 considering for instance the anticancer potency of Ru(η6-arene) compounds bearing ligands providing (N,N), (N,O) and (O,O) donor sets,8 or catalytic activity of Rh, Ir and Ru complexes containing 1,10-phenanthroline (phen) or its derivatives for the regeneration of NADH in the chemoenzymatic reduction of ketones.9 However, the knowledge on the aqueous solution chemistry of this type of half-sandwich organometallic compounds is still limited. Information about the stability, predominant forms at various concentrations and pH values, ratio of the active aqua and the chlorido species is strongly required for the understanding of their solution behavior. Determination of equilibrium constants for organometallic compounds is less abundant in the literature regarding the huge number of the synthesized structures. A panel of solution equilibrium studies of [Ru(η6-p-cymene)(X,Y)(Z)] complexes is reported by Buglyó et al.,12,13 while in the publications of Sadler et al. mostly pKa values were determined for [Ru(η6-arene)(X,Y)(H2O)] compounds and the hydrolysis of the chlorido complexes was also investigated in detail.6,14 Solution equilibrium constants for various bidentate (O,O),15,16 (O,N),16–18 (O,S)19 and (N,N)20 donor containing Rh(η5-pentamethylcyclopentadienyl) (Rh(η5-C5Me5)) coordination compounds were reported in our previous works. These results revealed that the chloride affinity of the [Rh(η5-C5Me5)(L)H2O)]2+/+ complexes seems to be a crucial factor, just like in case of analogous Ir(η5-C5Me5) and some Ru(η6-arene) compounds.6,21While the Rh(η5-C5Me5) complexes of the simplest bidentate (N,N) donor ethylenediamine and the aromatic diimine bpy exhibited only poor anticancer activity,7 the analogous complexes of phen,7 polypyridyl ligands7 and their various derivatives22 with more extended aromatic systems are reported to show remarkable cytotoxic properties in various human cancer cell lines. Due to the lack of solution equilibrium data on the latter complexes herein we investigate Rh(η5-C5Me5) complex of phen in addition to methylated derivatives of ethylenediamine. 2-Picolylamine was also involved as a representative of a mixed (N,N) donor ligand containing an aliphatic amine and an aromatic imine (Chart 1). The main aim of our study is to reveal correlations between complex architectures and thermodynamic data regarding their solution behavior.
 | ||
| Chart 1 Chemical structures of the ligands: N,N′-dimethylethylenediamine (dmen), N,N,N′,N′-tetramethylethylenediamine (tmeda), 2-picolylamine (pin) and 1,10-phenanthroline (phen) and the general formula of the prepared [Rh(η5-C5Me5)(L)(Cl)]+ complexes. | ||
Results and discussion
Synthesis and X-ray structures of the organometallic rhodium(III) complexes
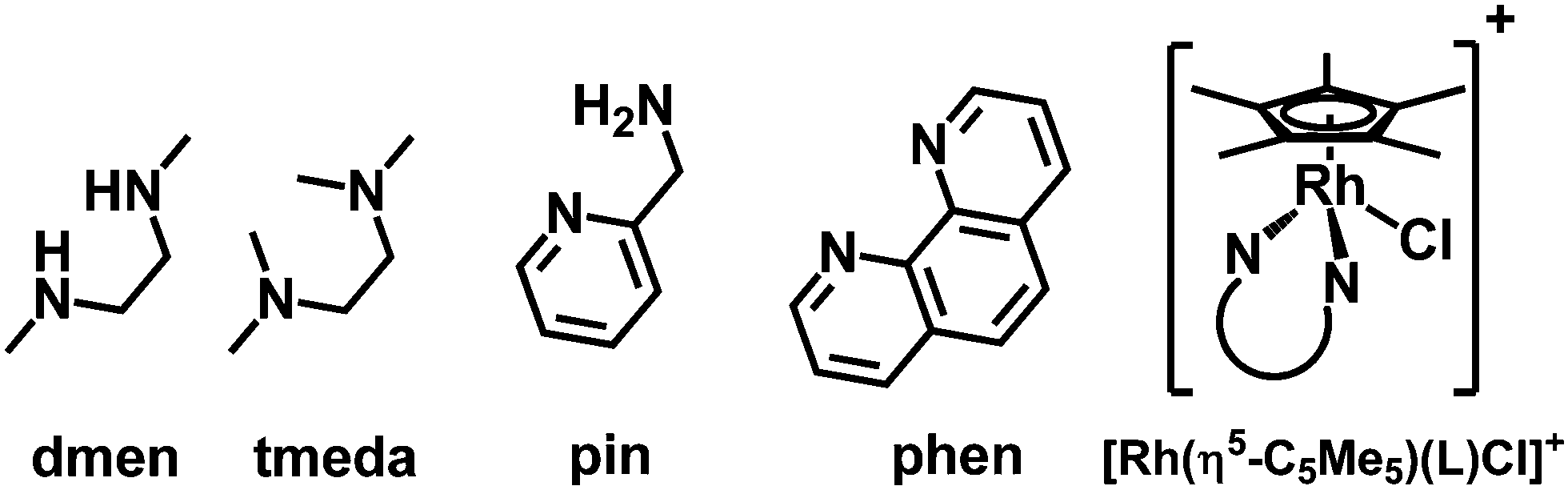
The rhodium(III) precursor [Rh(η5-C5Me5)(μ-Cl)Cl]2 used for the complex preparation was synthesized according to literature.23 The synthesis of [Rh(η5-C5Me5)(tmeda)Cl]Cl and [Rh(η5-C5Me5)(phen)Cl]Cl has been already reported,24,25 herein the complexes of dmen, tmeda, pin and phen were obtained following the established procedure reported by Scharwitz el al.,25 however the 2-picolylamine complex was prepared without the chloride elimination step. Pure compounds as [Rh(η5-C5Me5)(L)Cl]CF3SO3 (L = dmen, tmeda, phen) as triflate salt or [Rh(η5-C5Me5)(L)Cl]Cl (L = pin) with chloride as counterion were isolated from a CH3OH/CH2Cl2 solvent mixture in moderate to good yields (34–72%). The organometallic rhodium(III) complexes were characterized by means of standard analytical methods (1H NMR spectroscopy, elemental analysis and electrospray ionization mass spectrometry (ESI-MS)). Single crystals of [Rh(η5-C5Me5)(dmen)Cl]+ (1), [Rh(η5-C5Me5)(tmeda)Cl]+ (2) and [Rh(η5-C5Me5)(pin)Cl]+ (3) with CF3SO3− (dmen, tmeda) or Cl− (pin) counter anion were obtained by the slow evaporation method from a CH3OH/H2O mixture at room temperature. The X-ray structures of the phen complex with various counter ions are well-documented in the literature.25,26 The ORTEP representations of the complexes 1–3 are depicted in Fig. 1, 2 and Fig. S1 (ESI†). Crystallographic data are presented in Table S1 (ESI†), and selected bond lengths and angles are listed in Table 1. All complexes possess ‘piano stool’ configuration, whereby C5Me5− forms the seat and the chelating (N,N) ligand as well as the chlorido leaving group constitute the chair legs. Complexes 2·CF3SO3 and 3·Cl crystallize in the space group P121/n1, while complex 1·CF3SO3 is a representative of the space group P212121. The molecular structures of the studied complexes were directly compared to each other and to that of the [Rh(η5-C5Me5)(en)Cl]ClO4 complex determined in our former work (Table 1).20 Regarding the Rh-to-ring centroid distances in [Rh(η5-C5Me5)(en)Cl]ClO4 (1.763 Å), 1·CF3SO3 (1.778 Å) and 2·CF3SO3 (1.812 Å) we can conclude that it is increasing with the higher number of the methyl substituents. The bond lengths between Rh and the nitrogen donor atoms show a similar trend. However, not only these bond lengths represent considerable differences, as the methyl group-ring plane torsion angles become higher and higher in the order of the complexes of en, dmen and tmeda as well (Table 1). This observation is well-represented when the structures of [Rh(η5-C5Me5)(en)Cl]+ and 2 are superimposed (Fig. 2). It is clearly seen that the methyl groups of the C5Me5− moiety are out of the plane of the ring system in 2. Most probably the steric hindrance between the methyl groups of the arenyl ring and the tetramethylated ligand results in the elongated Rh–ring centroid, Rh–N distances and the bigger torsion angle (7.50°) in complex 2. Relatively long Rh–N bond lengths are also reported for the analogous [Ru(η5-C5Me5)(tmeda)Cl] and [Ir(η5-C5Me5)(tmeda)Cl]Cl complexes, in which 8.5° and 7.0° methyl group-ring plane torsion angles are calculated respectively based on the published data.10,27 Therefore, our findings predict a lower solution stability of 2 compared to the complex of ethylenediamine.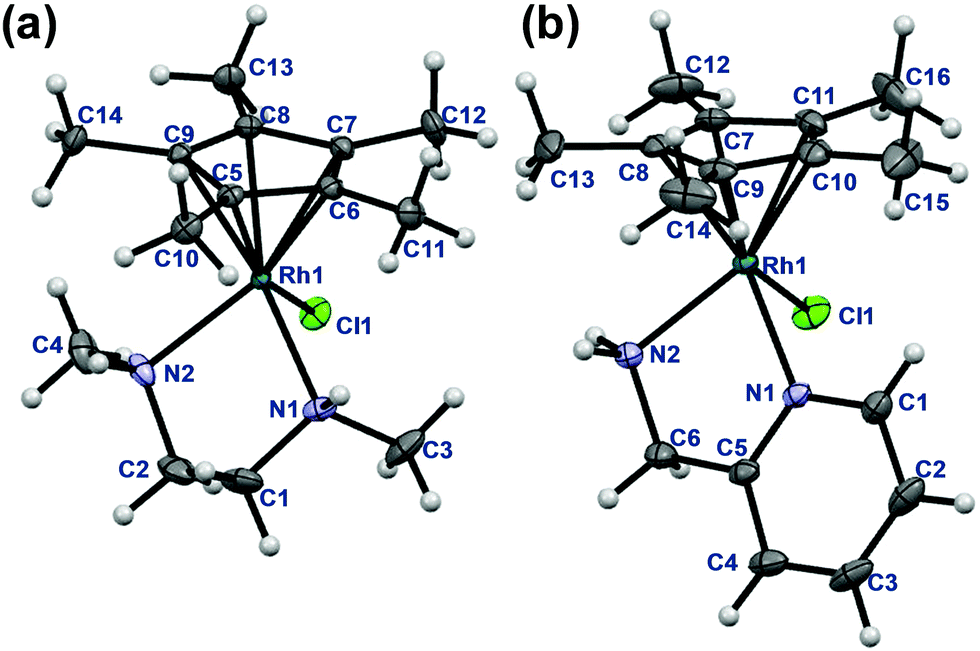 | ||
| Fig. 1 Molecular structures of the metal complex 1 (a) and 3 (b). Solvent molecules and counter ions are omitted for clarity. Displacement ellipsoids are drawn at 50% probability level. | ||
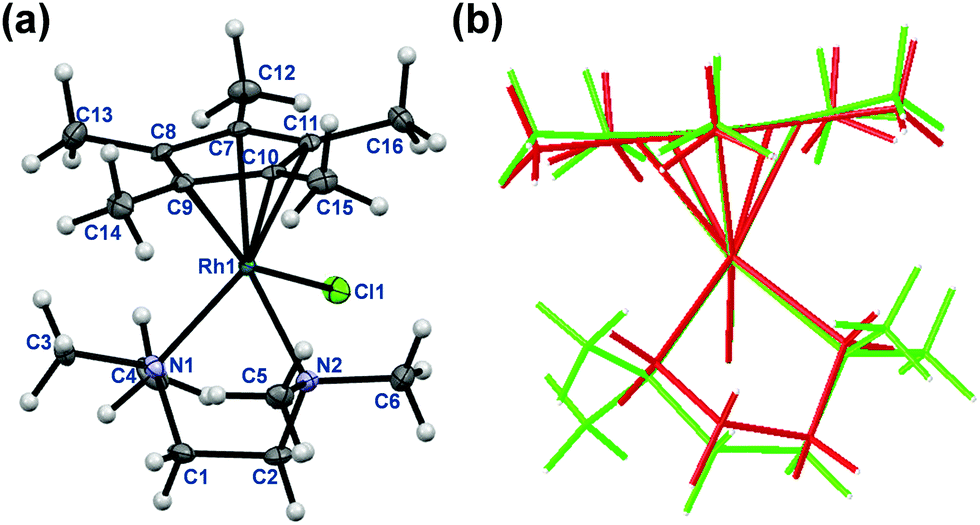 | ||
| Fig. 2 Molecular structure of 2. Solvent molecules and counter ions are omitted for clarity. Displacement ellipsoids are drawn at 50% probability level (a). Comparison of the molecular structure of complex 2 (coloured with green) with [Rh(η5-C5Me5)(en)(Cl)]+ (coloured with red) (b). | ||
| [Rh(η5-C5Me5) (en)(Cl)]ClO420 | 1·CF3SO3 | 2·CF3SO3 | 3·Cl | |
|---|---|---|---|---|
| Bond lengths (Å) | ||||
| Rh-ring centroid | 1.763 | 1.778 | 1.812 | 1.782 |
| Rh–N1 | 2.145 | 2.158(1) | 2.234(2) | 2.142(1) |
| Rh–N2 | 2.124 | 2.143(2) | 2.184(2) | 2.114(1) |
| Rh–Cl | 2.434 | 2.406(1) | 2.431(1) | 2.427(1) |
| Angles (°) | ||||
| N1–Rh–N2 | 80.23 | 81.02(6) | 80.36(7) | 77.47(4) |
| N1–Rh–Cl | 88.09 | 92.24(4) | 90.13(5) | 86.66(3) |
| N2–Rh–Cl | 85.41 | 88.16(4) | 87.74(5) | 89.04(3) |
| Torsion angles (°) | ||||
| CH3-ring plane | 2.146 | 3.27(15) | 7.50(18) | 3.93(13) |
| N1–C–C–N2 | 53.82 | 56.6(2) | 56.5(3) | 25.63(17) |
It is worth mentioning that a significant difference is also observed between the N1–C–C–N2 torsion angles in the case of the various (N,N) donor ligands. Compounds bearing only aliphatic amines (en, dmen, tmeda) have torsion angle falling in the range of 53.82–56.62°, while for the rigid bpy and phen fairly low torsion angles (0.00°, 0.24° respectively) were observed. This torsion angle for the complex of 2-picolylamine (3·Cl) falls between these extremities (25.63°).
Proton dissociation processes of the ligands and hydrolysis of the organometallic cation
Proton dissociation constants (pKa) of dmen, tmeda, pin and phen (Table 2) were determined herein by pH-potentiometry in a chloride-free medium and values are in good agreement with those reported in the literature29–31 when account is taken of the different ionic strengths. Notably, the tertiary diamine (tmeda) has significantly lower pKa values compared to the secondary (dmen) and primary diamine ethylenediamine. The pK (H2L2+) and pK (HL+) of 2-picolylamine are attributed to the deprotonation of the pyridinium and the primary amine nitrogens, respectively. In the case of phen only pKa of HL+ species could be determined in the studied pH range with adequate accuracy.K [ML]2+) and proton dissociation constants (pKa [ML]2+) of the Rh(η5-C5Me5) complexes formed with (N,N) donor bidentate ligands in chloride-free aqueous solutions determined by various methods; H2O/Cl− exchange constants (logK′) and conditional stability constants at physiological pH logK7.4′ for the [Rh(η5-C5Me5)(L)(H2O)]2+ complexes {T = 25 °C; I = 0.2 M (KNO3)}a
| Constants | enb | dmen | tmeda | pin | bpyb | phen |
|---|---|---|---|---|---|---|
|
a Uncertainties (SD) of the last digits are shown in parentheses. Hydrolysis products of the organometallic cations: logβ [(Rh(η5-C5Me5))2(OH)2(H2O)2]2+ = −8.53, logβ [(Rh(η5-C5Me5))2(OH)3]+ = −14.26 at I = 0.20 M (KNO3) taken from ref. 15.
b Data taken from ref. 20.
c Determined by pH-potentiometric titrations at pH 2.0–11.5.
d pK (H2L2+) = 7.12 and pK (HL+) = 10.05, I = 0.2 M (KCl) in ref. 29.
e pK (H2L2+) = 6.06 and pK (HL+) = 9.29, I = 0.2 M (KCl) in ref. 29.
f pK (H2L2+) = 2.14 and pK (HL+) = 8.57, I = 0.1 M (KNO3) in ref. 30.
g pK (H2L2+) = 1.90 and pK (HL+) = 4.96, I = 0.1 M (NaNO3) in ref. 31.
h Determined by UV-vis spectrophotometry at pH 2.0–5.3.
i Determined by 1H NMR spectroscopy at pH 2.0–11.5.
j For the [Rh(η5-C5Me5)(en)(H2O)]2+ + L ⇌ [Rh(η5-C5Me5)(L)(H2O)]2+ + en equilibrium determined at various total L concentrations by UV-vis.
k For the [Rh(η5-C5Me5)(L)(H2O)]2+ + Cl− ⇌ [Rh(η5-C5Me5)(L)Cl]+ + H2O equilibrium determined at various total chloride ion concentrations by UV-vis.
|
||||||
| pKa (H2L2+)c | 7.25 | 7.16(1)d | 5.95(2)e | 2.29(2)f | — | —g |
| pKa (HL+)c | 10.01 | 10.04(1)d | 9.25(1)e | 8.69(1)f | 4.41 | 4.92(1)g |
| logK [ML]2+ |
15.04 | 14.80(2)h | 7.40(10)i | 13.59(8)j | ≥12.95 | ≥13.80j |
| pKa [ML]2+i |
9.58 | Isomer (S,R): 8.61(9) | 8.42(3) | 8.48(3) | 8.61 | 8.58(2) |
| Isomer (R,S): 8.40(6) | ||||||
| logK7.4′ [ML]2+ |
12.20 | 11.99 | 5.53 | 12.28 | ≥12.95 | ≥13.80 |
| logK′ (H2O/Cl−)k |
2.14 | 2.60(1) | — | 2.43(1) | 2.58 | 2.92(1) |
The hydrolytic behavior of the aquated organometallic cation [Rh(η5-C5Me5)(H2O)3]2+ has been studied previously,28 and the overall stability constants were reported for the μ-hydroxido-bridged dinuclear rhodium(III) species [(Rh(η5-C5Me5))2(μ-OH)3]+, [(Rh(η5-C5Me5))2(μ-OH)2]2+) in our former work,15 and were used for the calculations.
Complex formation equilibria of [Rh(η5-C5Me5)(H2O)3]2+ with the selected (N,N) donor ligands
The complexation between [Rh(η5-C5Me5)(H2O)3]2+ (= M2+) and the studied (N,N) bidentate ligands always follows a fairly simple scheme in aqueous solution in the absence of chloride ions (Chart S1, ESI†), since only mono-ligand [Rh(η5-C5Me5)(L)(H2O)]2+ (= [ML]2+) and [Rh(η5-C5Me5)(L)(OH)]+ (= [ML(OH)]+) complexes are formed, similarly to the case of numerous analogous half-sandwich organorhodium compounds.15–20 Complex formation of [Rh(η5-C5Me5)(H2O)3]2+ with the ligands containing solely aliphatic nitrogen donor atoms (dmen, tmeda) was found to be a rather slow process that hindered the use of pH-potentiometric titrations. In order to overcome this problem, individual samples were prepared by the addition of various amounts of KOH under argon, and the actual pH, the 1H NMR and UV-vis spectra were measured only after 24 h. During this period the equilibrium could be reached assuredly based on the time-dependent measurements.The logK [ML]2+ constant of the dmen complex was determined from the UV-vis spectral changes in the pH range from 2.0 to 5.3 (Fig. S2, ESI†). The 1H NMR spectra recorded for the dmen complex reveal slow ligand-exchange processes on the NMR time scale (t1/2(obs) ∼ 1 ms) and as a consequence the peaks belonging to the free or bound metal fragment (and ligand) could be detected separately (Fig. 3). Based on the integrated peak areas of the C5Me5 protons in the unbound and bound fractions a logK [ML]2+ constant could be also calculated from data collected at pH < 7.5 (Table 2), that represents good agreement with the constant obtained spectrophotometrically. According to the 1H NMR spectra the bound dmen ligand can be found in two types of [ML]2+ complexes which are assumed to be isomers. The free and achiral ligand in the H2L2+ form has two singlet peaks of the CH2 (3.44 ppm) and CH3 (2.80 ppm) protons and they turn to be doublet of triplets and doublet, respectively in the metal-bound forms.
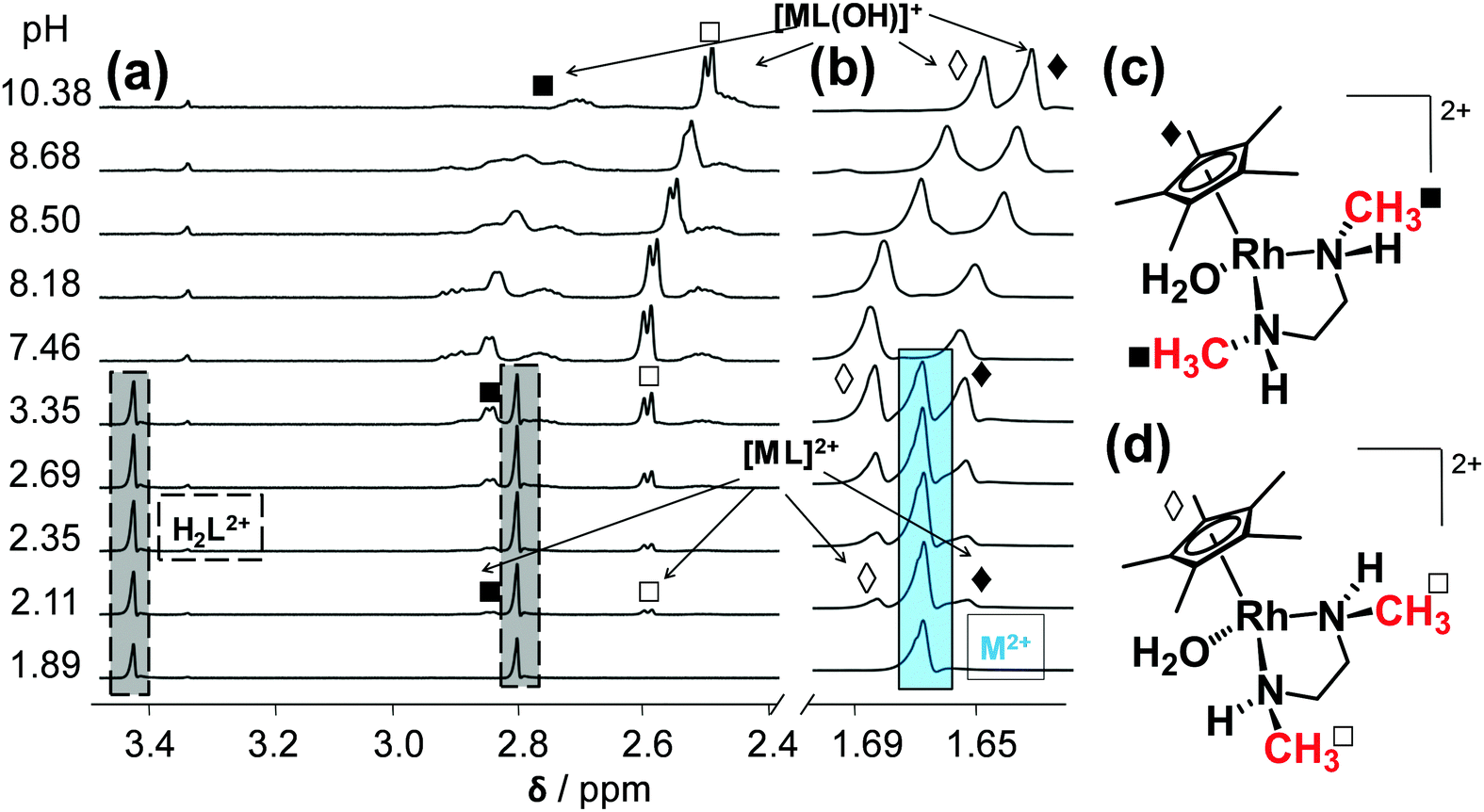 | ||
| Fig. 3
1H NMR spectra for the [Rh(η5-C5Me5)(H2O)3]2+–dmen (1:1) system recorded at the indicated pH values with peak assignation: peaks of dmen (a); peaks of C5Me5− (b) {cRh = cdmen = 1 mM; T = 25 °C; I = 0.20 M (KNO3); 10% D2O}. Structures of the (R,S) isomer (c) and the (S,R) isomer (d) of [Rh(η5-C5Me5)(dmen)(H2O)]2+. | ||
These secondary amine nitrogen atoms have three different substituents and when coordinating to Rh they become chirality centers, thus formation of four different isomers is possible. This phenomenon was also observed in the case of [Pt(dmen)Cl2] complexes and the (S,S′) and (R,R′) isomers crystallized from aqueous solution.32 Based on the 1H NMR spectra two isomers are formed and their ratio is ca. 1:1. The ratio of the doublets represents the ratio of the nitrogens in the different chemical environment and configuration. On the other hand the ratio of the methyl protons of the C5Me5 fragment of the two complexes is also ca. 1:1. One of the isomers is most probably the (R,S) complex that was crystallized from the solution (vide supra), while the other is assumed to be the (S,R) isomer. (Otherwise the ratio cannot be 1:1.) The peaks of the CH3 protons of the coordinated ligand and the C5Me5− moiety are found at higher and at lower chemical shift (δ) values, respectively in the (R,S) isomer as compared to the other isomer, as a results of the stronger steric hindrance between the Me groups in the (R,S) isomer. An upfield shift of all peaks belonging to both [ML]2+ isomers is observed in the basic pH range due to the fast exchange process between the aquated and the mixed hydroxido [ML(OH)]+ species. Therefore, pKa of the aqua isomers as microscopic constants could be determined on the basis of the pH-dependent δ values (Table 2). The spectra recorded undoubtedly reveal that neither the free organometallic ion nor the free ligand is present at pH > 5.3, which means that the dmen complexes do not suffer from decomposition at pH 7.4. The decomposition is negligible even at 1 μM concentration at this pH on the basis of the stability constants determined.
On the contrary unbound ligand and organometallic fragment are detected by 1H NMR spectroscopy in the whole pH range studied (2–11.5) in the [Rh(η5-C5Me5)(H2O)3]2+–tmeda (1:1) system even at 1 mM concentration (Fig. 4). Notably, only one kind of [ML]2+ complex is formed in the pH range from 4 to 10 reaching the maximum fraction (85%) at pH 7.0 (Fig. 4b). Based on these 1H NMR spectra logK [ML]2+ and pKa [ML]2+ constants were computed (Table 2). These data undoubtedly indicate the formation of complexes with much lower stability in the case of tmeda as compared to dmen (or en) as it was expected on the basis of the findings of the X-ray structure analysis (vide supra).
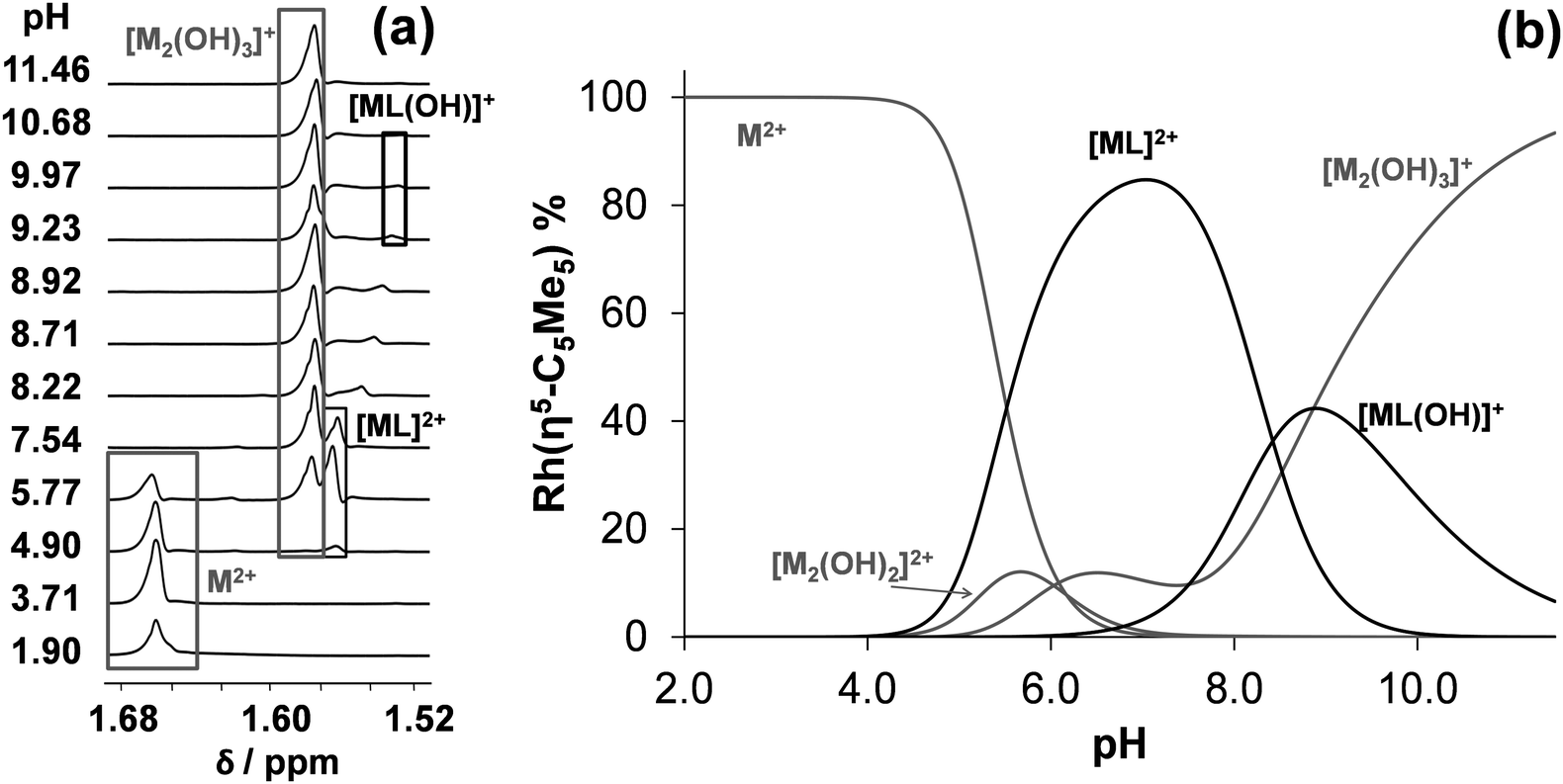 | ||
| Fig. 4 High-field region of the 1H NMR spectra for the [Rh(η5-C5Me5)(H2O)3]2+ (M2+)–tmeda (1:1) system recorded at the indicated pH values {cRh = ctmeda = 1 mM; T = 25 °C; I = 0.20 M (KNO3); 10% D2O} (a). Concentration distribution curves for the [Rh(η5-C5Me5)(H2O)3]2+–tmeda (1:1) systems calculated on the basis of the stability constants determined {cRh = ctmeda = 1 mM; T = 25 °C; I = 0.20 M (KNO3)} (b). | ||
The complex formation with the aromatic nitrogen containing ligands (pin, phen) was found to be fast, although only bound fractions of the ligands and the metal ion could be detected by 1H NMR titrations in the pH range 2–11.5 (Fig. S3 (ESI†) for pin complex). This is the consequence of the formation of complexes with outstandingly high solution stability. Based on the spectral changes only pKa [ML]2+ constants were computed (Table 2).
Thus, the stability constants for the [ML]2+ species were determined by ligand competition measurements using spectrophotometry. Ethylenediamine was chosen as competitor. Ligand phen or pin was added to the [Rh(η5-C5Me5)(en)Cl]+ complex and clear UV-vis spectral changes were observed due to the stepwise displacement of the originally metal-bound ethylenediamine (Fig. 5 and Fig. S4, ESI†). The logK [ML]2+ value for the 2-picolylamine complex (Table 2) could be calculated by deconvolution of the recorded spectra using the computer program PSEQUAD.33 However, only a lower limit for the phen complex could be estimated, as the displacement of ethylenediamine was quantitative. Representative concentration distribution curves for the [Rh(η5-C5Me5)(H2O)3]2+ – 2-picolylamine system were computed on the basis of the stability constants determined (Fig. S3b, ESI†). They exhibit the predominant formation of the [ML] complex up to pH 7.0. The direct comparison of the logK [ML]2+ values is not adequate, since the complex formation equilibrium is superimposed by other accompanying equilibria, such as (de)protonation of the ligands and hydrolysis of the organometallic cation. As only the ligands differ in this series (the metal ion is the same), conditional stability constants (logK7.4′ [ML]2+) were computed at pH 7.4 taking into consideration the different basicities of the ligands (Table 2). Ligands containing two aromatic nitrogen donors (phen, bpy) form the highest stability complexes, and the other ligands give the following trend: pin > en ∼ dmen ≫ tmeda.
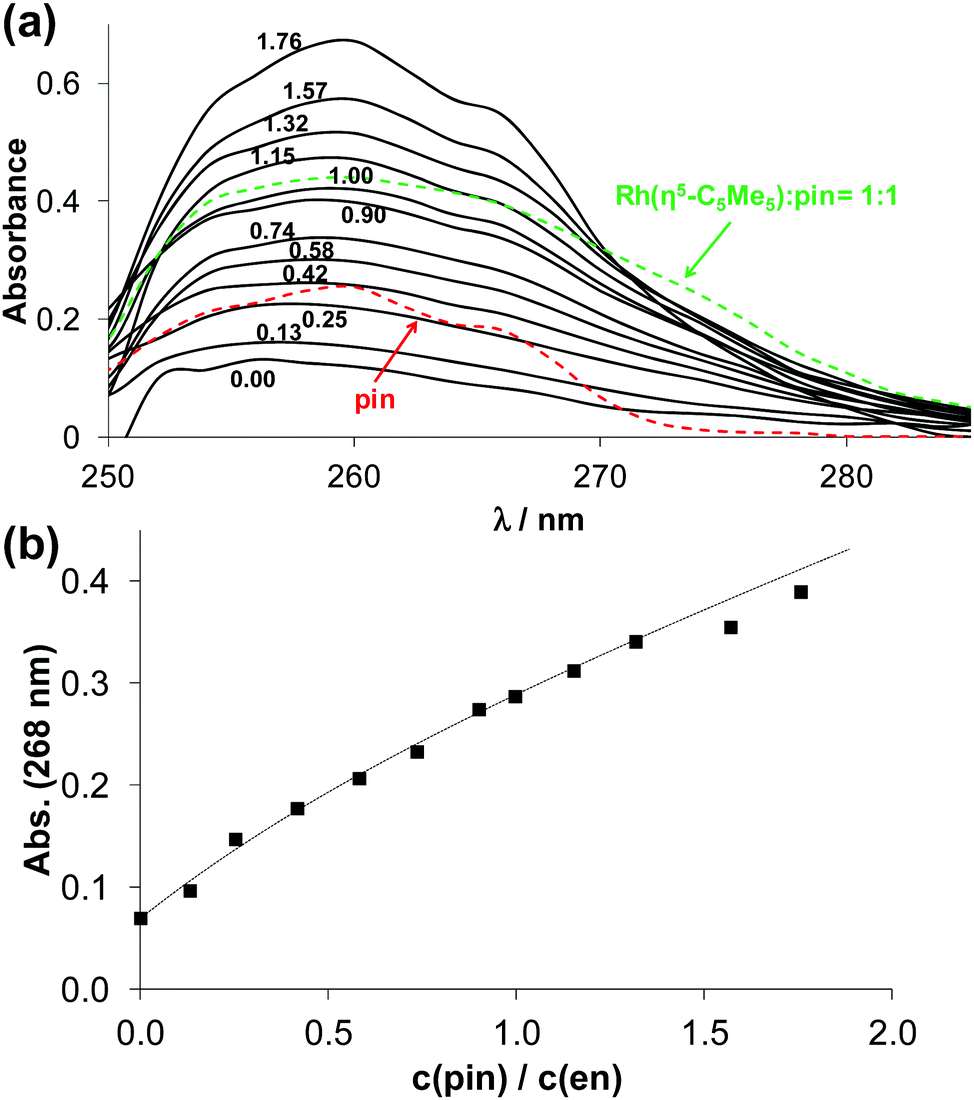 | ||
| Fig. 5 UV-vis spectra for the displacement study of [Rh(η5-C5Me5)(en)(H2O)]2+–pin (1:1) system (black solid lines). The numbers show the different c(pin)-to-c(en) ratios. The spectra of [Rh(η5-C5Me5)(pin)(H2O)]2+ and pin are shown with dashed lines (a). Absorbance values at 268 nm (■) plotted against the c(pin):c(en) ratio, dotted line shows the fitted spectral change (b); spectra are background subtracted {cRh = cen = 100 μM; I = 0.20 M KNO3, pH = 7.30, T = 25 °C, l = 1 cm}. | ||
Comparing the pKa [ML]2+ values of the [Rh(η5-C5Me5)(L)(H2O)]2+ complexes of en, dmen, tmeda, pin, bpy and phen (Table 2) it can be concluded that they fall into the range of 8.4–8.6 except to the complex of ethylenediamine (9.5820). These values indicate the formation of low fraction of mixed hydroxido species (6–9%) at pH 7.4 in the absence of chloride ions. However, the presence of the chloride ions generally results in higher pKa values15,16,20 thus even a smaller fraction of [ML(OH)]+ species at physiological pH.
Chloride ion affinity and correlations between equilibrium constants and crystallographic data
The Rh(η5-C5Me5) complexes of the studied bidentate (N,N) donor containing ligands (dmen, tmeda, pin, phen) have a chlorido ligand as a leaving group in their solid forms. In aqueous solution the chlorido ligand can be partly or completely exchanged to water (or OH−) depending on the concentration of the chloride ions and the pH. Aquation (Cl− → H2O exchange) is reported to be a crucial activation step for many anticancer metallodrugs such as cisplatin34 or half-sandwich organometallic compounds of the type [M(η6-arene)(X,Y)Cl] (M = Ru(II), Os(II)).6 In order to characterize the chloride ion affinity of these organorhodium complexes the following equilibrium process was monitored spectrophotometrically:| [Rh(η5-C5Me5)(L)(H2O)]2+ + Cl− ⇌ [Rh(η5-C5Me5)(L)(Cl)]+ + H2O. |
The chloride–water exchange process was studied at a pH value where the formation of the [ML]2+ complex is 100% (pH = 7.0–7.4). The reaction was found to be fast in all cases and takes place within a few minutes. The logK′ (H2O/Cl−) constants were calculated by the deconvolution of UV-vis spectra of the [Rh(η5-C5Me5)(L)(H2O)]2+ complexes recorded at various chloride ion concentrations. The displacement of H2O by Cl− results in characteristic spectral changes in the spectra as Fig. S5 (ESI†) shows for the [Rh(η5-C5Me5)(dmen)(H2O)]2+. In the case of the tmeda complex we could not determine this equilibrium constant since there is no appropriate condition at which the [Rh(η5-C5Me5)(tmeda)(H2O)]2+ complex forms predominantly due its low solution stability (vide supra). The obtained logK′ (H2O/Cl−) constants (2.1–2.9) are fairly high compared to the values of complexes formed with (O,O) bidentate ligands (e.g. deferiprone: 0.78,16 maltol: 1.1715). The higher logK′ (H2O/Cl−) constants indicate the higher chloride ion affinity of the complexes. As a consequence in the case of high logK′ (H2O/Cl−), the more difficult replacement of Cl− by water or donor atoms of proteins is feasible. In addition the complexes bearing the neutral (N,N) donor ligands are positively charged either in their aquated (2+) or chlorinated (+) forms resulting in their hydrophilic character. These two factors are not advantageous to the biological activity. The complexes of ethylenediamine, 2,2′-bipyridine are not cytotoxic (IC50 > 100 μM in human breast adenocarcinoma MCF-7 cell line7), on the contrary the compound [Rh(η5-C5Me5)(phen)(Cl)]CF3SO3 was found to be active (e.g. IC50 = 4.7 μM in MCF-7 cell line7). Notably, [Rh(η5-C5Me5)(L)Cl]+ complexes of polypyridyl ligands such as dipyrido-[3,2-f:2′,3′-h]quinoxaline (dpq) or dipyrido[3,2-a:2′,3′-c]phenazine (dppz) were reported to be similar or even more cytotoxic due to their intercalative binding into DNA.7
Analysis of the logK′ (H2O/Cl−) and pKa [ML]2+ constants being available in the literature for half-sandwich [Rh(η5-C5Me5)(XY)(H2O)]2+/+ complexes (where XY is a bidentate ligand, Table S2, ESI†) clearly reveals the strong correlation between these values as shown in Fig. 6. The coordinated ligands in the complexes are: deferiprone16 as (O,O) donor, 2-picolinic acid,16 6-methylpicolinic acid,17 quinoline-2-carboxylic acid,17 3-isoquinolinecarboxylic acid,17 8-hydroxyquinoline,18 8-hydroxyquinoline-5-sulfonate18 and 7-(1-piperidinylmethyl)-8-hydroxyquinoline18 as (O,N) donor and en,20 dmen, pin, bpy20 and phen as (N,N) donor. The higher logK′ (H2O/Cl−) is accompanied by a lower pKa [ML]2+ meaning the stronger tendency for the deprotonation of the coordinated water, thus higher OH− affinity of the complex. Since both the logK′ (H2O/Cl−) constants and the X-ray crystal structures of [Rh(η5-C5Me5)(XY)(Cl)]+/0 complexes of the same set of ligands listed above are reported in the literature (or determined in this work for some (N,N) donor bearing compounds), we examined their correspondence to cover a structure–property relationship. Different crystallographic parameters were involved in the analysis such as Rh–ring centroid distance, Rh-donor atom, Rh–Cl bond lengths, X–Rh–Y, X–Rh–Cl, Cl–Rh–Y angles, methyl group-ring plane torsion angle in addition to the charges of the [ML]2+/+ complexes (Table S3, ESI†). First of all we investigated which factors show a linear relationship with the logK′ (H2O/Cl−) constants. Then multiple linear regression approach was performed by Microsoft Excel. The logK′ (H2O/Cl−) constants were predicted as a function of the linear combination of a set of selected crystallographic parameters and were compared to the experimentally obtained values.
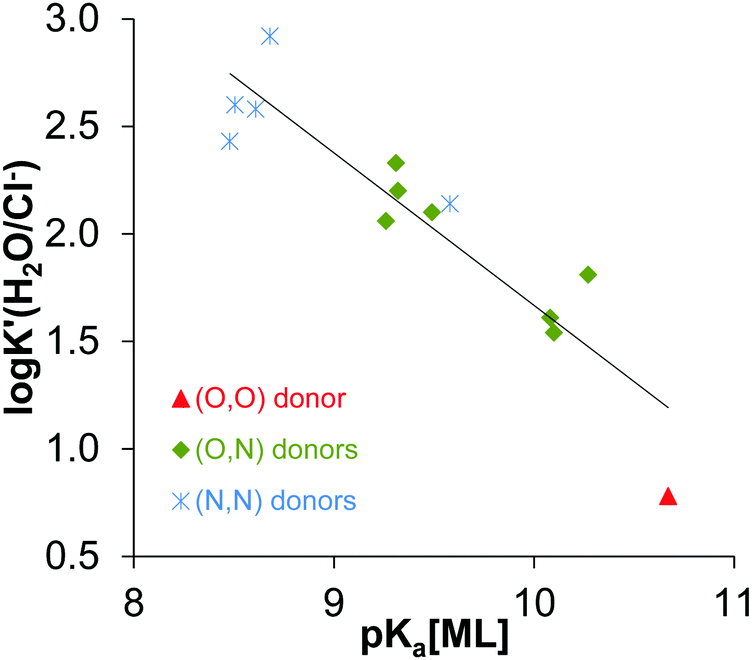 | ||
| Fig. 6 logK′ (H2O/Cl−) values vs. pKa (ML) for the Rh(η5-C5Me5) complexes containing various bidentate ligands with O/N/S donor atoms: R2 = 0.8403, logK′ (H2O/Cl−) = −0.7095 × pKa [ML] + 8.7623. The coordinated ligands in the complexes used in the correlation are: deferiprone16 as (O,O) donor, 2-picolinic acid,16 6-methylpicolinic acid,17 quinoline-2-carboxylic acid,17 3-isoquinolinecarboxylic acid,17 8-hydroxyquinoline,18 8-hydroxyquinoline-5-sulfonate18 and 7-(1-piperidinylmethyl)-8-hydroxyquinoline18 as (O,N) donors, en,20 dmen, pin, bpy20 and phen as (N,N) donors (see the constants collected in Table S2, ESI†). | ||
Among the various equations the following one gave the best-fitting straight line:
| calculated logK′ (H2O/Cl−) = 27.59 × distance(Rh–centroid) − 0.23 × angle(X–Rh–Y) − 0.23 × methyl group-ring plane torsion angle + 0.46 × charge of [ML] − 28.75. |
The calculated logK′ (H2O/Cl−) constants are plotted against the values determined spectrophotometrically in Fig. 7. Based on these findings we can conclude that the chloride affinity shows dependence on the Rh–centroid distance, X–Rh–Y angle and the methyl group-ring plane torsion angle. Based on this finding the logK′ (H2O/Cl−) for a novel [Rh(η5-C5Me5)(L)(Cl)] complex can be predicted based on the crystallographic data.
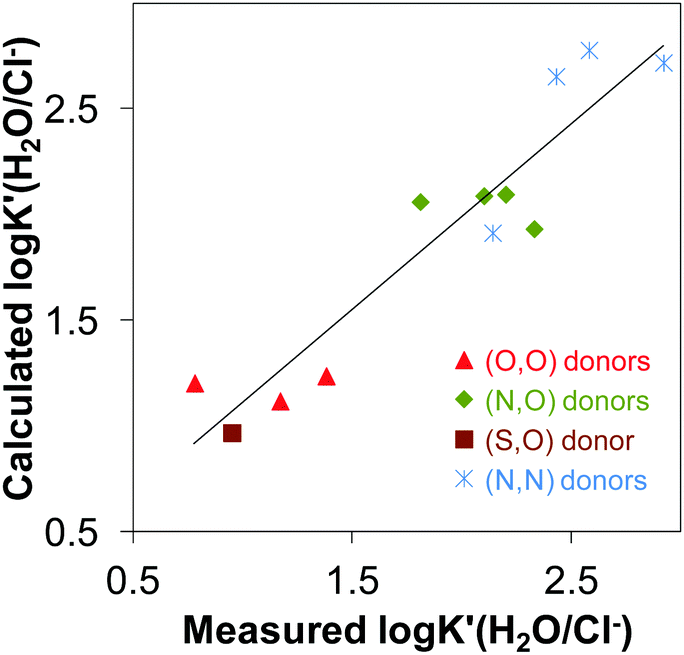 | ||
| Fig. 7 Multilinear regression between logK′(H2O/Cl−) vs. geometrical parameters: R2 = 0.8799; y = 27.59 × distance(Rh–centroid) − 0.23 × angle(X–Rh–X) − 0.23 × torsion angle(methyl group-ring plane) − 28.75. The coordinated ligands in the complexes used in the correlation are: deferiprone,16 maltol15,35 and allomaltol15 as (O,O) donors, 2-picolinic acid,16,35 6-methylpicolinic acid,17 quinoline-2-carboxylic acid,17 8-hydroxyquinoline18 as (O,N) donors, thiomaltol19 as (O,S) donor, en,20 pin, bpy20,25 and phen25 as (N,N) donors. | ||
Interaction of [Rh(η5-C5Me5)(L)(Cl)] complexes with DNA
DNA is a classical target for metallodrugs in general and was suggested for the complex [Rh(η5-C5Me5)(phen)(Cl)]+ as well.7 However, other primary targets such as proteins are also considered for anticancer half-sandwich Rh and Ru complexes. In order to compare the DNA binding affinity of [Rh(η5-C5Me5)(phen)(Z)] to that of other [Rh(η5-C5Me5)(XY)(Z)] complexes (Z = Cl− or H2O, charges omitted) ultrafiltration/UV-vis and fluorescence measurements were carried out.The binding of Rh(η5-C5Me5) complexes of deferiprone, 2-picolinic acid, quinoline-2-carboxylic acid, 3-isoquinolinecarboxylic acid, 8-hydroxyquinoline, en, dmen, tmeda, pin, bpy and phen towards DNA from calf thymus was studied by ultrafiltration/UV-vis quantification with a 10 kDa cutoff membrane filter. The binding was monitored at 1:1 complex-to-nucleotides ratio, at pH 7.4 and at 37 °C.
The chloride concentration of the samples was 4 mM according to cell nucleus. The low molecular mass (LMM) samples were analyzed by comparing their UV-vis spectra with the corresponding reference spectra yielding the fractions of the bound (and unbound) compounds (Fig. 8). Binding of [Rh(η5-C5Me5)(H2O)3]2+ was also involved (notably in the presence of chloride ions the aqua ligand is partly replaced by Cl−). Based on the recorded spectra for the LMM samples it could be concluded that these complexes do not suffer from decomposition during the DNA binding since no ligand release was observed. Comparing the bound metal complex fractions significant differences are seen. The fragment [Rh(η5-C5Me5)(H2O)3]2+ showed the strongest binding exceeding that of the intercalating ethidium bromide (EB). The Rh(η5-C5Me5) complex of 8-hydroxyquinoline exhibited the highest bound fraction among the studied [Rh(η5-C5Me5)(XY)(Z)] compounds, while not merely [Rh(η5-C5Me5)(phen)(Z)] but [Rh(η5-C5Me5)(en)(Z)] (without ligand with aromatic ring) also shows considerable binding. The binding behavior was further investigated by spectrofluorimetry in the case of [Rh(η5-C5Me5)(H2O)3]2+ (without ligand) and the Rh(η5-C5Me5) complexes of phen and ethylenediamine by the use of the fluorescent DNA probe EB. This compound has weak intrinsic fluorescence emission, but the adduct formation with DNA results in enhanced fluorescence intensity. Emission spectra were recorded for the DNA–EB system in the absence and in the presence of the metal complexes of phen and ethylenediamine, and the fraction of the unbound EB was obtained by the deconvolution of the spectra. Results are shown in Fig. S6 (ESI†). The free EB fraction is similar for the [Rh(η5-C5Me5)(H2O)3]2+ and the phen complex 4, while it is lower for the complex of ethylenediamine. However, the displacement of EB by these complexes does not mean clearly their intercalative binding mode as binding to nucleobase nitrogen of DNA was also suggested by Scharwitz et al.25 for the complexes of phen, bpy and ethylenediamine based on UV-vis absorption, melting temperature and viscosity measurements.
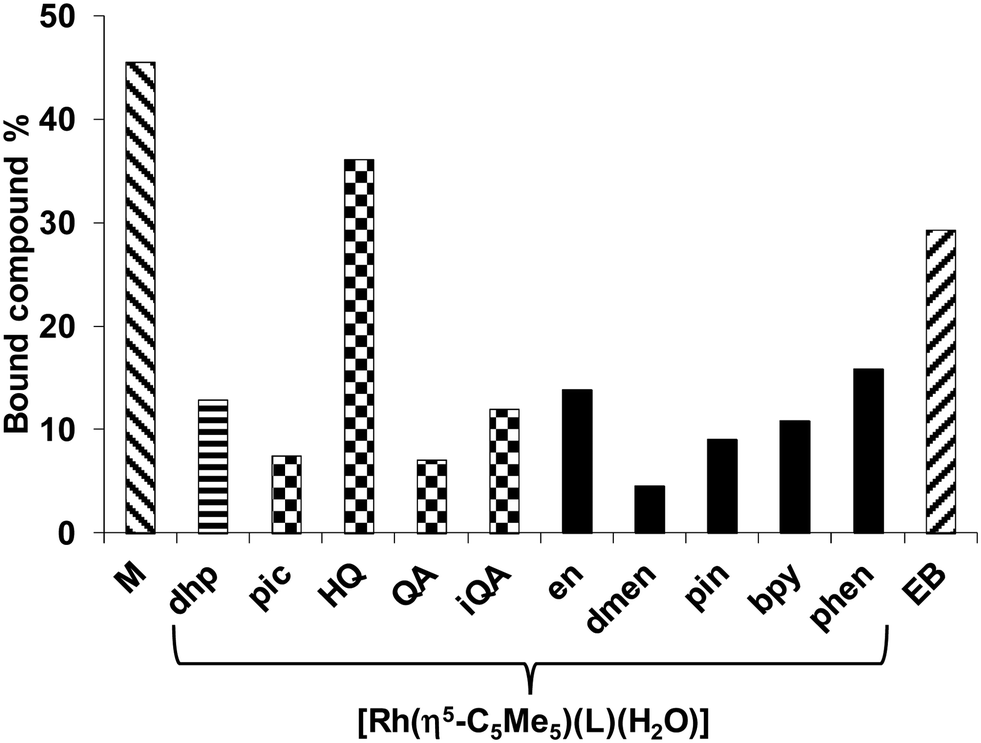 | ||
| Fig. 8 Bound [Rh(η5-C5Me5)(H2O)3]2+ fragment (M) without ligand, and its complexes of the general formula [Rh(η5-C5Me5)(L)(H2O)] (L = deferiprone (dhp), 2-picolinic acid (pic), 8-hydroxyquinoline (HQ), quinoline-2-carboxylic acid (QA), 3-isoquinolinecarboxylic acid (iQA), en, dmen, pin, bpy and phen respectively) and EB at 1:1 DNA nucleoside-to-compound ratio, measured by ultrafiltration-UV-vis method. {cCT-DNA = cRh = cL = 100 μM; pH = 7.40 (20 mM phosphate, 4 mM KCl); T = 37 °C; t = 24 h}. | ||
The hindrance of the EB binding might be a consequence of a structural distortion of the DNA due to the covalent (coordinative) binding of the studied Rh(η5-C5Me5) complexes to the donor atoms of the macromolecule. Therefore their binding to adenosine and guanosine was also compared using 1H NMR spectroscopy at 1:1 Rh:nucleoside ratio at pH 7.4 (Fig. 9).
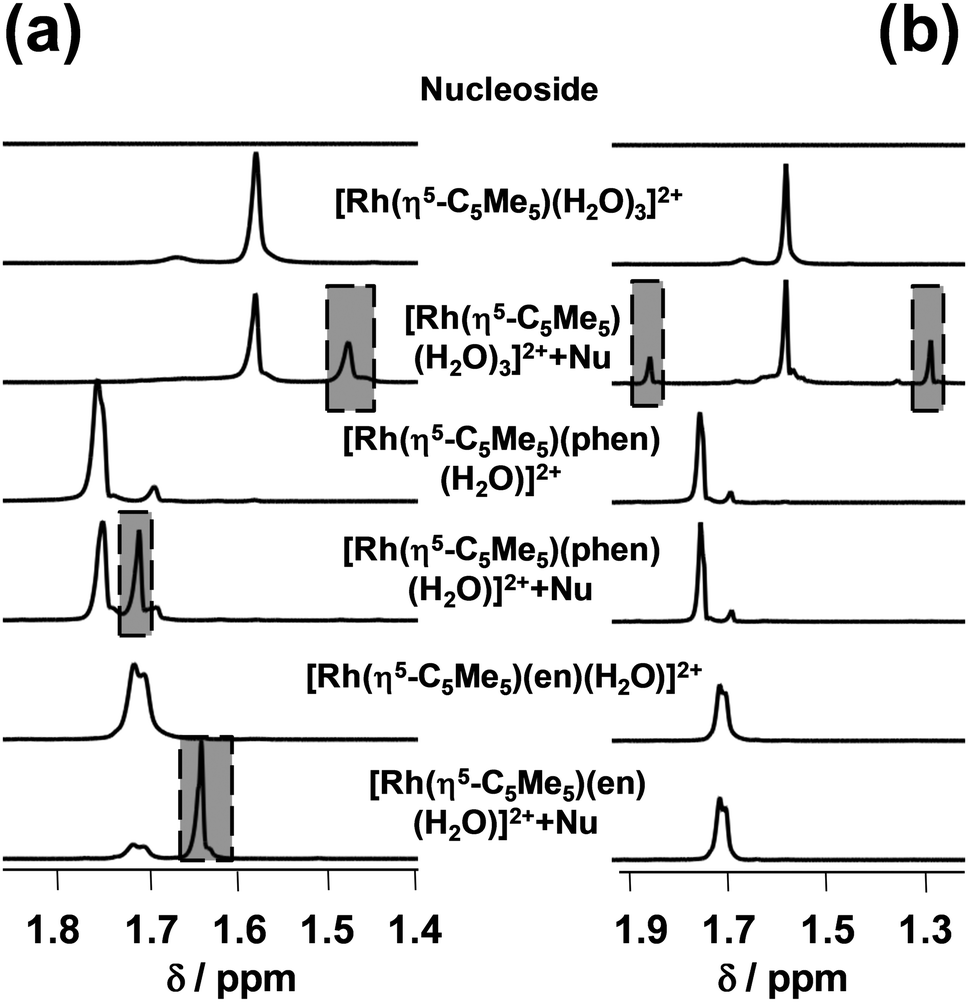 | ||
| Fig. 9 High-field region of 1H NMR spectra of the guanosine (a) and adenosine (b), [Rh(η5-C5Me5)(H2O)3]2+, [Rh(η5-C5Me5)(phen)(H2O)]2+, [Rh(η5-C5Me5)(en)(H2O)]2+ and their mixed systems. Abbreviations: M = [Rh(η5-C5Me5)]2+ and Nu = nucleoside {cadenosine = cguanosine = 1 mM; cRh = cphen = 1 mM; cCl− = 4 mM; pH = 7.40 (20 mM phosphate); T = 25 °C; t = 24 h}. | ||
We have found that only [Rh(η5-C5Me5)(H2O)3]2+ binds to adenosine (28%), while binding levels to guanosine reach 28%, 35% and 72% in the case of [Rh(η5-C5Me5)(H2O)3]2+, [Rh(η5-C5Me5)(phen)(Z)] and [Rh(η5-C5Me5)(en)(Z)] respectively. The hampered binding of the ethylenediamine complex to adenosine can be explained by the steric hindrance between the NH2 moieties of the ligand and the nucleoside (Chart S2, ESI†) as it was suggested for the analogous Ru(II)-containing RAED complexes by Sadler et al.6 Based on these results the binding of the studied Rh(η5-C5Me5) complexes to DNA via coordination of guanosine nitrogen is also feasible.
Conclusions
Metal complexes of various (N,N) donor containing ligands (dmen, tmeda, pin, phen) formed with [Rh(η5-C5Me5)(H2O)3]2+ organometallic cation were synthesized and characterized in solid phase and in aqueous solution.The structures of dmen, tmeda and pin complexes were determined by single-crystal X-ray diffraction showing a pseudo-octahedral ‘piano-stool’ geometry. Solution equilibrium processes were studied via a combined approach using 1H NMR spectroscopy, UV-vis spectrophotometry and pH-potentiometry and were compared to literature data of ethylenediamine and 2,2′-bipyridine. Complex formation with ligands possessing aliphatic nitrogens (dmen, tmeda) was found to be much slower compared to 2-picolylamine and phen.
Mono complexes with a general formula of [Rh(η5-C5Me5)(L)(H2O)]2+ are formed with significantly high solution stability except of tmeda, and decomposition was not observed even at low micromolar concentrations at physiological pH. The obtained stability trend is: phen, bpy > pin > en ∼ dmen ≫ tmeda. The low solution stability of the tmeda complex is reflected in its crystallographic data, namely longer Rh–ring centroid distance, Rh–N bond and larger methyl group-ring plane torsion angle were found as compared to [Rh(η5-C5Me5)(en)(Cl)]+. Deprotonation of the aqua complexes is fast, and moderate pKa [ML]2+ values (8.4–8.6) were obtained for dmen, pin and phen indicating the formation of low fraction of mixed hydroxido species [Rh(η5-C5Me5)(L)(OH)]+ at pH 7.4.
Based on the determined H2O/Cl− co-ligand exchange equilibrium constants the studied complexes possess high chloride ion affinity. The clear correlation was shown between the logK′ (H2O/Cl−) and pKa [ML]2+ constants for a series of Rh(η5-C5Me5) complexes bearing (O,O), (O,N) and (N,N) donor sets. On the other hand logK′ (H2O/Cl−) constants could be described foremost in the literature as a linear combination of a set of crystallographic parameters, that reveals a dependence of the chloride ion affinity of the complexes on the Rh–centroid distance, X–Rh–Y angle and the methyl group-ring plane torsion angle.
DNA binding of Rh(η5-C5Me5) complexes of various bidentate ligands including dmen, tmeda, pin and phen as well as [Rh(η5-C5Me5)(H2O)3]2+ cation was monitored by ultrafiltration and ethidium bromide displacement fluorescence experiments. Significant binding to DNA for [Rh(η5-C5Me5)(H2O)3]2+ and its complexes with 8-hydroxyquinoline, phen and ethylenediamine was detected by ultrafiltration. Competition with EB was also found for [Rh(η5-C5Me5)(H2O)3]2+ and the latter two complexes; however, it can be a result of DNA distortion (instead of intercalation) due to the covalent binding of the Rh(η5-C5Me5) fragment.
Experimental
Chemicals
All solvents were of analytical grade and used without further purification. Dmen, en, phen, pin, tmeda, [Rh(η5-C5Me5(μ-Cl)Cl]2, adenosine, guanosine, EB, DNA from calf thymus, KCl, KNO3, AgNO3, HCl, HNO3, KOH, KH-phthalate, 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), KH2PO4, NaH2PO4 and Na2HPO4 were purchased from Sigma-Aldrich in puriss quality. Milli-Q water was used for sample preparation. The exact concentration of the ligand stock solutions together with the proton dissociation constants were determined by pH-potentiometric titrations with the use of the computer program Hyperquad2013.36 The aqueous [Rh(η5-C5Me5)(H2O)3](NO3)2 stock solution was obtained by dissolving exact amounts of [Rh(η5-C5Me5(μ-Cl)Cl]2 in water followed by the removal of chloride ions by addition of equivalent amounts of AgNO3. The exact concentration of [Rh(η5-C5Me5)(H2O)3]2+ was determined by pH-potentiometric titrations employing stability constants for [(Rh(η5-C5Me5))2(μ-OH)i](4−i)+ (i = 2 or 3)15 complexes. Solutions of adenosine and guanosine were prepared on a weight-in-volume basis in a modified phosphate buffer (20 mM, pH 7.40) which contains 4 mM KCl and the concentration of the Cl− ion corresponds to that of the nucleus. Stock solution of DNA from calf thymus was dissolved in 20 mM phosphate buffer containing 4 mM KCl, pH 7.40 and it was filtered after 3 days, then the exact concentration (nucleobase concentration) and purity was estimated from its UV absorption: ε260nm(DNA) = 6600 M−1 cm−1,37A260nm/A280nm ∼ 1.8.pH-Potentiometric measurements
pH-Potentiometric measurements determining proton dissociation constants of ligands dmen, tmeda, pin and phen were carried out at 25.0 ± 0.1 °C in water and at a constant ionic strength of 0.20 M KNO3. The titrations were performed with a carbonate-free KOH solution (0.20 M). The exact concentrations of HNO3 and KOH solutions were determined by pH-potentiometric titrations. An Orion 710A pH-meter equipped with a Metrohm ‘double junction’ combined electrode (type 6.0255.100) and a Metrohm 665 Dosimat burette was used for the pH-potentiometric measurements. The volume resolution of the burette is 0.001 mL and its precision is 0.002 mL. The electrode system was calibrated to the pH = −log[H+] scale by means of blank titrations (strong acid vs. strong base: HNO3vs. KOH), as suggested by Irving et al.38 The average water ionization constant, pKw, was determined as 13.76 ± 0.01 at 25.0 °C, I = 0.20 M (KNO3), which is in accordance to literature.39 The reproducibility of the titration points included in the calculations was within 0.005 pH units. The pH-potentiometric titrations were performed in the pH range between 2.0 and 11.5. The initial volume of the samples was 10.0 mL. The ligand concentration was 1.0 mM. The goodness-of-fit measured in Hyperquad201336 by sigma (σ) represents the overall goodness-of-fit derived from the sum of squared residuals (calculated-experimental titration data). The model was accepted when σ was close to one (<1.5). The standard deviation of the logβ values of species included into the model was always lower than 0.1. Samples were degassed by bubbling purified argon through them for about 10 min prior to the measurements and the inert gas was also passed over the solutions during the titrations.
logβ values for the various hydroxido complexes [(Rh(η5-C5Me5))2(μ-OH)i](4−i)+ (i = 2 or i = 3) were calculated based on the pH-potentiometric titration data in the absence of chloride ions and were found to be in good agreement with our previously published data.15
Stability constants for MpLqHr complexes cannot be determined by pH-potentiometry because of several problems. In the case of dmen, complex formation was too slow to use pH-potentiometry. Also the dissociation of the tmeda complex was slow. [Rh(η5-C5Me5)(phen)(H2O)]2+ and [Rh(η5-C5Me5)(pin)(H2O)]2+ were formed > 90% at the starting pH value (∼2.0) as a result of high stability.
UV-vis spectrophotometric, 1H NMR and fluorometric measurements
A Hewlett Packard 8452A diode array spectrophotometer was used to record the UV-vis spectra in the interval 200–800 nm. The path length was 1 or 0.5 cm. Equilibrium constants (proton dissociation, stability constants and H2O/Cl− exchange constants) and the individual spectra of the species were calculated with the computer program PSEQUAD.33 The spectrophotometric titrations were performed in aqueous solution on samples containing the ligands with or without the organometallic cations and the concentration of the ligands was 100–200 μM. The organometallic cation was also titrated (200 μM). The metal-to-ligand ratio was 1:1 in the pH range from 2 to 11.5 at 25.0 ± 0.1 °C at an ionic strength of 0.20 M (KNO3). Measurements for 1:1 metal-to-ligand systems were also carried out by preparing individual samples in which KNO3 was partially or completely replaced by HNO3 and pH values, varying in the range ca. 0.7–2.0, were calculated from the strong acid content. In the case of the dmen and tmeda complexes the absorbance data were always recorded after 24 h waiting time. UV-vis spectra were used to investigate the H2O/Cl− exchange processes of complexes [Rh(η5-C5Me5)(L)(H2O)]2+ at 200 μM (dmen) or 100 μM (pin, phen) concentration and at pH 7.40 (using 20 mM phosphate buffer) as a function of chloride concentrations (0–100 mM).
1H NMR studies were carried out on a Bruker Ultrashield 500 Plus instrument. All 1H NMR spectra were recorded with the WATERGATE water suppression pulse scheme using DSS internal standard. 1H NMR spectra were recorded after 24 h waiting time. Stability constants for the complexes were calculated by the computer program PSEQUAD.33
Fluorescence spectra were recorded on a Hitachi-F4500 fluorimeter in 1 cm quartz cell at 25.0 ± 0.1 °C. All DNA-containing solutions were prepared in 20 mM phosphate buffer with 4 mM KCl, which mimics the chloride concentration of the nucleus. The concentration of DNA from calf thymus (as nucleobases) was 20 μM, 5 μM for ethidium bromide and the EB-to-metal ion/or metal complex ratio was varied between 1:10 and 1:50. The excitation wavelength was 510 nm and the emission was read in the range of 530–680 nm, where the absorption of the metal ion and the metal complex is negligible. All samples were incubated for 24 h.
Ultrafiltration-UV-vis measurements
Stock solutions of the Rh(η5-C5Me5) complexes (containing deferiprone, 2-picolinic acid, 6-methylpicolinic acid, quinoline-2-carboxylic acid, 3-isoquinolinecarboxylic acid, 8-hydroxyquinoline, 8-hydroxy-quinoline-5-sulfonate, 7-(1-piperidinylmethyl)-8-hydroxy-quinoline, en, bpy, dmen, pin, phen) were prepared by mixing the aqueous solutions of [Rh(η5-C5Me5)(H2O)3]2+ and the ligand at 1:1 ratio (cstock = 0.50–1.00 mM) in 20 mM phosphate buffer with 4 mM KCl. This kind of in situ preparation of the complexes was proved to be efficient at the indicated conditions (and at the proper incubation time for certain compounds).15–18,20 The DNA-containing samples were prepared in phosphate buffer (20 mM) containing 4 mM KCl. These samples were incubated for 24 h at 37.0 ± 0.1 °C. In the first series the DNA from calf thymus and metal complex concentration was 100–100 μM. Eppendorf Minispin Plus centrifuge and 10 kDa membrane filters (Millipore Amicon Ultra-0.5 centrifugal filter unit) were used. Samples were centrifuged for 10 min with 10000 rpm. UV-vis spectra of LMM fraction were recorded by a Hewlett Packard 8452A diode array spectrophotometer.
Preparation of metal complexes 1·CF3SO3, 2·CF3SO3, 3·Cl and 4·CF3SO3
Two equivalents of Ag(CF3SO3) were added to an acetone solution (10 mL) of [Rh(η5-C5Me5)(μ-Cl)Cl]2 (92.71 mg, 0.15 mmol) and stirred in the dark for 30 min. The formed AgCl precipitate was filtered off and solvent was removed under vacuum. The residue was dissolved in CH3OH/CH2Cl2 (1:1, 10 mL) and two equivalents of the bidentate ligand (0.3 mmol) were added. The reaction mixture was stirred for 2 h. After concentration to dryness, complexes 1·CF3SO3, 2·CF3SO3 and 4·CF3SO3 were isolated as orange solid.
In the case of 2-picolylamine there was no need for chloride ion abstraction. Two equivalents of pin (31 μL) was added to suspension of [Rh(η5-C5Me5)(μ-Cl)Cl]2 (92.71 mg, 0.15 mmol) in dichloromethane (30 mL). The mixture was stirred for 3 h at room temperature. Subsequent solvent removal under vacuum afforded 3·Cl as orange powder. The complexes were characterized by 1H NMR spectroscopy and elemental analysis in addition to X-ray crystallography. Elemental analysis of all compounds was performed with a Perkin-Elmer 2400 CHN Elemental Analyser (Perkin-Elmer, Waltham, MA) at the Microanalytical Laboratory of the University of Vienna. ESI-MS measurements were performed using a Micromass Q-TOF Premier (Waters MS Technologies) mass spectrometer equipped with electrospray ion source (Fig. S7, ESI†).
Single crystals suitable for X-ray diffraction experiment of compound 1·CF3SO3, 2·CF3SO3 and 3·Cl were grown from water/methanol solution mixture (1:1, 2.0 mL).
Chemical characterization of [Rh (η5-C5Me5)(dmen)Cl](CF3SO3), 1·CF3SO3
Yield = 76 mg (50%) anal. calc. for C15H27ClF3N2O3RhS·0.1H2O (512.6): C, 35.15; H, 5.35; N, 5.46; S, 6.26. Found: C, 35.13; H, 5.28; N, 5.55; S, 6.25. 1H NMR (500.10 MHz, CDCl3) (two isomers): δ = 1.76 (s, C5Me5); 1.78 (s, C5Me5); 2.48–2.53 (m, CH2); 2.68–2.69 (d, 3JH,H = 6 Hz, CH3); 2.76–2.77 (d, 3JH,H = 5.5 Hz, CH3); 2.83–2.90 (m, CH2); 2.93–2.94 (d, 3JH,H = 6 Hz, CH3); 3.45–3.55 (m, CH2); 5.64–5.73 (m, NH); 6.07–6.17 (m, NH) ppm. ESI-MS (m/z): [M–Cl–H]+ (C14H26N2Rh+, calc.: 325.1146) = 325.1108, [M]+ (C14H27ClN2Rh+, calculated: 361.0913) = 361.10805 and [M–Cl + CF3SO3]+ (C15H27F3O3N2RhS+, calculated: 475.0745) = 475.0735.Chemical characterization of [Rh(η5-C5Me5)(tmeda)Cl](CF3SO3), 2·CF3SO3
Yield = 72 mg (66%) anal. calc. for C17H31ClF3N2O3RhS·0.5 H2O (547.9): C, 37.27; H, 5.89; N, 5.11; S, 5.85. Found: C, 37.19; H, 5.77; N, 5.31; S, 5.79. 1H NMR (500.10 MHz, CDCl3): δ = 1.63 (s, 15H, C5Me5); 2.62–2.68 (m, 2H, CH2); 2.80–2.88 (m, 2H, CH2); 2.82 (s, 6H, CH3); 3.17 (s, 6H, CH3) ppm. ESI-MS (m/z): [M–Cl–H]+ (C16H30N2Rh+, calc.: 353.1459) = 353.1393, [M]+ (C16H31ClN2Rh+, calc.: 389.1226) = 389.0992 and [M–Cl + CF3SO3]+ (C17H31F3N2O3RhS+, calc.: 503.1058) = 503.1016.Chemical characterization of [Rh(η5-C5Me5)(pin)Cl](Cl), 3·Cl
Yield = 44 mg (34%) anal. calc. for C16H23Cl2N2Rh·0.5H2O (426.2): C, 45.09; H, 5.68; N, 6.57. Found: C, 45.26; H, 5.68; N, 6.62. 1H NMR (500.10 MHz, CDCl3): δ = 1.65 (s, 15H, C5Me5); 4.23–4.25 (d, 3JH,H = 9 Hz, 2H, CH2); 7.45–7.47 (d, 3JH,H = 8 Hz, 1H, CH); 7.50–7.52 (t, 3JH,H = 6 Hz, 1H, CH); 7.89–7.92 (t, 3JH,H = 8 Hz, 1H, CH); 8.59–8.61 (d, 3JH,H = 5 Hz, 1H, CH) ppm. ESI-MS (m/z): [M–Cl–H]+ (C16H22N2Rh+, calc.: 345.0833) = 345.0579 and [M]+ (C16H23ClN2Rh+, calc.: 381.0600) = 381.0326.Chemical characterization of [Rh(η5-C5Me5)(phen)Cl](CF3SO3), 4·CF3SO3
Yield = 131 mg (72%) anal. calc. for C23H23ClF3N2O3RhS (602.9): C, 45.82; H, 3.85; N, 4.65; S, 5.32. Found: C, 45.92; H, 3.83; N, 4.50; S, 5.27. 1H NMR (500.10 MHz, DMSO-d6): δ = 1.75 (s, 15H, C5Me5), 8.22–8.25 (dd, 3JH,H = 5 Hz, 3JH,H = 8 Hz, 2H, CH), 8.34 (s, 2H, CH), 8.97–8.99 (d, 3JH,H = 9 Hz, 2H, CH), 9.42–9.43 (d, 3JH,H = 6 Hz, 2H, CH) ppm. ESI-MS (m/z): [M–Cl–H]+ (C22H22N2Rh+, calc.: 417.0833) = 417.0721 [M]+ (C22H23ClN2Rh+, calc.: 453.0600) = 453.0298 and [M–Cl + CF3SO3]+ (C23H23F3O3N2RhS+, calc.: 567.0432) = 567.0347.Single-crystal X-ray structures analysis
The X-ray intensity data were measured on a Bruker D8 Venture diffractometer equipped with multilayer monochromator, Mo K/a INCOATEC micro focus sealed tube and Kryoflex cooling device. The structures were solved by direct methods and refined by full-matrix least-squares techniques. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were inserted at calculated positions and refined with a riding model. The following software was used: Bruker SAINT software package40 using a narrow-frame algorithm for frame integration, SADABS41 for absorption correction, OLEX242 for structure solution, refinement, molecular diagrams and graphical user-interface, Shelxle43 for refinement and graphical user-interface SHELXS-201344 for structure solution, SHELXL-201345 for refinement, Platon46 for symmetry check. The crystallographic data files for complexes (1·CF3SO3), (2·CF3SO3) and (3·Cl) have been deposited with the Cambridge Crystallographic Database as CCDC 1590516, 1590517 and 1590518.† Crystal data and structure refinement details for complexes 1–3 are given in Table S1 (ESI†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hungarian National Research Development and Innovation Office through the projects FK 124240, GINOP-2.3.2-15-2016-00038, the UNKP-17-4 & UNKP-17-3 New National Excellence Program of the Ministry of Human Capacities (E. A. E.; J. P. M.) and the Austrian-Hungarian Scientific & Technological Cooperation TET 15-1-2016-0024.References
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- H. A. Burris, S. Bakewell, J. Bendell, J. Infante, S. Jones, D. Spigel, G. J. Weiss, R. K. Ramanathan, A. Ogden and D. Von Hoff, ESMO Open, 2017, 1, e000154 CrossRef PubMed.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549–1560 CrossRef.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef PubMed.
- M. Pizarro, A. Habtemariam and P. J. Sadler, Top. Organomet. Chem., 2010, 32, 21–56 CrossRef.
- Y. Geldmacher, M. Oleszak and W. S. Sheldrick, Inorg. Chim. Acta, 2012, 393, 84–102 CrossRef.
- A. Habtemariam, M. Melchart, R. Fernandez, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef PubMed.
- J. Canivet, G. Süss-Fink and P. Stepnicka, Eur. J. Inorg. Chem., 2007, 4736–4742 CrossRef.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef PubMed.
- J. J. Soldevilla-Barreda, A. Habtemariam, I. Romero-Canelón and P. J. Sadler, J. Inorg. Biochem., 2015, 153, 322–333 CrossRef PubMed.
- P. Buglyó and E. Farkas, Dalton Trans., 2009, 8063–8070 RSC.
- L. Bíró, E. Farkas and P. Buglyó, Dalton Trans., 2010, 39, 10272–10278 RSC.
- A. L. Noffke, A. Habtemariam, A. M. Pizzaro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef PubMed.
- É. A. Enyedy, O. Dömötör, C. M. Hackl, A. Roller, M. S. Novak, M. A. Jakupec, B. K. Keppler and W. Kandioller, J. Coord. Chem., 2015, 68, 1583–1601 CrossRef.
- O. Dömötör, C. M. Hackl, K. Bali, A. Roller, M. Hejl, M. A. Jakupec, B. K. Keppler, W. Kandioller and É. A. Enyedy, J. Organomet. Chem., 2017, 846, 287–295 CrossRef.
- O. Dömötör, V. F. S. Pape, N. V. May, G. Szakács and É. A. Enyedy, Dalton Trans., 2017, 46, 4382–4396 RSC.
- C. M. Hackl, M. S. Legina, V. Pichler, M. Schmidlehner, A. Roller, O. Dömötör, E. A. Enyedy, M. A. Jakupec, W. Kandioller and B. K. Keppler, Chem. – Eur. J., 2016, 22, 17269–17281 CrossRef PubMed.
- É. A. Enyedy, J. P. Mészáros, O. Dömötör, C. M. Hackl, A. Roller, B. K. Keppler and W. Kandioller, J. Inorg. Biochem., 2015, 152, 93–103 CrossRef PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef PubMed.
- J. M. Cross, T. R. Blower, N. Gallagher, J. H. Gill, K. L. Rockley and J. W. Walton, ChemPlusChem, 2016, 81, 1276–1280 CrossRef.
- C. White, A. Yates, P. M. Maitlis and D. M. Heinekey, Inorg. Synth., 1992, 29, 228–234 Search PubMed.
- G. Garcia, G. Sánchez, I. Romero, I. Solano, M. D. Santana and G. Lopez, J. Organomet. Chem., 1991, 408, 241–246 CrossRef.
- M. A. Scharwitz, I. Ott, Y. Geldmacher, R. Gust and W. S. Sheldrick, J. Organomet. Chem., 2008, 693, 2299–2309 CrossRef.
- M. T. Youinou and R. Ziessel, J. Organomet. Chem., 1989, 363, 197–208 CrossRef.
- M. H. Wang, U. Englert and U. Kölle, J. Organomet. Chem., 1993, 453, 127–131 CrossRef.
- M. S. Eisen, A. Haskel, H. Chen, M. M. Olmstead, D. P. Smith, M. F. Maestre and R. H. Fish, Organometallics, 1995, 14, 2806–2812 CrossRef.
- I. Sóvágó and A. Gergely, Inorg. Chim. Acta, 1976, 20, 27–32 CrossRef.
- R. G. Lacoste and A. E. Martell, Inorg. Chem., 1964, 3, 881–884 CrossRef.
- S. Bandyopadhyay, G. N. Mukherjee and M. G. B. Drew, Inorg. Chim. Acta, 2005, 358, 3786–3798 CrossRef.
- E. M. Christensen, S. Oh, D. Oliver, D. E. Janzen and S. M. Drew, J. Chem. Crystallogr., 2014, 44, 236–242 CrossRef.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. L. Leggett, Plenum Press, New York, 1985, pp. 291–353 Search PubMed.
- R. B. Martin, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, VHCA & Wiley-VCH, Zürich, Switzerland, 1999, pp. 181–205 Search PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, J. Fawcett and D. R. Russell, J. Chem. Soc., Dalton Trans., 1995, 3709–3713 RSC.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef PubMed.
- S. R. Gallagher, in Current Protocols in Molecular Biology, ed. F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith and K. Struhl, Greene and Wiley-Interscience, New York, 1994, p. A-3D-1-14 Search PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef.
- SCQuery, The IUPAC Stability Constants Database, Academic Software (Version 5.5), Royal Society of Chemistry, 1993–2005.
- Bruker SAINT v8.32BA Copyright©2005–2017 Bruker AXS.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, SHELXS, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXL, University of Göttingen, Germany, 1996 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected equilibrium constants and X-ray diffraction data. CCDC 1590516–1590518. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj01681j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |