
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interactions of Schiff base compounds and their coordination complexes with the drug cisplatin†
József
Magyari
 a,
Berta
Barta Holló
a,
Ljiljana S.
Vojinović-Ješić
a,
Mirjana M.
Radanović
a,
Stevan
Armaković
b,
Sanja J.
Armaković
a,
Joseph
Molnár
c,
Annamária
Kincses
c,
Márió
Gajdács
c,
Gabriella
Spengler
c and
Katalin
Mészáros Szécsényi
*a
a,
Berta
Barta Holló
a,
Ljiljana S.
Vojinović-Ješić
a,
Mirjana M.
Radanović
a,
Stevan
Armaković
b,
Sanja J.
Armaković
a,
Joseph
Molnár
c,
Annamária
Kincses
c,
Márió
Gajdács
c,
Gabriella
Spengler
c and
Katalin
Mészáros Szécsényi
*a
aUniversity of Novi Sad, Faculty of Sciences, Department of Chemistry, Biochemistry and Environmental Protection, Trg Dositeja Obradovića 3, 21000, Novi Sad, Serbia. E-mail: mszk@uns.ac.rs; Tel: +381-64-230-2871
bUniversity of Novi Sad, Faculty of Sciences, Department of Physics, Trg Dositeja Obradovića 4, 21000, Novi Sad, Serbia
cUniversity of Szeged, Faculty of Medicine, Department of Medical Microbiology and Immunobiology, Dóm tér 10, Szeged H-6720, Hungary
First published on 8th March 2018
Abstract
There is a complex interplay between the structural and other physicochemical properties of new compounds and the molecules in living organisms. To understand the mechanism of the interactions at the molecular level, the correlations between the selected properties and their biological responses have to be examined. With this aim, in this paper, density functional theory (DFT) and LMP2 calculations were carried out for the 2-acetylpyridine-aminoguanidine ligand, L, and its copper(II) complexes containing different monoanionic ligands. In addition, several parameters, most frequently used for the prediction of drug-likeness of new compounds, were calculated. The influence of the compounds on the effectiveness of the reference chemotherapeutic drug cisplatin was determined in vitro, by comparison of their combination indices (CIs). The drug interactions between cisplatin and the earlier synthesized ligands L1 (bis(3-chloropyridazine-6-hydrazone)-2,6-diacetylpyridine) and L2 (bis(phthalazine-1-hydrazone)-2,6-diacetylpyridine) and their Co(III), Ni(II), Cu(II) and Zn(II) complexes, respectively, were also measured. The ligands L, L2, and L3, as well as their complexes, showed different interactions in combination with cisplatin from strong antagonism of L to strong synergism of 4-L1 and 4-L2. The experimental results and the calculated parameters were analyzed to evaluate their correlation with the measured interactions. The thermal stability of the L·2HCl ligand and its four copper(II) complexes was determined and the thermal stability data were correlated to selected calculated molecular descriptors.
Introduction
Schiff bases, as well as their complexes, represent a group of compounds showing a wide range of biological activities, such as anticancer and antibacterial properties. Earlier studies of the activity of 2-acetylpyridine-aminoguanidine and its coordination compounds with Cu(II) against bacteria and yeasts showed moderate to low activity to some bacterial species and very low activity toward fungi.1 On the other hand, both the ligand2–5 and copper(II)6,7 are expected to have other, significant biological functions. A copper(II) complex with a Schiff base-type ligand in interaction with human serum albumin increased its target ability to cancer cells.8 Extensive research is taking place exploring the biological role of copper complexes with Schiff bases,9–16 listing here only selected papers published in 2017. Special attention is paid to their bio-molecular interactions17 as well as their correlations with selected parameters like the acidity constant of the compounds and their role in selected biological processes.18 As tools for assessing bioavailability, often DFT19–22 computations or QSAR23 and docking24 studies are carried out.Malignant diseases represent one of the most important factors of morbidity and mortality worldwide, accounting for more than 8 million deaths in 2015, only preceded by the mortalities caused by cardiovascular diseases.25 Chemotherapy is a hallmark of the treatment of these malignancies, often combined with surgical interventions and radiotherapy.26 The emergence of cancers with a multidrug-resistant phenotype is an important obstacle to effective treatment, as these malignancies are found to be resistant to a variety of anticancer drugs of different mechanisms of action and structure.27 The dosage of most of the commercial chemotherapeutic drugs is limited because of their serious side effects. Hence, combinations of two or more drugs28,29 are often used in cancer treatment with the aim of decreasing the side effects and increasing the sensitivity of the cancer cells toward anticancer agents.30
Our recent research has been focused on the biological activity and the reversal of multidrug resistance of new Schiff bases and their coordination compounds with 3d metals. We tested the bis(3-chloropyridazine-6-hydrazone)-2,6-diacetylpyridine ligand, L1, and its Co(III), Ni(II), Cu(II) and Zn(II) complexes for antiproliferative effects and for the reversal of multidrug resistance (MDR) in selected tumor cell lines. The structures were theoretically investigated employing density functional theory (DFT) calculations. The calculated molecular electrostatic potential (MEP) and the average local ionization energy (ALIE) surfaces were used to correlate the effect of the compounds with the inhibition of multidrug resistance in cancer. The compounds showed a different, but increased inhibitory effect on ABC transporter ABCB1 or P-glycoprotein.31 Coordination compounds of the same metal centers with a similar ligand, bis(phthalazine-1-hydrazone)-2,6-diacetylpyridine, L2, exhibited strong to moderate antimicrobial activity against selected bacteria but had no effect on fungi, while the ligand itself was practically inactive.32
In this paper, DFT and LMP2 calculations were carried out for the 2-acetylpyridine-aminoguanidine ligand, L, and its copper(II) complexes containing different monoanionic ligands. In addition, several parameters, most frequently used for the prediction of drug-likeness of new compounds, were calculated. The influence of the compounds on the effectiveness of the reference chemotherapeutic drug cisplatin was determined in vitro, by comparison of their combination indices (CIs). The drug interactions between cisplatin and the earlier synthesized ligands L131 and L2,32 and their Co(III), Ni(II), Cu(II) and Zn(II) complexes, respectively, were also measured. The experimental results and the calculated parameters were analyzed to evaluate their correlation with the measured interactions. The thermal stability of the L·2HCl ligand and its four copper(II) complexes was determined and the thermal stability data were correlated to selected calculated molecular descriptors.
Results and discussion
DFT results
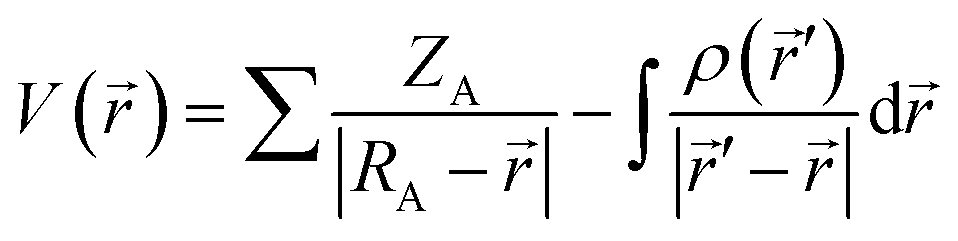 | (1) |
 | ||
| Fig. 1 MEP surfaces of L and its complexes. | ||
The results provided in Fig. 1 indicate that the lowest MEP value has been calculated in the case of the 4-L complex. Namely, in the vicinity of the nitrogen atom of the thiocyanate group of the 4-L complex, the lowest MEP value has a value of almost −57 kcal mol−1. A significantly low value of MEP in the case of the 4-L complex can be predicted for the sulfur atom S2, characterized by the yellow to reddish color in Fig. 1, corresponding to the MEP value of ca. −50 kcal mol−1, which is still lower than in the case of the lowest MEP values of other complexes. These results designate complex 4-L as the most sensitive towards electrophilic attacks, especially the nitrogen and sulfur atoms of its thiocyanate group. The highest MEP values have been calculated for the same complex. Namely, the hydrogen atoms of its NH2 and methyl groups are characterized by the purple color in Fig. 1, corresponding to MEP values of almost 65 kcal mol−1. Therefore, these molecule sites could be sensitive towards nucleophilic attacks. All other complex molecules are characterized by the lowest MEP values of around −45 kcal mol−1, while in the highest MEP values there are some differences. The ligand, L, is characterized by the lowest charge separation, with the lowest magnitudes of negative and positive MEP values. This low charge separation in the case of the L molecule is also confirmed by the lowest dipole moment (DP) of all the investigated structures in this work.
Although MEP surfaces are very useful for the determination of molecule sites with significant reactive properties, when it comes to the sensitivity towards electrophilic attacks, ALIE surfaces are often a better choice since they indicate the molecule sites where electrons are most easily removed from the molecule. Murray et al.38–40 have defined ALIE as a sum of orbital energies weighted by the orbital densities and used it initially to understand the chemical reactivity of aromatic systems. The equation for the calculation of ALIE I(r) is defined as:
 | (2) |
![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) ) denotes the electronic density of the i-th molecular orbital at the point , while εi is the orbital energy. ρi() is the total electronic density function. Representative ALIE surfaces of all investigated structures in this work are presented in Fig. 2. The range of the rainbow bar has been chosen according to the lowest and the highest ALIE values in case of the molecules investigated in this work.
) denotes the electronic density of the i-th molecular orbital at the point , while εi is the orbital energy. ρi() is the total electronic density function. Representative ALIE surfaces of all investigated structures in this work are presented in Fig. 2. The range of the rainbow bar has been chosen according to the lowest and the highest ALIE values in case of the molecules investigated in this work.
 | ||
| Fig. 2 ALIE surfaces of L and its complexes. | ||
Results of ALIE surfaces provided in Fig. 2 indicate that the lowest ALIE value has been calculated in the case of the 3-L complex. In this complex, the lowest ALIE values of ∼150 kcal mol−1 are located in the near vicinity of the bromine atom. Although structurally 1-L and 3-L differ in only one atom (3-L has a bromine atom instead of one chlorine atom), they differ significantly when it comes to the lowest ALIE values and therefore their sensitivity towards electrophilic attacks. Namely, the lowest ALIE value of 1-L has been calculated to be ∼165 kcal mol−1 and is located in the near vicinity of chlorine atoms. Although the 4-L complex is not recognized by the ALIE values as the most sensitive complex towards electrophilic attacks, the color distribution confirms the results obtained by the MEP values that the nitrogen atom and the sulfur atom of the thiocyanate group are the most sensitive atoms of 4-L towards electrophilic attacks. In general, the topology of the lowest/highest MEP and ALIE values agrees well for the structures investigated in this work.
Drug-likeness parameters
In recent decades, many useful computational descriptors and correlation methods41 have been developed in order to screen potential pharmaceutical candidates for practical applications in drug formulations. In this work, we have calculated several parameters that are most frequently used for the prediction of drug-likeness of new compounds. Drug-likeness parameters, usually addressed by the Lipinski's rule of five,42,43 have been summarized in Table 1.| HBDa | HBAb | Mass/g mol−1 | PSAc/Å2 | Refractivity/m3 mol−1 | Number of atoms | Rotatable bonds | |
|---|---|---|---|---|---|---|---|
| a Hydrogen bond donors. b Hydrogen bond acceptors. c Polar surface area. | |||||||
| L | 2 | 0 | 187.290 | 88.1300 | 53.0828 | 26 | 4 |
| 1-L | 0 | 0 | 311.662 | 58.9100 | 42.7780 | 27 | 0 |
| 2-L | 2 | 0 | 277.217 | 56.9700 | 45.2883 | 27 | 0 |
| 3-L | 0 | 0 | 311.662 | 59.9100 | 42.7780 | 27 | 0 |
| 4-L | 1 | 1 | 357.928 | 151.160 | 54.1696 | 31 | 0 |
According to standard criteria, it is desired that a potential drug candidate has less than 5 and 10 HBD and HBA, respectively. Concerning the molar refractivity, the desired range for prospective drug candidates is between 40 and 130 m3 mol−1 according to ref. 42. The number of atoms should be between 20 and 70, while the number of rotatable bonds should be less than 10. These conditions are fulfilled by all molecules investigated in this work. PSA should not take values higher than 140 Å2 and the results provided in Table 1 indicate that the 4-L molecule has just a slightly higher value for this descriptor.
However, one of the most important drug-likeness descriptors is related to the lipophilic character of the drug candidate. This property is usually measured by octanol/water partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P). There are several computational models to predict the lipophilicity descriptor and in this work, we have used the frequently employed clogP parameter,44–49 which turned out to be a great choice in comparison with the other models for the calculation of the logP parameter. Besides clogP, it is also very useful to estimate the aqueous solubility of a compound (clogS) which influences its absorption and distribution characteristics. Both the clogP and clogS values have been calculated by the DataWarrior software50 and listed in Table 2 along with the calculated dipole moments (DPs).
P). There are several computational models to predict the lipophilicity descriptor and in this work, we have used the frequently employed clogP parameter,44–49 which turned out to be a great choice in comparison with the other models for the calculation of the logP parameter. Besides clogP, it is also very useful to estimate the aqueous solubility of a compound (clogS) which influences its absorption and distribution characteristics. Both the clogP and clogS values have been calculated by the DataWarrior software50 and listed in Table 2 along with the calculated dipole moments (DPs).
logP, AlogP, clogS and DP values for all investigated molecules
|
clogP |
AlogP |
clogS |
DP | |
|---|---|---|---|---|
| L | −2.4842 | −0.59 | −1.195 | 5.37 |
| 1-L | −0.5715 | −1.16 | −3.403 | 9.42 |
| 2-L | −0.2394 | −1.16 | −1.829 | 8.78 |
| 3-L | −0.5715 | −2.75 | −0.011 | 9.70 |
| 4-L | 0.0592 | −3.55 | −3.197 | 15.58 |
According to the data,51 the highest percentage of traded drugs has clogP values in the range between 2 and 4. However, a significant number of commercially available drugs used in clinical practice have clogP values in the range between −1 and 0. The data provided in Table 2 indicate that all the complex molecules investigated in this work have clogP values in the range between −0.6 and 0, which makes them promising candidates for pharmaceutical purposes. Additionally, according to the same source,52 the highest number of traded drugs has clogS values in the range between −4 and −2. In this regard, it is particularly interesting to notice that complexes 4-L and 1-L have clogS values between −3.2 and −3.7, which completely corresponds to the desired range. At the same time, the clogS value of the 2-L complex is equal to −1.8, which is very close to the desired value of −2. Of all the complex molecules, the complex 3-L has the worst value of clogS parameter (−0.011). However, its clogS value, due to the mentioned facts, is still interesting.
Complexes as carriers of cisplatin
Cisplatin (cisp) belongs to a group of compounds widely and frequently used for the treatment of various cancerous diseases. In this work, we have considered the possibility that our newly synthesized complexes could serve as carriers of cisp, from both experimental and computational aspects.From the computational aspects, we have considered the interactions between the complexes and cisp by investigation of noncovalent interactions and binding energies. Ground state geometries of the systems consisting of the complex and cisp together with visualized noncovalent interactions between the complexes and cisp are presented in Fig. 3.
 | ||
| Fig. 3 Interactions of cisp with the complexes. | ||
According to the results shown in Fig. 3, binding between the complexes and cisp is based on electrostatic interactions, since no new bonds have been formed. This is of particular interest since the release of cisp could be achieved under easier conditions than, for example, if binding were based on chemical interactions.
Next, we refer to the binding energies between the complexes and cisp, which have been calculated at the LMP2 and DFT levels of theory (Fig. 4).
 | ||
| Fig. 4 Binding energies between the complexes and cisp. | ||
Binding energies have been calculated at both the LMP2 and DFT levels of theory. The LMP2 level of theory has been used since it is “immune” to the basis superposition error and it does not require the usage of counterpoise correction. On the other side the LMP2 level of theory is not available in many molecular modeling packages so we decided to calculate binding energies at the DFT/B3LYP-D3 level of theory as well, which is a level of theory that is readily available in all molecular modeling packages capable of dealing with DFT calculations and which is recommended in cases when noncovalent interactions take place.53–55 Although the LMP2 and DFT levels of theory provide expectedly different values of binding energies, the trends are completely in agreement. According to the binding energies, the strongest interaction occurs in the case of the system consisting of the 4-L complex and cisp, with the corresponding binding energy of −64 kcal mol−1 at the LMP2 level of theory (−61 kcal mol−1 at DFT level of theory). The 1-L and 3-L complexes have very similar binding energies (around −43 kcal mol−1), as calculated at the LMP2 level of theory (around −54 kcal mol−1 at DFT level of theory). The lowest binding energy has been calculated in the case of complex 2-L and cisp, with the corresponding binding energy of −14 kcal mol−1 as calculated at the LMP2 level of theory (−18 kcal mol−1 at the DFT level of theory).
The results shown in Table 2 indicate that the 4-L complex has the most representative clogP and clogS values of all the investigated complexes in this study. At the same time, this complex has the highest binding interaction with the cisp compound, indicating that it can not only permeate the barrier, but also serve as a carrier of cisp and have an interesting synergistic effect. Although the 2-L complex has the lowest binding energy with cisp, that binding energy is still high enough to be considered as a possible carrier of cisp. On the other side, the much lower binding energy with cisp would mean an easier release of cisp as well, which could be an important factor for the efficient practical application of this complex.
Determination of combination index
To see how the experimental results can be related to the calculated parameters, the combination indices (CIs) for the ligands and the corresponding coordination compounds were determined. Determination of the combination index is a useful, resource saving method for in vitro testing of the combination of two or more active compounds.56 In order to see how the Schiff base type ligands L, L1, and L2 and their complexes influence the effect of the standard cisplatin drug, dilutions of the compounds were mixed with cisp and applied on multidrug-resistant mouse T-lymphoma cells overexpressing the ABCB1 transporter.Table 3 lists the combination indices of L and its copper(II) complexes 1, 2, 3 and 4 along with the CIs of the L1 and L2 ligands and their Co(III), Ni(II), Cu(II) and Zn(II) complexes, respectively.31,32 The referent CI values are given in Table 5.
| Compound | Cisplatin | |
|---|---|---|
| CI (ED50) | Type of interaction | |
| L | 3.81 | Strong antagonism |
| 1-L | 2.05 | Antagonism |
| 2-L | 0.80 | Moderate synergism |
| 3-L | 1.08 | Additive effect |
| 4-L | 0.75 | Moderate synergism |
| L1 | 0.78 | Moderate synergism |
| 1-L1 | 1.79 | Antagonism |
| 2-L1 | 1.51 | Antagonism |
| 3-L1 | 0.84 | Moderate synergism |
| 4-L1 | 0.31 | Strong synergism |
| L2 | 0.37 | Synergism |
| 1-L2 | 2.52 | Antagonism |
| 2-L2 | 0.47 | Synergism |
| 3-L2 | 1.02 | Additive effect |
| 4-L2 | 0.26 | Strong synergism |
The ligand L (Scheme 1) and its coordination compounds in combination with cisp show all three effects (Table 3). Strong antagonism is characteristic for the ligand, L, and for complex 1 with two coordinated chlorides. 3 containing one coordinated chloride and one bromide shows an additive effect, while complexes 2 and 4 with one coordinated chlorido and thiocyanato ligand, respectively, show moderate synergism. These observations might be correlated with the role of the halido ligands and their position in the complexes. Complexes 1, 2 and 3 have square-pyramidal geometry while 4 has a square-planar arrangement around the central atom. In 1, with the chlorido ligands in the cis position, antagonism is detected. Compound 3 with the mixed chlorido and bromido ligands shows an additive effect. For 2 with a coordinated chloride ion (and MeOH in the solid state) and for the quasi square-pyramidal complex 4 with one coordinated thiocyanato ligand in the basal plane and the second one in the apical position, a moderate synergism is characteristic. As the molar conductivity data in N,N-dimethylformamide (DMF) solution refer to an electrolyte of 1:1 type for 2, while to 4 as a non-electrolyte,1 in solution both compounds have a similar geometry.
 | ||
| Scheme 1 Structure of the ligand L. | ||
The difference between the ligands L1 and L2 is that L1 is a bis(3-chloropyridazine-6-hydrazone) derivative of 2,6-diacetylpyridine while L2 is the bis(phthalazine-1-hydrazone)-2,6-diacetylpyridine (Scheme 2). Ligand L1 shows moderate synergism with cisp. Coordination compounds of Co(II) and 1-L1, and Ni(II) and 2-L1, show antagonism with cisp. The Cu(II) complex, 3-L1, displays moderate synergism with cisp, while the zinc(II) compound, 4-L1, exhibits strong synergism with cisp.
 | ||
| Scheme 2 Structures of the ligands L1 and L2. | ||
On the contrary, while screening reveals synergism between L2 (Scheme 2) and cisp, the Co(III) complex (1-L2) acts as an antagonist to cisp, 2-L2 with a Ni(II) center acts as a synergist, 3-L2 with Cu(II) shows an additive effect and the Zn(II) complex, 4-L2, displays strong synergism.
As all four complexes with L1 are ion-neutral complexes, with no additional ligands, their different behavior with cisp is due exclusively to the different central ions and/or geometry.31 On the other hand, complexes 1–3 with L2 contain chloride ions, too, but have different geometries. In 1-L2 the central Co(III) has an octahedral surrounding with two chloride ions in the trans position. The Ni(II) complex, 2-L2, adopts a square-planar structure and the chloride ion is not coordinated. In 3-L2 a square-pyramidal arrangement around Cu(II) is established with the chloride ion in the apical position. In addition, complexes 1-L2 and 3-L2 are non-electrolytes in DMF while 2-L2 is a 1:1 electrolyte,32 so their different action with cisp is expected.
The activity of cisplatin depends on the hydrolysis of its chloride ligands. This process in vivo is hindered in the bloodstream, with a stable chloride concentration of 100 mM. Inside the cell, the Cl− concentration is significantly lower (4–20 mM) and therefore assists the hydrolysis.57 Compounds containing chloride ions may alter the chloride concentration in the cellular media, and therefore could have an impact on the anticancer effects of cisplatin. Among the tested compounds ligands L and L2 crystallize with HCl; the counter-ion in complex 2-L2 is chloride, while in the coordination complexes 1L–3L and 1-L2 and 3-L2 the chloride ion is coordinated, so in their combination with cisp, the chloride ion may take part in the interactions. However, the M–Cl bond lengths vary in the range of 2.20–2.26 Å1,32 and thus it is very difficult to say anything about their actual involvement in the reactions.58
Apart from the role of the chloride ions, the targets of metal complexes in vivo are diverse. The primary targets are nucleic acids, redox processes, mitochondria and proteins.59 The protein binding of cisp is cc. 95% in vivo. Any compound which binds to plasma proteins (e.g. albumin) more competitively will proportionally increase the amount of free (pharmacologically active) drug in the system.60
Cisplatin exerts its activity by forming bifunctional intra- and interstrand crosslinks (platinating) through covalent bonds with DNA on its nucleophilic sites (the N7 atoms of adenine and guanine-purine nucleosides are the most nucleophilic), although it has been observed that only around 1% of the agent inside the cell is found in the nuclei of cells, and around 5–10% of cisp reacts with DNA, while much of the agent is involved in reactions with a diverse range of other biomolecules, such as protein-platinum complexes.59 Some hypotheses suggest that using combinational agents with cisplatin can affect the stability of the said DNA-crosslinks, sensitize or desensitize several biomolecules to the effect of the drug, and influence the amount of cisplatin penetrating the nucleus, thus modifying the extent of the potential anticancer effects by a considerable margin.61
Crosslinks in DNA lead to the termination of transcriptional processes and eventually cell death. However, according to recent literature data, more than one mechanism could have a role in the anticancer effects of platinum-based anticancer agents. The different reaction paths could potentially serve as an explanation for the interactions with Schiff base type compounds and their coordination complexes. A novel cell death mechanism, ferroptosis, has been implicated in the cell death caused by the platinum complexes. Ferroptosis is a type of regulated cell death (RCD) which is most predominantly induced by iron-dependent ROS production and subsequent lipid-peroxidation.62 In theory, cisplatin adversely affects iron-metabolism inside the cells, activating the pathways implicated. Since the metal complexes can probably modulate the homeostasis of metal ions inside the cells, their activity should influence the activity of cisp.
Evidence suggests that cisplatin affects the proteins of the signal transducer and activation of transcription (STAT) pathway, especially STAT3, which results in direct inhibition of the STAT signaling pathway.63 The platinum-based drugs inhibit de novo STAT protein phosphorylation, and if the Schiff base type compounds also exert STAT protein modulating activities, then this field could also present an area of potential pharmacological interactions. The upregulation of phospho-p53 was also observed in cisplatin-treated cells, which is another viable route to drug–drug interactions if the tested compounds possess such activity.64,65
Cisplatin can enter the target cells by passive transport as well as by relying on the CTR1 copper transporter. Compounds with the ability to regulate the expression and/or activity of the transporters will alter the pharmacological potency of the chemotherapeutic drug.66 As complex 4-L has the highest binding interaction with cisp, it definitely could serve as its carrier. Although the binding energy of 2-L is the lowest, it still may be a drug carrier. On the other hand, the lower binding energy means an easier release of cisp. Indeed, only these two compounds in the L series show a synergistic effect with cisp.
Being a metal complex itself, cisplatin can interact with a multitude of pharmacologically active molecules in many ways. Apart from steric interactions between the molecules (described in the article elsewhere), conditions may allow for the association of novel drug complexes or drug macrostructures in a biological environment.67 These macrostructures could have physicochemical properties not resembling the original parent molecules which, in turn, is observed in their different (enhanced or suppressed) biological activities.
The calculated parameters and clogP values for the L series well follow the trend of the determined IC values. Also, the correlation of the dipole moments shows a relatively good agreement with the IC data while the other calculated parameters cannot be unambiguously related to the measured effect. Copper(II) compounds from all three series show an additive effect or synergism with cisp except that with L2, while compounds with Co(III), Ni(II) and Zn(II) exhibit different types of interactions. While both the L1 and L2 ligands show a synergistic effect, strong antagonism was found for L. A highly probable explanation for this behavior is that the NH2 groups of the guanidine substituent of L may replace the chloride in cisp, in this way hindering its bioavailability. In an in vitro environment, the possible antagonism of L can also be explained by its coordination to the platinum, as cisp has good leaving NH3 ligands, forming thus a stable chelate complex which shows no activity.
Thermal data
The practical applicability of new compounds and materials, generally, depends to a large extent on their thermal properties.21,68–72 In addition, using isothermal titration calorimetry, the interactions between biomolecules can be measured.73 Sometimes, the thermal stability and biological activity of bioactive molecules may be correlated and partly explained.74 Therefore, the thermal decomposition of L and its complexes 1–4L was determined. The thermoanalytical data and a detailed description of the mechanism of the decomposition are given in the ESI.† Here only the most important data are discussed.The thermogravimetric curves of all the complexes are in agreement with the structural data. In compound 2-L the coordinated MeOH evaporates at a rather high temperature (118 °C onset), though its amount (Δmexp = 3.2%, Δmcalcd = 8.65%) is significantly less than the stoichiometric one, probably as a consequence of its evaporation accompanied by structural rearrangement of the lattice.75,76 In compound 1-L traces of water were found. Except for complex 4-L (DTG onset 159 °C), the desolvated ligand and its complexes are stable to above 200 °C, and therefore their thermal stability would have an insignificant effect on the eventual drug manufacturing.
While the thermal curves of the coordination compounds are in agreement with the structure and the composition determined by the X-ray structure analysis, the ligand, L, on the basis of the EGD data contains 2 molecules of crystal water, L·2HCl·2H2O (Δmexp = 12.6%, Δmcalcd = 12.59%) which evaporate at 120 °C. However, after the water evaporation, the ligand is stable up to 210 °C DTG onset.
Despite that the thermal stability of the compound and the corresponding calculated molecular descriptors are seemingly not related, Table 4 shows a relatively good Pearson correlation (PCC) coefficients between several parameters, calculated in this work and the DTG onset temperatures. Among others, it can be seen that the thermal stability of the compounds (DTG onset data) with enough statistical significance (p) nicely correlates with the dipole moments, AlogP and PSA. The highest correlation has been obtained in the case of DTG onset versus PSA parameters, with an inverse proportional character. Somewhat lower, but still significant is the correlation of the DTG onsets in the case of dipole moments and AlogP parameters. As AlogP and PSA are parameters related to pharmacokinetics, while dipole moments are related to charge distribution, it can be considered that the correlation of the DTG onsets with the dipole moments might be of particular importance. According to the data provided in Table 4, the DTG onset temperatures and the dipole moments are inversely proportional.
| Biology | DTG | DP |
AlogP |
PSA | Refractivity | ||
|---|---|---|---|---|---|---|---|
| Biology | PCC | 1.000 | −0.370 | 0.736 | −0.542 | 0.039 | −0.407 |
| p | 0.539 | 0.157 | 0.345 | 0.950 | 0.496 | ||
| DTG | PCC | −0.370 | 1.000 | − 0.889 | 0.885 | −0.925 | −0.603 |
| p | 0.539 | 0.044 | 0.046 | 0.024 | 0.282 | ||
| DP | PCC | 0.736 | −0.889 | 1.000 | −0.848 | 0.679 | 0.215 |
| p | 0.157 | 0.044 | 0.070 | 0.208 | 0.728 | ||
|
AlogP |
PCC | −0.542 | 0.885 | −0.848 | 1.000 | −0.690 | −0.324 |
| p | 0.345 | 0.046 | 0.070 | 0.198 | 0.595 | ||
| PSA | PCC | 0.039 | −0.925 | 0.679 | −0.690 | 1.000 | 0.854 |
| p | 0.950 | 0.024 | 0.208 | 0.198 | 0.065 | ||
| Refractivity | PCC | −0.407 | −0.603 | 0.215 | −0.324 | 0.854 | 1.000 |
| p | 0.496 | 0.282 | 0.728 | 0.595 | 0.065 | ||
Experimental
Synthesis
The details of the synthesis and the characterization of the ligand L·2HCl and its complexes [CuLCl2] (1), [CuL(Cl)MeOH]NO3 (2), CuL(Cl)Br (3) and [CuL(NCS)(SCN)] (4) are given in ref. 1. The synthesis and characterization of the ligand L1 and its coordination complexes with 3d metals have been described previously, together with their antitumor and multidrug resistance reversing activities.31 The synthesis and structures of the ligand L2 and its complexes are also published.32L1 and L2 here denote the bis(3-chloropyridazine-6-hydrazone)-2,6-diacetylpyridine and bis(phthalazine-1-hydrazone)-2,6-diacetylpyridine ligands and their complexes with Co(III), Ni(II), Cu(II) and Zn(II) are numbered as 1, 2, 3 and 4, respectively. The coordination structures of the solid compounds with L1 and L2 are as follows: [Co2(μ-L1–2H)2(μ-O2)]·4H2O, [Ni(L1–2H)], [Cu(L1–2H)]·H2O, [Zn(L1–2H)]·H2O and complexes [Co(L2–H)Cl2]·MeOH, [Ni(L2–H)]Cl·MeOH, [Cu(L2–H)Cl] and [Zn(L2–2H)]·H2O. To get the amount of compound needed for assays, the compounds were re-synthesized.Computational details
Dispersion-corrected density functional theory (DFT-D3)77,78 and local second-order Moller-Plesset perturbation theory (LMP2)79,80 calculations have been done by the Jaguar 9.6 program,81 as implemented in Schrödinger Materials Science Suite 2017-2.82 The DFT-D3 calculations were done with a dispersion corrected B3LYP83,84 hybrid exchange–correlation functional, namely the B3LYP-D3 functional. LACVP basis sets have been employed as well. The LMP2 calculations were done considering all atom pairs. Geometrical optimizations were performed without any constraints, with increased grid size and integral accuracy. In all cases, geometrical optimizations and vibrational analysis were conducted in order to assure that true ground states, characterized by the absence of imaginary frequencies, were identified. Noncovalent interactions were identified according to the studies in ref. 85 and 86. After obtaining ground state geometries, the MEP and ALIE values were obtained after single point energy calculations. The MEP and ALIE values were mapped to the electron density surface in order to identify reactive molecule areas. Interaction energies were obtained at the LMP2 level of theory, for which it is not needed to include counterpoise correction for basis superposition error.Biological measurements
The compounds used as reagents were: cisplatin (TEVA Pharmaceutical Company, Petah Tikva, Israel), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, St Louis, MO, USA), sodium dodecyl sulfate (SDS; Sigma) and dimethyl sulfoxide (DMSO; Sigma). The ligands and the complexes were dissolved in DMSO to obtain stock solutions. The concentration of DMSO was below 1% in all the experiments. All solutions were prepared on the day of the assay.Cell lines
L5178Y mouse T-cell lymphoma cells (PAR) (ECACC Cat. No. 87111908, obtained from FDA, Silver Spring, MD, USA) were transfected with pHa MDR1/A retrovirus, as previously described by Cornwell et al.87 The ABCB1-expressing cell line L5178Y (MDR) was selected by culturing the infected cells with colchicine. The L5178Y human ABCB1-transfected subline was cultured in McCoy's 5A medium (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% heat-inactivated horse serum (Sigma-Aldrich, St Louis, MO, USA), 2 mM L-glutamine (Sigma-Aldrich, St Louis, MO, USA) and a penicillin–streptomycin (Sigma-Aldrich, St Louis, MO, USA) mixture in concentrations of 100 U L−1 and 10 mg L−1, respectively. The dose of penicillin is defined in international units (IU or U); 1 mg of pure penicillin G potassium equals 1595 U.Checkerboard combination assay
A checkerboard microplate method was applied to study the effect of drug interactions between the Schiff bases and their coordination compounds and the reference chemotherapeutic drug cisplatin, thus to determine the CI for the compounds.56,58,88The assay was carried out using multidrug-resistant mouse T-lymphoma cells overexpressing the ABCB1 transporter. The starting concentration of cisplatin (10 μg mL−1; from a 0.5 mg mL−1 stock solution) was determined during preliminary experiments on multidrug-resistant mouse T-lymphoma cells and starting concentrations of the tested compounds were determined according to previous experimental results. The dilutions of cisplatin were made in a horizontal direction in 100 μL, and the dilutions of the tested compounds were made vertically in the microtiter plate in 50 μL volume. The cells were re-suspended in the culture medium and distributed into each well in 50 μL containing 6 × 103 cells each. The plates were incubated for 72 h at 37 °C in 5% CO2 atmosphere. The cell growth rate was determined after MTT staining. At the end of the incubation period, 20 μL of MTT (thiazolyl blue tetrazolium bromide, Sigma) solution (from a stock solution of 5 mg mL−1) were added to each well. After incubation at 37 °C for 4 h, 100 μL of sodium dodecyl sulfate (SDS) (Sigma) solution (10% in 0.01 M HCI) was added to each well and the plates were further incubated at 37 °C overnight. Optical density (OD) was measured at 540/630 nm with a Multiscan EX ELISA reader (Thermo Labsystems, Cheshire, WA, USA) as described elsewhere.89 Combination index (CI) values at 50% of the growth inhibition dose (ED50), were determined using CompuSyn software (ComboSyn, Inc., Paramus, NJ. 07652 USA) to plot four to five data points for each ratio. The CI values were calculated by means of the median-effect equation, according to the Chou-Talalay method 3–5, where CI < 1, CI = 1, and CI > 1 represent synergism, additive effect (or no interaction), and antagonism, respectively. The referent CI values are given in Table 5.88
| CI | Interaction |
|---|---|
| 0–0.1 | Very strong synergism |
| 0.1–0.3 | Strong synergism |
| 0.3–0.7 | Synergism |
| 0.7–0.85 | Moderate synergism |
| 0.85–0.9 | Slight synergism |
| 0.9–1.1 | Additive effect |
| 1.1–1.2 | Slight antagonism |
| 1.2–1.45 | Moderate antagonism |
| 1.45–3.3 | Antagonism |
| 3.3–10 | Strong antagonism |
| >10 | Very strong antagonism |
Thermal analysis
Thermal data and the detection of evolved gases (EGD) were measured using a TA Instruments SDT Q600 thermal analyzer coupled to a Hiden Analytical HPR-20/QIC mass spectrometer from room temperature to 550 °C at 10 °C min−1 heating rate in nitrogen carrier gas (flow rate: 50 cm3 min−1) with ∼4 mg sample mass. The sample holder and the reference were an alumina crucible and an empty alumina crucible. Selected ions between m/z = 1–120 were monitored in Multiple Ion Detection Mode (MID).Conclusions
In order to get a better insight into the mechanism of action of potentially bioactive Schiff-base type ligands and their coordination complexes with 3d metal ions in combination with the chemotherapeutic drug cisplatin, the combination indices (CIs) were determined by the checkerboard microplate method using L5178Y mouse T-cell lymphoma cells transfected with pHa MDR1/A retrovirus (MDR) with respect to the chemotherapeutic drug cisplatin reference. The tested compounds were 2-acetylpyridine-aminoguanidine and its copper(II) complexes containing additional monoanionic ligands, as well as bis(3-chloropyridazine-6-hydrazone)-2,6-diacetylpyridine and bis(phthalazine-1-hydrazone)-2,6-diacetylpyridine and their Co(III), Ni(II), Cu(II) and Zn(II) complexes.DFT and LMP2 calculations were carried out for the 2-acetylpyridine-aminoguanidine ligand, L, and its copper(II) complexes with different monoanionic ligands. The MEP and ALIE surfaces and drug-likeness parameters (PSA, the number of atoms and rotatable bonds as well as the clogP, clogS, and DP) were calculated for the compounds. In order to estimate the possibility of the compounds as drug carriers, the noncovalent interactions and binding energies were also calculated. On the basis of these calculations, practically all the compounds satisfy the criteria as potential drug candidates. The clogS values for 4-L and 1-L are in the optimal range, and complex 1-L shows an optimum value for clogP, too. The highest binding energy between the compound and cisplatin was found for the 4-L complex, while the lowest one belonged to the compound 2-L and cisp which is still high enough for 2-L to be a drug carrier, but having the advantage of easier drug release.
Compounds 4-L and 2-L show a synergistic effect with cisp, indicating their possible role as drug carriers. The clogP values well follow the trend of the determined IC values. The dipole moments are also in relatively good agreement with the IC data, while the other calculated parameters cannot be unambiguously related to the IC data.
Another interesting phenomenon was observed concerning the thermal stability of the compounds. Namely, the DTG onset data with enough statistical significance correlate with AlogP and PSA. The DTG onsets and the dipole moments are inversely proportional and the correlation between them is still significant. The relationship between the thermal stability and the biological activity of a series of compounds with a similar composition and structure to our knowledge has not yet been explored in the literature as it is associated with seemingly not related properties. However, the thermal stability is related to the binding energies of the molecules; therefore, it might be of interest in the future investigations of compound series with potential biological activity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthesis of the compounds was financed by the Ministry of Education, Science and Technological Development of Serbia, grant number ON172014. The computational part of this work was performed thanks to the support received from Schrödinger Inc. Part of this study was performed within the project funded by the Ministry of Education, Science and Technological Development of Serbia, grant number III41017. Gabriella Spengler was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. Annamária Kincses and Márió Gajdács were supported by the UNKP-17-3 New National Excellence Program of the Ministry of Human Capacities. This study was supported by the project GINOP-2.3.2-15-2016-00038.References
- L. S. Vojinović-Ješić, M. M. Radanović, M. V. Rodić, V. Živković-Radovanović, L. S. Jovanović and V. M. Leovac, Polyhedron, 2016, 117, 526–534 CrossRef.
- C. V. Garcia, G. L. Parrilha, B. L. Rodrigues, P. J. S. Barbeira, R. M. Clarke, T. Storr and H. Beraldo, Polyhedron, 2017, 124, 86–95 CrossRef CAS.
- S. S. Chourasiya, D. Kathuria, S. S. Nikam, A. Ramakrishnan, S. Khullar, S. K. Mandal, A. K. Chakraborti and P. V. Bharatam, J. Org. Chem., 2016, 81, 7574–7583 CrossRef CAS PubMed.
- Y. Izuhara, M. Nangaku, S. Takizawa, S. Takahashi, J. Shao, H. Oishi, H. Kobayashi, C. van Ypersele de Strihou and T. Miyata, Nephrol., Dial., Transplant., 2008, 23, 497–509 CrossRef CAS PubMed.
- H. Özgüneş and S. Sabuncuoglu, Turkiye Klinikleri J. Med. Sci., 2009, 29, 976–986 Search PubMed.
- J. Osredkar and N. Sustar, J. Clin. Toxicol., 2011, s3, 001 Search PubMed.
- E. A. Buvaylo, V. N. Kokozay, O. Y. Vassilyeva, B. W. Skelton, O. V. Nesterova and A. J. L. Pombeiro, Inorg. Chem. Commun., 2017, 78, 85–90 CrossRef CAS.
- W. Zhang and C.-H. Tung, ACS Appl. Mater. Interfaces, 2017, 9, 8547–8555 CAS.
- A. Azary, A. Bezaatpour, S. Zahri and M. Amiri, New J. Chem., 2017, 41, 12554–12561 RSC.
- Y. Jia, L. Lu, C. Yuan, S. Feng and M. Zhu, J. Inorg. Biochem., 2017, 170, 46–54 CrossRef CAS PubMed.
- E. Pahonţu, F. Julea, Y. Chumakov, P. Petrenco, T. Roşu and A. Gulea, J. Organomet. Chem., 2017, 836–837, 44–55 CrossRef.
- R.-K. Pan, J.-L. Song, G.-B. Li, S.-Q. Lin, S.-G. Liu and G.-Z. Yang, Transition Met. Chem., 2017, 42, 253–262 CrossRef CAS.
- A. K. Saini, M. Saraf, P. Kumari and S. M. Mobin, New J. Chem., 2018, 42, 3509–3518 RSC.
- B. K. Singh, P. Mishra, A. Prakash and N. Bhojak, Arabian J. Chem., 2017, 10, S472–S483 CrossRef CAS.
- K. Singh, Y. Kumar, P. Puri, C. Sharma and K. R. Aneja, Arabian J. Chem., 2017, 10, S978–S987 CrossRef CAS.
- N. H. Yarkandi, H. A. El-Ghamry and M. Gaber, Mater. Sci. Eng., C, 2017, 75, 1059–1067 CrossRef CAS PubMed.
- S. Adhikari, S. Lohar, B. Kumari, A. Banerjee, R. Bandopadhyay, J. S. Matalobos and D. Das, New J. Chem., 2016, 40, 10094–10099 RSC.
- M. S. El-Shahawi, W. Ahmad, G. I. Mohammed, Y. M. Moustafa, G. A. Al-Hazmi and A. A. El-Asmy, New J. Chem., 2017, 41, 4853–4861 RSC.
- J.-P. Costes, C. Duhayon, L. Vendier and A. J. Mota, New J. Chem., 2018, 42, 3683–3691 RSC.
- S. Banerjee, P. Ghorai, P. Brandão, D. Ghosh, S. Bhuiya, D. Chattopadhyay, S. Das and A. Saha, New J. Chem., 2018, 42, 246–259 RSC.
- O. E. Sherif and N. S. Abdel-Kader, Arabian J. Chem., 2015 DOI:10.1016/j.arabjc.2015.07.008.
- D. Biswal, N. R. Pramanik, S. Chakrabarti, M. G. B. Drew, B. Sarkar, M. R. Maurya, S. K. Mukherjee and P. Chowdhury, New J. Chem., 2017, 41, 4116–4137 RSC.
- S. Tahlan, P. Kumar, K. Ramasamy, V. Mani, R. K. Mishra, A. B. A. Majeed and B. Narasimhan, Arabian J. Chem., 2017, 10, S2009–S2017 CrossRef CAS.
- N. Biswas, S. Khanra, A. Sarkar, S. Bhattacharjee, D. Prasad Mandal, A. Chaudhuri, S. Chakraborty and C. Roy Choudhury, New J. Chem., 2017, 41, 12996–13011 RSC.
- Available at WHO, 2017. http://www.who.int/mediacentre/factsheets/fs297/en/.
- W. Stewart and P. Wild, World Cancer Report 2014, World Health Organization, Geneva, 2014 Search PubMed.
- J.-P. Gillet and M. M. Gottesman, Methods Mol. Biol., 2010, 596, 47–76 CAS.
- R. H. Berndsen, A. Weiss, U. K. Abdul, T. J. Wong, P. Meraldi, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Sci. Rep., 2017, 7, 43005 CrossRef CAS PubMed.
- D. Tunc, E. Dere, D. Karakas, B. Cevatemre, V. T. Yilmaz and E. Ulukaya, Bioorg. Med. Chem., 2017, 25, 1717–1723 CrossRef CAS PubMed.
- X. Su, C. Gao, F. Shi, X. Feng, L. Liu, D. Qu and C. Wang, Drug Delivery, 2017, 24, 10–19 CrossRef CAS PubMed.
- B. Barta Holló, J. Magyari, S. Armaković, G. A. Bogdanović, M. V. Rodić, S. J. Armaković, J. Molnár, G. Spengler, V. M. Leovac and K. Mészáros Szécsényi, New J. Chem., 2016, 40, 5885–5895 RSC.
- B. Holló, J. Magyari, V. Živković-Radovanović, G. Vučković, Z. D. Tomić, I. M. Szilágyi, G. Pokol and K. Mészáros Szécsényi, Polyhedron, 2014, 80, 142–150 CrossRef.
- S. Armaković, S. J. Armaković, J. P. Setrajčić and I. J. Setrajčić, J. Mol. Model., 2012, 18, 4491–4501 CrossRef PubMed.
- S. Armaković, S. J. Armaković, J. P. Šetrajčić and I. J. Šetrajčić, Chem. Phys. Lett., 2013, 578, 156–161 CrossRef.
- S. H. Rosline Sebastian, M. I. Attia, M. S. Almutairi, A. A. El-Emam, C. Y. Panicker and C. van Alsenoy, Spectrochim. Acta, Part A, 2014, 132, 295–304 CrossRef CAS PubMed.
- E. Kose, A. Atac, M. Karabacak, C. Karaca, M. Eskici and A. Karanfil, Spectrochim. Acta, Part A, 2012, 97, 435–448 CrossRef CAS PubMed.
- M. Chen, U. V. Waghmare, C. M. Friend and E. Kaxiras, J. Chem. Phys., 1998, 109, 6854–6860 CrossRef CAS.
- J. S. Murray, J. M. Seminario, P. Politzer and P. Sjoberg, Int. J. Quantum Chem., 1990, 38, 645–653 CrossRef.
- P. Politzer, F. Abu-Awwad and J. S. Murray, Int. J. Quantum Chem., 1998, 69, 607–613 CrossRef CAS.
- P. Sjoberg, J. S. Murray, T. Brinck and P. Politzer, Can. J. Chem., 1990, 68, 1440–1443 CrossRef CAS.
- M. I. Abou-Dobara, M. A. Diab, A. Z. El-Sonbati, A. A. El-Bindary and A. M. Barakat, Arabian J. Chem., 2017, 10, S1316–S1327 CrossRef CAS.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- A. Leo, P. Y. Jow, C. Silipo and C. Hansch, J. Med. Chem., 1975, 18, 865–868 CrossRef CAS PubMed.
- A. J. Leo, J. Pharm. Sci., 1987, 76, 166–168 CrossRef CAS PubMed.
- A. J. Leo, in Molecular design and modeling. Concepts and applications. Part A, Proteins, peptides and enzymes. Part B, Antibodies and antigens, nucleic acids, polysaccharides, and drugs, ed. J. J. Langone, Elsevier, 1992, vol. 202, pp. 544–591 Search PubMed.
- A. J. Leo, Chem. Rev., 1993, 93, 1281–1306 CrossRef CAS.
- A. J. Leo and D. Hoekman, Perspect. Drug Discovery Des., 2000, 18, 19–38 CrossRef CAS.
- Hydrophobic parameter: Measurement and calculation, in Molecular design and modeling. Concepts and applications. Part A, Proteins, peptides and enzymes. Part B, Antibodies and antigens, nucleic acids, polysaccharides, and drugs, ed. J. J. Langone and A. J. Leo, Elsevier, 1992, vol. 202, pp. 544–591 Search PubMed.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- http://www.openmolecules.org, clogP, available at: http://www.openmolecules.org/propertyexplorer/clogp.html, accessed 3 July 2017.
- www.openmolecules.org, clogS, available at: http://www.openmolecules.org/properties/properties.html#clogs, accessed3 July 2017.
- S. Armaković, S. J. Armaković and S. Koziel, Carbon, 2017, 111, 371–379 CrossRef.
- S. Armaković, S. J. Armaković, S. Pelemiš and D. Mirjanić, Phys. Chem. Chem. Phys., 2016, 18, 2859–2870 RSC.
- S. Armaković, S. J. Armaković, M. Vraneš, A. Tot and S. Gadžurić, J. Mol. Model., 2015, 21, 246 CrossRef PubMed.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- T. Lazarević, A. Rilak and Ž. D. Bugarčić, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef PubMed.
- T.-C. Chou and N. Martin, CompuSyn for Drug Combinations: PC Software and User's Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values, Paramus, NJ, 2005 Search PubMed.
- M. Marloye, G. Berger, M. Gelbcke and F. Dufrasne, Future Med. Chem., 2016, 8, 2263–2286 CrossRef CAS PubMed.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599–616 CrossRef PubMed.
- S. G. Davila-Manzanilla, Y. Figueroa-de-Paz, C. Mejia and L. Ruiz-Azuara, Eur. J. Med. Chem., 2017, 129, 266–274 CrossRef CAS PubMed.
- J. Guo, B. Xu, Q. Han, H. Zhou, Y. Xia, C. Gong, X. Dai, Z. Li and G. Wu, Cancer Res. Treat., 2017 DOI:10.4143/crt.2016.572.
- S. V. Hato, C. G. Figdor, S. Takahashi, A. E. Pen, A. Halilovic, K. F. Bol, A. Vasaturo, Y. Inoue, N. de Haas, D. Verweij, C. M. L. van Herpen, J. H. Kaanders, J. H. J. M. van Krieken, H. W. M. van Laarhoven, G. K. J. Hooijer, C. J. A. Punt, A. Asai, I. J. M. de Vries and W. J. Lesterhuis, Oncotarget, 2017, 8, 54434–54443 CrossRef PubMed.
- S. Chen, H. Hu, S. Miao, J. Zheng, Z. Xie and H. Zhao, Tumour Biol., 2017, 39, 1010428317705330 Search PubMed.
- Z. Chen, B. Sellergren and X. Shen, Front. Chem., 2017, 5, 60 CrossRef PubMed.
- M. C. Akerfeldt, C. M.-N. Tran, C. Shen, T. W. Hambley and E. J. New, J. Biol. Inorg. Chem., 2017, 22, 765–774 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, Y. Gou, M. Jiang, H. Khan, Z. Zhou, H. Liang and F. Yang, J. Inorg. Biochem., 2017, 172, 1–8 CrossRef CAS PubMed.
- M. Budiul, P. Albu, G. Vlase, V. Turcuş and T. Vlase, J. Therm. Anal. Calorim., 2017, 127, 555–564 CrossRef CAS.
- A. P. S. Matos, J. S. Costa, J. Boniatti, R. C. Seiceira, A. Pitaluga, D. L. Oliveira, A. L. Viçosa and C. Holandino, J. Therm. Anal. Calorim., 2017, 127, 1675–1682 CrossRef CAS.
- Ž. K. Jaćimović, G. Giester, M. Kosović, G. A. Bogdanović, S. B. Novaković, V. M. Leovac, N. Latinović, B. B. Holló and K. M. Szécsényi, J. Therm. Anal. Calorim., 2017, 127, 1501–1509 CrossRef.
- B. Barta Holló, K. Mészáros Szécsényi, M. Deli, L. Kiss, A. Kállay-Menyhárd, V. Živković-Radovanović and Z. D. Tomić, Struct. Chem., 2016, 27, 1121–1133 CrossRef.
- L. S. Vojinović-Ješić, L. S. Jovanović, V. M. Leovac, M. M. Radanović, M. V. Rodić, B. Barta Holló, K. Mészásaros Szécsényi and S. A. Ivković, Polyhedron, 2015, 101, 196–205 CrossRef.
- K. Gardikis, M. Signorelli, C. Ferrario, A. Schiraldi, M. G. Fortina, S. Hatziantoniou, C. Demetzos and D. Fessas, Int. J. Pharm., 2017, 516, 178–184 CrossRef CAS PubMed.
- S. Tanasković, M. Antonijević-Nikolić, B. Holló, B. Drazić, T. Stanojković, K. Mészáros Szécsényi and G. Vučković, J. Serb. Chem. Soc., 2014, 79, 1235–1247 CrossRef.
- S. Kumar and S. K. Mandal, CrystEngComm, 2015, 17, 8801–8806 RSC.
- M. Nagarathinam and J. J. Vittal, Angew. Chem., Int. Ed., 2006, 45, 4337–4341 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- P. Pulay and S. Saebø, Theor. Chim. Acta, 1986, 69, 357–368 CrossRef CAS.
- S. Saebo and P. Pulay, Annu. Rev. Phys. Chem., 1993, 44, 213–236 CrossRef CAS.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- Schrödinger LLC, Materials Science Suite 2017-2, Schrödinger LLC, New York, 2017.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- M. M. Cornwell, I. Pastan and M. M. Gottesman, J. Biol. Chem., 1987, 262, 2166–2170 CAS.
- C. T. Le, Austin. Biom. Biostat., 2014, 1(2), 6 Search PubMed , http://austinpublishinggroup.com/biometrics/fulltext/biometrics-v1-id1007.php.
- D. Takács, Á. Csonka, Á. Horváth, T. Windt, M. Gajdács, Z. Riedl, G. Hajós, L. Amaral, J. Molnár and G. Spengler, Anticancer Res., 2015, 35, 3245–3251 Search PubMed , http://www.ncbi.nlm.nih.gov/pubmed/26026084.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal data on the decomposition of the compounds for the 2-acetylpyridine-aminoguanidine ligand, and its copper(II) complexes. See DOI: 10.1039/c8nj00357b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |