DOI:
10.1039/C7NJ02953E
(Paper)
New J. Chem., 2018,
42, 363-371
Synthesis, characterization and in vitro screening of a nano-hydroxyapatite/chitosan/Euryale ferox nanoensemble – an inimitable approach for bone tissue engineering†
Received
9th August 2017
, Accepted 17th November 2017
First published on 17th November 2017
Abstract
In order to explore novel synthetic bone scaffolds as part of our ongoing research in the field of bone tissue engineering, we have synthesized a biomimmetic, osteoinductive, tricomposite scaffold incorporating Euryale ferox (EF) with nano-hydroxyapatite and chitosan. The surface morphology of the resultant nanocomposite was visualized via SEM and TEM studies which exhibited a rougher surface of n-HA/CS/EF compared to the n-HA/CS nanocomposite with the particle size ranging between 15 and 20 nm. The n-HA/CS/EF scaffold was degraded up to (16.9 ± 0.70)% in contrast to degradation of (7.4 ± 0.2)% for n-HA/CS. The in vitro SBF study exhibited excellent biomineralization capacity for n-HA/CS/EF which was further elucidated using the ARS staining procedure. The MTT assay illustrated significantly better cell viability compared to the n-HA/CS nanocomposite. A comparatively better antibacterial activity of n-HA/CS/EF was observed against a number of bacterial pathogens. The osteogenic differentiation test (ALP assay) assessed the osteoconductivity of the n-HA/CS/EF nanocomposite. Thus, the synthesized scaffold can be envisioned for potential use in reparation of bone defects.
1. Introduction
Natural bone is a highly vascularized tissue comprising hierarchically arranged inorganic–organic nanocomposite materials serving as a substructure for the appendage and functioning of skeletal muscles and other tissues.1,2 A major area of concern in orthopedics deals with bone fractures resulting in non-unions and deformities due to degenerative conditions where minimal tissue integrity remains in the defect site.3 According to the current survey, nearly 2.2 million patients annually utilize bone defect regeneration procedures worldwide costing more than 2.5 billion US dollars which is expected to increase two-fold by 2020 globally.4,5 Therefore, bone defects that cannot be regenerated via normal physiological conditions necessitate intervention in the form of artificial bone grafts. Autografts and allografts are the most frequently and preferably used methods.6,7 However, they have serious impediments in the form of donor site morbidity associated with autografts and disease transmission and immune rejection in the case of allografts, thus triggering the need for more desirable methods.8,9 Bone tissue engineering provides alternatives to autografts and allografts through the implementation of synthetic grafts leading to tissue regeneration. Nanocomposite biomaterials fabricated as synthetic bone scaffolds are inflating the current frontiers of bone tissue engineering. These scaffolds should accomplish certain criteria for their successful application such as the scaffold should be osteoconductive and serve as a template to facilitate bone formation, and it should stimulate osteoprogenitor cells to differentiate into osteoblasts.10,11 In addition, it should have a highly porous structure along with adequate mechanical properties, non toxicity, and controlled biodegradability.12,13
To date, different classes of materials, exclusively natural and synthetic polymers, have been explored extensively for scaffold fabrication. Inorganic–organic hybrid polymer nanocomposites form excellent bone grafts emerging from the synergistic effects between the components. Synthetic polymers of interest include polyesters such as polycaprolactone (PCL), poly(lactic acid) (PLA), poly(glycolic acid) (PGA), and poly(lactic-co-glycolic acid) (PLGA).14,15 However, these synthetic polymers encompass the major pitfall of insufficient cell adhesion and their hydrophobic surface impedes cell growth in a three-dimensional bone scaffold. Natural polymers probed are polysaccharides such as alginate and chitosan along with proteins, essentially collagen, gelatin and silk fibroin.16 Chitosan (CS), a cationic polysaccharide composed of D-glucosamine and N-acetyl-D-glucosamine residues, has received substantial interest in bone tissue engineering. It is considered to be the most pertinent biomaterial because of its high biocompatibility, biodegradability, non-antigenicity, and hydrophilicity along with its ability of chemical modification to generate desired distinctive properties.17–19 Synthetic ceramic materials based on calcium phosphate (CaP), particularly in the composition of tricalcium phosphate [TCP-Ca3(PO4)2], hydroxyapatite [HA-Ca10(PO4)6(OH)2] and biphasic calcium phosphates (BCP, a mixture of HA and TCP), have been combined with chitosan resulting in enhanced mechanical and biological properties.20,21 Hydroxyapatite, the major inorganic component of natural bone, has been extensively used because of its outstanding biocompatibility, osteoconductivity, bioactivity, non-toxicity, exceptionally slow degradation rate and direct bonding to the regenerated bone tissues making bone regeneration expeditious.22–24 However, due to brittleness and inflexibility it is difficult to shape hydroxyapatite, which necessitates the utilization of bioactive ceramic–polymer biocomposites. These biocomposites have gained phenomenal impetus for their bone analogue design with improved performances to meet specific clinical requirements.25 Different binary systems with hydroxyapatite and various polymers have been synthesized previously and at present, the ternary systems with enhanced properties incorporating multiple components are receiving enormous attention.26,27 We have recently synthesized and reported a few ternary systems incorporating β-cyclodextrin, starch and polyethylene glycol with nano-hydroxyapatite (n-HA) and chitosan which have proved to be efficacious for bone tissue engineering applications.28–30 In continuation of our previous work, we have emphasized the use of natural products considering their significant structural diversity of molecular frameworks which can be used for the synthesis in biological chemistry.31 Therefore, it was thought worthwhile to incorporate starchy kernels of Euryale ferox (EF) as a natural source in the ternary system involving nano-hydroxyapatite and chitosan in view of its physiologically active components and antioxidant activity which have been reported in medicines to cure various diseases, and for reducing inflammation, regulating blood pressure, and detoxification of the spleen.32,33 In addition, it is calcium rich, and therefore is employed in the treatment of arthritis, for osteoporosis patients and also in other joint problems. However, Euryale ferox in combination with n-HA/CS has not been reported in tissue engineering so far. Hence, considering its biological properties, we have explored the standalone applications of E. ferox in association with nano-hydroxyapatite and chitosan in order to enhance the physicochemical and biological properties of the scaffold for bone tissue engineering applications. We have synthesized the n-HA/CS and n-HA/CS/EF nanocomposites to compare their various thermal and physical properties and biocompatibility with a view to exploring their applicability as a novel scaffold in vitro using human osteoblasts (MG-63 cells).
2. Experimental
2.1 Materials and methods
3-(4,5-Dimethylthiazol-2-yl)-(2,5-diphenyl-tetrazoliumbromide) (MTT), chitosan (CS) with a degree of deacetylation >85%, phosphate buffered saline (PBS) (pH = 7.4) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Sigma-Aldrich (USA) and Invitrogen, USA, respectively. The kernels of Euryale ferox were commercially procured from the local market of Aligarh (India). Commercially available calcium nitrate tetrahydrate [Ca(NO3)2·4H2O] (99%), diammonium hydrogen phosphate (NH4)2HPO4 (DAHP) (99%), acetic acid CH3COOH (99.8%), sodium hydroxide NaOH (>97%), dimethyl sulphoxide (DMSO), sodium chloride (NaCl), sodium bicarbonate (NaHCO3), potassium chloride (KCl), dipotassium hydrogen phosphate trihydrate (K2HPO4·3H2O), magnesium chloride hexahydrate (MgCl2·6H2O), calcium chloride (CaCl2), sodium sulphate (Na2SO4), tri-(hydroxylmethyl)amino methane [TRIS], hydrochloric acid (HCl) and ammonia solution (25%) were obtained from Merck, Mumbai, India. The MG-63 cells were purchased from NCCS, Pune, India.
2.2 Synthesis of the n-HA/CS/EF nanocomposite
The synthesis of the n-HA/CS/EF nanocomposite was performed via a co-precipitation method. Firstly, individual solutions of CS, EF and n-HA were prepared. The solution of 2 g chitosan dissolved in 2 wt% aqueous acetic acid was added drop-wise to the n-HA solution prepared by dissolving n-HA (synthetic procedure of n-HA is discussed in the ESI†) in 2% aqueous orthophosphoric acid on sonication for 2 h. The mixture was thoroughly homogenized by continuous stirring for about 3 h at room temperature. The solution of EF obtained after sonication for 1 h was slowly added to the above mixture at constant stirring. Finally, the pH of the mixture was adjusted to ∼11 by adding 0.5 M NaOH solution which facilitates the nucleation of n-HA. The mixture was kept constantly stirring for 24 h. The resultant creamish white precipitate was kept for aging for another 24 h. The settled product was filtered repeatedly to attain a neutral pH. The synthesized product thus obtained was oven dried at 85 °C. The n-HA/CS nanocomposite was also synthesized accordingly to compare its various properties with that of the n-HA/CS/EF nanocomposite.
2.3 Morphology
The morphology, microstructure and dimensions of the composite scaffolds were observed by recording scanning electron microscopy (SEM) images at different magnifications using a JEOL-JAPAN Scanning Electron Microscope. The scaffolds were sputter coated with gold before observing them under SEM. The size of the particles was determined using transmission electron microscopy (TEM) images recorded on a Hitachi H-7500, Japan with an accelerating voltage of 120 kV.
2.4 Water uptake assay
The water uptake test was performed by comparing the weights of the dry scaffold (Wdry) and the scaffolds immersed in deionized (DI) water (Wwet) for 24 h and the absorbed water was calculated using eqn (1).| | | W(absorbed) = (Wwet − Wdry)/Wdry × 100 | (1) |
2.5 Physicochemical properties
The structural and functional properties of the n-HA/CS/EF and n-HA/CS nanocomposite samples were examined by Fourier transform infra-red spectroscopy (FTIR, Perkin Elmer – 2400 spectrometer), as KBr pellets in the frequency range of 4000–400 cm−1. The crystalline nature and size were recorded using an X-ray Diffractometer, Philips PW1710 with Cu Kα radiation at 1.540 Å in the range of 10°–80° at 40 kV. The compressive strength and compressive modulus of the synthesized nanocomposites were measured using a universal testing machine (Shimadzu with Instron Microtensile Tester, 5848). The compression test was performed at a crosshead speed of 10 mm min−1.
2.6 Biomineralization study
The in vitro bioactivity of the n-HA/CS/EF and n-HA/CS nanocomposite scaffolds was determined on pellets (6 mm in diameter and 2 mm in thickness) made from dried samples of the nanocomposites. The pellets were immersed in a beaker containing 20 ml of SBF solution prepared as described in the literature34 oscillating at 37 °C in a water bath which results in the soaking of the SBF solution. The pellets after soaking in SBF for different time intervals, i.e., 1, 2 and 4 weeks, were withdrawn, rinsed with DI water, dried and their SEM images were recorded.
2.7 Biodegradation
In vitro degradation study is another important property of nanocomposites to be investigated in order to examine the biopotential of the scaffold. The biodegradability of the scaffolds was estimated by placing them for 10 days in PBS containing lysozyme equivalent to the circulating levels of blood (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U L−1).35 Firstly, the dry samples of weight Wi (i = initial) were incubated at 37 °C and the changes in weight of the scaffold for 3, 7 and 10 days were recorded as Wf (f = final). The degradation percentage of the nanocomposite scaffolds was calculated by the following formula:
000 U L−1).35 Firstly, the dry samples of weight Wi (i = initial) were incubated at 37 °C and the changes in weight of the scaffold for 3, 7 and 10 days were recorded as Wf (f = final). The degradation percentage of the nanocomposite scaffolds was calculated by the following formula:| | | Biodegradation (%) = (Wi − Wf/Wi) × 100 | (2) |
2.8 Cell viability assay
For evaluating the viability of cells in the presence of the n-HA/CS/EF and n-HA/CS nanocomposites, the MTT assay was performed on human osteoblasts like MG-63 cells. Briefly, the MG-63 cells were seeded on the n-HA/CS/EF and n-HA/CS nanocomposites in 96-well culture plates with an initial density of 0.5 × 104 cells per well.36 The concentration of the nanocomposites was varied from 0 to 256 μg ml−1. The culture plates were incubated for 24 h at 37 °C in 5% CO2. The samples were then removed from the wells and washed with PBS. The viable cells which were adherent to the well walls were incubated with 0.5% MTT solution. After incubation for 4 h, the obtained formazan crystals were solubilized by adding 0.1% DMSO. The absorbance of each well was quantified at 570 nm with a UV spectrophotometer and cell viability was calculated.
2.9 Protein adsorption
The adsorption behavior of proteins on the n-HA/CS and n-HA/CS/EF nanocomposites was evaluated in order to elucidate cell attachment on the scaffolds which was studied as described previously.29 Briefly, the nanocomposites were washed with PBS, dried and incubated with 500 μl of fetal bovine serum (FBS-GIBCO) for 2 h at 37 °C. They were then rinsed with PBS to remove non-adherent proteins. Hereafter, the adsorbed proteins from the surface of the nanocomposites were removed by adding 1 ml aqueous solution of sodium dodecyl sulfate (SDS, 1 wt%). The protein concentration in the SDS solution analyzed by protein analysis kit (Micro BCA Protein Assay Kit, Pierce Biotechnology, IL, USA) quantified the protein adsorbed on the surface.
2.10 Antibacterial activity
The minimum inhibitory concentration (MIC) is the lowest concentration of an antimicrobial compound that will inhibit the visible growth of microorganisms after a specified time period. MICs are important in order to study the resistance of microorganisms against antimicrobial agents and also in the screening of activity of new antimicrobial agents.
For MIC estimation, the microdilution method was carried out on 4 common pathogenic strains namely S. aureus (ATCC 25923), E. coli (ATCC 25922), L. monocytogens (MTCC 657) and V. cholera (MTCC 3906). The values were determined on 96-well micro-dilution plates according to the protocols developed previously.37
The antibacterial potential of the n-HA/CS/EF nanocomposite was also evaluated using the agar well diffusion method (ESI†).
2.11 Biomineralization: ARS analysis
To quantify the mineralization process, alizarin red staining (ARS) was performed on the n-HA/CS and n-HA/CS/EF nanocomposites according to the procedure described by H. R. Pant et al., where a 40 mM solution of ARS was prepared by dissolving in deionized water and adjusting its pH to slightly acidic (∼4).38 It was stored in the dark after filtration to remove the undissolved particles. After the incubation for 10 days, the scaffolds were rinsed and fixed with 3.7% buffered formaldehyde for 30 min. The n-HA/CS and n-HA/CS/EF nanocomposites were stained with the previously prepared ARS solution for 30 min and then rinsed repeatedly until all the unbound dye was washed off. The scaffolds were treated with 50% acetic acid to extract the bound ARS which was pipetted out, diluted and the absorbance was measured at 550 nm.
2.12 Alkaline phosphatase activity
The osteogenic differentiation was determined by alkaline phosphatase activity (ALP) assay performed on the osteoblast cells from the femur bone shaft of Wistar rat. The cells were cultured in Dulbecco's modified Eagle's medium containing 15% FBS and 100 g ml−1 penicillin–streptomycin. The cultured cells were seeded on the n-HA/CS/EF nanocomposite in polystyrene 6-well dishes. The cultured medium was removed after 4 days and the wells were washed with PBS to eliminate the non-adherent cells. The adherent cells were cultured in 5% CO2/95% air atmosphere at 37 °C with the medium replenished once every 3 days. The cells were dissociated with trypsin/EDTA 0.05% and sub-cultured at a density of 5 × 105 cells per dish. ALP activity was assessed after 1, 3, 7 and 10 days of cell culture. 50 μl of Triton lysate was added to 0.012 M p-nitrophenyl phosphate in 0.05 M diethanolamine at pH 9.8 and incubated for 30 min at 37 °C followed by the addition of 50 μl stop solution (2.5 M NaOH).39p-Nitrophenol was used as a standard and the absorbance was measured at 405 nm. Alkaline phosphatase activity was expressed as total protein content (U g−1 protein).
2.13 RBC lysis
The experimental procedure is discussed in the ESI.†
2.14 Statistical analysis
Statistical analysis was performed using the Student's t-test with a significance level of p < 0.05. All the quantitative results were expressed as mean ± standard deviation with n = 3.
3. Results and discussion
3.1 FTIR
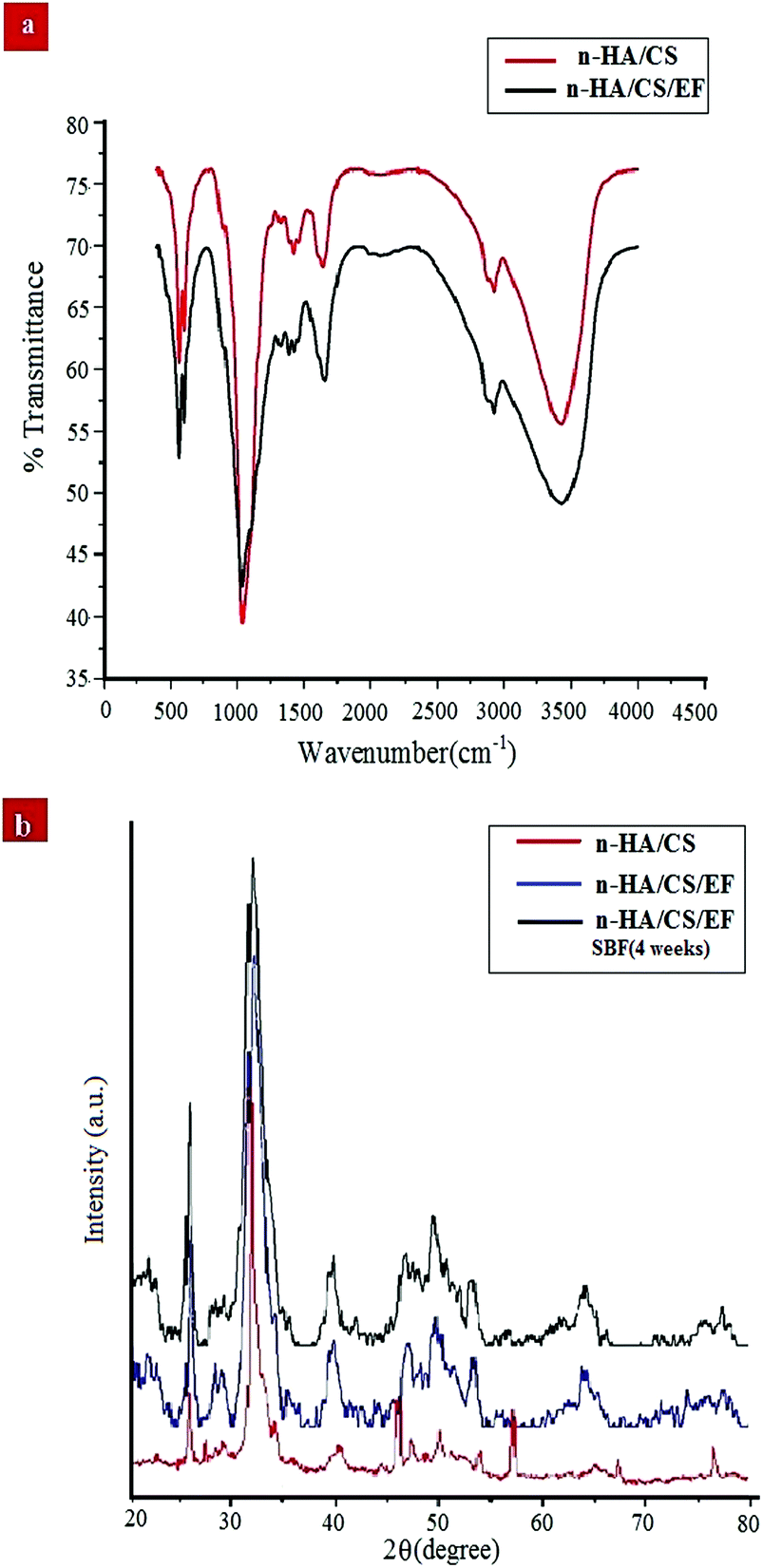
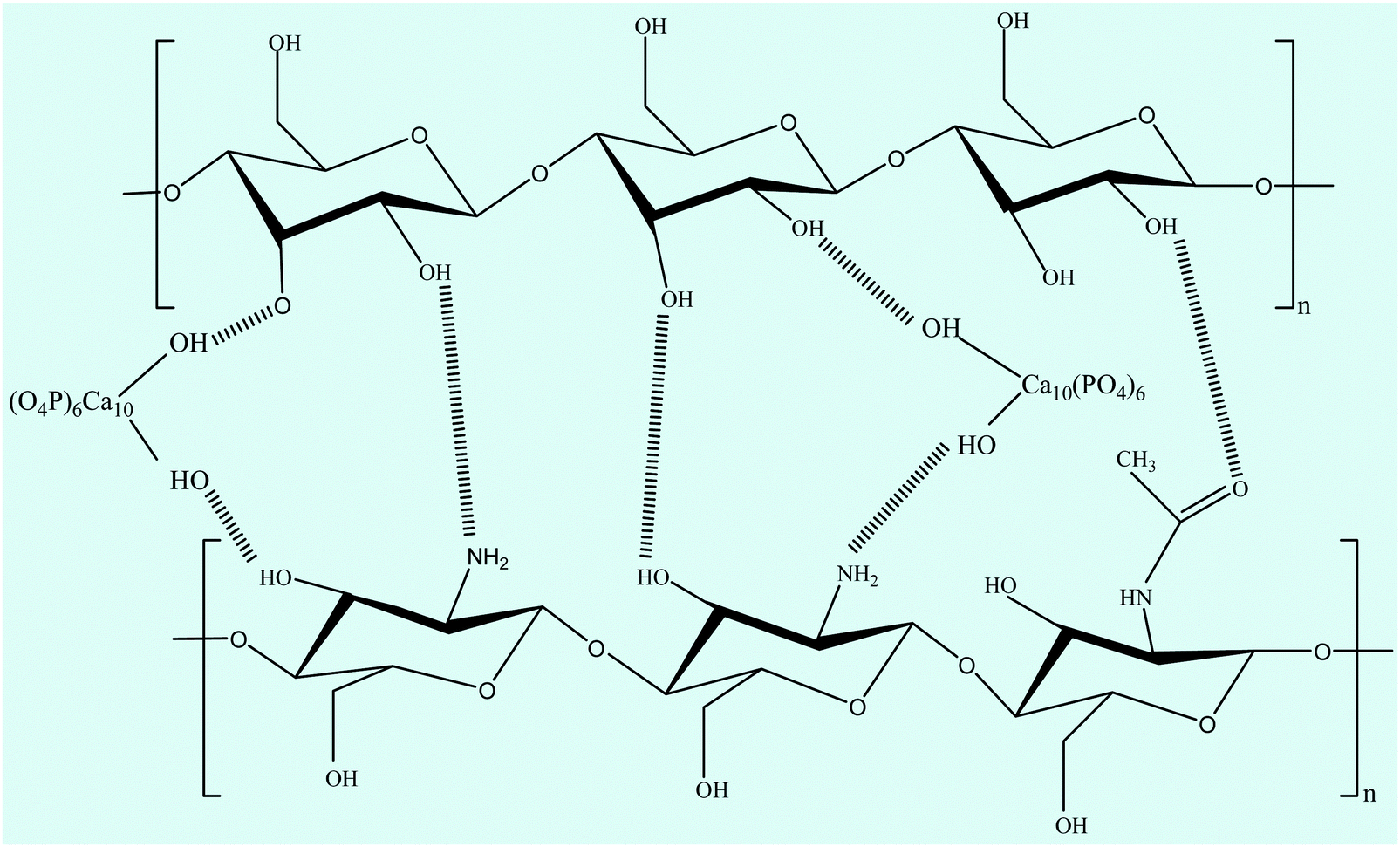
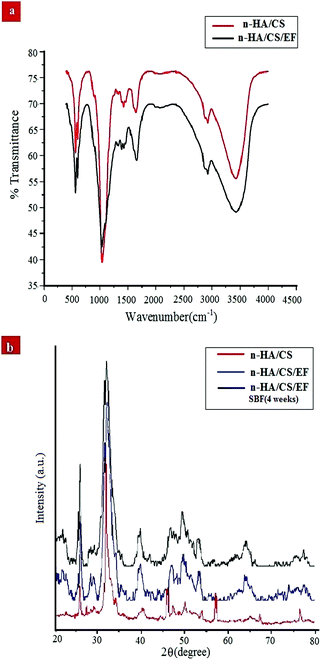
FTIR spectroscopy is one of the most conventionally used techniques to analyze the interaction of functional groups of different components and hence to interpret their structure. The FTIR spectra of the n-HA/CS and n-HA/CS/EF nanocomposites shown in Fig. 1(a) comprise characteristic peaks of different functional groups present. In general, for chitosan, the NH stretching vibration and OH stretching vibration overlap to give an absorption band at 3459 cm−1 while the characteristic band of hydroxyapatite appears at 3570 cm−1 corresponding to the OH stretching vibration.21 In the present study, the FTIR spectrum of the n-HA/CS nanocomposite displays the interaction between CS and n-HA with the appearance of the peak at 3433 cm−1. It shows that the shifted peak of CS at 3459 cm−1 may reasonably be due to the presence of n-HA [Fig. 1(a)]. The characteristic peaks of hydroxyapatite at 563 cm−1 and 603 cm−1 attributed to phosphate bending vibration confirmed the presence of n-HA in the chitosan matrix.40 The peak at 1035 cm−1 depicted the overlapping of the C–O–C stretching vibration of CS and the P–O stretching vibration of the phosphate group in n-HA while the bands at 1640 cm−1 and 1520 cm−1 were assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (amide I) and NH bending (amide II) of chitosan, respectively.41 The FTIR spectrum of n-HA/CS/EF exhibited all the characteristic peaks of n-HA and CS along with a slight modification due to the introduction of E. ferox. The broad peak at 3435 cm−1 may represent the interaction between the hydroxyl group of E. ferox and chitosan. The shifting of the peak from 1640 cm−1 in n-HA/CS to 1658 cm−1 in n-HA/CS/EF could be understood in terms of the possibility of H-bonding between the OH of E. ferox and the amino group of chitosan. A slight broadening in the peak at 1030 cm−1 may be due to the overlapping of the stretching mode of vibration of the phosphate group in n-HA and the stretching mode in amorphous starch of EF42,43 (Scheme 1). Thus, the FTIR spectra revealed the possible interactions among the various components of the synthesized n-HA/CS/EF nanocomposite.
O stretching (amide I) and NH bending (amide II) of chitosan, respectively.41 The FTIR spectrum of n-HA/CS/EF exhibited all the characteristic peaks of n-HA and CS along with a slight modification due to the introduction of E. ferox. The broad peak at 3435 cm−1 may represent the interaction between the hydroxyl group of E. ferox and chitosan. The shifting of the peak from 1640 cm−1 in n-HA/CS to 1658 cm−1 in n-HA/CS/EF could be understood in terms of the possibility of H-bonding between the OH of E. ferox and the amino group of chitosan. A slight broadening in the peak at 1030 cm−1 may be due to the overlapping of the stretching mode of vibration of the phosphate group in n-HA and the stretching mode in amorphous starch of EF42,43 (Scheme 1). Thus, the FTIR spectra revealed the possible interactions among the various components of the synthesized n-HA/CS/EF nanocomposite.
 |
| | Fig. 1 (a) FTIR spectra of the n-HA/CS and n-HA/CS/EF nanocomposites. (b) XRD patterns of the n-HA/CS, n-HA/CS/EF and n-HA/CS/EF-SBF (4 weeks) nanocomposites. | |
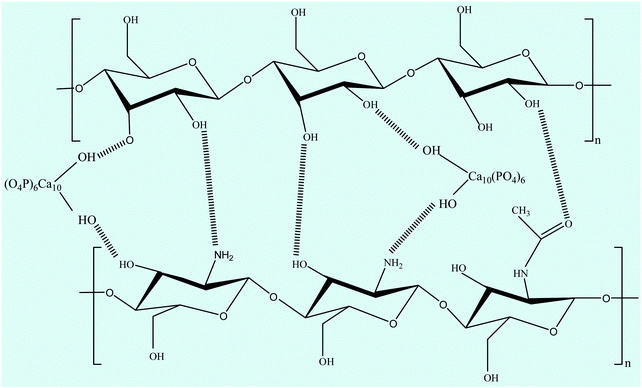 |
| | Scheme 1 Possible interaction between chitosan, nano-hydroxyapatite and the major constituent (starch) of E. ferox. | |
3.2 XRD
The XRD analysis of the n-HA/CS, n-HA/CS/EF and n-HA/CS/EF-SBF (4 weeks) nanocomposites in Fig. 1(b) exhibited characteristic crystalline peaks of n-HA at 2θ = 25.8, 29.4, 32.5, 39.8, etc. These peaks suggested the existence of nano hydroxyapatite which retained its crystallinity even in the nanocomposite. The appearance of a new peak at 2θ = 23 assured the presence of Euryale ferox in the n-HA/CS/EF nanocomposite.42 The average crystallite size of n-HA/CS, n-HA/CS/EF and n-HA/CS/EF-SBF (4 weeks) was calculated by the Scherer equation and was found to be 25.3 nm, 17.2 nm and 15.8 nm, respectively, comparable to that of the natural human bone.28 The reduction in size after SBF immersion may be due to the decreased crystallinity of n-HA resulting in slightly broader peaks.
3.3 Morphological study of the scaffolds
3.3.1 SEM.
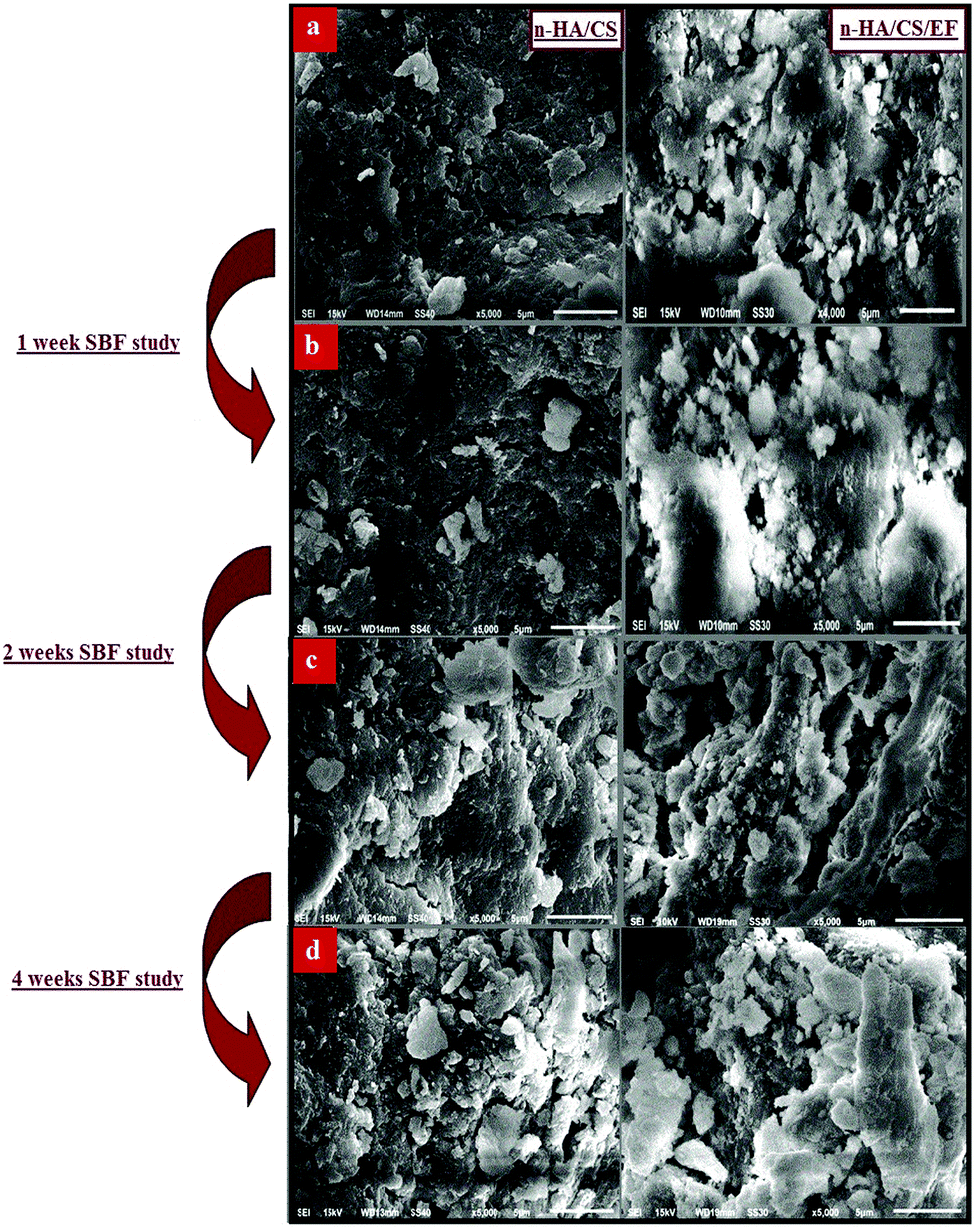
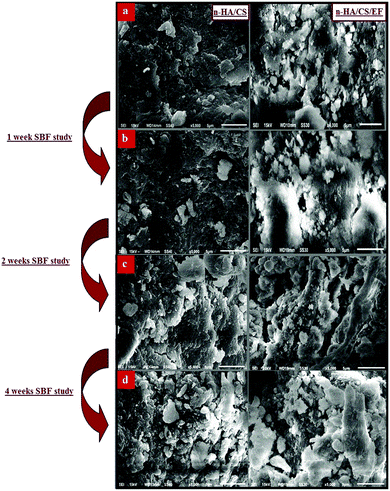
The investigation of the SEM images throws light on the morphological details of the nanocomposite. The SEM micrographs of the n-HA/CS/EF nanocomposite shown in Fig. 2(a) revealed a porous interconnected architecture having a high surface area to volume ratio which may possibly trigger off nutrient diffusion, neo vascularisation and migration into the cellular framework.44,45 The roughness of the n-HA/CS/EF nanocomposite as compared with the relatively smooth and compact surface of n-HA/CS promotes cell attachment and spreading, an established prerequisite for an ideal bone scaffold. The presence of Ca and P in the nanocomposite scaffolds was observed from the (Energy Dispersive Spectroscopy) EDX analysis (ESI†).
 |
| | Fig. 2 (a) SEM images of the n-HA/CS and n-HA/CS/EF nanocomposites and their respective SBF study after (b) 1 week, (c) 2 weeks and (d) 4 weeks. | |
3.3.2 TEM.
The TEM images of n-HA/CS/EF exhibited a markedly homogenous dispersal of particles contrary to the n-HA/CS nanocomposite which consist of severely agglomerated particles as shown in Fig. 3 implying that the addition of E. ferox in the n-HA/CS scaffold escalated the homogenous particle distribution and obstructed their aggregation. The average particle size of n-HA/CS was found to be (25 ± 1.5) nm as compared to an average particle size of (17 ± 1.3) nm for n-HA/CS/EF which seems to be in close proximity with that of human bone. The decrease in average particle size of the n-HA/CS/EF scaffold may be due to the presence of EF supporting its suitability to be used as a bone implant.
 |
| | Fig. 3 Transmission electron micrographs of the (a) n-HA/CS and (b) n-HA/CS/EF nanocomposites. | |
3.4
In vitro biomineralization study
Biomineralization of the n-HA/CS and n-HA/CS/EF scaffolds was studied by immersing them in SBF solution with a physiological ion concentration. The in vitro study is extremely essential to evaluate the bioactivity of the nanocomposite by the formation of hydroxyl carbonated apatite at the surface of the material.46 The biological behavior was examined by SEM micrographs after the SBF study for 1, 2 and 4 weeks. The comparative study of the SEM images revealed that a considerable biomimetic deposition was observed in the case of the ternary n-HA/CS/EF nanocomposite as compared to binary n-HA/CS [Fig. 2(b–d)]. Moreover, the SEM image of the n-HA/CS/EF scaffold kept for 30 days of incubation in the SBF solution showed a very thick coating of apatite layer indicating maximum mineralization contrary to the SEM image of n-HA/CS displaying scattered growth of the apatite layer under similar conditions, which was further supported by the EDX study [ESI†]. This may be explained in terms of increased bioactivity due to the inclusion of EF in the n-HA/CS scaffold which in turn results in the porous morphology of n-HA/CS/EF leading to apatite layer formation more rapidly. This apatite layer is necessary for the in vivo performance as the proliferation of osteoblast cells takes place on it, which differentiate to form extracellular matrix enabling the surrounding bone to come into direct contact with the surface apatite layer. Therefore, it can be concluded that the n-HA/CS/EF nanocomposite exhibited promising potential to be used as a synthetic bone graft.
3.5 Water absorption study
Water absorption test of a scaffold is an important study for the assessment of biomaterials for tissue engineering applications. It provides an estimation of the fluid uptake from the medium and transfer of nutrients and metabolites.47 There should be optimal water absorption as an excess amount of water uptake may lead to swelling and damage of the scaffold. The water uptake of n-HA/CS was found to be (31 ± 1.24)% as compared to the water uptake capacity of n-HA/CS/EF which had absorbed water up to (23 ± 1.56)% of its initial volume. The decrease in water uptake may be due to the presence of Ca and PO43− moieties of n-HA which interact with the NH2 or OH groups of chitosan and E. ferox, resulting in a decrease in the hydrophilicity of the scaffold. The above result revealed that the synthesized scaffold has optimal hydrophilicity suitable for its application as a bone implant.
3.6 Mechanical analysis
The mechanical strength of the nanocomposite is an essential criterion in selecting desirable bone scaffolds for bone tissue engineering applications. A comparative study of the compressive modulus of n-HA/CS and n-HA/CS/EF nanocomposites exhibited a compressive modulus of (52 ± 1.5) MPa and (72 ± 2.1) MPa, respectively. Moreover, the compressive strengths of n-HA/CS and n-HA/CS/EF were found to be (2 ± 0.32) MPa and (6 ± 0.14) MPa, respectively. The compressive strength of the n-HA/CS/EF nanocomposite was significantly higher than that of n-HA/CS but lies in the range of the compressive strength of human cancellous bone (2–12) MPa.
3.7
In vitro biodegradation study
A comparative study of the degradation rate of the nanocomposites was evaluated by immersing them in the PBS + lysozyme solution for 3, 7 and 10 days. A degradation of about (7 ± 0.4)% and (17 ± 0.70)% was observed for the n-HA/CS/EF nanocomposite on days 3 and 10 respectively as compared to degradation of (7 ± 0.2)% for the n-HA/CS nanocomposite after 10 days. The increase in the degradation rate of the n-HA/CS/EF nanocomposite may be explained in terms of breakdown of the glycosidic linkage of chitosan and E. ferox.48,49 Thus, the reported scaffold fulfilled the condition of maintaining the degradation rate and stability of the structure optimum for acceleration of cell growth on the scaffold.
3.8 Antibacterial activity
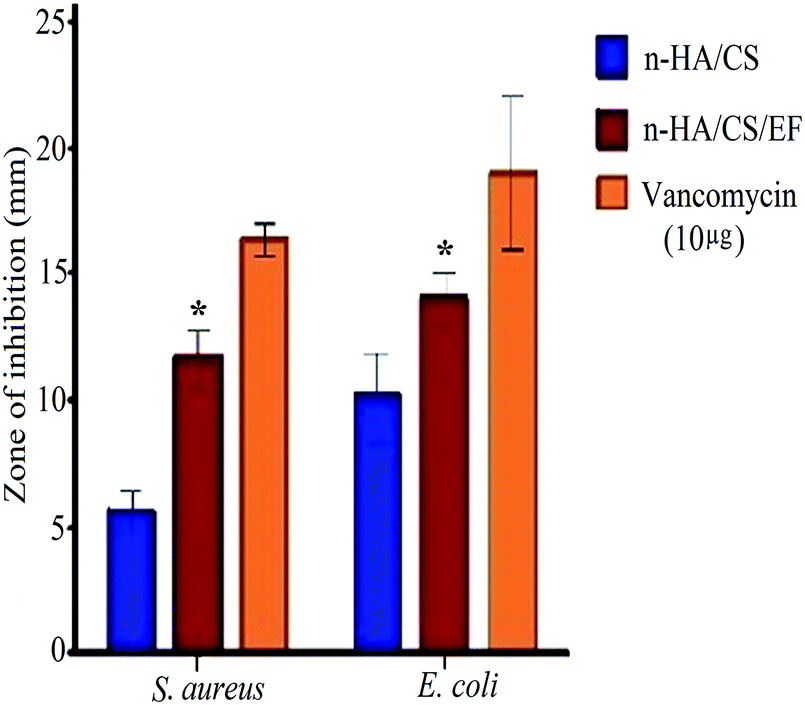
The synthesized nanocomposite was investigated for its antimicrobial activity against various Gram +ve and Gram −ve bacterial pathogens, including Staphylococcus aureus, Escherichia coli, Vibrio cholerae and Listeria monocytogenes by the broth dilution method [Table 1] and further evaluated on 2 strains by the agar diffusion method. Both the composites exhibited antibacterial activity but the MIC value for n-HA/CS/EF on the aforementioned bacteria was lower than that of n-HA/CS.29 The antibacterial activity of n-HA/CS was due to the characteristic antibacterial nature of chitosan. However, the significantly higher antibacterial activity of the n-HA/CS/EF nanocomposite may be due to the smaller particle size of n-HA/CS/EF enhancing its surface area. The results were further supported by the agar diffusion method with an increased zone of inhibition observed in the case of the n-HA/CS/EF nanocomposite [(14 ± 1) mm and (12 ± 1.15) mm against E. coli and S. aureus, respectively] compared to the values obtained in the case of the n-HA/CS nanocomposite40 [(9 ± 1.1) mm and (5 ± 1.19) mm against E. coli and S. aureus, respectively] as shown in Fig. 4.
Table 1 MIC values (μg ml−1) of the n-HA/CS and n-HA/CS/EF nanocomposites
| Nanocomposites |
Bacterial strains |
| Gram −ve |
Gram +ve |
|
|
V. cholera |
E. coli |
L. monocytogenes |
S. aureus |
| n-HA/CS |
256 |
1600 |
256 |
1000 |
| n-HA/CS/EF |
64 |
128 |
256 |
128 |
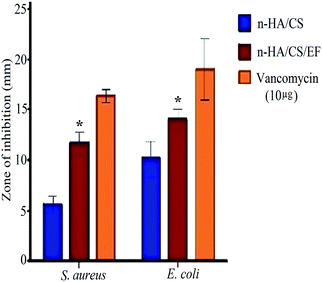 |
| | Fig. 4 Antibacterial activity of the n-HA/CS and n-HA/CS/EF nanocomposites. * statistical significance level by t-test (p < 0.05). | |
3.9 Cell viability assay
The biocompatibility of the scaffolds is indispensable for their use in the orthopedics as bone implants. Thus, a cytotoxicity study of the synthesized nanocomposites was performed with MG-63 cells based on the MTT assay. The MTT enters the cell where the dehydrogenase activity of mitochondria reduces it into a purple formazan compound. The assessment of cytotoxicity of the nanocomposite scaffolds was performed by incubating them for 24 h with varying concentrations (0–256 μg ml−1). It was observed from Fig. 5(a) that the n-HA/CS/EF nanocomposite exhibited considerably higher viability for the bone cells even at larger concentrations as compared to the n-HA/CS nanocomposite. Hence, the n-HA/CS/EF nanocomposite can be successfully incorporated as a bone scaffold owing to its higher biocompatibility which makes it a better host for the bone cells.
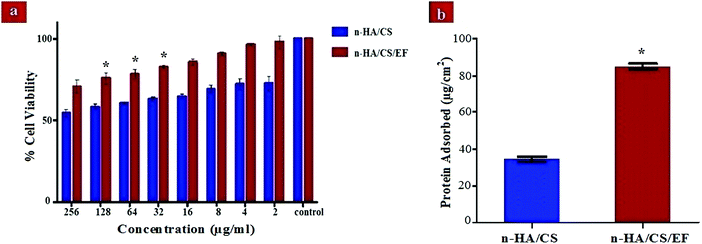 |
| | Fig. 5 (a) Cell viability of the n-HA/CS and n-HA/CS/EF nanocomposites on MG-63 cells. (b) Protein adsorbed on the n-HA/CS and n-HA/CS/EF nanocomposites. * statistical significance level by t-test (p < 0.05). | |
3.10 Protein adsorption
The analysis of the protein adsorption study on the surface of the n-HA/CS and n-HA/CS/EF nanocomposites is important to understand the cell behavior on these scaffolds and thus unveil their in vivo potential. The value of protein adsorption for n-HA/CS/EF (85 ± 1.34) μg cm−2 was remarkably greater than that of the n-HA/CS nanocomposite scaffold (34 ± 1.54) μg cm−2 [Fig. 5(b)]. The increased surface roughness and porosity of n-HA/CS/EF as revealed by SEM micrographs and the increased hydrophobicity enhanced its protein adsorption ability befitting its role as a bone scaffold.
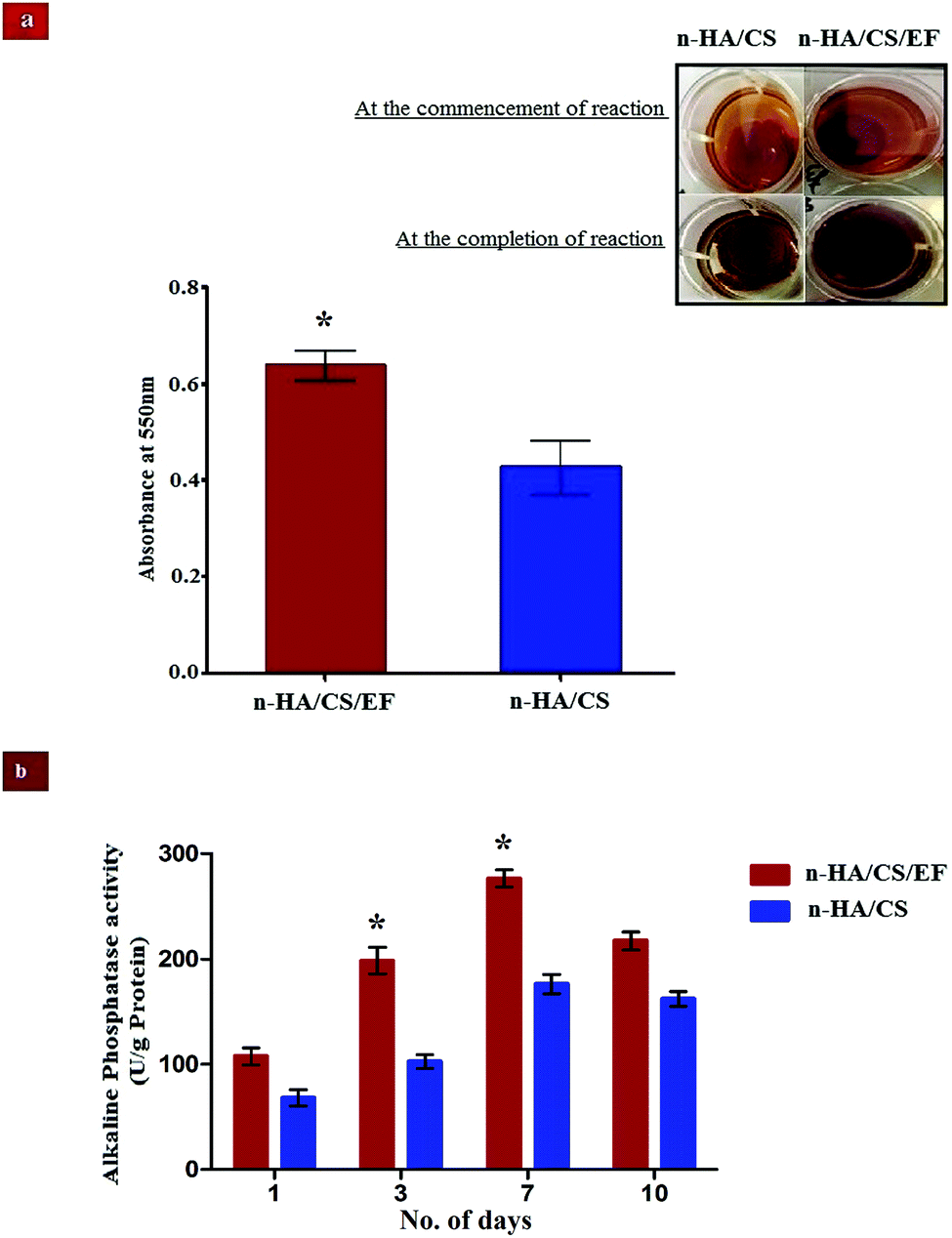
3.11 ARS study
The deposition of n-HA exhibits a crucial step in the production of a calcified extracellular matrix that can be quantified by staining with ARS which is an anthraquinone derivative forming a water insoluble salt with Ca.50 The higher degree of staining in n-HA/CS/EF as visualized from Fig. 6(a) exhibited remarkably increased mineralization relative to n-HA/CS which was also confirmed by SEM micrographs of n-HA/CS/EF showing greater apatite deposition on the surface during its in vitro SBF study. The increased nucleation sites of Ca2+ ions provided by the n-HA/CS/EF nanocomposite scaffold might be responsible for the enhanced deposition of calcified extracellular matrix analogous to bone.
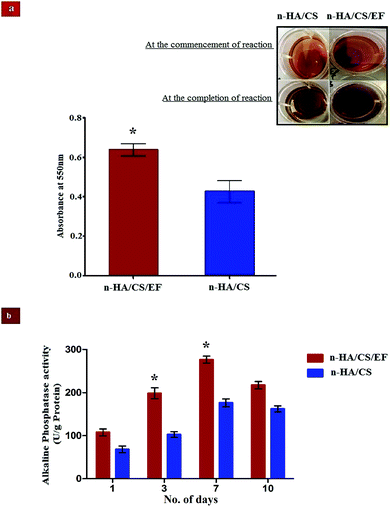 |
| | Fig. 6 (a) Mineralization of calcium compounds on the surface of the n-HA/CS and n-HA/CS/EF scaffolds. (b) ALP activity of osteoblast cells from the femur bone shaft of Wistar rats treated with n-HA/CS/EF and n-HA/CS nanocomposites for different time intervals. * statistical significance level by t-test (p < 0.05). | |
3.12 ALP activity
ALP analysis is widely used as a benchmark for osteoblastic cell differentiation. The ALP activity of the osteoblasts cell seeded on the n-HA/CS and n-HA/CS/EF nanocomposites was evaluated after 1, 3, 7 and 10 days. As shown in Fig. 6(b), a significantly higher ALP activity was observed for the n-HA/CS/EF nanocomposite as compared to the n-HA/CS nanocomposite. Furthermore, for n-HA/CS/EF the ALP level increased two-fold after 3 days as compared to the ALP level after 1 day. There was a further enhancement in the ALP level for 7 days and subsequently a reduction in the ALP value was observed for both the nanocomposites. The decrease in the ALP activity after 7 days exhibited the onset of the maturation phase of the n-HA/CS and n-HA/CS/EF nanocomposites.51 Thus, the n-HA/CS/EF nanocomposite may possibly act as an efficient substrate for cell proliferation and differentiation.
3.13 RBC lysis
A hemolytic assay was performed to determine the biosafety of the as-synthesized nanocomposite in the human body which might get exposed to the blood environment damaging the erythrocytes. The pre-incubation of our synthesized nanocomposite with healthy cells showed very little toxicity and caused limited lysis of cells. The percent lysis of n-HA/CS/EF was observed at various concentrations exhibiting hemolysis of less than 5% (ESI†). Thus, the synthesized nanocomposite was found to be hemocompatible with the human body.
4. Conclusion
The development of a smart and conducive scaffold that has an expeditious responsive mechanism along with phenomenal biodegradation and mechanical properties remains the most challenging strategy in the field of bone tissue engineering. To compliment this rapidly expanding area we have successfully synthesized an Euryale ferox (natural product) fabricated n-HA/CS nanocomposite, mimicking the nanoscale framework and chemical properties of natural bone. The SEM images revealed a porous, interconnected and rough surface in the n-HA/CS/EF nanocomposite compared to the n-HA/CS. Furthermore, the SBF studies of n-HA/CS/EF showed an increased nucleation of n-HA with greater deposition of an apatite layer compared to n-HA/CS suggesting better bioactivity. This was further supported by the ARS study which exhibited a high mineralization potential of the Ca2+ ions. The FTIR spectra of the n-HA/CS/EF nanocomposite revealed a significant intermolecular interaction between different components while the results of the XRD data indicated a decrease in size of n-HA/CS with the incorporation of E. ferox. The mechanically strong nanocomposite revealed distinguished cell growth and differentiation along with superior non-toxic nature corroborated from the results of the ALP and MTT assay. The superior bactericidal ability of the n-HA/CS/EF nanocomposite was attributed to its smaller particle size. Therefore, the conclusion drawn from the aforementioned studies deciphers the successful intervention of Euryale ferox into the biomimetic system, yielding promising constructs better than n-HA/CS scaffolds for bone tissue engineering.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the grant support from DRS-II, FIST and PURSE to the Department of Chemistry, A. M. U., Aligarh. The authors are thankful to the University Grants Commission for financial assistance.
References
- A. Teimouri, M. Azadi, R. Emadi, J. Lari and A. N. Chermahini, Polym. Degrad. Stab., 2015, 121, 18–29 CrossRef CAS.
- L. Wang and C. Li, Carbohydr. Polym., 2007, 68, 740–745 CrossRef CAS.
- S. K. L. Levengood and M. Zhang, J. Mater. Chem. B, 2014, 2, 3161 RSC.
- A. R. Amini, C. T. Laurencin and S. P. Nukavarapu, Crit. Rev. Biomed. Eng., 2012, 40, 363–408 CrossRef PubMed.
- P. Kerativitayanan, M. Tatullo, M. Khariton, P. Joshi, B. Perniconi and A. K. Gaharwar, ACS Biomater. Sci. Eng., 2017, 3, 590–600 CrossRef CAS.
- F. R. A. J. Rose and R. O. C. Oreffo, Biochem. Biophys. Res. Commun., 2002, 292, 1–7 CrossRef CAS PubMed.
- L. Chen, J. Hu, J. Ran, X. Shen and H. Tong, Int. J. Biol. Macromol., 2014, 65, 1–7 CrossRef CAS PubMed.
- S. Sai Nievethitha, N. Subhapradha, D. Saravanan, N. Selvamurugan, W. B. Tsai, N. Srinivasan, R. Murugesan and A. Moorthi, Int. J. Biol. Macromol., 2017, 98, 67–74 CrossRef CAS PubMed.
- A. Bhowmick, N. Pramanik, T. Mitra, A. Gnanamani, M. Das and P. P. Kundu, New J. Chem., 2017, 41, 7524–7530 RSC.
- B. Lowe, J. Venkatesan, S. Anil, M. S. Shim and S. K. Kim, Int. J. Biol. Macromol., 2016, 93, 1479–1487 CrossRef CAS PubMed.
- G. Chiara, F. Letizia, F. Lorenzo, S. Edoardo, S. Diego, S. Stefano, B. Eriberto and Z. Barbara, Int. J. Mol. Sci., 2012, 13, 737–757 CrossRef CAS PubMed.
- H.-T. Liao, K. T. Shalumon, K.-H. Chang, C. Sheu and J.-P. Chen, J. Mater. Chem. B, 2016, 4, 1827–1841 RSC.
- A. Bhowmick, N. Pramanik, T. Mitra, A. Gnanamani, M. Das and P. P. Kundu, New J. Chem., 2017, 41, 190–197 RSC.
- P. Jithendra, A. M. Rajam, T. Kalaivani, A. B. Mandal and C. Rose, ACS Appl. Mater. Interfaces, 2013, 5, 7291–7298 CAS.
- S.-S. Kim, M. Sun Park, O. Jeon, C. Yong Choi and B.-S. Kim, Biomaterials, 2006, 27, 1399–1409 CrossRef CAS PubMed.
- M. Swetha, K. Sahithi, A. Moorthi, N. Srinivasan, K. Ramasamy and N. Selvamurugan, Int. J. Biol. Macromol., 2010, 47, 1–4 CrossRef CAS PubMed.
- A. Ghaee, J. Nourmohammadi and P. Danesh, Carbohydr. Polym., 2017, 157, 695–703 CrossRef CAS PubMed.
- J. Venkatesan and S. K. Kim, Mar. Drugs, 2010, 8, 2252–2266 CrossRef CAS PubMed.
- S. Gholizadeh, F. Moztarzadeh, N. Haghighipour, L. Ghazizadeh, F. Baghbani, M. A. Shokrgozar and Z. Allahyari, Int. J. Biol. Macromol., 2017, 97, 365–372 CrossRef CAS PubMed.
- B. Torabinejad, J. Mohammadi-Rovshandeh, S. M. Davachi and A. Zamanian, Mater. Sci. Eng., C, 2014, 42, 199–210 CrossRef CAS PubMed.
- W. W. Thein-Han and R. D. K. Misra, Acta Biomater., 2009, 5, 1182–1197 CrossRef CAS PubMed.
- R. Arun
Kumar, A. Sivashanmugam, S. Deepthi, S. Iseki, K. P. Chennazhi, S. V. Nair and R. Jayakumar, ACS Appl. Mater. Interfaces, 2015, 7, 9399–9409 Search PubMed.
- P. T. Sudheesh Kumar, S. Srinivasan, V. K. Lakshmanan, H. Tamura, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2011, 85, 584–591 CrossRef CAS.
- M. Ribeiro, M. A. De Moraes, M. M. Beppu, M. P. Garcia, M. H. Fernandes, F. J. Monteiro and M. P. Ferraz, Eur. Polym. J., 2015, 67, 66–77 CrossRef CAS.
- C. Ao, Y. Niu, X. Zhang, X. He, W. Zhang and C. Lu, Int. J. Biol. Macromol., 2017, 97, 568–573 CrossRef CAS PubMed.
- C. Yang, S. Chen, J. Wang, T. Zhu, G. Xu, Z. Chen, X. Ma and W. Li, Appl. Surf. Sci., 2016, 362, 163–168 CrossRef CAS.
- N. Charernsriwilaiwat, P. Opanasopit, T. Rojanarata, T. Ngawhirunpat and P. Supaphol, Carbohydr. Polym., 2010, 81, 675–680 CrossRef CAS.
- M. Shakir, R. Jolly, M. S. Khan, N. E. Iram and H. M. Khan, Int. J. Biol. Macromol., 2015, 80, 282–292 CrossRef CAS PubMed.
- M. Shakir, R. Jolly, M. S. Khan, A. Rauf and S. Kazmi, Int. J. Biol. Macromol., 2016, 93, 276–289 CrossRef CAS PubMed.
- M. Shakir, R. Jolly, M. S. Khan, N. E. Iram, T. K. Sharma and S. I. Al-Resayes, Polym. Adv. Technol., 2015, 26, 41–48 CrossRef CAS.
- A. Harvey, Drug Discovery Today, 2000, 5, 294–300 CrossRef PubMed.
- S. E. Lee, E. M. Ju and J. H. Kim, Exp. Mol. Med., 2002, 34, 100–106 CrossRef CAS PubMed.
- C. W. Song, S. M. Wang, L. L. Zhou, F. F. Hou, K. J. Wang, Q. Bin Han, N. Li and Y. X. Cheng, J. Agric. Food Chem., 2011, 59, 1199–1204 CrossRef CAS PubMed.
- B. a Allo, A. S. Rizkalla and K. Mequanint, ACS Appl. Mater. Interfaces, 2012, 4, 3148–3156 CAS.
- S. Soumya, K. M. Sajesh, R. Jayakumar, S. V. Nair and K. P. Chennazhi, Carbohydr. Polym., 2012, 87, 1787–1795 CrossRef CAS.
- D. Gopi, S. Nithiya, E. Shinyjoy, D. Rajeswari and L. Kavitha, Ind. Eng. Chem. Res., 2014, 53, 7660–7669 CrossRef CAS.
- G. Sandri, M. C. Bonferoni, F. Ferrari, S. Rossi, C. Aguzzi, M. Mori, P. Grisoli, P. Cerezo, M. Tenci, C. Viseras and C. Caramella, Carbohydr. Polym., 2014, 102, 970–977 CrossRef CAS PubMed.
- H. R. Pant, P. Risal, C. H. Park, L. D. Tijing, Y. J. Jeong and C. S. Kim, Colloids Surf., B, 2013, 102, 152–157 CrossRef CAS PubMed.
- A. K. Jaiswal, H. Chhabra, S. S. Kadam, K. Londhe, V. P. Soni and J. R. Bellare, Mater. Sci. Eng., C, 2013, 33, 2926–2936 CrossRef CAS PubMed.
- S. Saravanan, S. Nethala, S. Pattnaik, A. Tripathi, A. Moorthi and N. Selvamurugan, Int. J. Biol. Macromol., 2011, 49, 188–193 CrossRef CAS PubMed.
- T. Bourtoom and M. S. Chinnan, LWT – Food Sci. Technol., 2008, 41, 1633–1641 CrossRef CAS.
- L. Zhao, J. Huang, J. Man, H. Huai, Y. Chen and C. Wei, Int. J. Food Prop., 2016, 19, 289–299 CrossRef CAS.
- M. Ji, H. Li, H. Guo, A. Xie, S. Wang, F. Huang, S. Li, Y. Shen and J. He, Carbohydr. Polym., 2016, 153, 124–132 CrossRef CAS PubMed.
- J. A. Sowjanya, J. Singh, T. Mohita, S. Sarvanan, A. Moorthi, N. Srinivasan and N. Selvamurugan, Colloids Surf., B, 2013, 109, 294–300 CrossRef CAS PubMed.
- H. H. Jin, D. H. Kim, T. W. Kim, K. K. Shin, J. S. Jung, H. C. Park and S. Y. Yoon, Int. J. Biol. Macromol., 2012, 51, 1079–1085 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- B. Nasri-Nasrabadi, M. Mehrasa, M. Rafienia, S. Bonakdar, T. Behzad and S. Gavanji, Carbohydr. Polym., 2014, 108, 232–238 CrossRef CAS PubMed.
- M. Shankar, N. Chaudhary and D. Singh, Int. J. Pharm. Biol. Arch., 2010, 1, 101–107 Search PubMed.
- T. Kean and M. Thanou, Adv. Drug Delivery Rev., 2010, 62, 3–11 CrossRef CAS PubMed.
- M. Wiens, T. A. Elkhooly, H. C. Schröder, T. H. A. Mohamed and W. E. G. Müller, Acta Biomater., 2014, 10, 4456–4464 CrossRef CAS PubMed.
- J. H. Kim, D. K. Kim, O. J. Lee, H. W. Ju, J. M. Lee, B. M. Moon, H. J. Park, D. W. Kim, J. H. Lee and C. H. Park, Int. J. Biol. Macromol., 2016, 82, 160–167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj02953e |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Sumbul
Mirza
a,
Reshma
Jolly
a,
Ahmar
Rauf
b and
Mohammad
Owais
b
*a,
Sumbul
Mirza
a,
Reshma
Jolly
a,
Ahmar
Rauf
b and
Mohammad
Owais
b