
NMR spectroscopy: a potent tool for studying monolayer-protected metal nanoclusters
G.
Salassa
 * and
T.
Bürgi
*
* and
T.
Bürgi
*
Department of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211 Geneva 4, Switzerland. E-mail: Giovanni.salasa@unige.ch; thomas.buergi@unige.ch
First published on 25th April 2018
Abstract
Monolayer protected metal clusters are currently in the focus of interest both for fundamental reasons and for their use in possible applications. In the past two decades the interest was mainly focused on the evolution of the structrue and properties as the clusters grow in size. The field profited tremednously from mass spectrometry and X-ray structure analysis. For future applications of monolayer protected clusters other properties like the interaction of the clusters with molecules will become important. Also, it has been realized more recently that these monolayer protected clusters are rather dynamic, which calls for techniques able to address this property. By discussing selected examples we demonstrate the power of nuclear magnetic resonance (NMR) spectroscopy to study the structure and the dynamics of clusters and their interacion with molecules (sensing). NMR spectroscopy is an abundant technique and has become very sophisticated. Future work in the field of monolayer protected clusters may greatly profit from this. We believe that NMR spectroscopy, although not yet used much in the field of monolayer protected clusters, has the potential to become a key technique complementary to mass spectrometry and X-ray structure determination.
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is today arguably the most powerful tool in chemical sciences for the identification and structure determination of molecules in solution. In contrast, for the characterization of nanoparticles NMR is much less used, due to several reasons. Metal NMR-active nuclei are rare and, in many cases, their low abundance causes sensitivity problems. In the case of the more employed nuclei (1H, 13C, 19F etc.), the NMR signals, originated from the organic molecule attached to the nanoparticle surface, are directly influenced by the size of the nanoparticle. The bigger the nanoparticles the broader the peaks become up to complete disappearance. Moreover, the information content in the peaks (multiplicity) becomes less pronounced (or lost) due to increased rigidity of the ligand on the surface. Despite these disadvantages, NMR can facilitate the investigation of nanoparticles as demonstrated by the increasing number of examples reported.1Monolayer-protected metal clusters, or metal nanoclusters2,3 (NCs) of a few tens of metal atoms have properties that are at the same time molecular and nanoparticle-like. In contrast to nanoparticles, clusters have a discrete molecule-like electronic structure. The surface chemistry (metal–ligand interface) is similar for clusters and nanoparticles. For monolayer-protected metal clusters NMR spectroscopy has been used in the past, which is the topic of this short review; however, compared to the numerous papers dealing with monolayer-protected clusters, NMR has not been extensively applied up to now. The two most important techniques in the field are mass spectrometry and X-ray crystallography. The latter technique has contributed tremendously to the field by revealing the structure of numerous monolayer-protected clusters in the crystal. However, X-ray crystallography has its limitations and it is here that NMR spectroscopy can serve as the most powerful complementary technique. NMR spectroscopy can provide structural information of the dissolved cluster, the latter state being important for many applications. The protecting ligands surrounding the cluster core bear a structure, which is “frozen” in the crystal. In solution the ligand can adopt conformations different from the ones in the solid state. In addition, the ligands are dynamic in solution, which is not captured by a crystal structure. NMR can furthermore be used to study reactions of and between clusters. Finally, many applications of monolayer-protected clusters rely on the (dynamic) nature of the ligand shell and its interaction with other molecules, an aspect where NMR spectroscopy offers powerful opportunities. All this comes at a price. The information contained in NMR spectra is not as direct as the information gained from a crystal structure.
By discussing selected NMR works we would like to convince the reader of the many potent opportunities that NMR spectroscopy offers to the cluster field. NMR is an abundant technique and many highly sophisticated experiments (pulse sequences) have been developed in the past, which can also serve the researchers in the field of monolayer-protected clusters.
NMR investigation of structural and dynamic properties in NCs
For more than twenty years NMR techniques have been applied for structure determination of proteins and biological macromolecules.4 Multidimensional NMR spectroscopy enables examination of the three-dimensional structures and dynamical interactions of proteins with accuracy comparable to single-crystal X-ray crystallography.5 The same approach could be applied for the structural investigation of metal nanoclusters in the solution phase, obtaining important information on the structure and dynamics of the organic monolayer protecting the cluster. An excellent example in this respect was reported by Häkkinen and co-workers where a full comprehension of the organic monolayer protecting Au102(pMBA)44 (pMBA = para mercapto benzoic acid) was achieved.6 The structure of this cluster was resolved in 2007 using single-crystal X-ray diffraction.7 Au102(pMBA)44 is characterized by a 79 gold atom core protected by 19 monomeric (SR)–Au–(SR) (SR: thiolate) and two dimeric staple units (SR)–Au–(SR)–Au–(SR), and the C2 overall symmetry observed creates 22 distinct ligand environments (Fig. 1a).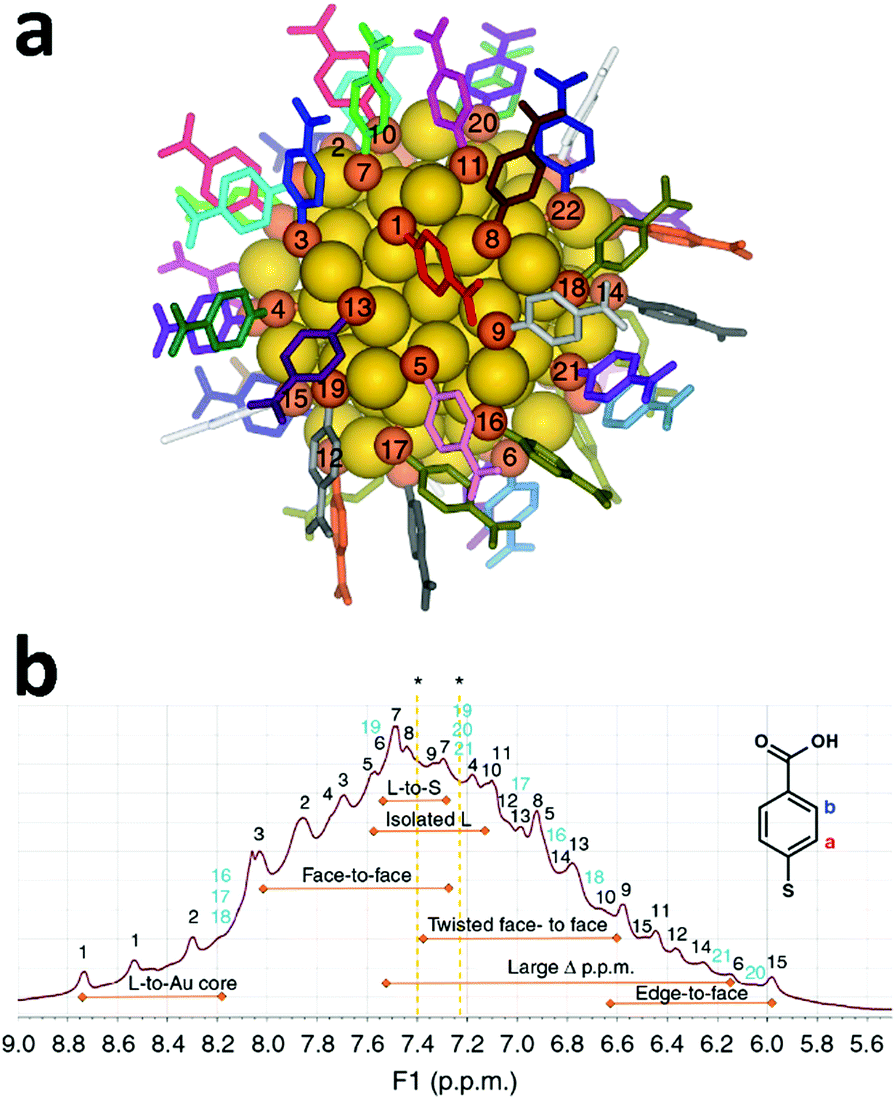 | ||
| Fig. 1 (a) Total structure of Au102(pMBA)44, with numbering and unique colouring of all 22 symmetry-unique ligands. Gold is yellow and sulphur is orange. (b) 1H NMR spectrum of the Au102(pMBA)44 cluster in 0.3 M D2O–NaOH with full assignment of the 22 symmetry-equivalent pMBA ligands based on the TOCSY spectrum. Strong correlation peaks have been marked with black and weaker signals in light blue (see the TOCSY spectrum in Fig. 2). Special ligand environments affecting the chemical shift shielding have been identified in the spectrum (orange double arrows). Asterisk (*) denotes chemical shifts of free pMBA at 7.40 and 7.23 ppm in 0.3 M D2O–NaOH. Reproduced with permission from ref. 6, Copyright 2014 Nature Publication Group. | ||
Each of the 22 ligands possesses a reduced mobility caused by their bond with the gold surface and the proximity with the neighbouring ligands. This effect, in concomitance with the electronic interaction with gold atoms and the other ligands, creates different local magnetic environments. 1H and 13C belonging to one of the 22 symmetry-equivalent ligands will experience a unique and distinguishable shielding compared to the other ligands. Therefore, if we examine the 1H-NMR spectra of Au102(pMBA)44, every ligand contributes with two equivalent pairs of aromatic protons giving rise to a broad envelope with 44 partially resolved peaks (Fig. 1b).
The full assignment of the 1H-NMR spectra to the 22 symmetry ligand environments was achieved employing multidimensional NMR spectroscopy combined with theoretical NMR calculations and supported by molecular dynamics simulations in water. Total correlated spectroscopy (TOCSY, Fig. 2a) allowed the determination of through-bond correlations and, therefore, enabled the separation of chemical shifts belonging to each pMBA ligand. Following heteronuclear single-quantum correlation spectroscopy HSQC was employed to correlate to which carbon the particular proton is attached. The combination of TOCSY and HSQC allowed the grouping and numbering of all 22 pMBA ligands. An initial assignment of the proton chemical shifts to their respective 22 pMBA ligands was achieved by comparison of the experimental TOCSY and HSQC correlation peaks to DFT-calculated proton and carbon chemical shifts for each of the symmetry-equivalent pMBA ligands based on the crystal structure of Au102(pMBA)44. Since the chemical shift is highly affected by the type of interactions (i.e. ligand-to-gold, ligand-to-sulphur, isolated ligand, face-to-face and edge-to-face aromatic interactions) that each ligand experiences, the DFT calculations were able to estimate (in some cases with high precision) the chemical shift shielding of each pMBA ligand based on its interaction with the other ligands and the cluster core.
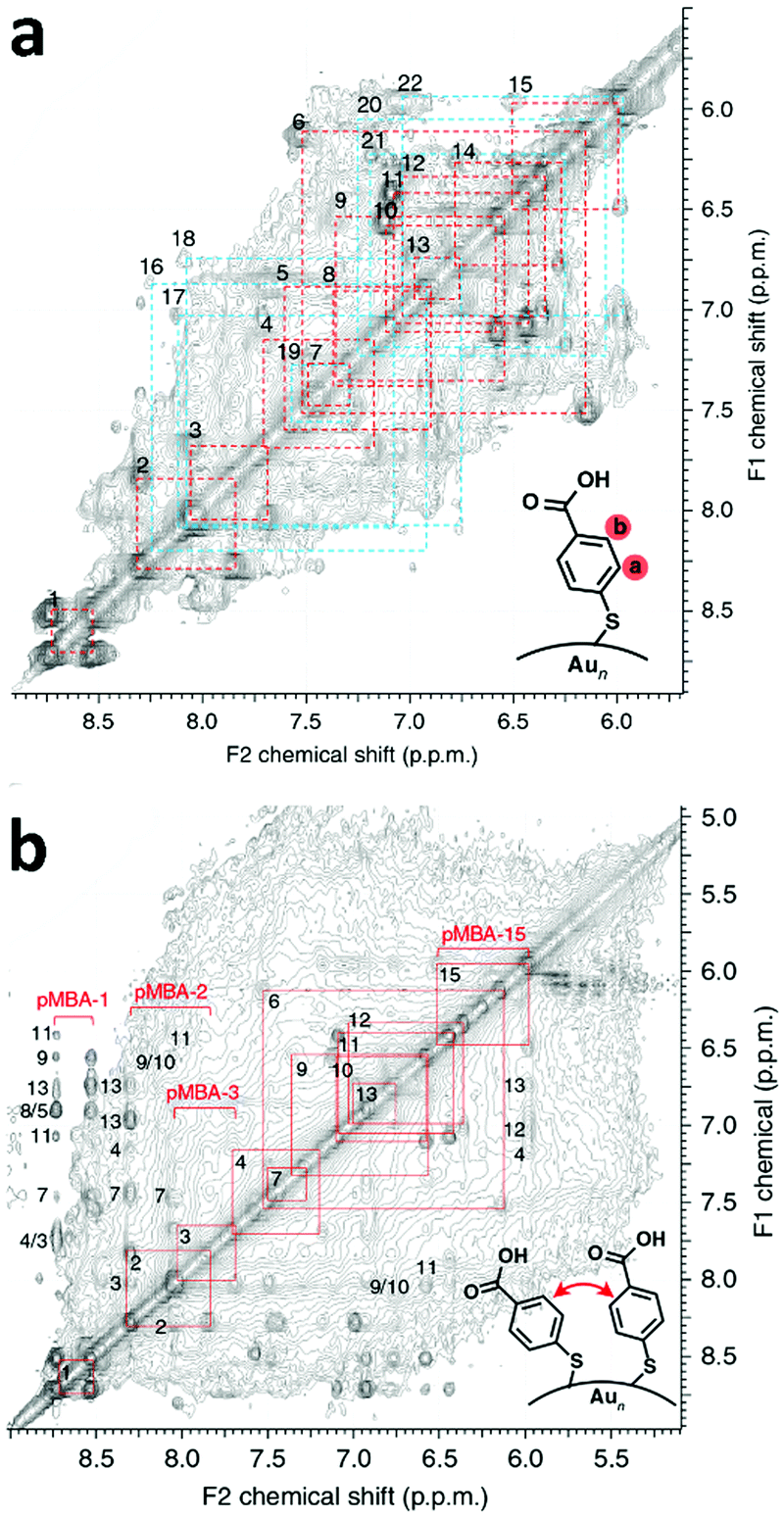 | ||
| Fig. 2 (a) TOCSY spectrum of Au102(pMBA)44 cluster in 0.3 M D2O–NaOH with full assignment of the 22 symmetry-equivalent pMBA ligands. Strong correlation peaks have been marked with red and weaker correlation peaks are marked with blue. (b) NOESY spectra of the Au102(pMBA)44 cluster in 0.3 M D2O–NaOH showing the assignment (red and blue squares) of the through-space correlation peaks between neighbouring pMBA ligands. Reproduced with permission from ref. 6, Copyright 2014 Nature Publication Group. | ||
However, this strategy allowed the assignment of only some of the 22 ligands. Nuclear Overhauser effect spectroscopy (NOESY) and rotating frame nuclear Overhauser effect spectroscopy (ROESY) were then employed by the authors to obtain through-space correlations, which disclosed information about the connectivity of neighbouring ligands within 5 Å proximity. NOESY (Fig. 2b) and ROESY spectra therefore afforded a connectivity map of neighbouring ligands, which enabled, together with the previous assignment, all 22 ligands to be fully revealed. The strongest NOESY and ROESY correlations were associated with sterically hindered ligands with low mobility. Molecular dynamics simulations were furthermore performed to confirm the different degrees of mobility and orientation dynamics of the ligands. A clear correlation between ligand mobility and chemical shift emerged. More static ligands gave stronger correlation peaks, whereas ligands that showed higher mobility in the MD simulation gave weaker correlation peaks (Fig. 2b).
The described example shows that a full assignment of the complicated 1H- and 13C-NMR spectra of Au102(pMBA)44 is possible, however, only by using a combination of multidimensional NMR methods, DFT calculations and MD simulations. More importantly, the authors showed that the different ligands bound at the surface of the nanocluster have their own intrinsic dynamical behaviour and different ligand–ligand interactions are taking place.
Another important aspect of gold nanoclusters that can be investigated with NMR spectroscopy is chirality. In 2011 Jin and co-workers reported an NMR study of the intrinsically chiral Au38(2PET)24 cluster (2PET = 2-phenylethanethiol). 2PET does not possess stereocenters.8 The chirality of Au38(2PET)24 originates from the dual-propeller-like distribution of the six Au2(SR)3 dimeric staples on the Au23 rod-shape core (Fig. 3). From X-ray structure determination a racemic pair was observed in the crystal with one enantiomer exhibiting clockwise (C-) and the other anticlockwise (A-) arrangements of the staples.9 Using chiral HPLC it was possible to separate the two enantiomers.10 Importantly the separation was reported only one year after the publication of Jin and co-workers, who at the time were seeking an alternative method to probe the chirality of Au38 in solution.
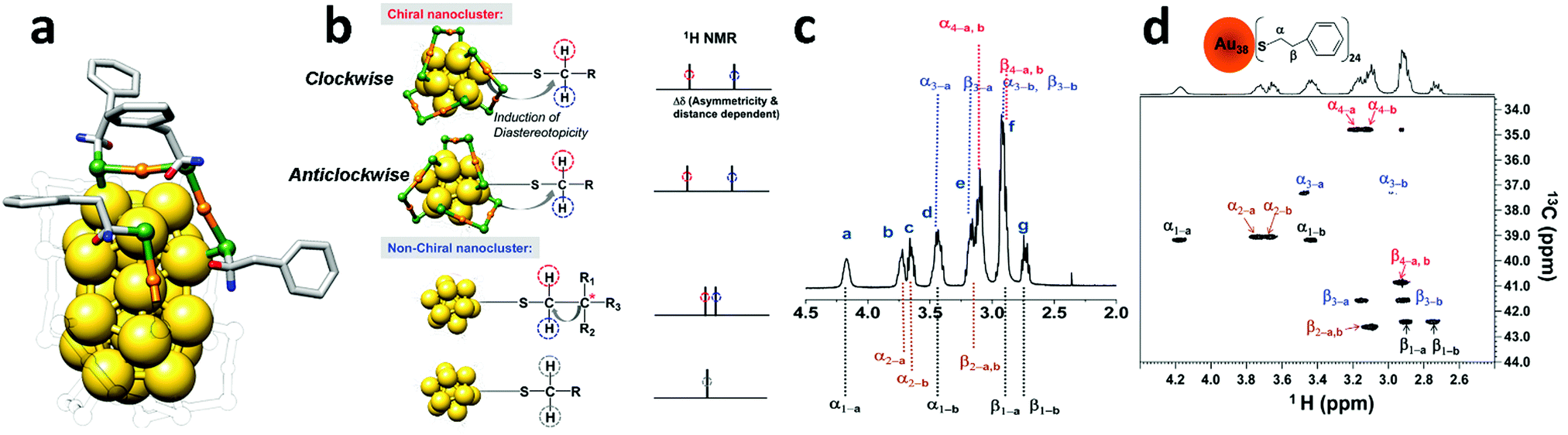 | ||
| Fig. 3 (a) Total structure of Au38(2PET)24, with solid colour for all 4 symmetry-unique ligands; the other ligands are transparent for clarity. Gold is yellow (core) and orange (staple), sulphur is green; the diastereotopic protons are in blue and red. (b) (from top to bottom) Diastereotopicity in the CH2 protons of the ligands on Au38(2PET)24 (A-,C-enantiomers). Diastereotopicity induced by a stereocenter in a non-chiral cluster and the normal CH2 protons of the ligands on a non-chiral cluster. (c) Full assignment of methylene protons in the 1H-NMR spectrum of Au38(2PET)24. (d) Two-dimensional 1H–13C-HSQC NMR of Au38(2PET)24 nanoclusters. The α-CH2 and β-CH2 are as labelled (see the top of panel d); 1–4 denote the four sets of symmetry-unique ligands; a and b represent each diastereotopic proton for each carbon (α- or β-CH2) in the four sets of ligands. Reprinted with permission from ref. 8. Copyright (2011) American Chemical Society. | ||
NMR spectroscopy is able to probe the chirality of nanoclusters without separation of enantiomers by using the diastereotopicity induced in the α-CH2 protons (next to sulphur) of the ligands bound to Au38. Diastereotopic protons are usually described as two protons belonging to the same CH2 group attached to (or in proximity to) a chiral stereocenter that do not show chemical equivalence in NMR spectroscopy.11 Jin and co-workers observed that Au38(2PET)24 nanoclusters exhibit a large chemical shift difference in the α-CH2 protons (Δδ of up to ∼0.8 ppm) of different ligands caused by the chiral distribution of the staples on the cluster surface. In agreement with the X-ray structure of Au38 (Fig. 3a) four different types of 2PET could be identified. The authors for the sake of comparison studied also the 1H-NMR of the non-chiral Au25(2PET)18 cluster in which its protons did not exhibit diastereotopicity. In the reported study 2D-NMR techniques such as COSY (COrrelated SpectroscopY) and HSQC (Fig. 3) were employed to determine the different spin systems of Au38(2PET)24. In other words, the authors were able to group all the 1H and 13C signals in four symmetry-equivalent ligand sets (Fig. 3a). However, the assignation with the corresponding NMR signal was not attained.
As seen in the previous examples, the cluster structure (symmetry), the type of ligand that surrounds its core and the presence of a chiral environment have an influence on the NMR spectra. Another important cluster property that can be analysed by NMR is the oxidation state. Murray and co-workers described how the 1H-NMR changes upon oxidation of Au25(2PET)18.12 It was demonstrated that this cluster behaves like a “simple” molecular redox species, changing its oxidation state from −1 to 0 and even +1.13
Au25 has a core composed of a Au13 icosahedron capped by six Au2(SR)3 staples. The 12 Au atoms in the staples can be considered as Au adatoms on the 12 faces of the Au13 core. According to this structure, the 18 capping ligands SR belong to either a family of 6 “outer” ligands ((SR)–Au–(SR)–Au–(SR)), where each sulphur atom is connected to two Au adatoms, or a family of 12 “inner” ligands ((SR)–Au–(SR)–Au–(SR)), where the sulphur atom is connected to one Au adatom and one core Au atom. Maran and co-workers reported a mixed DFT and NMR study in which they explained how the different oxidation states of Au25 affect the 1H- and 13C-NMR signals.14 They prepared [Au25(2PET)18]+ and [Au25(2PET)18]0 clusters by oxidizing [Au25(2PET)18]− with bis(pentafluorobenzoyl) peroxide, which reacts with the clusters according to an irreversible dissociative electron transfer. The NMR analysis of all three charge states revealed the presence of two families of ligands (symmetry ligand environments) in agreement with the X-ray structures.15,16 From the comparison between the -1 and the 0 charge states a remarkable chemical-shift difference was observed for the 12 inner ligands. This phenomenon is caused by the presence of an unpaired electron in [Au25(2PET)18]0. The paramagnetic behaviour mainly arises from the Fermi-contact term, as confirmed by DFT calculations of chemical shifts, and a clear correlation between the extent of spin delocalization and the nature of the ligands was established. Both 1H- and 13C- chemical-shift showed a significant down shift, as in the case of inner α-CH2 protons where the peak moves from 3.8 to ca. 25 ppm. The NMR behaviour of [Au25(2PET)18]+ was found to be quite similar to the one of [Au25(2PET)18]−, confirming the diamagnetic nature of the positive species.
NMR spectroscopy can also be applied to determine the size of nanoclusters by using NMR diffusion ordered spectroscopy (DOSY), which measures the diffusion coefficient. For a given solvent and temperature smaller particles or clusters diffuse faster. In particular for a spherical particle in a homogeneous solution, the hydrodynamic radius r can be calculated by using the Stokes–Einstein equation

Häkkinen and co-workers determined the hydrodynamic radius of three monodisperse 2PET-stabilized gold nanoclusters Au25(PET)18, Au38(PET)24, and Au144(PET)60 by DOSY.17 The obtained hydrodynamic diameters are in good agreement with the nanoclusters’ diameters in the respective crystal structures or from the theoretically predicted structures reported in the literature. Later the same group reported the hydrodynamic size of water-soluble Aum(pMBA)n clusters (m = 102, 144 and n = 44, 60) in aqueous media. The authors discovered that the hydrodynamic diameter of the pMBA− protected gold clusters was greatly influenced by the size and nature of the counter ion of the deprotonated pMBA− ligand. The DOSY and DFT calculations demonstrated that counter ions such as sodium and ammonium, residual acetic acid and even surrounding water molecules can affect very differently the nature and the hydrodynamic size of the Aum(pMBA)n clusters. These findings reveal important information of the surface chemistry of pMBA protected clusters which are very useful for the chemical modification or biological applications of water-soluble Aum(pMBA)n clusters. Such information is also hardly obtainable by mass spectrometry and X-ray crystallography.
NC's interactions with the environment monitored by NMR spectroscopy
NMR spectroscopy has unique potential to study the interaction of clusters with molecules and with the environment. This is of particular importance since such interactions play a crucial role for many possible applications. Nanoclusters and all nanoparticles in general, interact with the external environment through the organic monolayer. The type and strength of interactions that might occur are directly dependent on the molecular structure (i.e. functional groups and conformation) of the protecting ligands. These interactions together with the properties of the nanosized metal core can be employed in the fabrication of sensors.18 In the last five years Mancin and coworkers developed “NMR Chemosensing”, a very powerful sensing protocol for the identification of organic molecules in complex mixtures; it is based on the NMR magnetization transfer sequence and the recognition abilities of small nanoparticles.19–23 The authors used functionalized gold nanoparticles of around 1.8 nm, such dimension can still be considered in the range of nanocluster even though small polydispersity was observed. The lack of precise formula was not an indispensable requirement for the sensing protocol.NMR chemosensing utilizes the NMR sequences “NOE-pumping” (Fig. 4) which consists of two parts. First a diffusion filter is used to remove the magnetisation of all the molecular species present in the sample but not of the nanoparticles due to their lower diffusion coefficient. Secondly, the magnetisation is transferred via the NOE effect from the nanoparticle, to the interacting species (in a fast exchange regime). Therefore, only signals of the interacting analytes are displayed after the application of NOE-pumping and all the signals from the other species are filtered off. In one of the examples reported a final CPMGz filter (slow diffusing species like nanoparticles are filtered) was also applied to cancel the signal of the nanoparticles leaving only the spectrum of the interacting molecules.20
 | ||
| Fig. 4 Schematic representation of the NOE-pumping sequence employed in the NMR chemosensing protocol. A FID denotes the existence of observable signals after each pulse sequence block. The different sizes of the analytes represent the unbalance between populations of the free and bound states. Reprinted with permission from ref. 20. Copyright (2011) American Chemical Society. | ||
The other fundamental aspect in NMR chemosensing is the type of interactions employed to create the selective binding with the analyte. A variety of interactions were tested by Mancin and co-workers showing the broad range of applicability of these sensors. In the first example, nanoparticles protected with an amphiphilic ligand were employed for the detection of sodium salicylate in water and human urine.19 The identification of the analyte relied only on weak hydrophobic interactions between the ligand shell and the analyte. The detection limit was established to be only 2.5 mM. Lately, to improve the detection limit of this methodology, the authors decided to combine more than one non-covalent interaction (hydrophobic, electrostatic, metal–ligand coordination) thus improving the detection limit of salicylate to 0.5 mM.21
Nanoparticles functionalized with 18-crown-6-ether (supramolecular receptor) were also tested in the detection of biogenic protonated amines in complex mixtures. 18-Crown-6 is well known for its ability to bind ammonium and protonated primary amines by the formation of three NH+O hydrogen bonds with the oxygen atoms.20 In Fig. 5A is reported the NMR spectrum of a complex mixture of small aromatic molecules including α-aminoacid, and primary and secondary protonated amines in CD3OD.
 | ||
| Fig. 5 (A) 1H NMR spectrum of a mixture of compounds 2–8, each at 10 mM in CD3OD. (B) NOE pumping spectrum of the same mixture in the presence of 1-Au nanoparticles (29 μM). (C) The NOE pumping-CPMGz spectrum of the same mixture in the presence of 1-Au nanoparticles (29 μM) (* = nanoparticles’ residual signal, ° = residual solvents and impurities). Reprinted with permission from ref. 20. Copyright (2011) American Chemical Society. | ||
Several signals are present in the 1H NMR spectrum with some overlap, and assignment of the sets of signals arising from a single compound, even if well resolved, is not trivial. Moreover, the compounds in this mixture possess several functional groups which may potentially interact with the nanoparticle ligand shell via H-bonding. After the application of NOE-pumping in the presence of the functionalized nanoparticles, remarkably only the tyramine (2, primary amine) signals were observed, allowing its unambiguous identification (Fig. 5B). This challenging situation showed the chemical selectivity of the NMR chemosensor and its spectroscopic ability to discriminate overlapping signals at the same time.
The example discussed above impressively demonstrates the selectivity that can be introduced by judiciously choosing a NMR pulse sequence. A challenge of the approach, compared with other analytical techniques, is sensitivity. In the attempt to decrease the detection limit, the authors proposed the use of high affinity receptors. For example they employed a triazacyclononane (TACN) zinc complex (Zn2+) for the detection of phosphate molecules.22 Owing to the strong interaction between nanoparticle and analyte the system is no longer in the fast exchange range and the target molecule binding to the monolayer reduces its diffusion rate (becoming similar to the one of the nanoparticles). Therefore, with a simple application of a 1D NMR diffusion filter experiment (first part of NOE-pumping) it is possible to differentiate the strongly interacting molecule from non-interacting ones, whose signals are completely cancelled. With this approach, diphenyl phosphate was detected at micromolar concentration with good selectivity. The S/N ratio of the analyte signals is much greater here than in NOE-pumping experiments, because the main unwanted source of signal loss is limited to relaxation during the filtering sequence.
NC's reactivity monitored by NMR spectroscopy
NMR spectroscopy can be a powerful technique for monitoring reactions of monolayer protected gold nanoclusters in situ. The nanocluster reaction that has been studied the most is ligand exchange (or place exchange) reaction.3 It consists of the exchange of one protecting ligand with another ligand present in the solution. For gold nanoclusters the exchange of surface thiols is a largely used post-synthetic functionalization method. In the literature numerous studies were reported using different techniques to understand the reaction and the relevant reaction parameters. In a couple of those examples NMR spectroscopy was employed in order to gain kinetic and thermodynamic insights.24,25Pengo and co-worker presented a kinetic study on the ligand exchange reaction between [Au25(2PET)18]0 and para substituted arythiols.24 The kinetic experiments were performed by 1H NMR in benzene-d6 at 25 °C where a solution of clusters was mixed with 1.5 molar excess of arylthiol. The kinetic profiles were built by monitoring the time evolution of the integrals of the signals belonging to the protons β-Hout, the isochronous o-Hin, and the integrals of the β-H of 2PET during the exchange reaction. From the kinetic profiles it emerged that the “outer” position ((SR)–Au–(SR)–Au–(SR)) is more reactive than the “inner” ones ((SR)–Au–(SR)–Au–(SR)). However this difference in reactivity is influenced by the type of substituent on the phenyl ring. In particular the reaction rate of the exchange reaction at the outer position becomes bigger than the inner when a more electrodonating group is placed in the para position. On the other hand, for the case of thiophenol (Phen-SH) no significant difference in the rate constants was observed. The different selectivity for the two sites of the dimeric staples and its dependence on the electronic properties of the incoming ligands was an important finding that set the basis for a more efficient design of functionalized Au25 clusters.
1H-NMR was also employed by us in the study of the ligand exchange reaction of [Au25(SR)18]0 (where SR = 2PET or butanethiol).25 We monitored the evolution of the integrals related to entering and leaving thiols, and this in conjunction with MALDI mass spectra of the exchanged product allowed us to obtain information on the thermodynamics of this reaction. Ligand exchange reaction is affected by several factors like stability of the thiols in solution (solvation), the affinity of the sulphur to the gold cluster, and intermolecular interactions within the ligand layer. The number of exchanged ligands can then largely deviate from the statistical distribution (binomial) depending on the nature of the entering and exiting thiol. However, the situation changes when two [Au25(SR)18]0 clusters protected with different ligands are mixed together. An intercluster ligand exchange occurs leading to a distribution that is closer to statistical. This different behaviour is due to a different mechanism involved where the two clusters exchange ligands through a collision mechanism. The latter was confirmed also by in situ NMR studies where no trace of unbound ligand was detected. For the first time the existence of at least two mechanisms (the first via free thiols and the second through collisions between clusters) of ligand exchange for clusters was demonstrated. Even by using the same thiols, we found that the two mechanisms result in different equilibrium distributions. This recent discovery on gold nanoclusters and related systems is changing the general view on their nature. The ability to communicate between each other, exchanging metal atoms26,27 and/or thiols while maintaining their integrity, makes those clusters unique dynamic systems.
Conclusion
NMR spectroscopy offers many powerful possibilities to study monolayer protected clusters. NMR can be used to obtain structural and dynamic information on the (organic) ligands in the protecting layer. Total structure determination of monolayer protected clusters by NMR, in analogy to structure determination of proteins, seems difficult. In proteins a three-dimensional structure emerges from a linear (one dimensional) chain of connected monomers (amino acids). In contrast, the situation is more challenging for monolayer protected clusters, where the ligands are located on the surface (two dimensional) of a cluster core. However, in connection with an X-ray crystal structure (and theory) it is possible to assign the symmetry distinct ligands. Knowing that, it is, in principle, possible to study the effect of external parameters on specific regions (specific ligands) on the cluster surface. Such information is hardly obtainable by other methods.NMR spectroscopy is extremely powerful to study interactions of monolayer protected clusters or small nanoparticles with the environment and with other molecules. Using advanced pulse sequences (filters), which are already developed, one can introduce high selectivity in the detection scheme and thus drastically simplify complex spectra.
In general, NMR is not yet appreciated as a key method in the field of monolayer protected clusters. However, as we tried to highlight above, it gives molecular level information that other techniques are not able to provide. We are therefore convinced that NMR spectroscopy will become a central technique in the future work on monolayer protected clusters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the University of Geneva and the Swiss National Science Foundation (grant number 200020_172511) is kindly acknowledged. G. S. thanks EU Horizon 2020 Marie Skłodowska-Curie Action IF GOLDENSENS 747209 for financial support.Notes and references
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- K. Wüthrich, Nat. Struct. Biol., 2001, 8, 923–925 CrossRef PubMed.
- J. Cavanagh, W. J. Fairbrother, A. G. Palmer III, M. Rance and N. J. Skelton, Protein NMR Spectroscopy: Principles and Practice, Elsevier, 1995 Search PubMed.
- K. Salorinne, S. Malola, O. A. Wong, C. D. Rithner, X. Chen, C. J. Ackerson and H. Häkkinen, Nat. Commun., 2016, 7, 10401 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- H. Qian, M. Zhu, C. Gayathri, R. R. Gil and R. Jin, ACS Nano, 2011, 5, 8935–8942 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 25, 8280–8281 CrossRef PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- P. M. Nair and J. D. Roberts, J. Am. Chem. Soc., 1957, 79, 4565–4566 CrossRef CAS.
- J. F. Parker, J. P. Choi, W. Wang and R. W. Murray, J. Phys. Chem. C, 2008, 112, 13976–13981 CAS.
- J. F. Parker, C. A. Fields-Zinna and R. W. Murray, Acc. Chem. Res., 2010, 43, 1289–1296 CrossRef CAS PubMed.
- A. Venzo, S. Antonello, J. A. Gascón, I. Guryanov, R. D. Leapman, N. V. Perera, A. Sousa, M. Zamuner, A. Zanella and F. Maran, Anal. Chem., 2011, 83, 6355–6362 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt, R. W. Murray, K. Laboratories, V. Uni, C. Hill and N. Carolina, J. Am. Chem. Soc., 2008, 25, 3754–3755 CrossRef PubMed.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CAS.
- K. Salorinne, T. Lahtinen, J. Koivisto, E. Kalenius, M. Nissinen, M. Pettersson and H. Häkkinen, Anal. Chem., 2013, 85, 3489–3492 CrossRef CAS PubMed.
- E. Ertem, M. Diez-Castellnou, Q. K. Ong and F. Stellacci, Chem. Rec., 2017, 17, 1–11 CrossRef.
- B. Perrone, S. Springhetti, F. Ramadori, F. Rastrelli and F. Mancin, J. Am. Chem. Soc., 2013, 135, 11768–11771 CrossRef CAS PubMed.
- M.-V. Salvia, G. Salassa, F. Rastrelli and F. Mancin, J. Am. Chem. Soc., 2015, 137, 11399–11406 CrossRef CAS PubMed.
- M. V. Salvia, F. Ramadori, S. Springhetti, M. Diez-Castellnou, B. Perrone, F. Rastrelli and F. Mancin, J. Am. Chem. Soc., 2015, 137, 886–892 CrossRef CAS PubMed.
- M. Diez-Castellnou, M.-V. Salvia, S. Springhetti, F. Rastrelli and F. Mancin, Chem. – Eur. J., 2016, 22, 16957–16963 CrossRef CAS PubMed.
- L. Riccardi, L. Gabrielli, X. Sun, F. De Biasi, F. Rastrelli, F. Mancin and M. De Vivo, Chem, 2017, 3, 92–109 CAS.
- P. Pengo, C. Bazzo, M. Boccalon and L. Pasquato, Chem. Commun., 2015, 51, 3204–3207 RSC.
- G. Salassa, A. Sels, F. Mancin and T. Bürgi, ACS Nano, 2017, 11, 12609–12614 CrossRef CAS PubMed.
- B. Zhang, G. Salassa and T. Bürgi, Chem. Commun., 2016, 52, 9205–9207 RSC.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |