
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the copper(II) binding of asymmetrically functionalized tripodal peptides: solution equilibrium, structure, and enzyme mimicking†
Ágnes
Dancs
ab,
Katalin
Selmeczi
 b,
Nóra V.
May
c and
Tamás
Gajda
*a
b,
Nóra V.
May
c and
Tamás
Gajda
*a
aDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: gajda@chem.u-szeged.hu; Fax: +36-62-544340; Tel: +36-62-544435
bUniversité de Lorraine – CNRS, UMR 7053 L2CM, BP 70239, 54506 Vandœuvre-lès-Nancy, France
cInstitute of Organic Chemistry, Research Centre for Natural Sciences HAS, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
First published on 17th January 2018
Abstract
Our aim is to combine the preorganized structure of tripodal scaffolds and the advantageous metal-binding ability of histidine subunits. To this end, recently we have studied the copper(II) complexes of the tris(L-histidyl)-functionalized tren derivative, tren3his. Here we report the copper(II)-binding properties of the mono- and bis(L-histidyl)-functionalized tren ligands (tren1his (L1) and tren2his (L2)), and thus explore the impact of increasing histidine ‘density’ on the copper(II) binding of these tripodal peptides. Our solution equilibrium study was supplemented by several (UV-vis, CD, ESR, and NMR) spectroscopic and MS methods. The mono-His derivative L1 forms only mononuclear complexes. Above pH 4, the tren-like subunit is the main binding site, which is supplemented by an imidazole coordination. In the case of L2, both mono- and dinuclear copper(II) species are formed. In the acidic–neutral pH range, the highly stable bis-histamine-type binding mode dominates in the equimolar solution, while at a higher pH amide coordinated complexes are present. The bis-histamine coordination in CuHL2 creates a preorganized structure, which promotes the binding of a second metal at the tren-like binding site with {N−,Ntert,N−} coordination in Cu2H−1L2/Cu2H−2L2. Only the dinuclear Cu2H−2L2 complex was found to efficiently catalyze the oxidation of H2DTBC. The kinetic data resulted in a very high kcat/KM ratio (4360 M−1 s−1), which is due to the exceptionally strong substrate-binding ability of the dinuclear complex. However, the oxidation of the substrate within this adduct, which is a common feature of catalytically active dicopper(II) complexes, does not occur in the absence of dioxygen. This finding implies that the role of dioxygen is the oxidation of the substrate activated by the dinuclear complex. We assume a Cu(II)Cu(II)–catecholate–Cu(II)Cu(I)–semiquinone valence tautomer (VT) equilibrium. The semiquinone formed in small quantities reacts with dioxygen in a rate-determining step, which eventually results in DTBQ and H2O2 products. The Cu(II)–L2 complexes also exhibit efficient superoxide dismutase-like activity, supporting the versatility of tripodal peptide complexes in redox enzyme mimicking.
1. Introduction
Tripodal ligands are specially structured molecules containing several donor groups distributed among the three ‘arms’. They are extensively used in coordination chemistry as complexing agents for various transition metal ions. Metal complexes of tripodal ligands usually exhibit elevated thermodynamic stability due to the formation of fused chelate rings, which, depending on the denticity and symmetry of the ligand, may stabilize different coordination geometries. The targeted functionalization of tripodal ligands allows the fine-tuning of metal ion binding and also the development of additional functions, e.g. molecular recognition sites, additional metal binding or substrate binding sites, etc.Accordingly, tripodal ligands are applied in diverse fields of chemistry, such as metal sensing,1 metalloenzyme modelling,2–7 and stabilization of reactive species and intermediates8,9 and even for the development of molecular motors.10 Some tripodal ligands and/or their metal complexes show high tendency to self-assembly, creating metal–organic frameworks,11 metallocages with colorful compositions,12 hydrogels13 or other mesophase structures,14 thus expanding their application to supramolecular chemistry as well.
The advantages of peptide-type tripodal ligands in coordination chemistry have been recognized in the last few decades. The vast majority of the metal binding tripodal peptides studied are symmetrical, tri-substituted derivatives. They proved to be useful in e.g. the development of allosteric binding cavities in supramolecular assemblies,15 targeted chelation of Cu(I),16 mimicry of type-3 copper-binding sites,17 structural modeling of hydrolytic enzymes,6,18 and electrocatalytic water oxidation.7 So far, only a few examples can be found in the literature on the metal complexes of histidine-containing tripodal peptides,6,17,18 although histidine moieties are the major coordination sites in most metalloproteins.
Histidine-containing linear peptides are proved to be efficient metalloenzyme mimetics, exhibiting both hydrolytic19,20 and oxidative activities.21–26 These short peptides, however, are usually highly flexible, and the metal ion environments similar to the active site of native metalloenzymes are not stabilized. As a consequence, the metal-binding ability of linear peptides, along with their catalytic efficiency, is rather limited.
For these reasons, our aim was to combine the preorganized structure of tripodal scaffolds and the advantageous metal-binding ability of histidine(s). To this end, we have synthesized two histidine derivatives based on tris(2-aminoethyl)amine (tren) and nitrilotriacetic acid (nta) as tripodal platforms, containing N- and C-terminal histidines (tren3his and nta3his), respectively, and studied their coordination properties with copper(II) in details.27 The formation of both mono- and oligonuclear complexes confirmed the positive influence of tripodal platforms on metal coordination. Moreover, the oligonuclear complexes of both ligands exhibited efficient catechol oxidase-like activity.
In order to explore the impact of histidine ‘density’ on the coordination chemistry of these tripodal peptides, we have recently synthesized two new, asymmetrically functionalized His-ligands, tren1his and tren2his (Scheme 1), and described their zinc(II) binding properties.28 The three ligands in the trenXhis series (X = 1, 2, and 3) provide a number of donor sites for their zinc(II) complexes depending on the pH and metal-to-ligand ratios. The increasing level of functionalization favors the histamine-like coordination and allows the formation of oligonuclear complexes as well. Above pH 7 amide nitrogens and imidazolato-bridges also participate in zinc(II) binding, which results in a variety of structures.
 | ||
| Scheme 1 Schematic structures of the investigated ligands. | ||
Here, we report solution equilibrium and the structural as well as kinetic properties of copper(II) complexes formed with tren1his and tren2his (Scheme 1). Our aim was to explore how the increasing functionalization of tren arms affects the structure of copper(II) complexes and consequently their catalytic behaviors in catechol oxidation and superoxide dismutation.
2. Experimental section
2.1. Materials
Copper(II) perchlorate (Sigma-Aldrich) stock solution was standardized complexometrically. Potentiometric titrations have been carried out using 0.1 M standard NaOH solution (Sigma-Aldrich). Ethanol (VWR), 3,5-di-tert-butylcatechol (H2DTBC, Sigma-Aldrich), 2-(N-morpholino)ethanesulfonic acid (MES, Sigma-Aldrich), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma-Aldrich), N-cyclohexyl-2-aminoethanesulfonic acid (CHES, Alfa Aesar), catechol (Sigma-Aldrich), 4-nitrocatechol (H24NC, Sigma-Aldrich), sodium dihydrogen phosphate (NaH2PO4·2H2O, Sigma-Aldrich), disodium hydrogen phosphate (Na2HPO4·2H2O, Sigma-Aldrich), xanthine (Sigma-Aldrich), xanthine oxidase (Sigma-Aldrich) and nitroblue tetrazolium chloride (NBT, Alfa Aesar) were analytically pure chemicals and used without further purification. The ligands tren1his and tren2his were prepared as previously reported.282.2. Potentiometric measurements
The protonation and coordination equilibria were investigated by pH-potentiometric titrations in aqueous solutions (I = 0.1 M NaCl and T = 298.0 ± 0.1 K) under an argon atmosphere. The titrations were performed using a PC-controlled Dosimat 665 (Metrohm) automatic burette and an Orion 710A precision digital pH meter. The Metrohm micro-glass electrode (125 mm) was calibrated by the titration of hydrochloric acid and the data were fitted using the modified Nernst equation:29where JH and JOH are the fitting parameters representing the acidic and alkaline errors of the glass electrode, respectively (Kw = 10−13.75 is the autoprotolysis constant of water30). The parameters were calculated by the non-linear least squares method. The formation of the complexes was characterized by the general equilibrium process:


where M denotes the metal ion and L the fully deprotonated ligand molecule. The charges are omitted for simplicity, but can be easily calculated taking into account the composition of the protonated ligand at pH 2 ([H4L1]4+ and [H6L2]6+). The protonation constants of the ligands and the formation constants of the complexes were calculated using the PSEQUAD computer program31 from 4 and 4–7 independent titrations respectively (ca. 90 data points per titration). The metal ion concentrations varied between 1.5 and 3.0 × 10−3 M, using 2

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:2, 1:1 and 1:2 metal-to-ligand ratios.
:1, 3:2, 1:1 and 1:2 metal-to-ligand ratios.
2.3. UV-visible absorption (UV-vis) and circular dichroism (CD) spectroscopic measurements
UV-vis and CD spectra were recorded simultaneously on a BioLogic MOS-450 spectrometer (PMS-450 photomultiplier control unit, ALX-220 arc lamp power supply) using a quartz cuvette with a 1 cm optical pathlength. Data were collected in the wavelength interval of 300 to 820 nm at a resolution of 1 nm. Spectral changes were monitored between pH 2 and 11 at a 1:1 Cu(II)–L1 ratio and at 1:1 and 2:1 Cu(II)–L2 ratios. The metal ion concentration was 0.9 × 10−3 M and 1.6–3.2 × 10−3 M for Cu(II)–L1 and Cu(II)–L2 titrations, respectively. The pH-dependent UV-vis and CD spectra were processed together with the pH-potentiometric data using the PSEQUAD computer program,31 resulting in the determination of stability constants (logβ) as well as individual UV-vis and CD spectra of the Cu(II) complexes.
2.4. Electron paramagnetic resonance (EPR) measurements
The EPR spectra of the copper(II) complexes were recorded at 77 K, as individual pH-adjusted samples (I = 0.1 M NaCl), on a Bruker EMXplus 10/12 spectrometer (microwave frequency 9.39 GHz, microwave power 9.41 mW, modulation amplitude 5 G, and modulation frequency 100 kHz). The samples were prepared as aqueous solutions ([L1]tot = 1.56 × 10−3 M and [L2]tot = 1.70 × 10−3 M, and copper(II) perchlorate solution was added according to the relevant metal-to-ligand ratio). 100 μl volumes of the samples were added into the capillary, and then 20 μl of methanol was added to each of the samples in order to avoid water crystallization. The frozen solution EPR spectra were simulated using the “EPR” program,32 rhombic g-tensors (gx, gy, and gz) and copper hyperfine tensors (Ax, Ay, and Az), and the orientation-dependent linewidth parameters were fitted to set up the spectra. Nitrogen hyperfine couplings were not detected owing to the poor resolution of the spectra. Since the used copper(II) salt was a natural mixture of isotopes, all spectra were calculated as the sum of the spectra containing 63Cu and 65Cu weighted by their natural abundance. The copper and ligand coupling constants are given in units of gauss (1 G = 10−4 T).2.5. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric (MALDI-TOF MS) measurements
Molecular weight analysis of the formed complexes was executed by MALDI-TOF MS using a Bruker Reflex IV MALDI-TOF mass spectrometer. The samples were prepared in aqueous solutions at metal-to-ligand ratios of 1:1 (L1 and L2) and 2:1 (L2) by adjusting the desired pH using NaOH and HCl solutions. 1 μl of the sample was spotted onto the MALDI target plate and allowed to dry before adding 0.5 μl of the matrix solution (20 mg ml−1 2,5-dihydroxybenzoic acid (DHB) in water). Mass spectra were recorded in the positive reflectron mode (m/z range: 200–1500). External calibration affording ±100 ppm mass accuracy was performed using a polyethylene glycol standard (Aldrich). The DHB matrix was obtained from Sigma-Aldrich.
2.6. Oxidation of 3,5-di-tert-butylcatechol (H2DTBC)
All measurements were performed using a PerkinElmer Lambda 1050 or a Cary3E Varian spectrophotometer.According to the initial reaction rates (vi) method, the experimental data used for the calculations corresponded to ∼4% conversion of the initial substrate concentration. kobs values were determined under pseudo-first-order conditions from the slope of the increasing absorbance according to the following equation:
| vi = d[DTBQ]/dt = (ε400nm × l)−1 × (dA/dt) = kobs × [S]o |
Autooxidation of H2DTBC was taken into account in every experiment, resulting in kobs,corr values (kobs,corr = kobs − kobs,auto). The parameters of the Michaelis–Menten model (KM and kcat) were calculated by the non-linear least squares method. The reported kinetic data are averages of at least three parallel measurements.
2.7. Superoxide dismutase (SOD) activity
The SOD-like activity of the copper(II)–peptide complexes was studied at 298 K and at pH 7.4 (0.05 M phosphate buffer) by using the indirect method of nitroblue tetrazolium (NBT) reduction, according to reported procedures.22,24 The O2˙− radical was in situ generated using the xanthine/xanthine oxidase system, and the reduction of NBT was followed spectrophotometrically at 560 nm in 1 cm cuvettes using a PerkinElmer Lambda 1050 spectrophotometer. The quantity of xanthine oxidase was set to produce ΔA = 0.025–0.05 min−1, and xanthine concentration was 1 × 10−4 M in the reaction mixture. Kinetic tests were carried out in the presence of different amounts of copper(II) complexes from 0 to 1 × 10−6 M. The catalytic performance was established by calculating % inhibition, IC50 and k values (as reported in the literature36).3. Results and discussion
The protonation equilibria of both ligands have been investigated in details in our previous report28 by pH potentiometry and 1H NMR spectroscopy (their protonation constants are listed in the legend of Table 1).β) of L1 from H4L1 to HL1 are: 31.04, 26.01, 18.94, and 9.90; those of L2 from H6L2 to HL2 are: 36.46, 34.13, 29.51, 24.10, 17.28, and 9.5928
| tren1his (L1) | tren2his (L2) | |||
|---|---|---|---|---|
| logβ |
pK | logβ |
pK | |
| CuH3L | 30.735(2) | 3.197 | ||
| CuH2L | 27.181(6) | 3.951 | 27.538(1) | 4.984 |
| CuHL | 23.230(8) | 5.931 | 22.554(4) | 7.173 |
| CuL | 17.299(9) | 6.529 | 15.381(6) | 7.574 |
| CuH−1L | 10.77(1) | 7.807(5) | 11.077 | |
| CuH−2L | −3.27(1) | |||
| Cu2L | 21.906(4) | 4.741 | ||
| Cu2H−1L | 17.165(3) | 8.419 | ||
| Cu2H−2L | 8.746(4) | 9.368 | ||
| Cu2H−3L | −0.622(5) | |||
| Cu3H−1L2 | 36.72(2) | 7.38 | ||
| Cu3H−2L2 | 29.34(1) | |||
3.1. Copper(II) complexes of tren1his (L1)
The mono-histidyl-functionalized tren1his forms only mononuclear complexes in the presence of copper(II). The determined formation constants and some derived data are listed in Table 1, and the distribution curves of the copper(II)–L1 species are depicted in Fig. 1. The equilibrium constant for the reaction Cu2+ + H2L1 = CuH2L1 (logK = 8.24) as well as the individual UV-vis (λd–dmax ∼ 670 nm) and CD (λmax ∼ 720 nm) spectra (Fig. 2 and Table 2) of the CuH2L1 complex suggest histamine-like {Nim,αNH2} coordination in this species, similar to several HXX peptides.37,38
 | ||
| Fig. 1 Distribution of the species in the Cu(II)–L1 1:1 system ([L1]tot = [Cu(II)]tot = 0.001 M, I = 0.1 M NaCl, and T = 298 K). Secondary axis: measured CD intensities at 305 nm (red), 535 nm (green) and 635 nm (blue). | ||
 | ||
| Fig. 2 Individual molar UV-vis (A) and CD (B) spectra of the forming species in the Cu(II)–L1 1:1 system. | ||
| Complex | λ d–dmax (nm), ε (M−1 cm−1) | λ CDmax (nm), Δε (M−1 cm−1) | g o,calca |
|---|---|---|---|
| a g o,calc = (gx + gy + gz)/3, and anisotropic parameters and fitted spectra can be found in Table S1 and Fig. S5 (ESI). | |||
| CuH2L1 | ∼670, 60.6 | ∼720, 0.12 | |
| CuHL1 | 585, 118.6 | 535, 0.167 | 2.113 |
| 645, −0.188 | |||
| CuL1 | 615, 142.9 | 545, 0.076 | |
| 605, −0.016 | |||
| 695, 0.118 | |||
| CuH−1L1 | 632, 169.1 | 300, −1.02 | 2.112 |
| 635, 0.259 | |||
| CuH2L2 | 640, 53.1 | 305, −0.302 | |
| 635, 0.212 | |||
| CuHL2 | 635, 75.6 | 315, −0.616 | 2.096 |
| 675, 0.573 | |||
| CuL2 | 580, 88.0 | 675, 0.380 | |
| CuH−1L2 | 555, 156.3 | 315, 0.473 | 2.097 |
| 595, 0.223 | |||
| Cu2L2 | 580, 119.4 | 300, −0.884 | |
| 750, 0.263 | |||
| Cu2H−1L2 | 570, 161.1 | 300, 1.421 | 2.106 |
| 700, 0.246 | |||
| Cu2H−2L2 | 575, 168.4 | 310, 0.735 | |
| 515, −0.180 | |||
| 670, 0.347 | |||
| Cu2H−3L2 | 585, 205.1 | 300, −0.698 | 2.108 |
| 665, 0.307 | |||
| Cu2H−1L2–CuHL2 | 546, 11.3 | ||
| Cu2H−3L2–CuHL2 | 563, 147.7 | ||
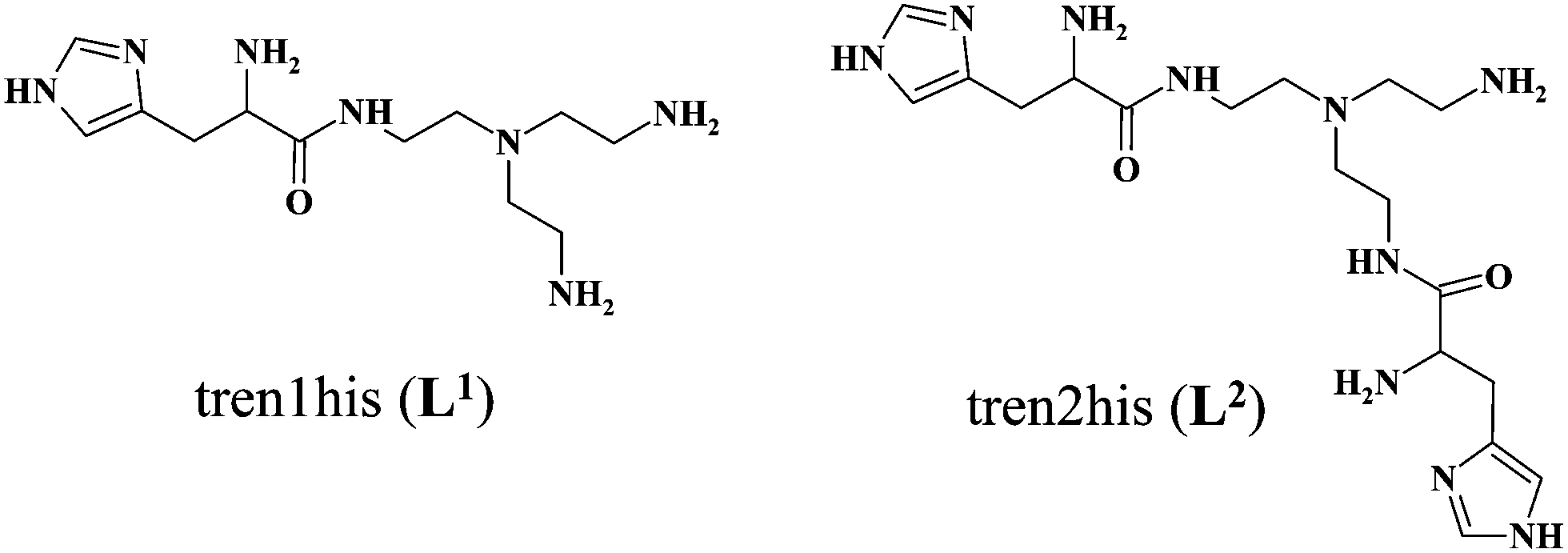
The formation of CuHL1, which is the dominant species around pH 5, induces remarkable changes in spectroscopic properties indicating a characteristically different coordination. In principle, several binding modes are possible in this species, but the intense CD spectrum (Fig. 2B) excludes the possibility of the sole coordination of the achiral tren-subunit, such as {NH2tren,Ntert,NH2tren}. In fact, the observed ‘inverse couplet’ on the CD spectrum of CuHL1 is unique and characteristic for the species in which chiral perturbation arises from a single histidine unit bound to copper(II) in a seven-membered {Nim,N−} chelate. This type of coordination results in a very similar ‘inverse CD couplet’ to the copper(II) complexes of Ac-HGG-NH2, Ac-HGGG-NH2, Ac-HGGGW-NH2 and several other analogs in the octarepeat domain of the prion protein, and therefore was studied in detail.38–40 Accordingly, in CuHL1 we suggest an {Nim,N−,Ntert,NH2tren} coordination (Scheme 2). The participation of the tertiary amino group in the coordination is the consequence of the enhanced stability of fused chelate rings. The 4N-coordination is also supported by the relatively high-energy d–d transitions (λd–dmax ∼ 585 nm) and the corresponding EPR parameters (see the ESI,† Table S1 and Fig. S5).
 | ||
| Scheme 2 Proposed structures of the main copper(II)–L1 complexes. | ||
Above pH 5, two overlapping deprotonations lead to CuH−1L1, which is the only species present in the solution above pH 8. UV-vis, CD and EPR spectra indicate a considerable rearrangement of the coordination sphere during these deprotonations: the d–d transitions are red-shifted by 47 nm and the g values are nearly unchanged, but Az is considerably decreased (178 G → 157 G, Table S1, ESI†). The ‘inverse couplet’ on the CD spectra also disappears during the CuHL1 → CuL1 → CuH−1L1 transformation. These facts indicate the coordination of a further N-donor in the axial position, and therefore the changes in the coordination geometry. Tren derivatives are known to prefer pentacoordinated structures, and the spectroscopic data are in accordance with the formation of the square pyramidal geometry. In one of the two possible structures, the equatorial binding sites of CuHL1 are retained, and the second tren-like NH2 group is coordinated in the apical position (Scheme 2), i.e. the αNH3+ deprotonates without metal ion assistance. Obviously, in the alternative structure, αNH2 may replace the imidazole ring. In order to support one of the above-mentioned binding modes, we recorded the 1H and 13C NMR spectra of L1 in the presence of copper(II). The 1H NMR spectra showed that the paramagnetic copper(II) induced only non-selective broadening of the signals. On the other hand, the 13C NMR spectra indicated a notable broadening of the signals corresponding to the imidazole moiety (C2H, C4, Fig. S6, ESI†) as compared to the αCH signal. This observation suggests the participation of the imidazole ring in the coordination, although the presence of the alternative structure involving the αNH2 cannot be ruled out.
3.2. Copper(II) complexes of tren2his (L2)
The ligand tren2his forms both mono- and oligonuclear copper(II) complexes, depending on the metal-to-ligand ratios, similar to the Zn(II)–tren2his system.28 The distribution curves of the detected species are depicted in Fig. 3, and their formation constants and the spectroscopic parameters of the main species are listed in Tables 1 and 2, respectively. | ||
| Fig. 3 Distribution of the species in Cu(II)–L2 1:1 (A) and 2:1 (B) systems ([L2]tot = 0.001 M, [Cu(II)]tot = 0.001 M and 0.002 M, respectively, I = 0.1 M NaCl, and T = 298 K). Secondary axis: measured CD intensities at 325 nm (A: green, B: blue) and 675 nm (orange). | ||
In an equimolar solution, only mononuclear complexes are formed. CuH2L2 is the first major species around pH 4, and its protonation state suggests 3N coordination. Indeed, the equilibrium constants for the process Cu2+ + H2L2 = CuH2L2 (logK = 10.26), the UV-vis spectrum of CuH2L2 (λd–dmax ∼ 640 nm), and its relatively weak positive CD band between 600 and 800 nm are in perfect agreement with {2Nim,NH2} coordinated complexes of several HXH37 or HXXH22 peptides.
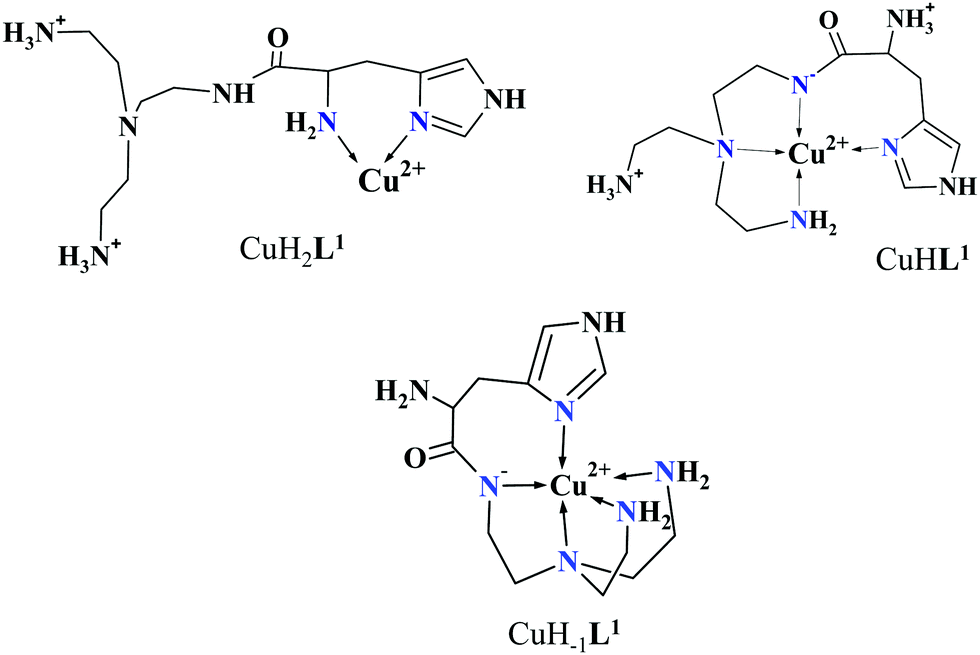
Approaching the neutral pH, the next deprotonation results in the formation of CuHL2. In this species, only the tren-like NH2 group is protonated, i.e. the metal ion has the highly stable bis-histamine-like {2Nim,2αNH2} coordination (Scheme 3). This binding mode is supported by spectral similarities to the analogous bis-histamine coordinated complexes: the detected d–d transitions (λd–dmax = 635 nm, Table 2) and CD spectra (an intense positive CD peak at 675 nm and a negative band around 300 nm) are nearly identical to those observed in Cu(II)-HisGly,41 Cu(II)-His-Xaa-Xaa,38 Cu(II)-N,N′-dihistidylethane-1,2-diamine42 and in the strongly related Cu(II)–tren3his systems.27
 | ||
| Scheme 3 Proposed structures for the main copper(II)–L2 complexes. | ||
The tren-like amino group in the bis-histamine coordinated structure of CuHL2 is too far from the metal ion to be axially coordinated. Nevertheless, the pK values of the subsequent two deprotonation steps are more than 2 units lower than the corresponding pK (= 9.59) in the free ligand. These facts and the fundamental spectroscopic changes (Fig. 5 and Fig. S5, ESI†) during the consecutive deprotonation steps (CuHL2 → CuL2 → CuH−1L2) indicate the rearrangement of the coordination sphere. The changes in the CD spectra (Fig. 5B) support different coordination modes of the histidyl unit(s) in CuHL2 and CuH−1L2. The important blue shift of the copper(II) d–d bands (635 → 555 nm, Fig. 5A) indicates a considerable increase in the ligand field strength around the metal ion, i.e. 4N coordination with at least one amide nitrogen. Indeed, since the free ligand is non-protonated around pH 10, the protonation state of the complex suggests one coordinated amide nitrogen. Considering the preferred formation of fused chelate rings as well, an {αNH2,N−,Ntert,NH2tren} coordination mode can be assumed in CuH−1L2 (Scheme 3). This donor set is very similar to that proposed above for CuHL1 (Scheme 2), except that of the αNH2/Nim switch. The preference of the αNH2 coordination over the imidazole ring in the present case is comprehensible, considering the more basic pH (the maximum formation of CuHL1/CuH−1L2 is at pH 5/9.5, respectively). A further support of the suggested coordination mode arises from the similarities of CD spectra observed for the CuHL1/CuH−1L2 pair, and those reported38 for the CuH−2L complexes of Ac-HGG-NH2/HGG peptides, where the chiral perturbation arises from the {Nim,N−}/{αNH2,N−} chelate rings, respectively.
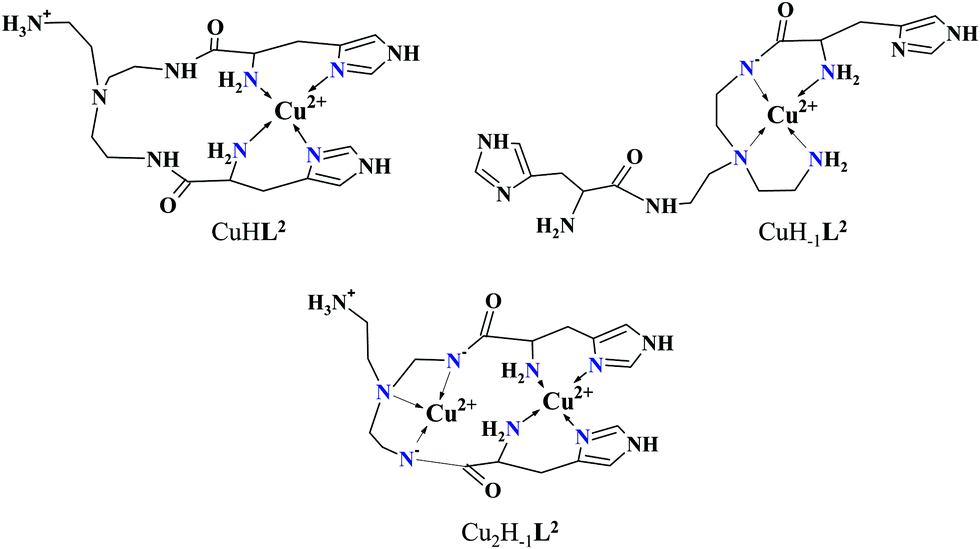
It must be pointed out that the proposed {αNH2,N−,Ntert,NH2tren} coordination mode in CuH−1L2 leaves a histidyl-functionalized arm free, with an unbound histamine-type binding site. As was previously observed in the case of the tren3his ligand, a free N-terminal His arm induces dimerization processes in the presence of excess metal ions via the formation of Cu3H−xL2-type complexes,27 due to the high stability offered by the bis-histamine-type binding for the third metal ion. Accordingly, the presence of trinuclear complex(es) was detected by MALDI-TOF MS (Fig. 4) at a copper(II)-to-ligand ratio of 3:2. The evaluation of our potentiometric data indicated the formation of two trinuclear species (Cu3H−1L22 and Cu3H−2L22), since their consideration was found to be necessary for the successful fitting of the titration curves performed at a copper(II)/L2 ratio of 3:2. However, these species do not form in significant quantities at 1:1 or 2:1 metal-to-ligand ratios, because of the high stability of both mono- and dinuclear complexes of L2, which were also detected by MALDI-TOF MS (Fig. 4).
 | ||
| Fig. 4 MALDI-TOF MS spectra measured at Cu(II)–L2 1:1 (A), 2:1 (B) and 3:2 (C) ratios. Inset: Calculated spectra of the corresponding complexes: [C18H31N10O2Cu2]+ (A), [C18H29N10O2Cu2]+ (B) and [C36H59N20O4Cu3]+ (C). | ||
The obtained isotopic distributions are in agreement with those calculated for the [CuIIH−1L2]+, [CuII3H−5L23]+ and [CuII2H−3L2]+ complexes with single positive charges. There is, however, a unit difference per metal ion in the m/z values between the measured and calculated spectra, which is due to the reduction of the copper(II) centers and the concomitant proton uptakes leading to [CuIL2]+, [CuI3H−2L23]+ and [CuI2H−1L2]+ species. The reduction of copper(II) during the ionization process of MALDI-TOF MS has been previously observed27 and proved43 by us and was also described in several other reports dealing with copper(II) complexes.44–46
The fact that in equimolar solutions the dinuclear complexes form only in small quantities indicates that L2 provides two coordination sites for copper(II) with rather different metal binding affinities. At a 2:1 metal-to-ligand ratio, the deprotonation of first appeared minor dinuclear species Cu2L2 results in the formation of Cu2H−1L2 (Fig. 3B), which predominates in the solution between pH 5 and 8. Taking into account that in the same pH range the bis-histamine-like {2Nim,2αNH2} coordination provides high stability in an equimolar solution, the conservation of this binding site in Cu2H−1L2 is safe to assume. In this manner, the four N-donors of the tren-like subunit are preorganized to bind a further metal ion. The protonation state of Cu2H−1L2 implies that the second copper(II) is bound to either an {N−,Ntert,NH2tren} or {N−,Ntert,N−} donor set (with a protonated tren-like amino group in the latter case, see Scheme 3). Assuming that the {2Nim,2αNH2} coordinated metal ion in Cu2H−1L2 has a similar component UV-vis spectrum as CuHL2, the (Cu2H−1L2–CuHL2) difference spectrum may be informative regarding the coordination environment around the second metal ion. This subtraction resulted in an ‘usual’ UV-vis spectrum (Fig. 5A) with a relatively high-energy d–d maximum (λd–dmax = 546 nm), which favors the {N−,Ntert,N−} coordination in the equatorial plane of the second copper(II). Although none of the above-mentioned two donor sets can be ruled out, the latter one may also explain the practically unchanged UV-vis and CD spectra during the next deprotonation, i.e. deprotonation of the tren-like NH3+ group without metal ion assistance. In fact, the processes Cu2H−1L2 → Cu2H−2L2 → Cu2H−3L2 are overlapped (Fig. 3B). During the last deprotonation, proton loss of either a coordinated water molecule or an imidazole ring can be taken into account.
 | ||
| Fig. 5 Individual molar UV-vis (A) and CD (B) spectra of the main species in the Cu(II)–L2 1:1 system. Black dashed line corresponds to the difference spectrum of species Cu2H−1L2 and CuHL2. | ||
3.3. Oxidation of 3,5-di-tert-butylcatechol (H2DTBC)
Our aim was to explore the effect of the increasing histidyl functionalization of tren arms on the catechol oxidase mimicking properties of the formed copper(II) complexes. Catechol oxidases are type-3 copper enzymes,47,48 therefore the copper(II) complexes of trenXhis (X = 1, 2, and 3) ligands are well suited for such biomimetic studies, and provide a decent possibility for the structure/nuclearity/activity comparison.In order to understand the activation of dioxygen by metalloenzymes, a large number of dinuclear copper(II) complexes, as biomimetic models of catechol oxidase, have been investigated.49–53 Nevertheless, very few studies report the catechol oxidase activity of copper(II)–peptide complexes,21,54,55 and for this purpose tripodal peptides have been studied only by us.27
The reaction was followed by the formation of the product 3,5-di-tert-butyl-o-benzoquinone (DTBQ) spectrophotometrically at 400 nm in a 50% EtOH/H2O solvent, in order to enhance the solubility of DTBQ. The observed catechol oxidase-like activity strongly depends on the applied ligand and the nuclearity of the complexes. Cu(II)–L1 complexes were found to be inactive. The mononuclear complexes of L2 showed only moderate activity, although the pH maximum falls advantageously in the neutral pH range (pHcorr ∼ 7.4, Fig. S7, ESI†). On the other hand, the dinuclear complexes of tren2his (L2) were found to be highly active. Therefore, a detailed kinetic study was performed only in the Cu(II)–L2 2:1 system.
The pH-rate constant profile for H2DTBC oxidation catalyzed by dinuclear copper(II) complexes is a maximum curve with a pH optimum around 8.7 (Fig. S7, ESI†). The oxidation of the substrate should proceed through the formation of a complex–DTBC2− ternary adduct.49 This is a pH-dependent process, thus the presence of the strong metal ion binder H2DTBC obviously alters the complex formation equilibria. In this manner, the pH-rate constant profile is not directly comparable with the speciation of the binary complexes. Nevertheless, Fig. S7 (ESI†) suggests that the observed catalytic activity is mainly related to the Cu2H−2L2 species. This was also supported by our CD measurements (Fig. 8B): the CD spectrum of the binary system at the pH optimum in a 50% EtOH/H2O solvent is very similar to that obtained in an aqueous solution for Cu2H−2L2, and significantly different, especially in the UV region, from that of Cu2H−3L2 (Fig. 6B).
 | ||
| Fig. 6 Individual molar UV-vis (A) and CD (B) spectra of the main species in the Cu(II)–L2 2:1 system. | ||
At the optimal pH, the initial rate of oxidation shows saturation kinetics above 40-fold excess of the substrate over the dinuclear complexes (Fig. 7). This is in accordance with the Michaelis–Menten enzyme kinetic model, indicating a fast pre-equilibrium between the substrate and the active complex before the subsequent rate-determining formation of the product. The non-linear regression of the kinetic data (solid line in Fig. 7) resulted in kcat = (0.157 ± 0.005) s−1 and KM = (0.036 ± 0.005) mM. The overall catalytic efficiency (kcat/KM) of the dicopper(II)–L2 system at pH 8.7 is very high (kcat/KM = 4360 M−1 s−1). To the best of our knowledge, only two dicopper(II) complexes were reported to have a higher efficiency.50,52 The key feature of this high catalytic efficiency seems to be the exceptionally strong substrate binding to the dinuclear species, as indicated by the very low KM value, since kcat is in the range generally observed for active dicopper(II) complexes.56
 | ||
| Fig. 7 Dependence of the initial reaction rates on H2DTBC concentrations promoted by the Cu(II)–L2 2:1 system (in EtOH/H2O 50/50%, pHcorr = 8.7, [complex]tot = 5.3 × 10−6 M). | ||
In order to gain further insights into the catalysis, reaction rates were also measured as a function of the concentration of dinuclear complexes and dioxygen under pseudo-first order conditions (Fig. S8, ESI†). In both cases, the first-order dependence was observed, similarly to many previously investigated catecholase model systems.51,57–59 As a consequence of the Michaelis–Menten model, the observed pseudo-second order rate constant (k′, the slope of the straight line in Fig. S8, ESI†) should be equal to kcat/(KM + [S]). The value calculated from the Michaelis–Menten parameters (kcalc′ = 272 M−1 s−1) is, indeed, relatively in good agreement with the measured one (k′ = 154.4 M−1 s−1).
3.4. Substrate binding study
Since the KM value obtained from the saturation kinetic data indicated impressively high substrate-binding affinity for the dinuclear complexes, we wanted to verify it by an independent method, and to collect further data on the complex–substrate adduct. To this end, spectrophotometric and CD measurements have been performed as a function of substrate concentration under inert conditions (argon atmosphere), in order to prevent the catalytic oxidation of H2DTBC (Fig. 8). Upon addition of increasing amount of H2DTBC, two overlapping bands appear in the UV-vis spectra between 350 and 450 nm, while the d–d band of Cu(II) shifts towards higher energies (from ∼580 to 564 nm, Fig. 8A). Neither the strong absorbance of DTBQ at 400 nm (which would have resulted in an absorbance of ∼1, within the applied experimental conditions) nor the reduction of copper(II), i.e. decrease of the d–d band intensity, was detected (Fig. 8). Consequently, the stoichiometric oxidation of the substrate by the copper(II) centers does not occur under anaerobic conditions. The absorption bands between 350 and 450 nm can be attributed to the CT band of the copper(II)-bound catecholate,57,60–62 overlapped with the absorbance of the free (unbound) mono-deprotonated HDTBC− present in small quantities (pKH2DTBC = 10.35 in 50/50% MeOH/H2O medium63), causing a continuous increase in absorbance at higher H2DTBC concentrations.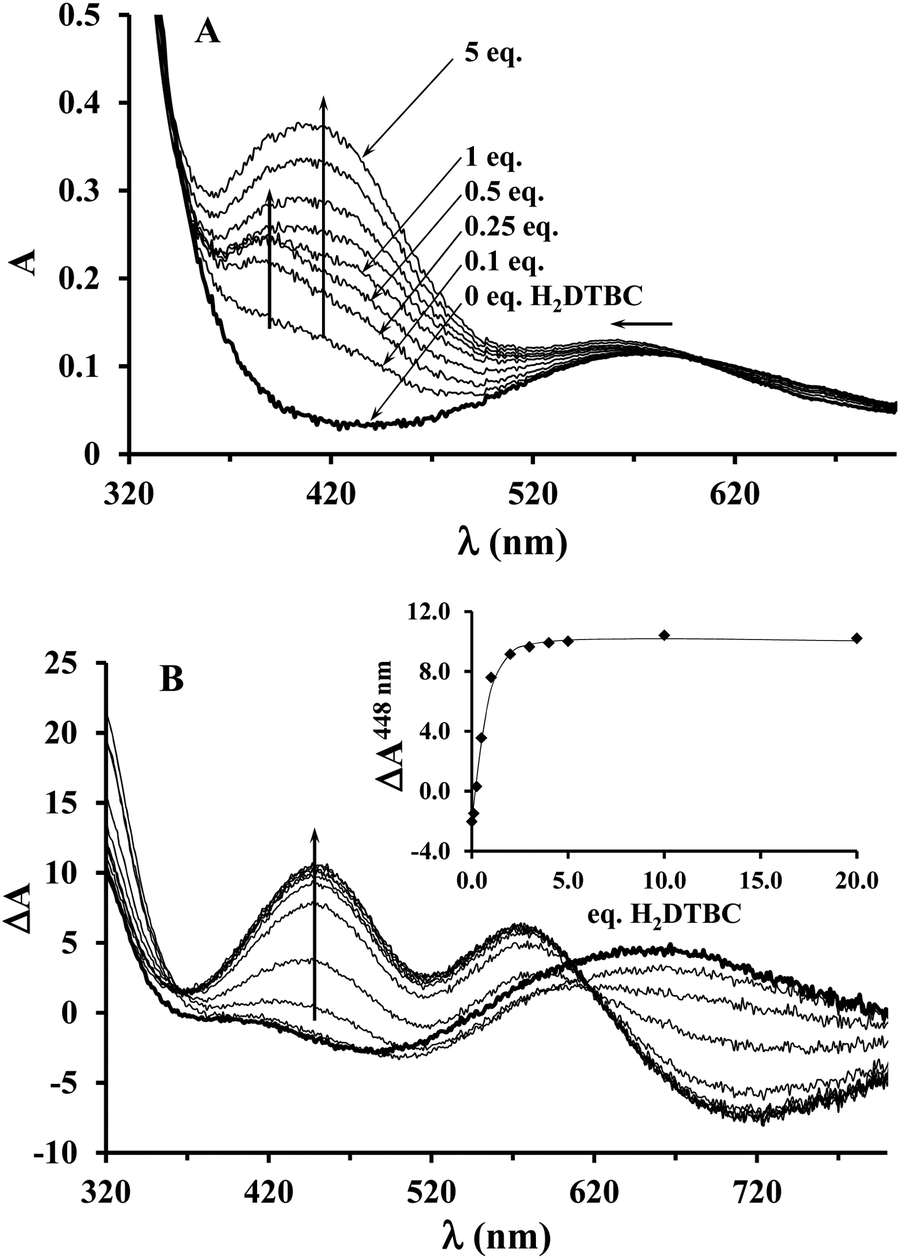 | ||
| Fig. 8 UV-vis (A) and CD spectra (B) of the Cu(II)–L2 2:1 system recorded under inert (argon) conditions, upon addition of 0–20 eq. H2DTBC (in EtOH/H2O 50/50%, pHcorr = 8.7, [complex]tot = 5.3 × 10−4 M). Inset: CD intensities at 448 nm as a function of added H2DTBC equivalents. | ||
The CD spectra, reflecting exclusively the environment of the metal ions, also show profound changes upon addition of H2DTBC (Fig. 8B), indicating important rearrangement of the coordination sphere upon substrate binding. Since the chiral perturbation arises exclusively from the L2 ligand, the CD spectra directly prove the formation of the Cu(II)–L2–substrate adduct.
The development of the characteristic CD band at 448 nm (Fig. 8B inset) allowed estimating the apparent stability constant (Kapp) of the complex–substrate adduct according to the equilibrium process Cu2HxL2 + H2DTBC = Cu2HyL2(DTBC2−). The non-linear regression of the data resulted in Kapp = 14200 M−1, which is in good agreement with the results of our saturation kinetic measurements (1/KM = 27624 M−1, since the Michaelis constant corresponds to the dissociation constant of the above-mentioned adduct). It is worth mentioning that this extraordinary binding ability is not restricted to H2DTBC; similarly high Kapp values (>20000 M−1) were determined for catechol and 4-nitrocatechol as well by similar CD measurements (Fig. S9, ESI†).
The observation that the two-electron oxidation of H2DTBC does not occur under anaerobic conditions is rare among dinuclear complexes, which are catalytically active in the presence of dioxygen,27,51,62 but typical for catechol oxidase mimicking mononuclear complexes.49,56 This feature clearly emphasizes that dioxygen is directly involved in the oxidation of the substrate, i.e. the mechanism is different from that proposed for the native enzyme.47,48 Although two-electron transfer from the copper(II) ions to the substrate does not occur in the present case, the metal centers are obviously involved in the activation of the substrate. Although, even zinc(II) is able to activate H2DTBC by coordination,64 in the case of copper(II) complexes the presence of a Cu(II)Cu(II)–catecholate/Cu(II)Cu(I)–semiquinone valence tautomerism (VT) is safe to assume (Scheme 4).57,65–67
 | ||
| Scheme 4 A proposed mechanism for the catalytic oxidation of 3,5-di-tert-butylcatechol by the dicopper(II) complex of L2. | ||
A similar VT was reported for a dicopper(II) complex with η1:η1 bridged catecholate as well.51 The absorption band observed between 350 and 450 nm (Fig. 8A) may correspond to both bidentate and η1:η1 bridged coordination of catecholate; however, the observed very low KM favors the formation of the catecholate bridge. The above-mentioned VT equilibrium is strongly shifted toward the initial state, since neither appreciable decrease of the d–d band intensity nor the EPR spectrum of the semiquinone radical was observed under anaerobic conditions. Nevertheless, the semiquinone formed in small quantities reacts with dioxygen in a rate-determining step, resulting in the formation of oxygenated radical intermediate(s).56 After a subsequent intramolecular electron transfer DTBQ and H2O2 products are released, regenerating the dicopper(II) complex and allowing the coordination of a new substrate (Scheme 4). This mechanism is similar to that proposed for the copper(II) complexes of the tris-histidyl analog tren3his,27 although the presently investigated system is more active (see Table 3).
3.5. Superoxide dismutase (SOD) activity
The Cu(II)–tren2his system, together with the previously investigated copper(II)–tren3his complexes,27 demonstrated strong ability for dioxygen activation via radical formation. Such oxidative reactions may involve the formation of a superoxide radical as well. Copper(II) complexes of His-containing linear peptides are well-known superoxide dismutase mimicking compounds.22–25,68–70 Therefore, we also studied the superoxide-dismutating ability of copper(II) complexes formed with our tripodal His-peptides, thus gaining deeper insights into their redox enzyme mimicking properties.The SOD activity of the copper(II)–L1 and –L2 systems has been characterized according to the modified McCord–Fridovich method,71 at a pH of 7.4. Similar to the catechol oxidase mimetic study, the copper(II) complexes of L1 were found to be completely inactive, implying the low accessibility of the copper(II) ion, unambiguously due to the saturated and highly stable coordination sphere in CuH−1L1.
The inhibition curves in the presence of copper(II)–L2 systems are depicted in Fig. 9, and the obtained kinetic parameters (IC50 and k) are presented in Table 4, together with that of the native Cu,Zn-SOD and free copper(II) as comparison.
 | ||
| Fig. 9 Inhibition of the NBT–superoxide reaction in the presence of Cu(II)–L2 1:1 (✦) and 2:1 (□) systems at pH 7.4 (in phosphate buffer, [NBT] = 2 × 10−4 M and T = 298 K). | ||
| Complex | IC50 (μM) | k (M−1 s−1) | Ref. |
|---|---|---|---|
| a k calculated as kNBT × [NBT]/IC50.36 b Ligands and their copper(II) complexes were studied in our previous work.27 | |||
| Cu,Zn-SOD (pH 6.8) | 0.0045 | 3.3 × 108 | 21 |
| Cu(II)–L2 2:1 (pH 7.4) |
0.13 | 4.6 × 107 | This work |
| Cu(II)–nta3hisb 1:1 (pH 7.4) |
0.13 | 2.3 × 107 | This work |
| Cu(II)–tren3hisb 1:1 (pH 7.4) |
0.17 | 1.8 × 107 | This work |
| Cu(II)–L2 1:1 (pH 7.4) |
0.20 | 3.0 × 107 | This work |
| Cu(HPO4) (pH 7.4) | 1.06 | 6.2 × 106 | 69 |
Both mono- and dinuclear copper(II)–L2 complexes were found to be able to dismutate the superoxide radical. At 0.1–1 μM L2 concentration and at pH 7.4, CuL2/CuH−1L2 and Cu2H−1L2 complexes are the major species at 1:1 and 2:1 metal-to-ligand ratios, respectively (the concentration of the free copper(II) ion is negligible). The obtained IC50 values are similar for all copper(II) complexes of tripodal peptides (Table 4). These values indicate the highly efficient superoxide dismutating ability, although the best SOD mimicking copper(II)–peptide complexes, containing exclusively imidazole coordinated metal centers, exhibit a 2–3-fold higher activity (IC50 = 0.084 and 0.044 μM).25
4. Conclusions
In this study, we extended our knowledge on the coordination chemistry of N-terminal histidine-containing tripodal peptides. Copper(II) complexes of the mono- and bis-functionalized tren derivatives, tren1his (L1) and tren2his (L2), respectively, are presented, focusing on solution equilibrium and catechol oxidase mimicking. The mono-His derivative L1 forms only mononuclear complexes. Around pH 5, the CuHL1 complex predominates in the solution with the {Nim,N−,Ntert,NH2tren} type coordination. Above pH 8, CuH−1L1 is the only species in which the copper(II) ion has the {Nim,N−,Ntert,2NH2tren} binding mode, i.e. the tren-like coordination is supplemented by an imidazole ring. In the case of L2, both mono- and dinuclear copper(II) species can be identified. In the acidic–neutral pH range, the highly stable bis-histamine-type binding mode dominates in the equimolar solution, while in basic pH amide coordinated complexes are present. The bis-histamine coordination in CuHL2 creates a preorganized structure which promotes the binding of a second metal at the tren-like binding site with {N−,Ntert,N−} coordination in Cu2H−1L2 and Cu2H−2L2.Only the Cu2H−2L2 complex was found to efficiently catalyze the oxidation of H2DTBC. The kinetic data resulted in a very high kcat/KM ratio (4360 M−1 s−1), which is due to the exceptionally strong substrate-binding ability of the dinuclear complex. Independent UV-vis and CD measurements confirmed the formation of a highly stable complex–substrate ternary adduct, bearing significantly different spectroscopic features as compared with the binary copper(II) complex. However, the oxidation of the substrate within this adduct, which is a common feature of catalytically active dicopper(II) complexes, does not occur in the absence of dioxygen. This finding implies that the role of dioxygen is the oxidation of the substrate activated by the dinuclear complex, and not the oxidation of the copper(I) species, as it was proposed for the native enzyme and for many dinuclear model compounds. We assume a Cu(II)Cu(II)–catecholate–Cu(II)Cu(I)–semiquinone valence tautomer (VT) equilibrium. The semiquinone formed in small quantities reacts with dioxygen in a rate-determining step, which eventually results in products DTBQ and H2O2. The Cu(II)–L2 complexes also possess efficient superoxide dismutase-like activity, supporting the versatility of tripodal peptide complexes in redox enzyme mimicking.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the National Research, Development and Innovation Office-NKFIH through projects GINOP-2.3.2-15-2016-00038, OTKA 101541 and TÉT_15-1-2016-0031. The authors are grateful to G. Krier and L. Vernex-Loset for MALDI-TOF MS measurements, A. Merlin for EPR measurements and S. Parant for technical help at Université de Lorraine.References
- Z. Dai and J. W. Canary, New J. Chem., 2007, 31, 1708–1718 RSC.
- M. Suzuki, Acc. Chem. Res., 2007, 40, 609–617 CrossRef CAS PubMed.
- N. Burzlaff, Adv. Inorg. Chem., 2008, 60, 101–165 CrossRef CAS.
- G. Parkin, New J. Chem., 2007, 31, 1996–2014 RSC.
- A. Szorcsik, F. Matyuska, A. Bényei, N. V. Nagy, R. K. Szilágyi and T. Gajda, Dalton Trans., 2016, 45, 14998–15012 RSC.
- M. M. Ibrahim and G. A. M. Mersal, J. Inorg. Biochem., 2010, 104, 1195–1204 CrossRef CAS PubMed.
- L. Szyrwiel, D. Lukacs, D. F. Sranko, Z. Kerner, A. Kotynia, J. Brasun, B. Setner, Z. Szewczuk, K. Malec and J. S. Pap, RSC Adv., 2017, 7, 24657–24666 RSC.
- W. A. Chomitz and J. Arnold, Chem. – Eur. J., 2009, 15, 2020–2030 CrossRef CAS PubMed.
- X. Hu and K. Meyer, J. Organomet. Chem., 2005, 690, 5474–5484 CrossRef CAS.
- G. Rapenne, Inorg. Chim. Acta, 2009, 362, 4276–4283 CrossRef CAS.
- F. Matyuska, A. Szorcsik, N. V. May, Á. Dancs, É. Kováts, A. Bényei and T. Gajda, Dalton Trans., 2017, 46, 8626–8642 RSC.
- S. Tartaggia, O. De Lucchi, A. Gambaro, R. Zangrando, F. Fabris and A. Scarso, Chem. – Eur. J., 2013, 19, 5701–5714 CrossRef CAS PubMed.
- K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663–1667 CrossRef CAS PubMed.
- A. Timme, R. Kress, R. Q. Albuquerque and H.-W. Schmidt, Chem. – Eur. J., 2012, 18, 8329–8339 CrossRef CAS PubMed.
- S. Blanc, P. Yakirevitch, E. Leize, M. Meyer, J. Libman, A. Van Dorsselaer, A.-M. Albrecht-Gary and A. Shanzer, J. Am. Chem. Soc., 1997, 119, 4934–4944 CrossRef CAS.
- A.-S. Jullien, C. Gateau, I. Kieffer, D. Testemale and P. Delangle, Inorg. Chem., 2013, 52, 9954–9961 CrossRef CAS PubMed.
- H. B. Albada, F. Soulimani, B. M. Weckhuysen and R. M. J. Liskamp, Chem. Commun., 2007, 4895–4897, 10.1039/B709400K.
- M. Gelinsky, R. Vogler and H. Vahrenkamp, Inorg. Chem., 2002, 41, 2560–2564 CrossRef CAS PubMed.
- D. Árus, N. V. Nagy, Á. Dancs, A. Jancsó, R. Berkecz and T. Gajda, J. Inorg. Biochem., 2013, 126, 61–69 CrossRef PubMed.
- E. Kopera, A. Krężel, A. M. Protas, A. Belczyk, A. Bonna, A. Wysłouch-Cieszyńska, J. Poznański and W. Bal, Inorg. Chem., 2010, 49, 6636–6645 CrossRef CAS PubMed.
- A. Jancsó, Z. Paksi, N. Jakab, B. Gyurcsik, A. Rockenbauer and T. Gajda, Dalton Trans., 2005, 3187–3194, 10.1039/B507655B.
- D. Árus, A. Jancsó, D. Szunyogh, F. Matyuska, N. V. Nagy, E. Hoffmann, T. Körtvélyesi and T. Gajda, J. Inorg. Biochem., 2012, 106, 10–18 CrossRef PubMed.
- N. I. Jakab, A. Jancsó, T. Gajda, B. Gyurcsik and A. Rockenbauer, J. Inorg. Biochem., 2008, 102, 1438–1448 CrossRef CAS PubMed.
- Z. Paksi, A. Jancsó, F. Pacello, N. Nagy, A. Battistoni and T. Gajda, J. Inorg. Biochem., 2008, 102, 1700–1710 CrossRef CAS PubMed.
- S. Timári, R. Cerea and K. Várnagy, J. Inorg. Biochem., 2011, 105, 1009–1017 CrossRef PubMed.
- Y. Jin and J. A. Cowan, J. Am. Chem. Soc., 2005, 127, 8408–8415 CrossRef CAS PubMed.
- Á. Dancs, N. V. May, K. Selmeczi, Z. Darula, A. Szorcsik, F. Matyuska, T. Páli and T. Gajda, New J. Chem., 2017, 41, 808–823 RSC.
- Á. Dancs, K. Selmeczi, I. Bányai, Z. Darula and T. Gajda, Inorg. Chim. Acta, 2017 DOI:10.1016/j.ica.2017.06.049.
- F. J. C. Rosotti and H. Rosotti, The determination of stability constants, McGraw-Hill Book Co., New York, 1962 Search PubMed.
- E. Högfeldt, Stability Constants of Metal-Ion Complexes, Part A. Inorganic Ligands, Pergamon, New York, 1982 Search PubMed.
- L. Zékány, I. Nagypál and G. Peintler, PSEQUAD for chemical equilibria, Technical Software Distributors, Baltimore, MD, 1991 Search PubMed.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- R. G. Bates, M. Paabo and R. A. Robinson, J. Phys. Chem., 1963, 67, 1833–1838 CrossRef CAS.
- L. G. Hepler, E. M. Woolley and D. G. Hurkot, J. Phys. Chem., 1970, 74, 3908–3913 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor and Francis, 88th edn, 2007–2008 Search PubMed.
- S. Durot, C. Policar, F. Cisnetti, F. Lambert, J.-P. Renault, G. Pelosi, G. Blain, H. Korri-Youssoufi and J.-P. Mahy, Eur. J. Inorg. Chem., 2005, 3513–3523 CrossRef CAS.
- A. Myari, G. Malandrinos, Y. Deligiannakis, J. C. Plakatouras, N. Hadjiliadis, Z. Nagy and I. Sóvágó, J. Inorg. Biochem., 2001, 85, 253–261 CrossRef CAS PubMed.
- N. I. Jakab, B. Gyurcsik, T. Körtvélyesi, I. Vosekalna, J. Jensen and E. Larsen, J. Inorg. Biochem., 2007, 101, 1376–1385 CrossRef CAS PubMed.
- A. P. Garnett and J. H. Viles, J. Biol. Chem., 2003, 278, 6795–6802 CrossRef CAS PubMed.
- E. Aronoff-Spencer, C. S. Burns, N. I. Avdievich, G. J. Gerfen, J. Peisach, W. E. Antholine, H. L. Ball, F. E. Cohen, S. B. Prusiner and G. L. Millhauser, Biochemistry, 2000, 39, 13760–13771 CrossRef CAS PubMed.
- G. Facchin, M. H. Torre, E. Kremer, E. J. Baran, A. Mombrú, H. Pardo, M. P. Araujo, A. A. Batista and A. J. Costa-Filho, Inorg. Chim. Acta, 2003, 355, 408–413 CrossRef CAS.
- I. Török, T. Gajda, B. Gyurcsik, G. K. Tóth and A. Péter, J. Chem. Soc., Dalton Trans., 1998, 1205–1212, 10.1039/A707408E.
- F. Matyuska, N. V. May, A. Benyei and T. Gajda, New J. Chem., 2017, 41, 11647–11660 RSC.
- P. Lubal, M. r. Kývala, P. Hermann, J. Holubová, J. Rohovec, J. Havel and I. Lukeš, Polyhedron, 2001, 20, 47–55 CrossRef CAS.
- G. Greiner, L. Seyfarth, W. Poppitz, R. Witter, U. Sternberg and S. Reissmann, Lett. Pept. Sci., 2000, 7, 133–141 CAS.
- K. Ősz, K. Várnagy, H. Süli-Vargha, D. Sanna, G. Micera and I. Sóvágó, Dalton Trans., 2003, 2009–2016, 10.1039/B211413E.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Mol. Biol., 1998, 5, 1084–1090 CAS.
- N. Hakulinen, C. Gasparetti, H. Kaljunen, K. Kruus and J. Rouvinen, J. Biol. Inorg. Chem., 2013, 18, 917–929 CrossRef CAS PubMed.
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083–7093 CrossRef CAS PubMed.
- I. A. Koval, K. Selmeczi, C. Belle, C. Philouze, E. Saint-Aman, I. Gautier-Luneau, A. M. Schuitema, M. van Vliet, P. Gamez, O. Roubeau, M. Lüken, B. Krebs, M. Lutz, A. L. Spek, J.-L. Pierre and J. Reedijk, Chem. – Eur. J., 2006, 12, 6138–6150 CrossRef CAS PubMed.
- E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbì, Inorg. Chem., 1998, 37, 553–562 CrossRef CAS PubMed.
- E. C. M. Ording-Wenker, M. A. Siegler, M. Lutz and E. Bouwman, Dalton Trans., 2015, 44, 12196–12209 RSC.
- W. M. Tay, A. I. Hanafy, A. Angerhofer and L.-J. Ming, Bioorg. Med. Chem. Lett., 2009, 19, 6709–6712 CrossRef CAS PubMed.
- S. Dell'Acqua, V. Pirota, C. Anzani, M. M. Rocco, S. Nicolis, D. Valensin, E. Monzani and L. Casella, Metallomics, 2015, 7, 1091–1102 RSC.
- S. K. Dey and A. Mukherjee, Coord. Chem. Rev., 2016, 310, 80–115 CrossRef CAS.
- K. Masahito, K. Tomohisa, K. Koji, T. Yoshimitsu, I. Shinobu and K. Shosuke, Bull. Chem. Soc. Jpn., 2003, 76, 1957–1964 CrossRef.
- J. Ackermann, S. Buchler and F. Meyer, C. R. Chim., 2007, 10, 421–432 CrossRef CAS.
- A. Granata, E. Monzani and L. Casella, J. Biol. Inorg. Chem., 2004, 9, 903–913 CrossRef CAS PubMed.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2004, 1061–1072, 10.1039/B315811J.
- D. G. Brown, W. J. Hughes and G. Knerr, Inorg. Chim. Acta, 1980, 46, 123–126 CrossRef CAS.
- S. Torelli, C. Belle, S. Hamman, J.-L. Pierre and E. Saint-Aman, Inorg. Chem., 2002, 41, 3983–3989 CrossRef CAS PubMed.
- C. A. Tyson and A. E. Martell, J. Am. Chem. Soc., 1968, 90, 3379–3386 CrossRef CAS.
- J. Kaizer, J. Pap, G. Speier, L. Párkányi, L. Korecz and A. Rockenbauer, J. Inorg. Biochem., 2002, 91, 190–198 CrossRef CAS PubMed.
- G. Speier, Z. Tyeklár, P. Tóth, E. Speier, S. Tisza, A. Rockenbauer, A. M. Whalen, N. Alkire and C. G. Pierpont, Inorg. Chem., 2001, 40, 5653–5659 CrossRef CAS PubMed.
- J. Kaizer, T. Csay, G. Speier and M. Giorgi, J. Mol. Catal. A: Chem., 2010, 329, 71–76 CrossRef CAS.
- M. J. Gajewska, W.-M. Ching, Y.-S. Wen and C.-H. Hung, Dalton Trans., 2014, 43, 14726–14736 RSC.
- G. Facchin, N. Veiga, M. G. Kramer, A. A. Batista, K. Várnagy, E. Farkas, V. Moreno and M. H. Torre, J. Inorg. Biochem., 2016, 162, 52–61 CrossRef CAS PubMed.
- L. L. Costanzo, G. de Guidi, S. Giuffrida, E. Rizzarelli and G. Vecchio, J. Inorg. Biochem., 1993, 50, 273–281 CrossRef CAS PubMed.
- B. Bóka, A. Myari, I. Sóvágó and N. Hadjiliadis, J. Inorg. Biochem., 2004, 98, 113–122 CrossRef.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data (including measured UV-vis, CD, NMR and EPR spectra of copper(II) complexes, along with further kinetic and substrate-binding experimental data). See DOI: 10.1039/c7nj04716a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |