
Computational approaches to cell–nanomaterial interactions: keeping balance between therapeutic efficiency and cytotoxicity
Hong-ming
Ding
a and
Yu-qiang
Ma
 *ab
*ab
aCenter for Soft Condensed Matter Physics and Interdisciplinary Research, Soochow University, Suzhou 215006, China
bNational Laboratory of Solid State Microstructures and Department of Physics, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China. E-mail: myqiang@nju.edu.cn
First published on 3rd October 2017
Abstract
Owing to their unique properties, nanomaterials have been widely used in biomedicine since they have obvious inherent advantages over traditional ones. However, nanomaterials may also cause dysfunction in proteins, genes and cells, resulting in cytotoxic and genotoxic responses. Recently, more and more attention has been paid to these potential toxicities of nanomaterials, especially to the risks of nanomaterials to human health and safety. Therefore, when using nanomaterials for biomedical applications, it is of great importance to keep the balance between therapeutic efficiency and cytotoxicity (i.e., increase the therapeutic efficiency as well as decrease the potential toxicity). This requires a deeper understanding of the interactions between various types of nanomaterials and biological systems at the nano/bio interface. In this review, from the point of view of theoretical researchers, we will present the current status regarding the physical mechanism of cytotoxicity caused by nanomaterials, mainly based on recent simulation results. In addition, the strategies for minimizing the nanotoxicity naturally and artificially will also be discussed in detail. Furthermore, we should notice that toxicity is not always bad for clinical use since causing the death of specific cells is the main way of treating disease. Enhancing the targeting ability of nanomaterials to diseased cells and minimizing their side effects on normal cells will always be hugely challenging issues in nanomedicine. By combining the latest computational studies with some experimental verifications, we will provide special insights into recent advances regarding these problems, especially for the design of novel environment-responsive nanomaterials.
 Hong-ming Ding | Dr Hong-ming Ding received his BS degree in condensed matter physics in 2010 and PhD degree in biophysics in 2015 from Nanjing University. He is currently an associate professor at the Center for Soft Condensed Matter Physics and Interdisciplinary Research in Soochow University. His main research interests include understanding the interactions of nanoparticles with proteins and cell membranes at the molecular level, as well as designing new types of nanomaterials for drug/gene delivery through computer simulations and phenomenological theories. |
 Yu-qiang Ma | Prof. Yu-qiang Ma received his PhD degree in physics from Nanjing University in 1993. In 1995, he became a professor at the Department of Physics of Nanjing University, and currently he is also the Director of the Center for Soft Condensed Matter Physics and Interdisciplinary Research in Soochow University. Prof. Ma's research interests are in the area of soft matter and biophysics. His recent projects include cell–nanomaterial interactions, phase transitions and dynamics in complex fluids, and nonequilibrium self-organization in living soft matter, such as the cellular cytoskeleton and self-propelled particles. |
1 Introduction
Nanomaterials (NMs) are defined as materials with at least one external dimension in the size range from approximately 1–100 nanometers.1 Nowadays, with the rapid development of nanotechnology, nanomaterials have been widely used in many fields of science and technology. As one of the most promising applications, the usage of nanomaterials in biomedical fields2,3 (e.g., drug/gene delivery,4,5 biosensing,6 bioimaging,7 and tissue engineering8) has recently received tremendous attention, due to their inherent advantages like appropriate size, high surface area to volume ratio, and unique electrical and magnetic behavior (over traditional ones).9However, recently an increasing number of critics have raised concerns regarding the potential risks of nanomaterials to human health and safety.10,11 These problems (e.g., potential toxicity and immunogenicity) may block the further clinical use of nanomaterials in biomedicine. Thus, it is imperative to have a better understanding of nanomaterial–biosystem interactions12–15 and develop in vitro and in vivo safety assessment approaches that relate the nanomaterial properties to the underlying mechanisms of biological injury, pathophysiology of dosimetry, disease, and exposure of humans and environmental life forms.11 Over the last decade, much effort has been made by experimental researchers on this topic and there has recently been great progress regarding the possible mechanism of cytotoxicity and related problems. On the basis of significant experimental studies, there have also been quite a few excellent reviews summarized by experimental researchers,16–23 which provide more comprehensive prospects of nanotoxicity as well as nanomedicine.
Moreover, with the present advance in computer technology and simulation methods, molecular simulations (including molecular dynamics (MD) simulations,24 Monte Carlo (MC) simulations,24 Brownian dynamics (BD) simulations,25 and dissipative particle dynamics (DPD) simulations26) have also been widely used to investigate the related problems, like interactions of nanomaterials with cell membranes27–30 and biomolecules.31–33 As an important complementarity of experiments, computer simulations may reveal a more detailed molecular mechanism regarding the cytotoxicity of nanomaterials from physical insights. However, to the best of our knowledge, there exist no comprehensive reviews that have summarized this topic from the computational view, though there have been many computational literature studies about this topic, like how nanoparticles affect the functions of cell membranes, proteins, and DNA etc.34–38 More importantly, it is of essential necessity to bridge the gap between simulations and experiments and combine these two methods to better understand the above issues.
In this review, we will firstly present the state-of-the-art progress regarding the physical mechanism of cell–nanoparticle interactions, especially regarding the impact (and/or side effect) of nanoparticles (NPs) on the properties and functions of lipid membranes as well as other biomolecules (e.g., proteins and DNA), which is mainly based on simulation results. Then from the view of theoretical researchers, how to minimize and make best use of the toxicity, as well as how to keep a balance between therapeutic efficiency and cytotoxicity will be discussed in detail. Finally, a summary and perspective will be given.
2 Nanomaterials used in biomedicine and their molecular modeling
2.1 Inorganic, organic and hybrid nanomaterials
Owing to great progress in materials science and biotechnology, various types of nanomaterials with different sizes, shapes and surface properties have been synthesized, and a broad range of nanocarriers have been used in biomedicine.39 Typically, they can be divided into organic and inorganic ones (Fig. 1).40 The organic nanomaterials mainly include liposomes,41 polymersomes (usually made of copolymers),42 dendrimers,43 and polymeric nanoparticles44 (cargos like DNA are encapsulated in the polymer matrix or absorbed on the surface). In addition, recently the assembly of natural biomacromolecules (e.g., specific proteins and DNA molecules) that are believed to have a better bio-compatibility has also been widely used in the biomedical fields.45,46 The inorganic nanomaterials mainly consist of magnetic nanoparticles47 (made up of magnetic elements such as iron, nickel, cobalt, and their chemical compounds), quantum dots (QDs)48 (colloidal nano-sized crystals, comprising atoms of elements from groups II B to VI A (e.g., Cd, Zn, Se, Te) or III B to V A (e.g., In, P, As)), metal nanoparticles (like gold nanoparticles49 and silver nanoparticles50), carbon-based nanoparticles (mainly including fullerenes,51 carbon nanotubes (CNTs)52 and graphene53), and silica nanoparticles.54 | ||
| Fig. 1 Schematic illustration of different types of nanomaterials used in biomedicine. Reproduced with permission from ref. 60 (Copyright 2011 Nature Publication Group), and ref. 63 (Copyright 2011 Royal Society of Chemistry). | ||
We should notice that there exist obvious differences between inorganic and organic nanomaterials for biomedical use. For example, the driving forces like covalent bonds and metallic bonds for the formation of inorganic nanoparticles can be very strong, leading to the stable property of inorganic nanomaterials in biomedical use. In contrast, organic nanomaterials are usually self-assembled (the dendrimer is one exception) by the basic elements (e.g., copolymers, lipids, and proteins) due to weak interactions like electrostatic interaction, hydrophobic interaction, and hydrogen bonds.40 Thus, they may have a dynamic or tunable character, which makes organic nanoparticles much more simple for the encapsulation (or release) of drugs or other entities. However, depending on their unique features and physicochemical properties, inorganic nanoparticles can have specific uses in biomedical applications. For example, magnetic nanoparticles can be used as contrast agents for magnetic resonance imaging (MRI) due to their large magnetic moment;55 QDs appear as a promising probe for long-term in vivo and in vitro experiments due to their high photostability;56 colloidal gold can be employed as a contrast agent in electron microscopy owing to its high atomic number;57 CNTs can be employed in tissue engineering scaffolds because of their incredible strength with extreme flexibility.58
Moreover, in order to synthesize nanomaterials with multiple biomedical functions, hybrid nanomaterials composed of two or more different materials have recently received great interest,59 which include the organic–organic hybrid ones (e.g., liposomes coated by different types of polymers and ligands60), inorganic–inorganic hybrid ones (e.g., iron oxide nanoparticles with silica-coating61), and organic–inorganic hybrid ones (e.g., using polymers or lipids as matrix to load inorganic nanoparticles62). We should notice that the final structure of the hybrid nanomaterial will probably be core/shell type, and can either exhibit characteristics of the two original materials or even new properties.63
2.2 Molecular modeling of different nanomaterials and biomolecules
With the rapid development of computing technology, typically there have been four types of computational methods with the time scale from fs to ms and length scale from Å to tens of μm: ab initio quantum mechanics (QM) calculations, all-atom (AA) molecular dynamics simulations, coarse-grained (CG) molecular simulations, and continuum mechanics calculations (see Fig. 2).64 For QM calculations, though the calculation accuracy is very high, the time and length of the system are greatly limited; while for continuum mechanics calculation, though the time and length scales are the largest, most (even all) of the molecular details are lost. In contrast, the balance between calculation accuracy and computing cost is well kept in all-atom and coarse-grained molecular simulations, thus these two methods are widely used in modeling the interactions among nanoparticles, biomolecules, and cell membranes. Moreover, recently there have been some attempts to combine two of the above methods (e.g., QM/AA molecular simulation65 and AA/CG molecular simulation66), which we think may better keep the balance between computing accuracy and cost. | ||
| Fig. 2 Schematic illustration of computational methods for studying nanoparticle–biosystem interactions across a range of time- and length-scales. Reproduced with permission from ref. 64 (Copyright 2013 Multidisciplinary Digital Publishing Institute). | ||
Now many types of nanoparticles can be modeled in all-atom molecular dynamics simulations. CHARMM (Chemistry at HARvard Macromolecular Mechanics),67 AMBER (Assisted Model Building with Energy Refinement),68 GROMOS (Groningen Molecular Simulation),69 and OPLS (Optimized Potentials for Liquid Simulations)70 are the four common atomistic force fields used for biomolecule modeling, where most of the existing biomolecules like peptides, lipids, DNA, and RNA can be simulated. Additionally, some common polymers can be also modeled in all-atom simulations. For example, based on the CHARMM General Force Field,71 many types of molecules as well as drug or drug-like molecules can be built, and thus organic nanoparticles composed of the above basic elements can now be well modeled. In contrast, the present atomistic force field for inorganic nanoparticles seems to be not complete. There does not exist standard data for many inorganic nanoparticles like iron oxide NPs and CdSe QDs in the above force fields, although the currently developed INTERFACE force field has covered metals like Al, Ag, Au, Cu, Ni, Pb, Pd, and Pt and the silicates, sulfates, apatites etc.72,73 Moreover, even for some inorganic nanoparticles like silver nanoparticles74 and carbon-based nanoparticles75 that have been included, the parameter in the force field is still not accurate enough to model all the biomolecule–nanoparticle systems. For example, as Heinz et al.76,77 pointed out, since the common all-atom force fields do not reproduce cation–π and π-stacking interactions explicitly, there could exist some differences between the quantum mechanics and common atomistic simulation methods when modeling the interaction of aromatic molecules with carbon nanotubes or graphenes.
Though one can refer to the ab initio quantum mechanics calculations, the length and time scales are greatly reduced. Actually, the length and time scales of all-atom simulations, where the length scale is about tens of nm and the time scale is on the order of hundreds of ns, may have already greatly limited the better investigation of some biological processes (e.g., endocytosis). In order to model a much larger system for a longer time, the coarse-grained method, in which small groups of atoms are treated as single beads, should be used. Besides, one can still alter the hydrophilic/hydrophobic property, charge property, and even the specific property (e.g., receptor–ligand interaction) of the basic beads,78,79 thus different types of nanomaterials and biomolecules can be simulated in coarse-grained simulations. Actually, modeling nanoparticle–biosystem interactions has become a booming field in the last two decades, where Martini coarse-grained molecular dynamics simulations80 and dissipative particle dynamics simulations26 have been widely used. To better understand these problems (especially how to model nanoparticles as well as biomolecules at different molecular levels), we refer readers to the latest reviews.29,81–83
3 Effect of nanomaterials on cell membranes
Cell membranes act as the first barrier between the intracellular and extracellular environments, and the cellular functions also greatly depend on the properties of the membrane.84 In order to be delivered into the cell interior, nanoparticles must firstly interact with the cell membrane, whose properties can be altered by the presence of nanoparticles. And the impacts of nanoparticles on cell membranes are related to many factors like the properties and amount of nanoparticles.3.1 Effect of nanoparticle penetration on cell membranes
Typically, when nanoparticles insert into the lipid membrane, they can affect the properties of membranes like lipid tail order parameter, membrane thickness, area per lipid, permeation ability, and gel/liquid phases. Wong-Ekkabut et al.37 used coarse-grained molecular simulation to investigate the effect of fullerenes on the lipid bilayer. Their results showed that the insertion of fullerenes into the membrane interior may have some effects on the properties of the lipid bilayer, e.g., enlarging the area per lipid, thickening the lipid bilayer and decreasing the lipid diffusion coefficient. But since one single fullerene is very small, the fullerenes could only induce modest increase of lipid membrane softness and are unlikely to cause severe damage to the membrane even under high concentrations. However, when nanoparticles become larger, they will contact with more lipid molecules. Since there may exist a hydrophilicity/hydrophobicity mismatch, the lipids may be redistributed, which leads to an obvious change in the properties of the lipid membrane.85 In addition, when compared with the lipids far away from the nanoparticle, the properties of the lipids around the nanoparticle can be very different. For example, Mhashal et al.86 investigated the properties of lipid molecules in different regions through all-atom MD simulations, and found that the lipids in the short-range region (within the interaction range) are mostly affected (Fig. 3a), leading to a large deformation of lipid structure and slow dynamics of lipid molecules; in contrast, lipids in the long-range region (not directly interacting with nanoparticles) also get slightly perturbed, which gives rise to an increase in the local ordering of the lipid domains and a decrease in the fluidity of the lipid membrane. Moreover, Mhashal et al.86,87 also studied the size effect of gold nanoparticles on the fluidity of lipid membranes, and concluded that NPs with different sizes have a similar effect on the bilayer while the magnitude of structural ordering and fluidity differs. Such a size-dependent effect was also observed by Dallavalle et al.88 through DPD simulations, where they found that small graphene sheets do not affect the order of the phospholipids while larger graphene sheets can wreak havoc with the membrane and create a patch of upturned phospholipids. | ||
| Fig. 3 Penetration of nanoparticles into cell membranes and their effects on the membrane properties. (a) Left panel: Schematic representation of the short range (SR), long range (LR) and buffer region, right panel: Scd order parameters of lipid chains, where square, triangle and circle symbols represent short range, buffer region and long range lipids, respectively. Reproduced with permission from ref. 85, Copyright 2016 Public Library of Science. (b) Variation of the area per lipid (left) and order parameter (right) during the main phase transition process for the lipid bilayer encapsulating smooth nanoparticles under different surface decorations. Reproduced with permission from ref. 91, Copyright 2014 Springer-Verlag. (c) Final snapshots of interactions between the charged dendrimers (left: G3, middle: G5, right: G7) and lipid membranes. Reproduced with permission from ref. 100, Copyright 2009 American Chemical Society. | ||
Apart from the size effect, the surface properties of nanoparticles may also have great impacts on cell membranes.89–92 For example, by using Martini coarse-grained simulation, Lin et al.91 found that a hydrophobic nanoparticle with the size of 3.0 nm can decrease the order parameter and increase the area per lipid obviously (Fig. 3b). By further investigating the effect of surface properties of nanoparticles, it was found that increasing the surface roughness of nanoparticles can increase the main phase transition temperature of the lipid membrane, while increasing the density of surface hydrophobic ligands will decrease the main phase transition temperature in a nonlinear way. Thus they concluded that the phase transition temperature of the lipid membrane can be controlled by varying the type of encapsulating hydrophobic nanoparticles.91 Such surface property effects were also investigated by Feng et al.93via all-atom molecular dynamic simulations. It was found that the insertion of carbon nanotubes into the lipid membrane will make the lipid tails around the carbon nanotubes more ordered than the rest of the lipid tails, which in turn increases the order parameter of the lipid tails and thickens the membrane. Importantly, by decorating different numbers of hydroxyl groups onto the CNT surface, they found that there exists one type of carbon nanotube (its hydrophicity/hydrophilicity is in line with the lipid bilayer) that has the least effect on the membrane.
The above studies mainly focus on the effect of spontaneous penetration of nanoparticles on the membrane properties. In some cases, one may add a force onto the nanoparticle to help its translocation.94,95 Though this may be beneficial for increasing the translocation efficiency of nanoparticles, the damage to lipid membranes can become much more evident. As Murad et al.96 showed, the active translocation of bare nanoparticles (i.e., adding external force or velocity on the nanoparticle) can have a great influence on the membrane properties, i.e., except for the change of lipid order parameter and membrane thickness, there also exist transient pores in the membrane, which cause water permeation, ion transportation across the membrane, and the flip-flop of lipid molecules. Additionally, they also found that this effect is size-dependent and will become more obvious with the increase of the size of the nanoparticles. More recently, Murad et al.97 also found that there may exist the loss of lipid molecules from the bilayer during the active translocation of ligand-coated nanoparticles. Such similar results were also found by Arai et al.98 through DPD simulations, where their results indicated that a vesicle cell under collision with the nanoparticle can be severely “damaged” when the initial velocity of the nanoparticle is beyond a critical value. Furthermore, even though the external force does not directly push the nanoparticles across the membrane, the damage may still be severe. For example, Yue et al.99 used DPD simulations to systematically investigate the interaction between lipid membranes and rotating NPs (due to external torque). In some cases, the rotation may promote the cellular uptake of nanoparticles. However, since the membrane may undergo a local distortion around the nanoparticle, if the rotation exceeds a critical value, the local membrane distortion can develop into a mechanical rupture in some cases. Generally, all these simulation studies revealed that the dynamic process of nanoparticles across cell membranes is an important factor (on affecting membrane property). Thus one must be very careful when using external energy or force to promote the translocation of nanoparticles.
The pore or leakage of membranes is usually transient during the insertion (or translocation) of hydrophobic (or hydrophilic) nanoparticles. After the translocation, it will be spontaneously closed. Actually, the insertion or weak adsorption due to hydrophobic interaction is thought to be not strong enough to destroy the lipid membranes (in the absence of external forces). In contrast, charged (especially positively charged) nanoparticles like dendrimers and polyelectrolyte matrixes can induce the formation of pores in the membrane due to electrostatic interactions. For example, Yan et al.100 found that when the membrane is tensionless, a generation 3 (G3) dendrimer only acts on one side of the membrane and causes almost no defects, while a generation 5 (G5) dendrimer gradually inserts into the membrane and leads to the formation of a very small pore. With the increase of membrane tension (or the area per lipid Al), more dendrimer beads penetrate into the bilayer membranes, and the rupture of the membrane and the formation of a big hole occur (Fig. 3c) when Al is larger than 1.44 nm2 (while in the absence of the dendrimer, the membrane is still stable). Generally, the pore formation is determined by the tension of the local region around the dendrimers. In this sense, though the initial membrane is tensionless, when there are many dendrimers adsorbing (or inserting) onto the membrane101 or the size of the dendrimer is very large,102 the membrane will also become greatly stressed, which may also lead to the obvious leakage of membranes. Such pore formation phenomena were also found by other theoretical researchers using different types of nanomaterials like charged rigid nanoparticles (or nanoparticles decorated with charged ligands), peptides, and linear polyelectrolytes.34,103,104 Generally, the pore is usually permanent under these situations, and thus charged nanoparticles can have non-negligible side effects on cell membranes, and may even cause cell death. So when using these cationic nanoparticles for therapeutic use (e.g., gene delivery), it is of great importance to decrease their severe damage on normal cells as much as possible.
3.2 Effect of nanoparticle engulfment on cell membranes
It is well known that endocytosis is also an important pathway of nanoparticles crossing cell membranes.105 Theoretically speaking, the whole successful endocytosis process may have very little effect on the cell membrane (and the receptors can also be recycled). However, if the membrane is stressed, the short-range specific interaction between nanoparticles and cell membranes during endocytosis can also lead to pore formation in the membrane.106 The reason is similar to the case of charged nanoparticles, namely when the local tension becomes very large, it may facilitate the rupture of the membrane nearby.In addition to the direct damage to cells, there may exist indirect damage caused by nanoparticles. For example, when the engulfment of nanoparticles by specific cells (e.g., the phagocytic cells in the immune system) can occur but cannot be completely finished (i.e., frustrated endocytosis), this may cause the release of inflammatory mediators to induce cell death,107 and may even lead to the death of other cells nearby. Recently, the underlying physical mechanism of frustrated endocytosis of nanoparticles has been extensively studied. By combining simulations and experiments, Shi et al.108 investigated the interactions between cylindrical one-dimensional nanomaterials (e.g., functioned nanotube) and cell membranes, and found that nanotubes with end caps or shells at their tips, will rotate a lot (driven by asymmetric elastic strain at the tube-bilayer interface) during the engulfment, and enter the cell in a near-vertical manner (Fig. 4a). However, when the nanotube is very long, the specific receptor–ligand interaction cannot balance out the extremely large bending energy originating from the membrane wrapping the nanotubes. Thus they concluded that the tip-entry mode provides a natural mechanical explanation for incomplete endocytosis or frustrated phagocytosis. This reason (i.e., the increased bending energy of cell membranes) can be also used to explain the frustrated endocytosis of oxidized graphene nanosheets. But different from the tip entry and long length of nanotubes, the laying down of graphene nanosheets on the cell membrane and its sharp edge are two main reasons for the larger bending energy.109,110
 | ||
| Fig. 4 Frustrated endocytosis of different types of nanoparticles. (a) Time evolution of simulation snapshots of a nanotube interacting with a cell membrane at an initial angle of 15°. Reproduced with permission from ref. 108, Copyright 2011 Nature Publication Group. (b) Left panel: Schematic illustration of interaction of vesicles with rigid and soft particles; right panel: elastic energy as a vesicle wraps around a particle with increasing wrapping degree f for different size ratios a/R and bending rigidity ratios. Reproduced with permission from ref. 112, Copyright 2016 American Chemical Society. (c) Time evolution of simulation snapshots of a ligand-coated nanoparticle interacting with a cell membrane. Reproduced with permission from ref. 115, Copyright 2012 Elsevier. | ||
Furthermore, Yi et al. used the Helfrich theory to explain why soft particles are much harder to be fully wrapped by lipid bilayers111 and vesicles112 than rigid ones. The main reason is that there exists the additional bending energy of the soft nanoparticle itself (except for that of cell membranes). From the kinetic view, when wrapping around a soft particle, the membrane does not deform initially, but it needs to catch up to almost the same configuration at full wrapping (Fig. 4b). This means a more abrupt rise in elastic energy at the later stage of wrapping, and consequently, very large adhesion energy would be required to balance out the more rapid rise in elastic energy. A similar phenomenon was also observed by Yue et al.113 and Shi et al.114 through DPD simulations. Moreover, Ding et al.115 investigated the receptor-mediated endocytosis of ligand-coated nanoparticles by using dissipative particle dynamics simulations, and raised one new mechanism of frustrated endocytosis. It was found that there may exist the spontaneous lack of ligands during the engulfment process (Fig. 4c). This is due to the fact that the ligands on the nanoparticles tend to interact with more receptors on the membrane so that some ligands may become stretched and get far away from their initial orientation (i.e., the radial axis), leading to the nonuniform distribution of ligands on the particle surface. And the lack of ligands on the upper surface will suppress further engulfment of nanoparticles.
In general, the above nanomaterials such as long nanotubes, large graphene sheets, and very soft nanoparticles should be used very carefully for real applications, even though these nanomaterials may have their own advantageous characters.
Furthermore, we should notice that the discussion on the cell membrane–nanoparticle interactions in this section is in the absence of proteins. Actually, many experimental studies have indicated that the uptake of nanoparticles and its impact on cells could be totally different in the presence of proteins, indicating that the proteins play important roles in the cellular delivery efficiency and cytotoxicity.116–118 We will discuss this in detail in Sections 5 and 6.
4 Effect of nanomaterials on functional biomolecules
Nanoparticles will also interact with various types of biomolecules in the body (e.g., protein, DNA, and RNA), which may greatly affect their biological functions.Previous experimental results have shown that the most severe damage to biomolecules mainly originates from reactive oxidative species (ROS).119 But it should be noted that moderate levels of ROS are not harmful. Instead, they play important roles in the modulation of proliferative response, gene expression, signal transduction, and protein redox regulation.21 Since nanoparticles can generate ROS in many ways (e.g., prooxidant functional groups on the reactive surface of NPs, active redox cycling on the surface of NPs due to transition metal-based NPs120), the entrance of NPs into cells may greatly increase the level of ROS. As a result, the excessive intracellular ROS can react with cellular macromolecules including DNA and proteins, and disturb the homeostasis of the intracellular milieu.21 Therefore, it is of essential importance to avoid the generation of ROS when using nanoparticles for biomedical applications. However, this topic seems to be far beyond the scope of the present review since the cytotoxicity caused by ROS belongs to biochemical aspects. Moreover, we should notice that though ROS are thought of as causing the most important and fatal damage to cells (especially mitochondrion), it is not the only route to causing cytotoxicity and there still exist some other routes, where the biological function of biomolecules may also be greatly destroyed by their physical interaction with nanoparticles. In this section, we will discuss the recent computational results of the effect of nanomaterials on the structure and bio-function of biomolecules from physical insights.
4.1 Effect of nanomaterials on proteins out of the cell membrane
Proteins are the most abundant biomolecules in living bodies. When nanoparticles are inserted into the body, they will unavoidably interact with different types of proteins. This will not only alter the physicochemical property of the nanoparticle itself,121 but also have a non-negligible effect on the molecular structure of the protein.122 In this subsection, we will mainly discuss the latter issue, while the former one will be discussed in detail in the next section.The carbon-based nanoparticles (e.g., fullerenes, carbon nanotubes, and graphene) are one of the most widely used nanomaterials in biomedicine. Recently, their interactions with proteins have been extensively studied. It has been realized that the hydrophobic interaction and the π–π stacking between carbon rings and aromatic residues are the main driving forces in carbon-based nanoparticle–protein interactions.123 But it should be noted that due to their different sizes and shapes (especially their distinct contacting surface curvatures), there may exist some differences of the driving force for the interaction of proteins with fullerenes, carbon nanotubes and graphene in some cases. Taking protein villin headpiece (HP35) as an example, Zuo et al.124 found that the π–π stacking interaction is the dominant driving force for the binding of HP35 onto graphene, where aromatic residues F35, W23, and F10 play a central role by forming π–π stacking (Fig. 5a). This is different from the interactions of HP35 with single-walled carbon nanotubes (SWCNTs) and C60, where the hydrophobic interactions between aliphatic residues of HP35 and SWCNT/C60 (especially C60) may be more important for the binding (Fig. 5b). Besides, it was also found that HP35 loses most of its native secondary and tertiary structures after adsorption onto graphene, indicating that the presence of nanoparticles can lead to a change in the molecular structure of proteins and in turn may affect their bio-functions.124 A similar phenomenon was observed by Sengupta et al.125,126 using the Aβ peptide, where it was found that the structure of Aβ peptide greatly changed upon adsorption onto the surface of single-walled carbon nanotubes. More importantly, by introducing multiple Aβ peptides into the simulation system, they further demonstrated that the intrinsic self-assembly propensity of Aβ was indeed hindered by the adsorption (on the CNTs).127 Besides, they also pointed out that although Aβ dimers had a marked weakening upon surface adsorption on the SWCNTs, they were still capable of growing into trimeric assemblies. These results may help better use self-assembling peptide/protein amyloids in nanomedicine.128
 | ||
| Fig. 5 Interactions between functional proteins and different types of nanoparticles. (a) Time evolution of simulation snapshots of HP35 adsorbing onto graphene. (b) Boxplot for the interaction energy among each residue of HP35 with C60, SWCNTs, and graphene. Reproduced with permission from ref. 124, Copyright 2011 American Chemical Society. (c) Time evolution of simulation snapshots of graphene nanosheets inserting into a protein dimer. Reproduced with permission from ref. 170, Copyright 2015 American Chemical Society. | ||
The disruption of protein structure by nanoparticles is not the only way to induce dysfunction of proteins; there also exist other ways of causing the loss of the original function of proteins. By using large-scale all-atom simulations, Zuo et al.35 systematically investigated the interactions between several proteins with WW domains and carbon nanotubes, and found that carbon nanotubes can plug into the hydrophobic core of the proteins to form stable complexes. As a result, specific ligands (e.g., PRM with a sequence GTPPPPYTVG) can no longer bind into the WW domain of the proteins. A similar phenomenon was also reported by Durdagi et al.129 through molecular docking and MD simulations, where it was observed that the fullerene can bind and suppress the activity of enzymes as exemplified by HIV-1 protease (HIV-1P). More recently, Lu et al.130 also showed that graphene can also have such abilities. Since the surface area of graphene nanosheets is much larger, it can even interrupt the protein–protein interaction or recognition (Fig. 5c), which may disrupt the cellular metabolism and cause the mortality of cells.
Additionally, other types of nanomaterials like MoS2/C2N,131,132 gold,133–136 silver,135,137 silica (or silicon),138,139 hydroxyapatite,140,141 and polymeric nanoparticles142 can also affect the conformation of proteins. Due to their distinct physicochemical properties from carbon-based nanoparticles, the driving forces may change a lot, i.e., in addition to the hydrophobic interactions (π–π stacking interaction may not occur here), there exist some other driving forces to induce binding, like electrostatic interaction142 and Au–thiol coordination bonds.143 More importantly, unlike the carbon-based nanomaterials, there exist many other factors that can affect the interaction of proteins/peptides with these nanomaterials. For example, by using all atom molecular simulations, Heinz et al.144,145 observed that the pH may affect the binding behaviors of peptides on the silica surface, and more recently they also found that the binding of peptides can be facet-selective in the case of metal nanoparticles.146,147 In addition, depending on the shape of the nanoparticles, there still exist two different ways of nanoparticles altering the protein function. For example, Brancolini et al.133 found that small gold nanoparticles can bind the protein to form persistent complexes. This binding of nanoparticles is able to block the active sites of domains from binding to another protein, thus leading to potential inhibition of the fibrillation activity. In contrast, when proteins adsorb onto a silver or gold substrate, their conformation may become close to the surface for the best-binding so that their space structures are totally altered.135 Furthermore, the size of nanoparticles also has a great impact, especially in the case of ultrasmall nanoparticles. As observed by Boselli et al.148 experimentally, a small change in nanoparticle size may induce distinct effects on the protein composition and structure. The underlying mechanism of this phenomenon deserves to be well investigated in future simulation studies.
4.2 Effect of nanomaterials on membrane proteins
Except for the proteins in the extracellular or intracellular environments, the nanoparticles may also interact with some membrane proteins inserting or throughout the cell membrane. But different from the above issue, the nanoparticles may just interact with some specific parts of membrane proteins because most parts of these proteins are “protected” by the membrane.The ion channel is one typical membrane protein, which can establish a resting membrane potential, shape the action potential and other electrical signals, control the flow of ions across secretory and epithelial cells, and regulate cell volume etc.149 It has been realized that many organic molecules can act as ion channel blockers. For example, Lidocaine and Novocaine can block the sodium ion channels, while dendrotoxin and iberiotoxin can block the potassium channels. Recently, more and more evidence has shown that nanoparticles with the appropriate size and hydrophobicity may also be capable of blocking ion channels, where fullerenes and CNTs are the most studied ones. By using atomistic molecular dynamics simulations, Kraszewski et al.150 demonstrated that C60 may indeed effectively hinder the function of K+ channels, since there are a variety of specific binding sites depending on the structure and properties of the channel. Recently, Calvaresi et al.38 further pointed out that C60 may block K+ channels with two mechanisms, i.e., a low affinity blockage from the extracellular side (Fig. 6a), and an open channel blockage from the intracellular side (Fig. 6b), where the intracellular binding site has an extremely high affinity for C60 and the molecular determinants of binding are conserved in the entire family of K+ channels. Moreover, by modifying the fullerene, Hilder et al.151 found that the fullerene derivatives (i.e., [Lys6]–C84) can be used to selectively block the specific ion channel, namely [Lys6]–C84 is capable of blocking the sodium channel, hNav1.7 with strong affinity (2.7 nM) while it can only weakly bind to hNav1.7 (14.5 mM) and hNav1.4 (3.7 mM). Furthermore, carbon nanotubes can be also used to block ion channels.152,153 By using the molecular docking method, Park et al.153 found that SWCNTs may be more effective than spherical fullerenes due to their large size. The shape of SWCNTs is also of great importance (Fig. 6c–e), and it was observed that due to its extended interaction with the selectivity filter, an open ended SWCNT is more efficient in blocking K+ channels when compared with a capped SWCNT.
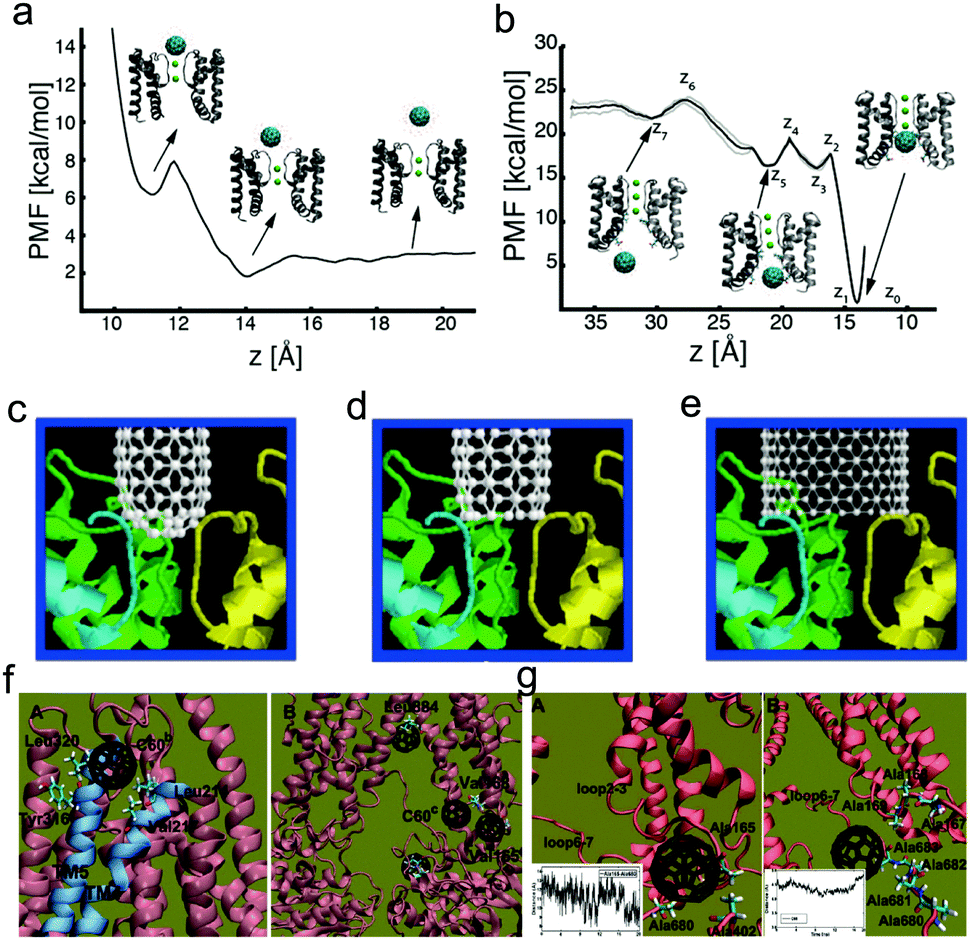 | ||
| Fig. 6 Interactions between membrane proteins and different types of nanoparticles. (a) Potential of mean force (PMF) for the movement of C60 from the extracellular entrance of the selectivity filter to the extracellular compartment; (b) PMF for the displacement of C60 from the intracellular side of the selectivity filter in the intracellular cavity of the MthK channel to the intracellular solution. Reproduced with permission from ref. 38, Copyright 2015 American Chemical Society. Docking simulation of capped SWCNTs with the size of 0.9 nm (c), open-ended SWCNTs with the size of 0.9 nm (d), and open-ended SWCNTs with the size of 1.3 nm (e) interacting with K+ channels. Reproduced with permission from ref. 153, Copyright 2003 American Society of Biochemistry and Molecular Biology. (f and g) Typical simulation snapshots of interactions of P-glycoprotein with C60. Reproduced with permission from ref. 155, Copyright 2012 Royal Society of Chemistry. | ||
There also exist other membrane proteins with specific biological functions. For example, P-glycoprotein, as one of the most important multidrug resistance proteins, pumps many foreign substances out of cells. Recently, Zhang et al.154 took paclitaxel and doxorubicin as an example, and used atomistic molecular dynamics simulations to answer the problem of why P-glycoprotein is able to recognize and transport a broad-range of structurally different drugs. Their results showed that van der Waals interactions (especially the interaction between the drugs and the inner residues) play a significant role in approaching the putative binding site. However, Xu et al.155 found that though C60 is also hydrophobic and has a similar size, it can avoid recognition by the P-glycoprotein efflux pump (Fig. 6f), which is mainly due to its more rigid property over hydrophobic drugs. Similar results were also observed by Shityakov et al.156 using molecular docking, and they also found that carbon nanotubes cannot effectively block P-glycoprotein, either.
4.3 Effect of nanomaterials on nucleic acids
Nucleic acids, including DNA and RNA, are made from monomers known as nucleotides. Each nucleotide has three components, i.e., a 5-carbon sugar, a phosphate group, and a nitrogenous base. Together with proteins, nucleic acids are the most important biological macromolecules, and are found in abundance in all living things. Thus when utilizing nanomaterials for biomedical applications, it is of great importance to minimize the genotoxicity of nanomaterials. In addition, due to the rapid development of gene delivery,157 nucleic acids have also been used for disease treatment. To achieve high loading efficiency and maintain their medical functions (e.g., altering the expression of existing genes, producing cytotoxic proteins or prodrug-activating enzymes), it is necessary to protect the transfected nucleic acids from damage by the external environment as well as the nanocarriers (that translocate nucleic acids into the interior of specific cells). Generally, all these issues require a better understanding of the molecular mechanism of interactions between DNA/RNA and nanoparticles, especially the effect of nanoparticles on the structures of DNA/RNA molecules.Since the size of fullerenes is close to that of the grooves of DNA and RNA, they can bind into these sites, inducing conformational changes in their inherent structures.158 By using all-atom MD simulations, Zhao et al.159 investigated the interaction of C60 with single-strand (ss) and double-strand (ds) DNA, and found that despite the hydrophobic nature of C60, fullerenes can strongly bind to nucleotides, where their interaction strength is much larger than that between two C60 molecules in water. Their results also showed that C60 can bind to ssDNA and deform the nucleotides significantly (Fig. 7a), while C60 can penetrate into the double helix of dsDNA from the end, form stable hybrids (Fig. 7b), and frustrate the hydrogen bonds between end-group base-pairs in the nucleotide. More interestingly, when the DNA molecule is damaged (e.g., a gap is created by removing a piece of the nucleotide from one helix), fullerenes can stably occupy the damaged site (Fig. 7c), indicating that C60 may negatively impact the self-repairing process of dsDNA. A similar effect of C60 on DNA was also investigated by Xu et al.160 Additionally, they also revealed the difference of the interaction mechanism between DNA and RNA. It was found that C60 binds with the minor grooves of DNA and triggers unwinding and disruption of the DNA helix, indicating that C60 can potentially inhibit DNA replication and induce potential side effects. In contrast, C60 only binds to the major grooves of the RNA helix, which stabilizes the RNA structure or transforms the configuration from stretch to curl.
 | ||
| Fig. 7 Interactions between nucleic acids and different types of nanoparticles. (a) Binding of C60 with single-strand DNA; (b) binding of C60 with double-strand DNA; (c) C60 occupies the defect site of the DNA. Reproduced with permission from ref. 159, Copyright 2005 Elsevier. (d) Final simulation snapshots of interaction between SWCNTs and DNA and RNA; (e) radial distribution of phosphate groups of nucleic acid strands at distance r from the central axis of SWCNTs; (f) contact areas between SWCNTs and nucleic acid strands; (g) interaction energies between SWCNTs and nucleic acid strands. Reproduced with permission from ref. 163, Copyright 2015 American Chemical Society. | ||
Carbon nanotubes and graphene can also induce the adsorption of RNA/DNA molecules and influence their molecular structures.161 By using molecular simulations, Zhao et al.162 found that the hydrophobic end groups of DNA are attracted to the hydrophobic surface of uncharged SWCNTs, while the hydrophilic backbone of DNA does not bind to the uncharged SWCNTs. Moreover, the effect of SWCNTs on RNA and DNA is also different. As Landry et al.163 pointed out, during the binding, the DNA strand appears to be more flexible as its nucleotides act independently while the RNA strand appears more rigid (Fig. 7d and e). These differences lead to a stronger binding of ssDNA to SWCNTs than ssRNA and contribute to lesser conformational stability of ssRNA on SWCNTs in comparison to ssDNA (Fig. 7f and g). It should be noted that the driving forces of interaction between the naked carbon-based nanomaterials and nucleic acids are hydrophobic interaction and π–π stacking, which is similar to the case of proteins. When the carbon-based nanomaterials are decorated with some chemical group, the electrostatic interactions can also contribute a lot to their binding affinity with the contacting basepair.164 Besides, there also exist some differences on the final conformation of DNA on the surface, i.e., the DNA molecule can both stand up and lie flat on the surface of graphene,162 while it just stands up on a graphene oxide (GO) surface with its axis perpendicular to the surface of GO.164
In gene delivery, cationic nanoparticles, especially cationic polymers, are mostly used for condensing the DNA or RNA molecules, where the electrostatic interaction may become more important. By using all-atom MD simulations, Sun et al.33,165 systematically investigated the assembly mechanism of polyethylenimine (PEI) and DNA (or RNA). They found that PEI can condense DNA through two mechanisms: polyion bridging and electrostatic screening of the DNA charges. And depending on the molecular structure and protonation state of PEI, their interaction with DNA molecules is very different. Moreover, no obvious structure change of DNA and RNA molecules was found in their simulations, which is mainly due to the flexible structure of PEI molecules. In this sense, the PEI molecules will not affect the medical functions of nucleic molecules in gene therapy. However, because of their positively charged properties, they may also induce the aggregation of DNA originally in the cell nucleus so they may have some impact on the chromatin structures.
The above discussions mainly focus on the side effects of nanomaterials on cellular structure and function. In order to better use nanomaterials in biomedicine, it is of great importance to reduce the potential toxicity of nanomaterials. However, since causing the death of diseased or foreign cells is the main mechanism in medical therapy, the cytotoxicity is not harmful in many cases. Thus the most efficient and ideal way is to utilize the toxicity to kill specific cells while minimizing the potential toxicity to normal or healthy ones. Actually, this is a hugely challenging problem in biomedicine and nanotechnology, which is attracting more and more attention from both computational and experimental researchers. In the next sections, we will discuss these problems in detail on the basis of recent computational results and some significant experimental studies.
5 Natural and artificial strategies to reduce the cytotoxicity of nanomaterials
5.1 Natural defense: serum proteins and immune response
There are various types of serum proteins in the blood. Serum albumin comprises 55% of blood proteins, and is a major contributor to maintaining the osmotic pressure of plasma and assisting the transportation of lipids and steroid hormones. Globulins make up 37% of blood proteins and transport ions, hormones, and lipids. Fibrinogen accounts for 7% of blood proteins, and the conversion of fibrinogen to insoluble fibrin is essential for blood clotting. The remaining plasma proteins (1%) are regulatory ones, such as enzymes, proenzymes, and hormones. When nanoparticles are inserted into the blood, the serum proteins can bind to the surface of the nanoparticles and form a biological coating around the nanoparticle, which is known as the protein corona.166 Since the physicochemical properties of nanoparticles are greatly altered by the protein corona, their biological identity, including their effect on the cells and biomolecules, will become totally distinct (from their synthetic identity).167,168By combining experiments and simulations, Ge et al.169 found that different types of serum proteins including bovine fibrinogen (BFG), gamma globulin (lg), transferrin (Tf), and bovine serum albumin (BSA) can adsorb onto the surface of SWCNTs due to π–π stacking and hydrophobic interaction, with a competitive order BFG > Ig > Tf > BSA (Fig. 8a and b). More importantly, they also revealed that the competitive binding of blood proteins on SWCNT surfaces can greatly alter their interactions with cells, and in turn result in much reduced cytotoxicity for the protein-coated SWCNTs (Fig. 8c and d). More recently, a similar system was also investigated by Chong et al.170 By combining experimental (atomic force microscope, fluorescence spectroscopy, and surface plasmon resonance) and simulation-based (molecular dynamics) approaches, they systematically investigated the adsorption of four high-abundance blood proteins onto GO and reduced GO (rGO). The driving forces for the adsorption of blood proteins as well as the binding affinity orders are approximately the same (Fig. 8e–g) as that in the previous case of SWCNTs,169 and their results also showed that the cytotoxicity of graphene nanosheets could be reduced in the presence of blood proteins. Besides, GO was observed to exhibit a greatly enhanced protein adsorption capacity over one-dimensional SWCNTs, which results in markedly less cytotoxicity than protein-coated SWCNTs. Generally, the reduced cytotoxicity mainly originates from the following two reasons. On one hand, the adsorption of serum protein can reduce the generation of ROS by the carbon-based nanomaterials. On the other hand, it makes the nanomaterials more hydrophilic and greatly decreases the two driving forces mentioned above, which can weaken the direct impact of the nanoparticle–serum protein complex on other functional biomolecules. In this sense, one can simply infer that the destructive extraction of phospholipids from lipid membranes by graphene nanosheets36 as well as the insertion of graphene nanosheets into the cell membrane36,171 may also be weakened in the presence of serum protein, since the strong hydrophobic interaction between graphene and lipid tails is greatly reduced. Actually, Ding et al.172 recently used dissipative particle dynamics simulation to show that the insertion of hydrophobic nanoparticles into the lipid membrane interior is blocked in the presence of human serum albumin (HSA) (Fig. 8h and i). More recently, Duan et al.173 also revealed that the adsorption of BSA can weaken the interaction of graphene with the lipid bilayer and reduce the damage of the lipid bilayer due to the reduction of the available hydrophobic surface area plus an unfavorable steric effect by using all-atom molecular dynamics simulations.
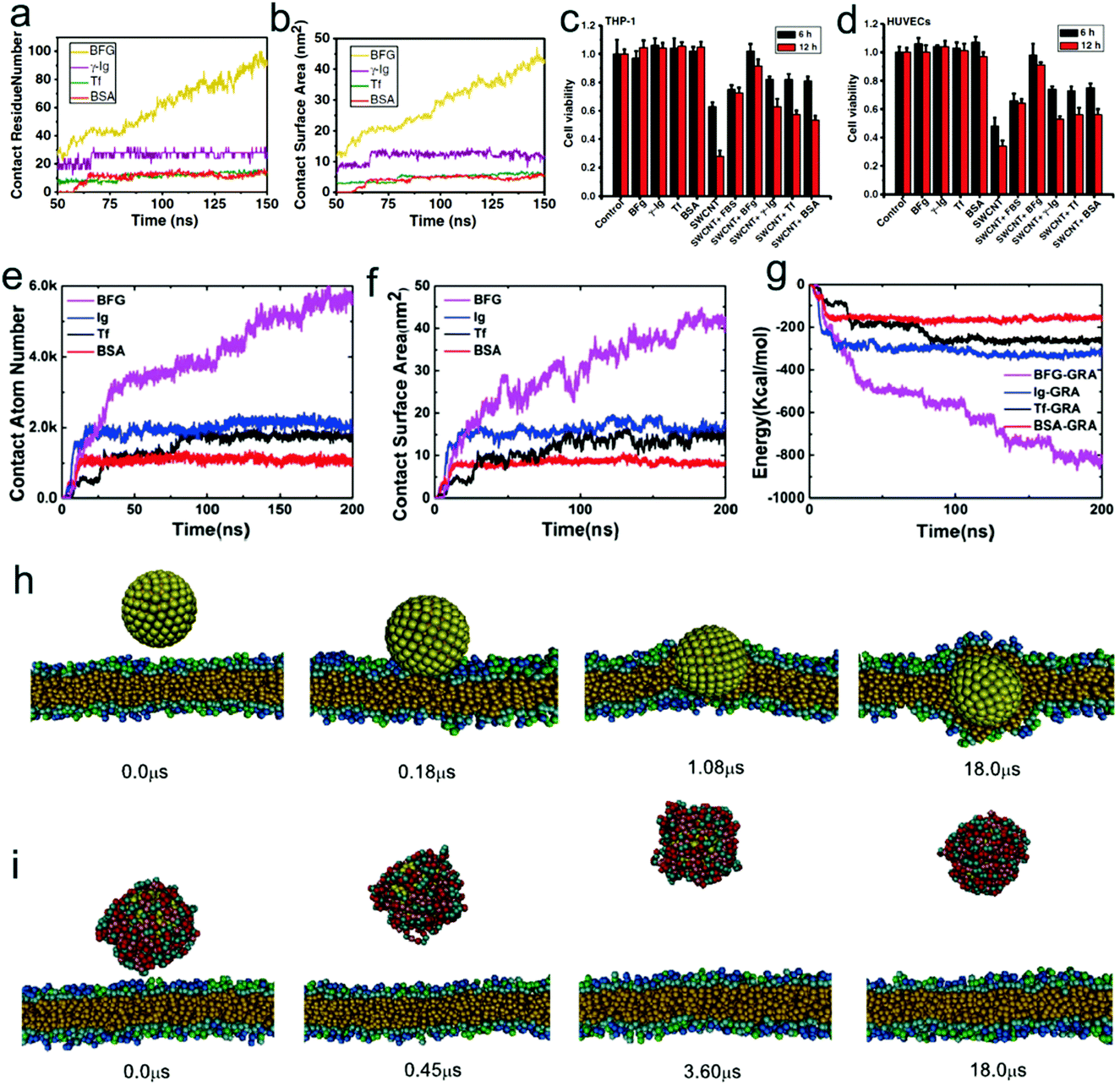 | ||
| Fig. 8 Effect of adsorption of serum proteins on the cytotoxicity and bio-behavior of nanoparticles. Simulation results of contact residue number (a) and contact surface area (b) of various serum proteins in adsorption of SWCNTs as a function of time. Experimental results of differential cytotoxicity of THP-1 (c) and HUVEC cells (d) in 30 μg mL−1 SWCNTs with/without protein coatings after treatment for 6 and 12 h. Reproduced with permission from ref. 169, Copyright 2011 National Academy of Sciences, USA. Time-series of the adsorption descriptors in MD simulations: (e) the contact atom number; (f) the contact surface area; and (g) the pair interaction energy between the proteins and graphene. Reproduced with permission from ref. 170, Copyright 2015 American Chemical Society. Time evolution of simulation snapshots of interactions between hydrophobic nanoparticles (with the radius of 3 nm) and cell membranes in the absence (h) and presence (i) of serum proteins. Reproduced with permission from ref. 172, Copyright 2014 Elsevier. | ||
Although the “acute” cytotoxicity of nanoparticles is greatly reduced by their interactions with blood proteins, it is very complicated to conclude whether coating serum proteins onto the nanoparticle surface has a positive or negative effect on the bio-systems in vivo. Actually, many studies indicate that nanoparticles covered with blood proteins are more visible to phagocytic cells, and may be removed from the bloodstream.174 To some extent, the clearance is useful for protecting the normal tissues in the body from damage by foreign objects. However, the clearance may greatly decrease the delivery efficiency of nanoparticles into diseased tissues. To make best use of proteins in the serum, on one hand, it is of great necessity to better identify the spatial location of proteins, their functional motifs, and their binding sites,175,176 which can be very important in molecular description of the biological identity of nanoparticles; on the other hand, since different types of proteins may lead to distinct bio-responses of nanoparticles, it is of great importance to well control the composition of the protein corona by tuning the physicochemical properties of nanoparticles.177
5.2 Artificial strategy: hydrophilic and stealth coating
The protein corona seems to be the natural coating to regulate the interaction between nanomaterials and biomolecules as well as cells. Over the last three decades, different types of surface decorations (i.e., artificial coatings) have also been synthesized experimentally to alter the physicochemical properties of bare nanoparticles. Actually, although the protein corona is what the cells see, the underlying surface ligands evidently play an important role in shaping and defining the physical characteristics of the corona, which ultimately impacts the cellular response.178 Therefore, surface functionalization of nanoparticles is an important aspect in tailoring NPs for specific therapeutic purposes.179 And we should notice that the surface functionalization of NPs is a very broad topic, for example, in nanoparticle design for drug delivery, it at least includes three types of coatings, namely the stealth coating, active-targeting coating, and environment-responsive coating.180 In this subsection, we will just focus on the effect of surface-decorated non-specific and non-responsive polymers on the interactions of nanoparticles with biomolecules and cell membranes.Polyethylene glycol (PEG) is a well-investigated polymer for the covalent modification of biological macromolecules and nanoparticles in many biomedical applications. PEGylation, known as covalently attaching PEG to therapeutic agents or nanoparticles, is firstly applied with small drug molecules181 and biomacromoles182 (e.g., proteins and enzymes) to overcome their shortcomings like low solubility, high toxicity, and nonspecific bio-distribution. Additionally, over the last three decades, it has been widely used in nanoparticle design, especially for the purpose of reducing the clearance of NPs from the circulation and protecting NPs from the immune system.183,184 Because of the high hydrophilicity of PEG (especially its ability of attracting large amounts of water molecules by hydrogen-bonding to its ether groups), when coating PEG onto the hydrophobic nanoparticle surface, the nanoparticle will become less hydrophobic (and even hydrophilic), and the hydrophobic interaction or π–π stacking between bare nanoparticles and protein hydrophobic residues or lipid tails will be very weak. For example, by combining simulations and experiments, Dragneva et al.185 found that significant conformational changes on immune-reactive sites in the D-domain of fibrinogen may occur when the proteins were adsorbed onto pure graphene surfaces while PEGylated graphene does not induce significant conformational changes due to the weakly hydrophobic interaction. A similar phenomenon was also observed in a PEGylated-nanoparticle–lipid membrane system. By using computer simulations, Li et al.186 found that the insertion of hydrophobic nanoparticles into the lipid membrane was blocked when they were coated with PEG molecules. Actually, even when adding an external force on the nanoparticles, less negative impact on the lipid membrane in the presence of PEGylation was observed.187
Moreover, since PEG is also charge-neutral, the electrostatic interaction between PEG-coated charged nanoparticles and negative serum proteins will become weak, leading to low affinity of protein adsorption. Meanwhile, the disruption of the negatively charged cell membrane by charged nanoparticles (e.g., dendrimers) is weakened when they are decorated with long PEG (Fig. 9a).188 Thus the cytotoxicity of charged NPs may be reduced. These results are consistent with the experimental findings, i.e., the PEGylation can indeed reduce the toxicity of nanoparticles (Fig. 9b).189 Generally, the coating of PEG onto the nanoparticle surface can not only weaken the interaction of nanoparticles with serum proteins and specific cellular receptors, but also block the insertion of nanoparticles into the lipid membrane as well as reduce the generation of ROS,190 thus it can greatly decrease the possibility of clearance by the immune system (to prolong the circulation time) and reduce the side effects of nanoparticles on the bio-systems.191 Nevertheless, the accelerated blood clearance (ABC) phenomenon192 (i.e., the intravenous injection of PEGylated liposomes may significantly alter the pharmacokinetic behavior of a second dose of PEGylated liposomes, when this second dose is administered after a several-day interval) may greatly limit their in vivo biomedical applications. Therefore, many other types of hydrophilic polymers like polyglycerol, polyoxazoline, and polysaccharide were also synthesized and have been used for biomedical applications in recent years.193 For example, chitosan, as one typical polysaccharide, has shown very excellent biocompatibility, admirable biodegradability with ecological safety, and low toxicity with versatile biological activities,194 which provide great opportunities for further development of nanoparticle design.
Zwitterionic polymers, containing both cationic and anionic groups (Fig. 9c), with their zeta potential usually close to zero, can also greatly reduce the rate and/or extent of non-specific adsorption of proteins to the nanoparticle surface.195,196 Due to their different molecular structures, zwitterionic materials possess highly charged groups, whereas PEG materials possess hydroxyl and ether groups.197 Therefore, there may exist some differences between zwitterionic polymers and PEG in some aspects like hydration and protein effect. For example, by using all-atom molecular simulations, Shao et al.198 compared the two polymers from a microscopic view. They calculated the hydration free energies of short zwitterionic chains and oligo-(ethylene glycol) (OEG) chains with the free energy perturbation method, and found that the hydration free energy of zwitterionic chains is much lower than that of nonionic OEG chains, indicating that zwitterionic materials exhibit stronger hydration. Moreover, and importantly, they also pointed out the difference between the different types of zwitterionic chains, i.e., sulfobetaine (SB) moieties attract more water molecules than carboxybetaine (CB) moieties, while the latter attracts individual water molecules more strongly (Fig. 9d and e). More recently, Ding et al.199 also compared the difference of protein resistance and cellular delivery between zwitterionic polymer and PEG from the mesoscopic view. By using DPD simulations, it was found that although the two types of polymers are charge-neutral and can both reduce the protein adsorption, there exist some differences between their ability of protein resistance, namely the charge mismatch of the charged parts between zwitterionic polymers and proteins (that induces the weakly repulsive electrostatic interaction) could make zwitterionic polymers more resistant to proteins. Moreover, both of the coating polymers may also greatly decrease the cellular uptake efficiency of nanoparticles. Nevertheless, and importantly, since the zwitterionic polymers may become positively charged under low pH environments, the nanoparticles can attach onto the cell membrane more firmly than those coated with PEG, which can further enhance the active targeting of nanoparticles. These results can give a reasonable explanation for the experimental observation of high uptake of zwitterionic polymer-coated nanoparticles by cancer cells.200 Generally, due to their easy preparation, chemical stability, low cost, and especially high protein-resistance ability, zwitterionic polymers may serve as a very promising next-generation biomaterial for a wide range of biomedical and engineering applications.
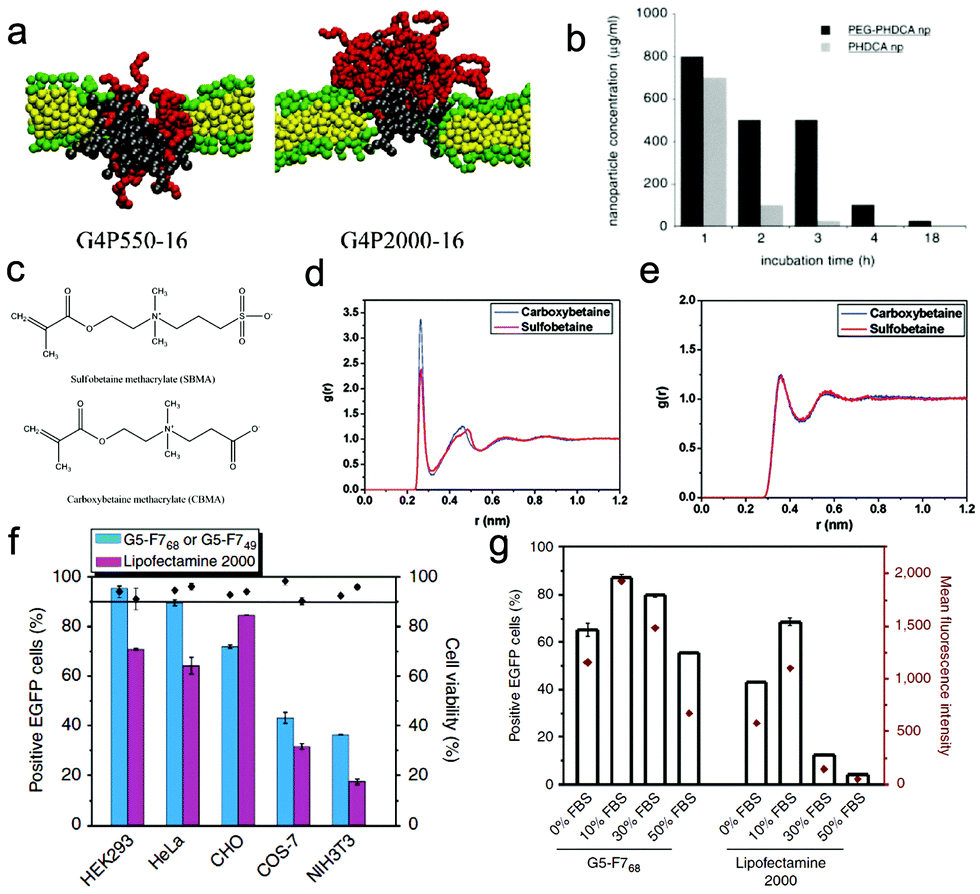 | ||
| Fig. 9 Effect of surface stealth coating on the cytotoxicity and bio-behavior of nanoparticles. (a) Final simulation snapshots of the PEGylated dendrimer-induced pores in lipid bilayers (left: G4P550-16, right: G4P2000-16). Reproduced with permission from ref. 188, Copyright 2011 American Chemical Society. (b) Concentration of PEG-PHDCA or PHDCA nanoparticles inducing 50% of cell death (IC50) as a function of the incubation time. Reproduced with permission from ref. 189, Copyright 1999 Elsevier. (c) Chemical structures of two different types of zwitterionic polymers; radial distribution functions between water molecules and certain atoms of the solute: (d) the oxygen atoms of the betaine molecules; (e) the carbon atoms in the methyl groups linked to the nitrogen atom of the betaine molecules. Reproduced with permission from ref. 198, Copyright 2010 American Chemical Society. (f) EGFP expression and cell viability in five cell lines transfected by fluorinated dendrimers and Lipofectamine 2000; (g) serum-resistance abilities of fluorinated dendrimers and Lipofectamine 2000 on EGFP gene transfection. Reproduced with permission from ref. 201, Copyright 2014 Nature Publication Group. | ||
Apart from the two types of polymers mentioned above, recently fluorinated polymers have also been used in nanoparticle design for drug/gene delivery. For example, Chen et al.201 took dendrimers as an example, and reported that decorating dendrimers with fluorinated polymers not only reduces their cytotoxicity (Fig. 9f), but also results in excellent serum resistance and a high gene transfection efficacy even in medium containing 50% FBS (Fig. 9g). This is mainly due to the fact that fluorination can resist protein adsorption, enhance the cellular uptake of the dendrimer/DNA polyplexes and facilitate their endosomal escape. Interestingly, our previous simulation results may give some hints for the enhanced cellular uptake behaviors, i.e., the frustrated engulfment may be avoided in the presence of fluorination.115 However, the underlying molecular mechanism for other processes is not completely revealed, and requires deep investigation.
6 Keeping the balance between therapeutic efficiency and cytotoxicity
Though artificial and natural coatings may minimize the potential toxicity of nanomaterials, they could be more useful if one can use the toxicity to diseased tissue. Actually, there have been some experiments that take advantage of the cytotoxicity in biomedical applications (e.g., antibacterial202 and antitumor203). Generally, the best way of using nanomaterials is to enhance the targeting ability to diseased cells and minimize the side effects on normal ones. Thus, recently more and more attention has been paid to the design of novel nanomaterials to increase the amount of nanoparticles in the targeted or diseased tissues and decrease the accumulation of nanoparticles (containing drug molecules) in the normal or healthy tissues.2046.1 Design strategy of active-targeting nanomaterials
It has been realized that tumor vasculature may have defective architecture with pore sizes ranging from 100 to 780 nm, allowing for extravasation of nanoparticles within this size range. Besides, tumor tissues usually lack effective lymphatic drainage, which prevents efficient removal of nanoparticles from tumors. These phenomena are known as the enhanced permeability and retention (EPR) effect,205,206 which results in the accumulation of nanoparticles at the tumor site. In order to enhance the passive targeting ability, nanoparticles should have the proper size (probably within the range of 20–100 nm). Moreover, nanoparticles also need to circulate for a prolonged period in the blood. In this sense, as we discussed before, a stealth coating like PEG and zwitterionic polymers can be decorated on the nanoparticle surface to avoid the adsorption of some types of serum proteins. However, the EPR effect is not very effective, i.e., the size-dependence, slow time frame, and variability among different tumors greatly limit its usefulness.180 And in order to better target the diseased tissues, together with the EPR effect, an active-targeting strategy, i.e., decorating with ligands that have specificity for receptors over-expressed on cancer cells (and normally or minimally expressed on healthy cells), should also be used in real applications.Typically, the commonly used ligands can be divided into four types, i.e., antibodies, peptides, aptamers, and other small molecules.207 Antibodies have very high selectivity and binding affinity due to the presence of two epitope binding sites in a single molecule.208 So when decorated onto the nanoparticle surface, they can greatly enhance the binding of nanoparticles to specific over-expressed targets in diseased cells. Peptides are also thought to be well suited for NP targeting because they are small, easy to synthesize and typically non-immunogenic, and have high avidity.209 Aptamers are single-stranded DNA or RNA oligonucleotides (typically 20–100 nucleotides in length) that fold into specific structures and bind to targeting molecules with high affinity.210 The folate receptor is a highly selective tumor marker over-expressed in greater than 90% of ovarian carcinomas. Folic acid (i.e., folate), a high affinity ligand of the folate receptor, retains its receptor binding properties when derivatized via its γ-carboxyl.211
Although a great many experimental studies have indicated that the coating of ligands onto the nanoparticle surface can indeed increase the uptake efficiency of the nanoparticles by cancer cells and improve their antitumor efficacy, it is still of great importance to make better use of the coating ligands. Actually, apart from the ligand density effect (nanoparticles with higher coverage of ligands are easier to take up),115 recently more and more computational studies have indicated that the surface distribution of ligands could also greatly affect the cellular uptake. By using DPD simulations, Schubertova et al.212 found that the speed of membrane wrapping was the fastest for sphere surface-functionalized nanoparticles with homogeneous ligand distributions. In contrast, when the ligand was distributed unhomogeneously, the speed could become very slow, and the nanoparticles may not be fully wrapped once there exist ligand-free patches on the nanoparticle surface. A similar phenomenon was also observed by Xue et al.213 using rod-like nanoparticles. Experimentally, they found that rod-like nanoparticles with smooth, abacus-like (i.e., beads-on-wires), and helical surface patterns can be prepared by the cooperative self-assembly of poly(γ-benzyl-L-glutamate)-block-poly(ethylene glycol) (PBLG-b-PEG) block copolymers and PBLG homopolymers. Additionally, combining the simulation and experiments, it was found that the smooth and helical ligand-coated nanoparticles were wrapped more quickly than the abacus-like ligand-coated nanoparticles (Fig. 10a–c). More importantly, they also found that the entry pathway could be also different under different surface distribution of ligands, e.g., helical NPs can enter the plasma membrane via a unique gyroscope-like pathway, and abacus-like NPs exhibit a tip entry pathway.
 | ||
| Fig. 10 (a) Schematic illustration of rod-like nanoparticles with different surface structures. (b) Experimental results of the normalized fluorescence intensity of the three NPs as a function of time; (c) simulation results of the wrapping percentage of a single NP as a function of time. Reproduced with permission from ref. 213, Copyright 2017 Wiley-VCH. (d) Time evolution of the order parameter for dual-ligand-coated nanoparticles with a ligand length of 5 (orange) and rigid nanoparticles (navy). (e) Time sequence of snapshots of interactions between dual-ligand coated Janus NPs and cell membranes. Reproduced with permission from ref. 218, Copyright 2017 Royal Society of Chemistry. | ||
Moreover, the strength of ligand–receptor interaction also plays an important role in the active targeting. Theoretically speaking, a strong ligand–receptor interaction is always beneficial for the uptake of nanoparticles. However, in the case of selective uptake, the strong interaction may not be the optimal choice since the uptake of nanoparticles by normal cells should be prevented. By using Monte Carlo simulations, Wang et al.214 found that for a range of surface-functionalized nanoparticles with monodisperse tethers, the desire for selective targeting of high receptor density at the adsorption onset can be achieved by decreasing the ligand–receptor binding energy or ligand density. In addition, they also implied that a bimodal tether distribution with shorter ligated tethers and longer nonfunctional tethers can be very beneficial in achieving selective nanoparticle adsorption. More recently, by using theory and simulation, Curk et al.215 also concluded that it is not a good idea to aim for very strong binding between the individual ligands on the surface-functionalized nanoparticles and the receptors on the cell. Instead, the individual ligand–receptor binding strength needs to be weak to achieve the selective targeting, namely the effective strength of the ligand–receptor interaction should be on the order of the thermal energy kBT. Besides, they also proposed that the concentration of the ligands on the nanoparticle should closely match the composition of the receptors on the target cell surface.
Since cancer cells typically over-express multiple receptors on their surface, more than one type of ligand can be decorated onto the nanoparticle surface to increase the targeting ability of NPs and enhance the delivery of NPs into cancer cells.216 However, some experiments also indicated that dual-ligand targeting cannot increase the delivery efficiency obviously.217 Thus the underlying molecular mechanism of cellular uptake of dual-ligand-coated NPs urgently needs to be revealed. Recently, by using dissipative particle dynamics simulations, Xia et al.218 systematically study the effect of the surface distribution and physicochemical properties of dual ligands on the cellular uptake of nanoparticles. It is found that the spontaneous rearrangement of dual ligands (from random to patterned distribution) on the nanoparticle surface can enhance the cellular uptake of nanoparticles. In contrast, since the short length of ligands may restrict the ligand rearrangement, nanoparticles coated with short dual ligands cannot be fully wrapped by cell membranes (Fig. 10d). In addition, it was also found that the physicochemical properties of ligands, such as the length mismatch between the dual ligands and the non-specific interaction among the ligands, may also have some negative effects on cellular uptake. To make best use of dual ligands, they demonstrated that NPs with a Janus distribution of dual ligands on their surface can be a good choice (Fig. 10e).
Though single or multiple types of ligands can increase the adhesion affinity or uptake efficiency of cancer cells, the targeting ability may be greatly influenced when used in vivo since the in vivo environment is so complicated and there are plenty of serum proteins in the blood. For example, in a recent experimental study, Salvati et al.118 found that the proteins in the media can shield transferrin conjugated on the nanoparticle surface from binding to both its targeting receptors on cells and soluble transferrin receptors (Fig. 11a and b). Thus the targeting ability of such functionalized nanoparticles may disappear when they are placed in a biological environment. Actually, in biology, most of the biological activity is driven by recognition of the surface-bound molecules by receptors. Thus, when the surface of nanoparticles is surrounded by the proteins, its recognition can be greatly changed and it may be “mistaken” by the cells under different situations.219 In order to avoid these undesired results, as we mentioned before, different types of hydrophilic polymers can be decorated together with specific ligands. However, during the decoration, one should still be very careful; despite their protein resistance ability, the hydrophilic polymers may also greatly affect the active targeting ability of nanoparticles. Recently, Dai et al.220 experimentally found that longer PEG coated on the receptor-targeting nanoparticle surface can also greatly prevent targeting recognition. Thus they concluded that the PEG length should be in appropriate regions, i.e., the length of the backfilled PEG molecules must be shorter than the length of the ligand linker (Fig. 11c and d). Recently, we used computer simulations to investigate the above phenomena in experiments. Our results show that the protein corona can change the interaction modes of hydrophobic nanoparticles and enhance the interaction of charged nanoparticles with macrophage cell membranes, while it may also cause the failure of insertion of hydrophobic nanoparticles and the loss of targeting specificity of charged nanoparticles with cancer cell membranes.172 To resist the adsorption of serum proteins, hydrophilic or zwitterionic polymers were also introduced in the simulation.199 Indeed it was found that though the decoration of charge-neutral polymers onto the nanoparticle surface can decrease the adsorption of proteins onto NPs, it may also block the insertion of nanoparticles and weaken the adsorption of charged nanoparticles onto the membrane. More importantly, by decorating antibodies onto the nanoparticle surface, we provided design maps for optimal surface decoration, and showed that protein resistance and active targeting can be simultaneously achieved by proper decoration (Fig. 11e and f), which was consistent with experimental findings.220
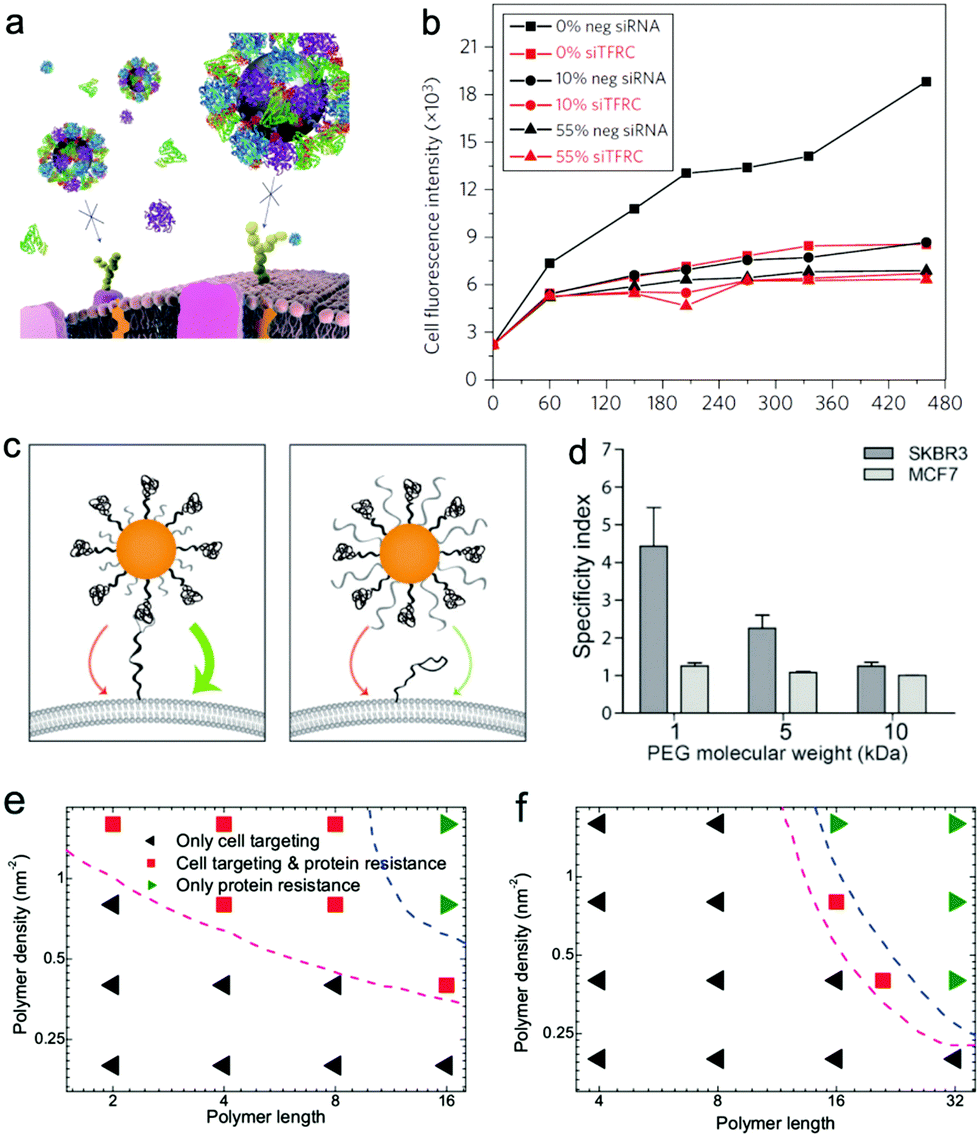 | ||
| Fig. 11 (a) Schematic representation of loss of targeting for Tf-conjugated nanoparticles in the presence of FBS proteins; (b) A549 cells are silenced for 72 h with a negative silencer control (neg siRNA) and for the transferrin receptor (siTFRC), before exposure to nanoparticles. Median cell fluorescence intensity obtained by flow cytometry from A549 cells exposed to 50 μg mL−1 PEGylated human Tf particles in serum-free MEM (0%), complete medium (10%) and MEM supplemented with 55% serum (55%). Reproduced with permission from ref. 118, Copyright 2013 Nature Publication Group. (c) Schematic representation of PEG length-dependent active targeting; (d) binding specificity of herceptin-conjugated gold nanoparticles with 1, 5, and 10 kDa PEG backfill incubated with SKBR3 and MCF7 cells post exposure to human serum. Reproduced with permission from ref. 220, Copyright 2014 Wiley-VCH. Phase diagrams describing the protein resistance and active targeting ability of polymer-coated nanoparticles on the polymer length–polymer density plane. (e) Hydrophobic nanoparticles with the size of 6 nm; (f) positively charged nanoparticle with the size of 10 nm. Reproduced with permission from ref. 199, Copyright 2016 Macmillan Publishers Ltd. | ||
6.2 Design strategy of stimulus-responsive nanomaterials
Notice that though targeted receptors are over-expressed in diseased cells, these receptors are also expressed to some degree on many types of healthy ones. As a result, the nanoparticles coated with specific ligands can still be endocytosed by normal cells. In addition, the active targeting may just increase the efficiency of cellular uptake and have less impact on the drastic increase of nanoparticle accumulation in tumors. In order to achieve higher delivery efficiency, in the last two decades, more and more attention has been paid to the design of stimulus-responsive nanoparticles to make best use of the difference between normal tissues and diseased ones.39,221Typically, the activation of stimulus-responsive nanoparticles can be divided into two types, i.e., the endogenous activation and the exogenous one.222
Endogenous activation uses the biochemical difference between the different micro-environments in the body to trigger the therapeutic activity. Particularly and importantly, tumors present multiple environments (e.g., pH, enzyme expression, and hypoxia) that are distinct from healthy tissues,222 which can be used for direct tumor targeting and precisely deliver drugs at the molecular level to the tumor site.
pH is one of the most widely used endogenous activations. The choice of using pH-sensitivity is mainly because there exist obvious (though not very sharp) pH differences between normal tissues, blood (pH ∼ 7.4) and tumors (pH ∼ 6.5).223 Due to the change in pH, the surface charge of nanoparticles may change from charge-neutral to positively charged, which can enhance the attachment of drug-containing nanoparticles onto negatively charged cancer cells,224 where a highly negatively charged cell membrane is another character of cancer cells. Recently, Ding et al.225 reported one new type of pH-responsive nanoparticle delivery system by using dissipative particle dynamics simulations. Differing from the main methods in experiments mentioned above, the key idea in the simulations is to use external pH-sensitive negatively charged polymers to “coat” and “uncoat” on the positively charged nanoparticle (e.g., gold nanoparticle decorated with short amino-groups) surface under different pH environments, which in turn can regulate the cellular uptake of nanoparticles. Interestingly, we also showed that the cellular uptake exhibits triply pH-responsive behavior (Fig. 12a): for lower and higher pH, the nanoparticles can be taken up by cell membranes, while for pH in the middle range, the endocytosis is blocked. This may give the designed delivery system some advantages in multi-pH bio-systems. In addition, the pH can also be different in the intracellular organelles (from extracellular environments). For example, the pH in the late endosome is about 6.0 and the pH in the lysosome is about 5.0. These differences have also been widely used for intracellular drug release.226,227 Usually, the responsive part is introduced as the linker that connects the drug to the polymer backbone, where the linker is degradable under acidic environments (e.g., pH ∼ 5.0).228 In addition, pH-responsive micelles (assembled by pH-sensitive polymers) containing drug molecules are another common pH-triggered drug release system. For example, by using DPD simulations, Wang et al.229 investigated the stability of DOX-loaded micelles prepared from block polymers His–Lys conjugated with docosahexaenoic acid under different pH conditions. When pH > 6.0, the micelles self-assembled from the polymers were dense and compact, with the drugs entrapped well in the micellar core. When pH > 6.0, the micelles shows a structural transformation from dense to swollen conformation, leading to an increased particle size and accelerated DOX release from micelles, which is also dependent on the ratio of His in the polymer (see Fig. 12b and c). Similar release behavior was reported by Luo et al.230 using pH-sensitive PAE-g-PEGLA vesicles, where they also proposed that the hybrid vesicle formed by a mixture of two copolymers can be used to control the release rate of DOX by combining MD and DPD simulations.
 | ||
| Fig. 12 Stimuli-responsive nanoparticles for drug delivery. (a) Snapshots of the nanoparticle–pH-sensitive polymer complex interacting with cell membranes under different pH conditions. Reproduced with permission from ref. 225, Copyright 2013 Macmillan Publishers Ltd. Kinetic micelle formation from (column 1) the homogeneous state (pH > 6.0) and the structural transformation of the micelles from (column 2) pH > 6.0 to (column 3) pH < 6.0. (b) The number of histidine residue is 10; (c) the number of histidine residue is 0. Reproduced with permission from ref. 229, Copyright 2015 American Chemical Society. (d) Simulation snapshots illustrating the release from swollen gel capsules of small nanoparticles in the presence/absence of microrods. (e) Cumulative fraction of released nanoparticles versus time. The squares, circles, triangles, crosses, and diamonds are for initially equilibrated capsules, swelling capsules, deswelling capsules, and deswelling capsules with one rod and two rods, respectively. Reproduced with permission from ref. 243, Copyright 2012 American Chemical Society. | ||
As the tumor mass increases, the interior of the tumor grows farther away from the existing blood supply, which may lead to a hypoxic microenvironment in tumors.231 Thus hypoxia is a primary target in the development of stimulus-responsive delivery because it is rarely found in normal tissues. For example, Thambi et al.232 designed one type of hypoxia-responsive nanoparticle (HR-NP), where a hydrophobically modified 2-nitroimidazole derivative was conjugated to the backbone of carboxymethyl dextran (CM-Dex) and DOX was effectively encapsulated into the interior. The in vitro results showed that DOX-loaded HR-NPs induce higher toxicity to hypoxic cells than to normoxic cells; and the in vivo results further demonstrated that HR-NPs were selectively accumulated at the hypoxic tumor tissues. Tumor environments also contain different types of enzymes from those observed in normal tissues. For example, the matrix metalloproteinase (MMP) is usually over-expressed in tumor tissues.221 For example, Wong et al.233 engineered a 100 nm nanoparticle with a core composed of gelatin and a surface covered with quantum dots. Enzymes like MMP-2 in the tumor environment can degrade the cores of 100 nm gelatin nanoparticles, and induce the release of smaller 10 nm nanoparticles from the original 100 nm nanoparticles. Such size change was efficient and effective in the enhancement of deeper penetration into tumors. However, to the best of our knowledge, there do not exist any computer simulations on the above topic, probably due to the complex and the large size of the tumor environment system. Presently, there only exist some phenomenological theories to qualitatively explain the experimental results. For example, Xue et al.234 developed a non-equilibrium thermodynamic theory to study the chemo-mechanical behaviors of tumor extracellular matrix (ECM). It was found that the osmosis is primarily responsible for the compression resistance of the ECM and well-designed ECM degradation can effectively modify the tumor microenvironment for improved efficiency of cancer therapy.
Exogenous activation uses external stimuli such as light, magnetic field, ultrasonics, and temperature to trigger the onset of therapy.222 The advantage of these activations is their temporal and spatial control over the triggering event.
The usage of light-responsive nanomaterials in biomedicine is usually by introducing a linker that can be cleaved upon irradiation with light of a certain wavelength.235 Such photo-cleavable polymers involve pyrenylmethyl esters, o-nitrobenzyl esters, and esters of (diethylamino)methylcoumarinyl.236 Additionally, the external light may also destabilize the micelle nanocarriers by changing the hydrophobic/hydrophilic properties of the copolymers, inducing the swelling of the micellar core to allow the release of the drug molecules. This property is very similar to the nanomaterials containing thermo-responsive polymers, where their hydrophobic/hydrophilic property can sensitively respond to the external temperature within a very narrow range. Poly(N-isopropylacrylamide) (PNIPAm) is a representative thermo-responsive polymer since it has a very sharp coil–globule transition, and copolymerization of other types of polymers onto PNIPAm has been widely used for drug carrier design.237 Furthermore, in order to activate the thermal stimulus, other external methods are needed to alter the temperature in the targeted tissues. For example, near-infrared light can be used to excite gold nanorods inside the tumor to generate heat that can trigger localized drug release.238
Ultrasound, with its frequency higher than the upper audible limit of human hearing, can also be used as an external trigger since it is non-invasive and allows spatial and temporal control. For example, Di et al.239 developed a novel ultrasound-triggered controlled insulin delivery system based on an injectable nano-network, where the nano-network can be effectively triggered to release insulin upon focused ultrasound system (FUS)-mediated administration. Due to its non-specificity, this system may be extended to deliver other drugs, therapeutic proteins, and nucleic acids etc. Similar to ultrasound, a magnetic field can also be used to control the timing and dose of repeatedly released drug molecules from nanoparticles. For example, Kong et al.240 designed one type of nanocapsule containing intentionally trapped magnetic nanoparticles and anticancer drugs. The nanocapsules can penetrate into the interior of tumors and allow a controlled on–off switchable release of the drug cargo via a remote radio frequency magnetic field. And both the in vitro and in vivo results showed that these nanocapsules can be enriched near mouse breast tumors and are effective in reducing tumor cell growth. However, these two methods may introduce additional energy to the system, and as we mentioned before, the external force or torsion on nanoparticles may have some negative impact. Thus one must be very careful to avoid potential side effects when using these methods.
Since these external stimuli like light and ultrasound are not easy to be included in the molecular simulations, presently it is very difficult to directly simulate the process of drug-release or cellular delivery of these responsive nanomaterials. In order to qualitatively or semi-quantitatively model these external stimuli, there are some attempts in simulations. For example, an external force,94 torque99 or momentum241 is usually used to model a magnetic field or ultrasound acting on the nanomaterials. Additionally, by adopting a reversible reaction between nanoparticles and ligands, Ding et al.242 used DPD simulations to design one new type of nanoparticle that can spontaneously penetrate through membranes. The dynamic bonds between nanoparticles and coating ligands may be broken when they were inserted into the cell membrane, thus the designed nanoparticles seem like light-responsive nanoparticles to some extent. A similar modeling method was used by Masould et al.243 to report one type of external stimuli-responsive nanocapsule, where they supposed that the bond length may be changed under external stimuli. On this basis, they found that the nanocapsules allow steady diffusive release of small nanoparticles and polymer chains, whereas gel deswelling causes burst-like discharge of solutes driven by an outward flow of the solvent enclosed within a shrinking capsule, and they also demonstrated that this hydrodynamic release can be regulated by introducing rigid microscopic rods in the capsule interior (see Fig. 12d and e). Generally, although the external stimuli cannot be completely included in simulations, computational work with some assumptions may still give some hints for future experimental design of novel responsive nanomaterials.
7 Summary and perspective
Given the great promise that nanomaterials hold for biomedical applications, a comprehensive understanding of cell–nanomaterial interactions, especially their potential side effect on cells and bio-systems, is becoming more and more important. In this review, we have illustrated the physical mechanism of the effect of nanomaterials on lipid membranes as well as other functional biomolecules on the basis of recent computational studies. Additionally, the role of natural and artificial coatings in regulating the cytotoxicity are discussed in detail. Furthermore, we also highlight the latest advances in the design of new and novel nanomaterials for achieving highly targeted and controllable delivery.Despite the impressive progress on the above issues, there still exist several hugely challenging problems waiting to be solved in the future. Firstly, there exist obvious differences between short-term toxic effects and long-term toxic effects of nanomaterials on bio-systems.244 Though recently the long-term toxic effects of nanomaterials have been studied by some groups,245,246 more attention should be paid to the underlying mechanism of long-term toxicity at the molecular, cellular and organism levels. Secondly, as we mentioned in the main text, there still exist many problems like interaction of different stimuli-responsive nanoparticles with biosystems that the simulations cannot well answer presently, which requires the improvement of present computational techniques and high-performance computing ability, especially the development of all-atom or coarse-grained force fields for the complex nano-biosystems under different situations. Actually, even for the well-studied system like the nanoparticle–membrane or nanoparticle–protein system, presently the simulation results can only be used to explain some or a small part of the experimental results, and there still exists a big gap between the simulations and experiments. To overcome this gap, deeper and more frequent cooperation between theoretical and experimental researchers is urgently needed. Generally, the balance between therapeutic efficiency and cytotoxicity should be well kept when using nanomaterials in real applications, which needs multidisciplinary research studies among physics, chemistry, materials science, medicine, biology, and environmental sciences.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21604060, 91427302 and 11474155).References
- P. Moriarty, Rep. Prog. Phys., 2001, 64, 297–381 CrossRef CAS.
- F. Caruso, T. Hyeon and V. M. Rotello, Chem. Soc. Rev., 2012, 41, 2537–2538 RSC.
- G. Y. Chen, I. Roy, C. H. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- T. M. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. X. Yang and Y. N. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. G. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- J. J. Xu, W. W. Zhao, S. P. Song, C. H. Fan and H. Y. Chen, Chem. Soc. Rev., 2014, 43, 1601–1611 RSC.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- S. Mitragotri, D. G. Anderson, X. Y. Chen, E. K. Chow, D. Ho, A. V. Kabanov, J. M. Karp, K. Kataoka, C. A. Mirkin, S. H. Petrosko, J. J. Shi, M. M. Stevens, S. H. Sun, S. Teoh, S. S. Venkatraman, Y. N. Xia, S. T. Wang, Z. Gu and C. J. Xu, ACS Nano, 2015, 9, 6644–6654 CrossRef CAS PubMed.
- A. E. Nel, E. Nasser, H. Godwin, D. Avery, T. Bahadori, L. Bergeson, E. Beryt, J. C. Bonner, D. Boverhof, J. Carter, V. Castranova, J. R. Deshazo, S. M. Mussain, A. B. Kane, F. Klaessig, E. Kuempel, M. Lafranconi, R. Landsiedel, T. Malloy, M. B. Miller, J. Morris, K. Moss, G. Oberdorster, K. Pinkerton, R. C. Pleus, J. A. Shatkin, R. Thomas, T. Tolaymat, A. Wang and J. Wong, ACS Nano, 2013, 7, 6422–6433 CrossRef CAS PubMed.
- A. E. Nel, Y. L. Zhao and L. Madler, Acc. Chem. Res., 2013, 46, 605–606 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- Q. X. Mu, G. B. Jiang, L. X. Chen, H. Y. Zhou, D. Fourches, A. Tropsha and B. Yan, Chem. Rev., 2014, 114, 7740–7781 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- P. Decuzzi, ACS Nano, 2016, 10, 8133–8138 CrossRef CAS PubMed.
- A. E. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- S. T. Kim, K. Saha, C. Kim and V. M. Rotello, Acc. Chem. Res., 2013, 46, 681–691 CrossRef CAS PubMed.
- A. Nel, T. Xia, H. Meng, X. Wang, S. J. Lin, Z. X. Ji and H. Y. Zhang, Acc. Chem. Res., 2013, 46, 607–621 CrossRef CAS PubMed.
- M. T. Zhu, G. J. Nie, H. Meng, T. Xia, A. E. Nel and Y. L. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- Y. L. Wu, N. Putcha, K. W. Ng, D. T. Leong, C. T. Lim, S. C. J. Loo and X. D. Chen, Acc. Chem. Res., 2013, 46, 782–791 CrossRef CAS PubMed.
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC.
- Y. Zhang, Y. H. Bai, J. B. Jia, N. N. Gao, Y. Li, R. N. Zhang, G. B. Jiang and B. Yan, Chem. Soc. Rev., 2014, 43, 3762–3809 RSC.
- S. J. Soenen, W. J. Parak, J. Rejman and B. Manshian, Chem. Rev., 2015, 115, 2109–2135 CrossRef CAS PubMed.
- D. Frenkel and B. Smit, Understanding Molecular Simulations: From Algorithms to Applications, Elsevier, Singapore, 2002 Search PubMed.
- B. Dunweg and W. Paul, Int. J. Mod. Phys. C, 1991, 2, 817–827 CrossRef.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- Z. G. Qu, X. C. He, M. Lin, B. Y. Sha, X. H. Shi, T. J. Lu and F. Xu, Nanomedicine, 2013, 8, 995–1011 CrossRef CAS PubMed.
- A. H. Bahrami, M. Raatz, J. Agudo-Canalejo, R. Michel, E. M. Curtis, C. K. Hall, M. Gradzielski, R. Lipowsky and T. R. Weikl, Adv. Colloid Interface Sci., 2014, 208, 214–224 CrossRef CAS PubMed.
- H. M. Ding and Y. Q. Ma, Small, 2015, 11, 1055–1071 CrossRef CAS PubMed.
- S. L. Zhang, H. J. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- N. Yanamala, V. E. Kagan and A. A. Shevedova, Adv. Drug Delivery Rev., 2013, 65, 2070–2077 CrossRef CAS PubMed.
- C. A. Jimenez-Cruz, S. G. Kang and R. H. Zhou, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2014, 6, 329–343 CrossRef CAS PubMed.
- D. Meneksedag-Erol, T. Tang and H. Uludag, Biomaterials, 2014, 35, 7068–7076 CrossRef CAS PubMed.
- J. Q. Lin, H. W. Zhang, Z. Chen and Y. G. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS PubMed.
- G. H. Zuo, Q. Huang, G. H. Wei, R. H. Zhou and H. P. Fang, ACS Nano, 2010, 4, 7508–7514 CrossRef CAS PubMed.
- Y. S. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. R. Liu, Q. Huang, C. H. Fan, H. P. Fang and R. H. Zhou, Nat. Nanotechnol., 2013, 8, 594–601 CrossRef CAS PubMed.
- J. W. Ekkabut, S. Baoukina, W. Triampo, I. Tang, D. P. Tieleman and L. Monticelli, Nat. Nanotechnol., 2008, 3, 363–368 CrossRef PubMed.
- M. Calvaresi, S. Furini, C. Demene, A. Bottoni and F. Zerbetto, ACS Nano, 2015, 9, 4827–4834 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. M. de la Fuente and V. Grazu, Nanobiotechnology: Inorganic Nanoparticles Vs Organic Nanoparticles, Front. Nanosci., 2012, 4, 35–80 Search PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CAS.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS PubMed.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- J. Li, C. H. Fan, H. Pei, J. Y. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- Y. Pan, X. W. Du, F. Zhao and B. Xu, Chem. Soc. Rev., 2012, 41, 2912–2942 RSC.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- X. H. Huang, P. K. Jain, I. H. EI-Sayed and M. A. EI-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Metraux, J. I. Culter and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS PubMed.
- S. J. Guo and S. J. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 RSC.
- Z. X. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- C. Sun, J. S. H. Lee and M. Q. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- A. M. Smith, H. W. Duan, A. M. Mohs and S. M. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- B. S. Harrison and A. Atala, Biomaterials, 2007, 28, 344–353 CrossRef CAS PubMed.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696–753 RSC.
- C. E. Ashley, E. C. Carnes, G. K. Philips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. M. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chacherian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–398 CrossRef CAS PubMed.
- F. Wang, G. M. Pauletti, J. T. Wang, J. M. Zhang, R. C. Ewing, Y. L. Wang and D. L. Shi, Adv. Mater., 2013, 25, 3485–3489 CrossRef CAS PubMed.
- B. Y. Shi, X. Du, J. Chen, L. B. Fu, M. Morsch, A. Lee, Y. Liu, N. Cole and R. Chung, Small, 2017, 13, 1603966 CrossRef PubMed.
- L. L. Zhou, J. J. Yuan and Y. Wei, J. Mater. Chem., 2011, 21, 2823–2840 RSC.
- R. Bradley and R. Ravi Radhakrishnan, Polymer, 2013, 5, 890–936 Search PubMed.
- A. K. Jana, M. K. Tiwari, K. Vanka and N. Sengupta, Phys. Chem. Chem. Phys., 2016, 18, 5910–5924 RSC.
- G. Yang, H. M. Ding, Z. Kochovski, R. T. Hu, Y. Lu, Y. Q. Ma, G. S. Chen and M. Jiang, Angew. Chem., Int. Ed., 2017, 56, 10691–10695 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wirkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, L. Vorobyov and A. D. Mackerell Jr, J. Comput. Chem., 2010, 31, 671–690 CAS.
- H. Heinz, T.-J. Lin, R. K. Mishra and F. S. Emami, Langmuir, 2013, 29, 1754–1765 CrossRef CAS PubMed.
- H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CAS.
- A. Kyrychenko, O. M. Korsun, I. I. Gubin, S. M. Kovalenko and O. N. Kalugin, J. Phys. Chem. C, 2015, 119, 7888–7899 CAS.
- S. J. Stuart, A. B. Tutein and J. A. Harrison, J. Chem. Phys., 2000, 112, 6472–6486 CrossRef CAS.
- H. Heinz and H. Ramezani-Dakhel, Chem. Soc. Rev., 2016, 45, 412–448 RSC.
- H. Heinz, C. Pramanik, O. Heinz, Y. F. Ding, R. K. Mishra, D. Marchon, R. J. Flatt, I. Estrela-Lopis, J. Llop, S. Moya and R. F. Ziolo, Surf. Sci. Rep., 2017, 72, 1–58 CrossRef CAS.
- H. M. Ding and Y. Q. Ma, Nanoscale, 2012, 4, 1116–1122 RSC.
- H. M. Ding and Y. Q. Ma, Biomaterials, 2013, 34, 8401–8407 CrossRef CAS PubMed.
- S. J. Marrink, A. H. de Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS.
- K. Meier, A. Choutko, J. Dolenc, A. P. Eichenberger, S. Riniker and W. F. van Gunsteren, Angew. Chem., Int. Ed., 2013, 52, 2820–2834 CrossRef CAS PubMed.
- H. I. Ingolfsson, C. A. Lopez, J. Uusitalo, D. H. de Jong, S. M. Gopal, X. Periole and S. J. Marrink, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 225–248 CrossRef CAS PubMed.
- M. G. Saunders and G. A. Voth, Annu. Rev. Biophys., 2013, 42, 73–93 CrossRef CAS PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular biology of the cell, Garland Science, Oxford, 4th edn, 2002 Search PubMed.
- R. Gupta and B. Rai, Sci. Rep., 2017, 7, 45292 CrossRef CAS PubMed.
- A. R. Mhashal and S. Roy, PLoS One, 2014, 9, e114152 Search PubMed.
- A. R. Mhashal and S. Roy, ChemPhysChem, 2016, 17, 3504–3514 CrossRef CAS PubMed.
- M. Dallavalle, M. Calvaresi, A. Bottoni, M. Melle-Franco and F. Zerbetto, ACS Appl. Mater. Interfaces, 2015, 7, 4406–4414 CAS.
- Y. Liu, B. Peng, S. Sohrabi and Y. L. Liu, Langmuir, 2016, 32, 10136–10143 CrossRef CAS PubMed.
- R. C. V. Lehn and A. Alexander-Katz, J. Phys. Chem. B, 2014, 118, 12586–12598 CrossRef PubMed.
- X. B. Lin and N. Gu, Nano Res., 2014, 7, 1195–1204 CrossRef CAS.
- X. B. Lin, C. L. Wang, M. Wang, K. Fang and N. Gu, J. Phys. Chem. C, 2012, 116, 17960–17968 CAS.
- J. W. Feng, H. M. Ding and Y. Q. Ma, J. Chem. Phys., 2014, 141, 094901 CrossRef PubMed.
- K. Yang and Y. Q. Ma, Nat. Nanotechnol., 2010, 5, 579–583 CrossRef CAS PubMed.
- K. Shimizu, H. Nakamura and S. Watano, Nanoscale, 2016, 8, 11897–11906 RSC.
- B. Song, H. J. Yuan, S. V. Pham, C. J. Jameson and S. Murad, Langmuir, 2012, 28, 16989–17000 CrossRef CAS PubMed.
- P. A. Oroskar, C. J. Jameson and S. Murad, Langmuir, 2015, 31, 1074–1085 CrossRef CAS PubMed.
- N. Arai, K. Yasuoka and X. C. Zeng, Nanoscale, 2013, 5, 9089–9100 RSC.
- T. T. Yue, X. R. Zhang and F. Huang, Soft Matter, 2015, 11, 456–465 RSC.
- L. T. Yan and X. B. Yu, ACS Nano, 2009, 3, 2171–2176 CrossRef CAS PubMed.
- H. Lee and R. G. Larson, J. Phys. Chem. B, 2008, 112, 7778–7784 CrossRef CAS PubMed.
- L. Q. Xie, W. D. Tian and Y. Q. Ma, Soft Matter, 2013, 9, 9319–9325 RSC.
- C. L. Ting and Z. G. Wang, Soft Matter, 2012, 8, 12066–12071 RSC.
- U. Kwolek, D. Jamroz, M. Janiczek, M. Nowakowska, P. Wydro and M. Kepczynski, Langmuir, 2016, 32, 5004–5018 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- T. T. Yue and X. R. Zhang, Soft Matter, 2011, 7, 9104–9112 RSC.
- K. Unfried, C. Albrecht, L. O. Klotz, A. V. Mikecz, S. G. Beck and R. P. F. Schins, Nanotoxicology, 2007, 1, 52–71 CrossRef CAS.
- X. H. Shi, A. V. D. Bussche, R. H. Hurt, A. B. Kane and H. J. Gao, Nat. Nanotechnol., 2011, 6, 714–719 CrossRef CAS PubMed.
- J. Mao, P. Y. Chen, J. S. Liang, R. H. Guo and L. T. Yan, ACS Nano, 2016, 10, 1493–1502 CrossRef CAS PubMed.
- X. Yi and H. J. Gao, Nanoscale, 2015, 7, 5457–5467 RSC.
- X. Yi, X. H. Shi and H. J. Gao, Phys. Rev. Lett., 2011, 107, 098101 CrossRef PubMed.
- X. Yi and H. J. Gao, Langmuir, 2016, 32, 13252–13260 CrossRef CAS PubMed.
- T. T. Yue and X. R. Zhang, Soft Matter, 2013, 9, 559–569 RSC.
- J. S. Sun, L. Zhang, J. L. Wang, Q. Feng, D. B. Liu, Q. F. Yin, D. Y. Z. Xu, Y. J. Wei, B. Q. Ding, X. H. Shi and X. Y. Jiang, Adv. Mater., 2015, 27, 1402–1407 CrossRef CAS PubMed.
- H. M. Ding and Y. Q. Ma, Biomaterials, 2012, 33, 5798–5802 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. A. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- A. Manke, L. Y. Wang and Y. Rojanasakul, BioMed Res. Int., 2013, 2013, 942916 Search PubMed.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, Chem. Soc. Rev., 2015, 44, 6094–6121 RSC.
- A. A. Shemetov, I. Nabiev and A. Sukhanova, ACS Nano, 2012, 6, 4585–4602 CrossRef CAS PubMed.
- G. H. Zuo, S. G. Kang, P. Xiu, Y. L. Zhao and R. H. Zhou, Small, 2013, 9, 1546–1556 CrossRef CAS PubMed.
- G. H. Zuo, X. Zhou, Q. Huang, H. P. Fang and R. H. Zhou, J. Phys. Chem. C, 2011, 115, 23323–23328 CAS.
- A. K. Jana, J. C. Jose and N. Sengupta, Phys. Chem. Chem. Phys., 2013, 15, 837–844 RSC.
- A. K. Jana and N. Sengupta, Biophys. Chem., 2013, 184, 108–115 CrossRef CAS PubMed.
- A. K. Jana and N. Sengupta, Soft Matter, 2015, 11, 269–279 RSC.
- G. Wei, Z. Q. Su, N. P. Reynolds, P. Arosio, L. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- S. Durdagi, Bioorg. Med. Chem. Lett., 2008, 18, 6283–6289 CrossRef CAS PubMed.
- B. Q. Luan, T. Huynh, L. Zhao and R. H. Zhou, ACS Nano, 2015, 9, 663–669 CrossRef CAS PubMed.
- Z. L. Gu, Z. X. Yang, S. G. Kang, J. R. Yang., J. D. Luo and R. H. Zhou, Sci. Rep., 2016, 6, 28252 CrossRef CAS PubMed.
- B. Y. Li, W. F. Li, J. M. Perez-Aguilar and R. H. Zhou, Small, 2017, 13, 1603685 CrossRef PubMed.
- G. Brancolini, D. Toroz and S. Corni, Nanoscale, 2014, 6, 7903–7911 RSC.
- G. Brancolini, A. Corazza, M. Vuano, F. Fogolari, M. C. Mimmi, V. Bellotti, M. Stoppini, S. Corni and G. Esposito, ACS Nano, 2015, 9, 2600–2613 CrossRef CAS PubMed.
- J. P. P. Hernandez, Z. H. Tang, Z. E. Hughes, Y. Li, M. T. Swihart, P. N. Prasad, T. R. Walsh and M. R. Knecht, Chem. Mater., 2014, 26, 4960–4969 CrossRef.
- L. B. Wright, P. M. Rodger, S. Corni and T. R. Walsh, J. Chem. Theory Comput., 2013, 9, 1616–1630 CrossRef CAS PubMed.
- F. Ding, S. Radic, R. Chen, P. Y. Chen, N. K. Geitner, J. M. Brown and P. C. Ke, Nanoscale, 2013, 5, 9162–9169 RSC.
- S. K. Ramakrishnan, M. Martin, T. Cloitre, L. Firlej and C. Gergley, J. Chem. Inf. Model., 2014, 54, 2117–2126 CrossRef CAS PubMed.
- N. Hildebrand, S. Koppen, L. Derr, K. Li, M. Koleini, K. Rezwan and L. C. Ciacchi, J. Phys. Chem. C, 2015, 119, 7295–7307 CAS.
- Y. Yang, Q. Cui and N. Sahai, Langmuir, 2010, 26, 9848–9859 CrossRef CAS PubMed.
- T.-J. Lin and H. Heinz, J. Phys. Chem. C, 2016, 120, 4975–4992 CAS.
- R. Domenech, O. Abian, R. Bocanegra, J. Correa, A. S. Herves, R. Riguera, M. G. Mateu, E. F. Meia, A. V. Campoy and J. L. Neira, Biomacromolecules, 2010, 11, 2069–2078 CrossRef CAS PubMed.
- L. M. Wang, J. Y. Li, J. Pan, X. M. Jiang, Y. L. Li, Y. F. Li, Y. Qu, Y. L. Zhao, X. C. Wu and C. Y. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- F. S. Emami, V. Puddu, R. J. Berry, V. Varshney, S. V. Patwardhan, C. C. Perry and H. Heinz, Chem. Mater., 2014, 26, 5725–5734 CrossRef CAS.
- S. V. Patwardhan, F. S. Emami, R. J. Berry, S. E. Jones, R. R. Naik, O. Deschaume, H. Heinz and C. C. Perry, J. Am. Chem. Soc., 2012, 134, 6244–6256 CrossRef CAS PubMed.
- H. Ramezani-Dakhel, N. M. Bedford, T. J. Woehl, M. R. Knecht, R. R. Naik and H. Heinz, Nanoscale, 2017, 9, 8401–8409 RSC.
- H. Ramezani-Dakhel, L. Y. Ruan, Y. Huang and H. Heinz, Adv. Funct. Mater., 2015, 25, 1374–1384 CrossRef CAS.
- L. Boselli, E. Polo, V. Castagnola and K. A. Dawson, Angew. Chem., Int. Ed., 2017, 56, 4215–4218 CrossRef CAS PubMed.
- C. Maffeo, S. Bhattacharya, J. Yoo, D. Wells and A. Aksimentiev, Chem. Rev., 2012, 112, 6250–6284 CrossRef CAS PubMed.
- S. Kraszewski, M. Tarek, W. Treptow and C. Ramseyer, ACS Nano, 2010, 4, 4158–4164 CrossRef CAS PubMed.
- T. A. Hilder, A. Robinson and S. H. Chung, ACS Chem. Neurosci., 2017, 8, 1747–1755 CrossRef CAS PubMed.
- J. Gao, L. M. Wang, S. G. Kang, L. N. Zhao, M. J. Ji, C. Y. Chen, Y. L. Zhao, R. H. Zhou and J. Y. Li, Nanoscale, 2014, 6, 12828–12837 RSC.
- K. H. Park, M. Chhowalla, Z. Iqbal and F. Sesti, J. Biol. Chem., 2003, 278, 50212–50216 CrossRef CAS PubMed.
- J. Q. Zhang, D. B. Li, T. Y. Sun, L. J. Liang and Q. Wang, Soft Matter, 2015, 11, 6633–6641 RSC.
- X. Xu, R. B. Li, M. Ma, X. Wang, Y. H. Wang and H. F. Zou, Soft Matter, 2012, 8, 2915–2923 RSC.
- S. Shityakov and C. Forster, Int. J. Comput. Biol. Drug Des., 2013, 6, 343–357 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Z. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- D. Roxbury, A. Jagota and J. Mittal, J. Phys. Chem. B, 2013, 117, 132–140 CrossRef CAS PubMed.
- X. C. Zhao, A. Striolo and P. T. Cummings, Biophys. J., 2005, 89, 3856–3862 CrossRef CAS PubMed.
- X. Xu, X. Wang, Y. Li, Y. H. Wang and L. Yang, Nucleic Acids Res., 2012, 40, 7622–7632 CrossRef CAS PubMed.
- S. Ghosh and R. Chakrabarti, J. Phys. Chem. C, 2016, 120, 22681–22693 CAS.
- X. C. Zhao, J. Phys. Chem. C, 2011, 115, 6181–6189 CAS.
- M. P. Landry, L. Vukovic, S. Kruss, G. Bisker, A. M. Landry, S. Islam, R. Jain, K. Schulten and M. S. Strano, J. Phys. Chem. C, 2015, 119, 10048–10058 CAS.
- J. L. Chen, L. Chen, Y. Wang and S. D. Chen, J. Phys. D: Appl. Phys., 2014, 47, 505401 CrossRef.
- C. B. Sun, T. Tang and H. Uludag, Biomaterials, 2013, 34, 2822–2833 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- M. Mahmoudi, N. Bertrand, H. Zope and O. C. Farokhzad, Nano Today, 2016, 11, 817–832 CrossRef CAS.
- C. C. Ge, J. F. Du, L. N. Zhao, L. M. Wang, Y. Liu, D. H. Li, Y. L. Yang, R. H. Zhou, Y. L. Zhao, Z. F. Chai and C. Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- Y. Chong, C. C. Ge, Z. X. Yang, J. A. Garate, Z. L. Gu, J. K. Weber, J. J. Lu and R. H. Zhou, ACS Nano, 2015, 9, 5713–5724 CrossRef CAS PubMed.
- Y. F. Li, H. Y. Yuan, A. von de Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. J. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef CAS PubMed.
- H. M. Ding and Y. Q. Ma, Biomaterials, 2014, 35, 8703–8710 CrossRef CAS PubMed.
- G. X. Duan, S. G. Kang, X. Tian, J. A. Garate, L. Zhao, C. C. Ge and R. H. Zhou, Nanoscale, 2015, 7, 15214–15224 RSC.
- S. M. Nie, Nanomedicine, 2010, 5, 523–528 CrossRef PubMed.
- P. M. Kelly, C. Aberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Z. Krpetic and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- M. C. L. Giudice, L. M. Herda, E. Polo and K. A. Dawson, Nat. Commun., 2016, 7, 13475 CrossRef PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- S. Khan, A. Gupta and C. K. Nandi, J. Phys. Chem. Lett., 2013, 4, 3747–3752 CrossRef CAS.
- R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Chem. Soc. Rev., 2012, 41, 2539–2544 RSC.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- W. J. Li, P. Zhan, E. D. Clercq, H. X. Lou and X. Y. Liu, Prog. Polym. Sci., 2013, 38, 421–444 CrossRef CAS.
- Z. Amoozgar and Y. Yeo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 219–233 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- N. Dragneva, O. Rubel and W. B. Floriano, J. Chem. Inf. Model., 2016, 56, 706–720 CrossRef CAS PubMed.
- Y. Li and Y. Hu, RSC Adv., 2014, 4, 51022–51031 RSC.
- P. A. Oroskar, C. J. Jameson and S. Murad, Langmuir, 2016, 32, 7541–7555 CrossRef CAS PubMed.
- H. Lee and R. G. Larson, J. Phys. Chem. C, 2011, 115, 5316–5322 CAS.
- M. T. Peracchia, E. Fattal, D. Desmaele, M. Besnard, J. P. Noel, J. M. Gomis, M. Appel, J. d'Angelo and P. Couvreur, J. Controlled Release, 1999, 60, 121–128 CrossRef CAS PubMed.
- Y. B. Zhang, Y. Xu, Z. G. Li, T. Chen, S. M. Lantz, P. C. Howard, M. G. Paule, W. Slikker Jr, F. Watanabe, T. Mustafa, A. S. Biris and S. F. Ali, ACS Nano, 2011, 5, 7020–7033 CrossRef CAS PubMed.
- Y. J. Li, L. Z. Feng, X. Z. Shi, X. J. Wang, Y. L. Yang, K. Yang, T. Liu, G. B. Yang and Z. Liu, Small, 2014, 10, 1544–1554 CrossRef CAS PubMed.
- T. Ishida, M. Harada, X. Y. Wang, M. Ichihara, K. Irimura and H. Kiwada, J. Controlled Release, 2005, 105, 305–317 CrossRef CAS PubMed.
- A. A. Barba, G. Lamberti, C. Sardo, B. Dapas, M. Abrami, M. Grassi, R. Farra, F. Tonon, G. Forte, F. Musiani, M. Licciardi, G. Pozzato, F. Zanconati, B. Scaggiante, G. Grassi and G. Cavallaro, Curr. Drug Metab., 2015, 16, 1–20 Search PubMed.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Prog. Polym. Sci., 2011, 36, 981–1014 CrossRef CAS.
- S. Y. Jiang and Z. Q. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- K. P. Garcia, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516–2529 CrossRef CAS PubMed.
- Q. Shao and S. Y. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- Q. Shao, Y. He, A. D. White and S. Y. Jiang, J. Phys. Chem. B, 2010, 114, 16625–16631 CrossRef CAS PubMed.
- H. M. Ding and Y. Q. Ma, Sci. Rep., 2016, 6, 26783 CrossRef CAS PubMed.
- T. Mizuhara, K. Saha, D. F. Moyano, C. S. Kim, B. Yan and Y. K. Kim, Angew. Chem., Int. Ed., 2015, 54, 6567–6570 CrossRef CAS PubMed.
- M. M. Wang, H. M. Liu and Y. Y. Chen, Nat. Commun., 2014, 5, 3053 Search PubMed.
- W. B. Hu, C. Peng, W. J. Luo, M. Lv, X. M. Li, D. Li, Q. Huang and C. H. Fan, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS PubMed.
- Y. Liu, C. Y. Chen, P. X. Qian, X. F. Lu, B. Y. Sun, X. Zhang, L. M. Wang, X. F. Gao, H. Li, Z. Y. Chen, J. L. Tang, W. J. Zhang, J. Q. Dong, R. Bai, P. E. Lobie, Q. F. Wu, S. L. Liu, H. F. Zhang, F. Zhao, M. S. Wicha, T. Zhu and Y. L. Zhao, Nat. Commun., 2015, 6, 5988 CrossRef CAS PubMed.
- S. M. Nie, Y. Xing, G. J. Kim and J. W. Simons, Annu. Rev. Biomed. Eng., 2007, 9, 257–288 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed.
- E. Ruoslahti, Adv. Mater., 2012, 24, 3747–3756 CrossRef CAS PubMed.
- H. T. Ma, J. P. Liu, M. M. Ali, M. A. I. Mahmood, L. Labanieh, M. R. Lu, S. M. Iqbal, Q. Zhang, W. A. Zhao and Y. Wan, Chem. Soc. Rev., 2015, 44, 1240–1256 RSC.
- B. A. J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef PubMed.
- V. Schubertova, F. J. Martinez-Veracoecheab and R. Vacha, Soft Matter, 2015, 11, 2726–2730 RSC.
- J. X. Xue, Z. Guan, J. P. Lin, C. H. Cai, W. J. Zhang and X. Q. Jiang, Small, 2017, 13, 1604214 CrossRef PubMed.
- S. H. Wang and E. E. Dormidontova, Phys. Rev. Lett., 2012, 109, 238102 CrossRef PubMed.
- T. Curk, J. Dobnikar and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7210–7215 CrossRef CAS PubMed.
- B. B. Wang, C. V. Galliford and P. S. Low, Nanomedicine, 2014, 9, 313–330 CrossRef CAS PubMed.
- R. R. Sawant, A. M. Jhaveri, A. Koshkaryev, F. Qureshi and V. P. Torchilin, J. Drug Targeting, 2013, 21, 630–638 CrossRef CAS PubMed.
- Q. S. Xia, H. M. Ding and Y. Q. Ma, Nanoscale, 2017, 9, 8982–8989 RSC.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. L. Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, ACS Nano, 2017, 11, 1884–1893 CrossRef CAS PubMed.
- Q. Dai, C. Walkey and W. C. W. Chan, Angew. Chem., Int. Ed., 2014, 53, 5093–5096 CrossRef CAS PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- C. S. Kim, B. Duncan, B. Creran and V. M. Rotello, Nano Today, 2013, 8, 439–447 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- X. Z. Yang, J. Z. Du, S. Dou, C. Q. Mao, H. Y. Long and J. Wang, ACS Nano, 2012, 6, 771–781 CrossRef CAS PubMed.
- H. M. Ding and Y. Q. Ma, Sci. Rep., 2013, 3, 2804 CrossRef PubMed.
- F. H. Meng, Y. N. Zhong, R. Cheng, C. Deng and Z. Y. Zhong, Nanomedicine, 2014, 9, 487–499 CrossRef CAS PubMed.
- K. Liang, G. K. Such, A. P. R. Johnston, Z. Y. Zhu, H. Ejima, J. J. Richardson, J. W. Cui and F. Caruso, Adv. Mater., 2014, 26, 1901–1905 CrossRef CAS PubMed.
- W. W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- Y. Wang, Q. Y. Li, X. B. Liu, C. Y. Zhang, Z. M. Wu and X. D. Guo, ACS Appl. Mater. Interfaces, 2015, 7, 25592–25600 CAS.
- Z. L. Luo, Y. Li, B. B. Wang and J. W. Jiang, Macromolecules, 2016, 49, 6084–6094 CrossRef CAS.
- R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS PubMed.
- T. Thambi, V. G. Deepahani, H. Y. Yoon, H. S. Han, S. H. Kim, S. Y. Son, D. G. Jo, C. H. Ahn, Y. D. Suh, K. Kim, I. C. Kwon, D. S. Lee and J. H. Park, Biomaterials, 2014, 35, 1735–1743 CrossRef CAS PubMed.
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popovic, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426–2431 CrossRef CAS PubMed.
- S. L. Xue, B. Li, X. Q. Feng and H. J. Gao, J. Mech. Phys. Solids, 2017, 104, 32–56 CrossRef CAS.
- L. Cheng, C. Wang, L. Z. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS PubMed.
- J. F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC.
- S. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909 CrossRef CAS.
- A. Agarwal, M. A. Mackey, M. A. El-Sayed and R. V. Bellamkonda, ACS Nano, 2011, 5, 4919–4926 CrossRef CAS PubMed.
- J. Di, J. Price, X. Gu, X. N. Jiang, Y. Jing and Z. Gu, Adv. Healthcare Mater., 2014, 3, 811–816 CrossRef CAS PubMed.
- S. D. Kong, W. Z. Zhang, J. H. Lee, K. Brammer, R. Lal, M. Karin and S. H. Jin, Nano Lett., 2010, 10, 3651–3657 Search PubMed.
- X. M. Lu, B. Yuan, X. R. Zhang, K. Yang and Y. Q. Ma, Appl. Phys. Lett., 2017, 110, 023701 CrossRef.
- H. M. Ding, W. D. Tian and Y. Q. Ma, ACS Nano, 2012, 6, 1230–1238 CrossRef CAS PubMed.
- H. Masoud and A. Alexeev, ACS Nano, 2012, 6, 212–219 CrossRef CAS PubMed.
- S. Fraga, A. Brandao, M. E. Soares, T. Morais, J. A. Duarte, L. Pereira, L. Soares, C. Neves, E. Pereira, M. D. L. Bastos and H. Carmo, J. Nanomed. Nanotechnol., 2014, 10, 1757–1766 CrossRef CAS PubMed.
- N. Chen, H. Wang, Q. Huang, J. Lo, J. Yan, D. N. He, C. H. Fan and H. Y. Song, Small, 2014, 10, 3603–3611 CrossRef CAS PubMed.
- J. K. Tabi, Y. Javed, L. Lartigue, J. Volatron, D. Elgrabli, I. Marangon, G. Pugliese, B. Caron, A. Figurrola, N. Luciani, T. Pellegrino, D. Alloyeau and F. Gazeau, ACS Nano, 2015, 9, 7925–7939 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |