
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically assisted flexible lanthanide upconversion luminescence sensing of heavy metal contamination with high sensitivity and selectivity†
Yuen-Ting
Wong‡
,
Sin-Yi
Pang‡
,
Ming-Kiu
Tsang
 ,
Yan
Liu
,
Haitao
Huang
,
Siu-Fung
Yu
and
Jianhua
Hao
*
,
Yan
Liu
,
Haitao
Huang
,
Siu-Fung
Yu
and
Jianhua
Hao
*
Department of Applied Physics, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, China. E-mail: jh.hao@polyu.edu.hk
First published on 20th August 2018
Abstract
Heavy metal contamination in water can pose lethal threats to public health; therefore it is highly desired to develop a rapid and sensitive sensor for monitoring water quality. Owing to their superior optical features, upconversion nanoparticles (UCNPs) are widely explored to detect metal ions based on resonance energy transfer to dye quenchers. However, these schemes heavily rely on the optical properties of the molecules, which limits the flexibility of the probe design. Herein, a flexible carbon fiber cloth/UCNP composite probe was fabricated for sensing copper(II) (Cu2+) ions and an electrochemical (E-chem) technique was implemented for the first time to enhance its sensing performance. By applying 0.3 V on the composite probe, Cu2+ ions can be effectively accumulated through oxidation, yielding a remarkable improvement in the selectivity and sensitivity. A more outstanding detection limit of the sensor was achieved at 82 ppb under the E-chem assistance, with 300-fold enhancement compared to the detection without the E-chem effect. This sensing approach can be an alternative to molecular quenchers and open up new possibilities for simple, rapid and portable sensing of metal ions.
Introduction
Heavy metal contamination in water has aroused increasing public awareness of public health owing to the rapid industrial development. Copper(II) (Cu2+) ions are one of the common metal contaminants due to their widespread use in industrial activities. Although Cu2+ ions are essential co-factors for different metabolic activities in the human body, imbalanced uptake of these ions in the long term can increase the risk of neurological problems and organ disorders by dyshomeostasis.1,2 Much effort has thus been devoted to developing highly sensitive and selective techniques, such as inductively coupled plasma mass spectrometry and atomic emission spectrometry,3,4 as well as atomic absorption spectrometry.5 Unfortunately, these methods require expensive and sophisticated equipment, specialists for operation, and tedious sample preparation and analytical processes, which are challenging for rapid and on-site monitoring. Optical sensing has emerged as a simple, sensitive and reliable method.1 Upconversion luminescent (UCL) sensors employing upconversion nanoparticles (UCNPs) are particularly interesting due to their large anti-Stokes shift, and narrow and sharp emissions as well as the absence of autofluorescence for an improved signal-to-noise ratio.6 In addition, their excellent photo-stability and bio-compatibility make these nanophosphors vital in biosensing7–9 and bioimaging.10–13Numerous reports have revealed the good sensitivity and selectivity of UCL sensors towards various metal ions such as Zn2+,8 Cu2+,14–16 Hg2+,17 and Pb2+ ions.18 Most strategies were established based on the luminescence resonance energy transfer (LRET) phenomenon, by which metal-responsive dyes played important roles in quenching UCL for ratiometric luminescence sensing while promoting the selectivity. However, these sensors rely heavily on the optical properties, conjugation efficiency and stability of molecules. In particular, absorption bands of the quenchers must overlap with upconversion emissions of the UCNPs to realize effective LRET. It should be noted that energy gap modulation for molecular quenchers can be challenging and this may limit the flexibility of the sensor design.19 Another barrier is to functionalize organic molecules, which allows chemical conjugation onto UCNPs to enhance conjugation efficiency and minimize leakage. In spite of improved optical responses and stability, the functionalization may complicate the design and synthesis of dyes. This paves an attractive way to establish an UCL sensor without using molecular quenchers or chelating agents.
Heavy metal ions have a pronounced quenching effect on emissions from lanthanide ions through efficient electron transfer.19–21 A recent study has revealed that Cu2+ ions could quench 95% of the UCL of N,N-di(2-picolyl)ethylenediamine (DPEA)-modified NaYF4@NaYF4:Yb,Tm@NaYF4 UCNPs without involving the LRET process.19 Although the concept of heavy metal ion-induced deactivation has been successfully employed to monitor biothiols in living tissues, it still required DPEA molecules to anchor Cu2+ ions on the probe surface. Considering that the lack of metal-chelating molecules deteriorates the UCL sensing performance,20 the choice of phosphors and other techniques becomes critical to overcome this limitation. NaGdF4:Yb/Tm@NaGdF4:Eu core@shell UCNPs are a potential candidate for sensing Cu2+ ions. On the one hand, the characteristic red emissions of Eu3+ ions can be effectively deactivated by Cu2+ ions via electron transfer or/and LRET to intensify optical responses. A thin layer of a Eu3+-silica downconverting matrix was utilized for Cu2+ ion sensing with detection limit down to 50 ppb.21 On the other hand, a Gd3+-doped host is not only able to promote the hexagonal crystal phase,22 but also allow an energy migration-mediated (EMU) mechanism to achieve the most efficient UCL of Eu3+ ions.23
Porous composite materials have drawn much attention because the pores provide large loading capacity and surface area as reaction sites,24–26 while a composite exhibits multi-functional properties for widespread applications.27,28 More importantly, inspired by the outstanding selectivity of electrochemical (E-chem) sensors,29 we introduced an E-chem technique for UCL probes for the sake of better Cu2+ ion accumulation on the probes via redox reactions. Therefore, the sensing performance can be improved as illustrated in Fig. 1. As a proof of concept, a composite probe was fabricated by electrostatic adsorption of core@shell UCNPs onto a flexible carbon fiber cloth (CFC), denoted as CFC-UCNP probe. CFC is extensively utilized in energy storage applications because of its low cost, good mechanical properties, chemical stability and electrical conductivity.30,31 Furthermore, its porous structure offers large surface area for accommodating the nanophosphors and supporting E-chem reactions, which make CFCs attractive as an E-chem-assisted UCL sensing platform. It should be mentioned that the feature of low energy excitation in a UCL sensor can avoid background interference from the ultraviolet absorption of carbon-based electrodes. In addition to its multi-functional properties, the excellent flexibility and easy processability of the composite probe enable easily handled fabrication and sensing applications.28
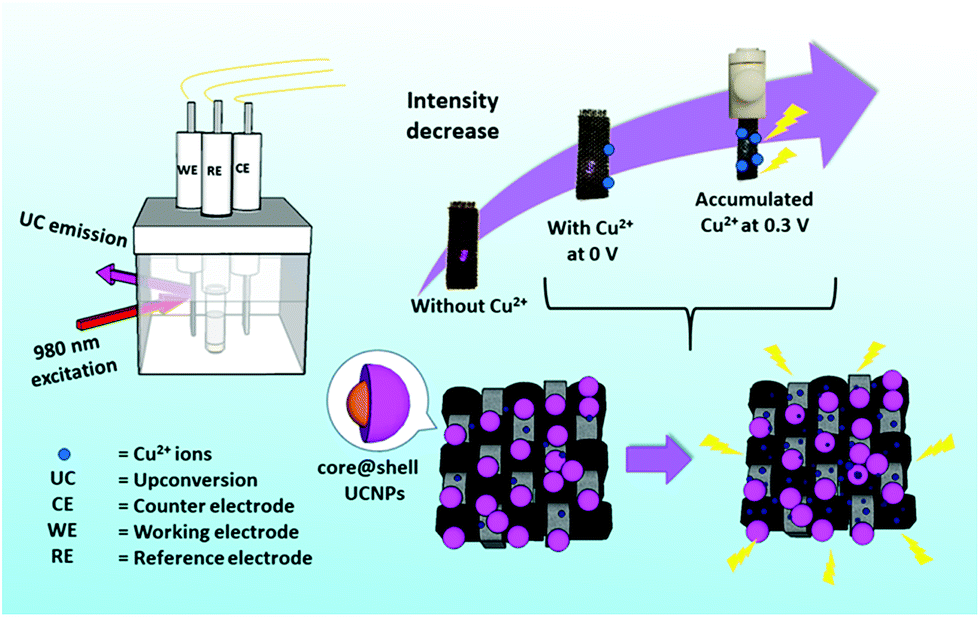 | ||
| Fig. 1 A schematic diagram showing the CFC-UCNP probe for Cu2+ ion sensing under the E-chem assistance. | ||
The sensitivity and selectivity of the CFC-UCNP probe were greatly improved after applying a low oxidation potential of 0.3 V. Notably, its limit of detection (LOD) was drastically lowered from 25.8 ppm to 82 ppb. To the best of our knowledge, this is the first time that an E-chem technique has been introduced to improve UCL sensing of Cu2+ ions. It is noteworthy that the LOD of this sensing approach is lower than the tolerance levels of 2 ppm and 1.3 ppm suggested by the World Health Organization (WHO)32 and the United States Environmental Protection Agency (USEPA),33 respectively. Therefore, this proposed E-chem-assisted sensing by flexible CFC-UCNP probes has potential for rapid and portable sensing of Cu2+ ions in drinking water. Moreover, it can be possibly extended for sensing other heavy metal ions by using an appropriate applied voltage and electrolyte.
Materials and methods
Materials
Gadolinium(III) acetate hydrate (Gd(CH3CO2)3·xH2O, 99.9%), ytterbium(III) acetate hydrate (Yb(CH3CO2)3·4H2O, 99.95%), thulium(III) acetate hydrate (Tm(CH3CO2)3·xH2O, 99.9%), europium(III) acetate hydrate (Eu(CH3CO2)3·xH2O, 99.9%), sodium hydroxide pellets (NaOH, 98%), ammonium fluoride (NH4F, 99.99%), 1-octadecene (1-ODE, 90%), oleic acid (OA, 90%), methanol (99.8%), anhydrous ethanol (EtOH), acetone (99.9%), cyclohexane (99.5%), hydrochloric acid (HCl, 37%) and nitric acid (HNO3, 70%), sodium chloride (NaCl, 99%), copper(II) sulfate (CuSO4, 99.99%), cobalt(II) sulphate heptahydrate (CoSO4·7H2O, 99%), nickel(II) sulphate hexahydrate (NiSO4·6H2O, 98%), calcium(II) chloride (CaCl2, 96%), barium(II) chloride (BaCl2, 99.9%), zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O, 98%), lead(II) nitrate (Pb(NO3)2, 99%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, 98%), chromium(III) nitrate nonahydrate (Cr(NO3)3·9H2O, 99.99%), and manganese(II) acetate tetrahydrate (Mn(CH3CO2)2·4H2O, 99%) were purchased from Sigma-Aldrich. CFCs were purchased from CeTech (W0S1002). All materials were used without further purification unless mentioned specifically.Synthesis of NaGdF4:49%Yb:1% Tm core UCNPs
The OA-capped UCNPs were synthesised by the co-precipitation route.34 0.2 M aqueous solutions of Ln(CH3CO2)3 (Ln = Gd, Yb and Tm) were mixed with 4 mL OA and 6 mL 1-ODE in a two-necked flask. The mixture was heated at 150 °C for 40 min under vigorous magnetic stirring. After cooling the solution to 50 °C, a methanolic mixture of NH4F (1.3 mmol) and NaOH (1 mmol) was quickly added and heated at 50 °C for 1 h. The temperature was then increased to 100 °C to remove the methanol residue and the colloidal solution was finally heated to 290 °C under argon protection for 90 min. The core UCNPs were collected by centrifugation and washed with EtOH several times. They were dispersed in cyclohexane for further use.Synthesis of NaGdF4:49% Yb:1% Tm@NaGdF4:15% Eu core@shell UCNPs
The growth of the Eu-doped shell on the core UCNPs was achieved by a route similar to the one above.34 Briefly, 0.2 M aqueous solutions of Ln(CH3CO2) (Ln = Gd and Eu) were heated with 4 mL OA and 6 mL 1-ODE at 150 °C for 40 min. The core UCNPs dispersed in cyclohexane were added to the mixture under stirring at 50 °C. Subsequently, a methanolic mixture containing NH4F and NaOH was quickly added. The colloidal solution was maintained at 50 °C for 1 h and then degassed by raising the temperature to 100 °C. After heating the colloidal solution at 290 °C for 90 min with argon gas purging, the core@shell nanoparticles were collected by centrifugation, followed by washing with EtOH several times and re-dispersion in cyclohexane for further use.Preparation of ligand free core@shell UCNPs
The OA ligands of core@shell UCNPs were removed by a modified acid treatment.35 Briefly, core@shell UCNPs were firstly dispersed in aqueous solution, whose pH was tuned to 4 by adding 0.1 M HCl. After stirring for 2 h, cyclohexane was added to the colloidal solution, followed by ultra-sonication and oil extraction for a few times to remove free OA. The colloidal solution was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 40 min and purified by repeated washing with EtOH and D.I. water.
000 rpm for 40 min and purified by repeated washing with EtOH and D.I. water.
Fabrication of the CFC-UCNP probe
The CFCs were oxidized by concentrated HNO3 to induce negative surface charges for more efficient electrostatic adsorption of the nanophosphors.36 Simply, the organic impurities on the CFC surface were removed by ultra-sonication with acetone and EtOH. Then, the dried CFCs were refluxed in 10 mL concentrated HNO3 at 100 °C for 5 h. The CFCs were finally washed with D.I. water several times to remove any acid residues. To conjugate ligand free core@shell UCNPs on an acid-treated CFC, the cloth (0.5 cm × 2 cm) was immersed in 40 mg mL−1 of the prepared UCNPs with shaking for 6 h, followed by repeated washing with deionized (D.I.) water, ultra-sonication and air-drying.Characterization
A JEOL-2100F transmission electron microscope (TEM) equipped with an Oxford Instruments energy dispersive X-ray (EDX) system was operated at 200 kV to characterize the morphology of the UCNPs. The crystal structure of the core@shell UCNPs was also characterized by high resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED). The samples used in TEM were prepared on holey carbon coated 400 mesh copper grids. Powder X-ray diffraction (XRD) patterns were recorded to confirm the crystal structure of the UCNPs using a Rigaku SmartLab 9 kW (Rigaku, Japan) using Cu Kα radiation (λ = 0.15406 nm). The surface properties of the UCNPs were studied by using a PerkinElmer Spectrum 100 Fourier-transform infrared spectroscopy (FTIR) spectrometer (PerkinElmer Inc., USA) equipped with a deuterated triglycine sulphate detector and KBr beam splitter assembly. UCL spectra were recorded and lifetime measurements performed using an FLS920 (Edinburgh Instruments) equipped with a continuous wave 980 nm diode laser excitation source and pulsed laser modulator. An external voltage DC source (JC1803A) purchased from Hang Zhou Jingce Electronics Co., Ltd was used for the photoluminescence experiments, which were conducted in 1 M NaCl electrolyte unless mentioned specifically. Cyclic voltammetry (CV) experiments were performed using a Solartron Analytical 1400 cell test system connected to a standard three-electrode system in 1 M NaCl electrolyte, which contained the as-prepared CFC-UCNP probe as the working electrode, a 1 × 1 cm2 platinum plate as the counter electrode and a saturated calomel reference electrode.Results and discussion
Characterization of the UCNPs and CFC-UCNP probe
All crystalline OA-capped UCNPs were synthesized via a co-precipitation route.34 The TEM images (Fig. S1†) indicate that the as-prepared UCNPs were mono-disperse. The epitaxial growth of the Eu3+-doped shell changed the shape of the nanophosphors from spherical to hexagonal, associated with an average size increase by 8.92 nm to yield 43.8 nm core@shell UCNPs. The XRD patterns (Fig. S2†) of the as-prepared UCNPs are in accord with the standard pattern of hexagonal NaGdF4 crystals (JCPDS#27-0699), suggesting the absence of phase change in the crystals after shell growth. In addition, the lattice spacing of 5.2 Å in the HRTEM image of the core@shell UCNPs (Fig. 2a) was indexed to the (100) plane of hexagonal NaGdF4 crystals. These structural characterization results are consistent with the SAED pattern of the core@shell UCNPs (Fig. 2b), further confirming their hexagonal crystal phase. Acid treatment played a role in removing OA surface ligands to endow the core@shell UCNPs with good water dispersibility and positive surface charges.35 Their naked surface is verified by the disappearance of infrared absorption peaks at around 2900 cm−1 and 1500 cm−1, characteristic of C–H and –COO stretching vibrations, respectively (Fig. S3†). Moreover, a broad band centered at 3435 cm−1, corresponding to –OH vibration, becomes more obvious for the ligand free core@shell UCNPs.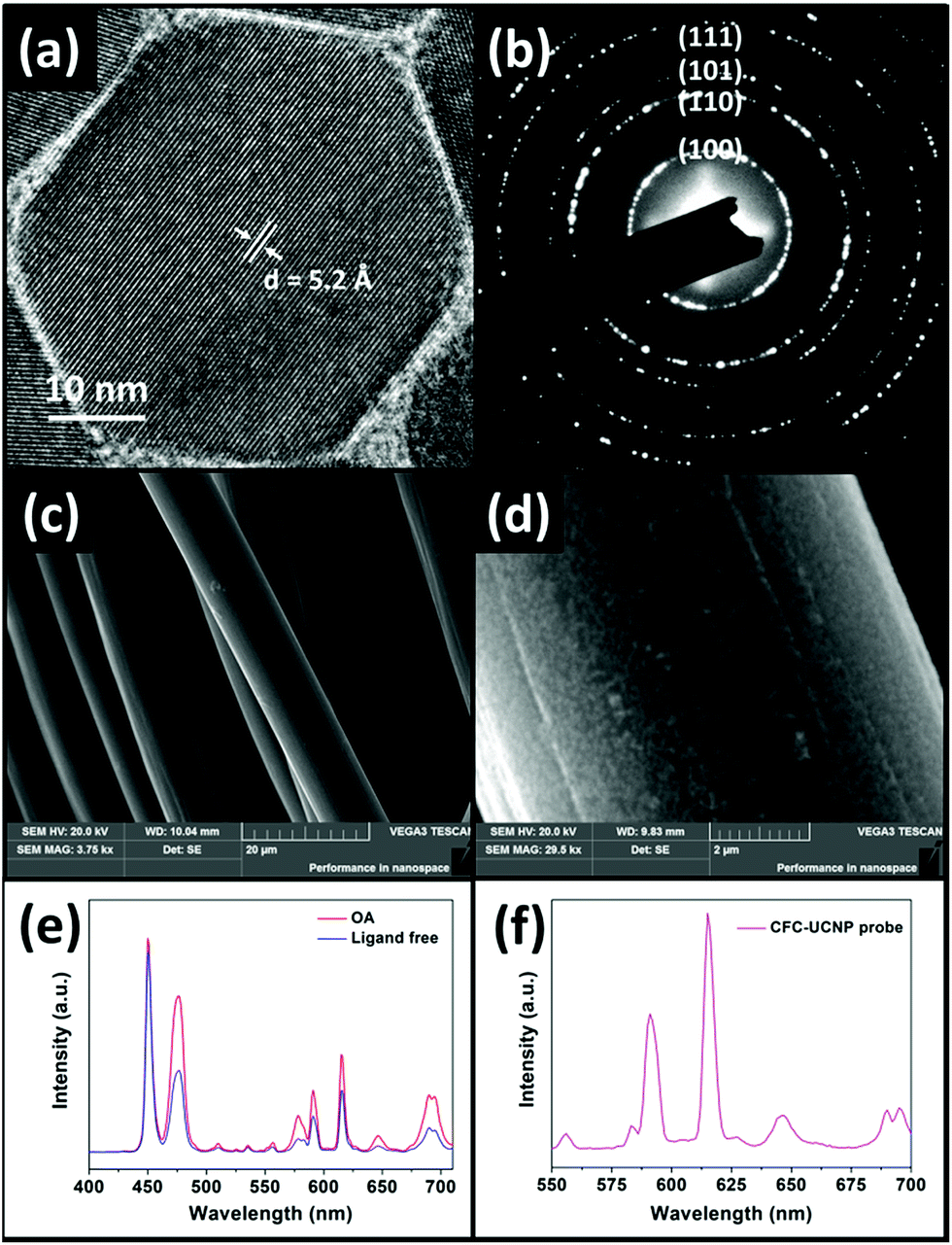 | ||
| Fig. 2 (a) HRTEM and (b) SAED images of the core@shell UCNPs. SEM images of (c) CFC and (d) the CFC-UCNP probe. UCL spectra of (e) OA-capped and ligand free core@shell UCNPs and (f) the CFC-UCNP probe under 980 nm excitation. | ||
The OA-capped core@shell UCNPs exhibited strong purple UCL upon 980 nm excitation, with the appearance of new emission peaks at 578, 591, 615 and 690 nm compared to the blue-emitting core UCNPs (Fig. S4a and b†). These peaks are the characteristic UCL of Eu3+ ions responsible for the transitions of 5D1 → 7F3 and 5D0 → 7FJ (J = 1, 2 and 4 respectively).23 Fig. S4c† depicts the EMU mechanism of the core@shell UCNPs. Briefly, 980 nm photons sensitized by Yb3+ ions are accumulated in the 1I6 excited state of Tm3+ ions via successive Yb3+–Tm3+ energy transfer. The energy is bridged to Eu3+ ions in the shell layer by the high-lying 6P7/2 state in Gd3+ ions which gives rise to the red emissions. It is worth noting that –OH surface quenchers on the naked nanophosphors greatly weakened the UCL at 615 nm by 1.6-fold compared to OA-capped nanophosphors (Fig. 2e).35 These optical observations prove the successful preparation of ligand free core@shell UCNPs, and are consistent with the aforementioned FTIR characterization. The CFC-UCNP probe was fabricated by 6 h electrostatic adsorption of the ligand free nanophosphors onto an acid-treated CFC (Fig. S5a†). Notably, the characteristic purple UCL of Tm3+/Eu3+ ions on the CFC-UCNP probe could be observed by the naked eye under 980 nm laser irradiation, while two major UCL peaks of the composite probe at 591 and 615 nm are not altered (Fig. 2f). By comparing the 615 nm emission of the initial and residual core@shell UCNPs used for the adsorption process, about 1.5 mg of them was accommodated into the cloth (Fig. S5b†). The increased roughness on carbon fibers observed in the SEM images (Fig. 2c and d and S6a–d†) results from the substantial attachment of spot-like core@shell UCNPs. Furthermore, the EDX spectra (Fig. S6e and f†) evidence the presence of the constituent elements of core@shell UCNPs in the CFC-UCNP probe but not in the CFC.
UCL sensing without E-chem assistance
NaCl (aq.) serves as an electrolyte necessary for E-chem reactions and chloride ions are able to stabilize Cu2+ ions in the form of copper(I) (Cu+) chloride complexes.37 This is evidenced by a red shifted absorption band of CuSO4 in concentrated NaCl (aq.), together with a color change from blue to green (Fig. S7a†). Since the absorption bands of Cu+ and Cu2+ ions overlap with the 615 and 690 nm emissions of Eu3+ ions, LRET possibly takes place once the metal ions are in close proximity to the core@shell UCNPs. As a result, UCL deactivation for the metal ion sensing can be achieved by electron transfer and LRET processes.UCL sensing assays supported on a porous substrate can improve the sensitivity and selectivity.26 As a proof of concept, Cu2+ ion sensing was firstly studied with ligand free core@shell UCNPs in D.I. water and 1 M NaCl (aq.), respectively (Fig. S8†). Unfortunately, optical sensing could not be effectively accomplished because UCL is very susceptible to an environment with high ionic strength and various surface quenchers.35 In contrast to solution-based sensing, emission of the CFC-UCNP probe at 615 nm was expectedly quenched with increasing Cu2+ ions (Fig. 3a). Therefore, the porous structure of CFC not only protects the nanophosphors from a complicated environment, but also likely concentrates them to enhance the optical responses.
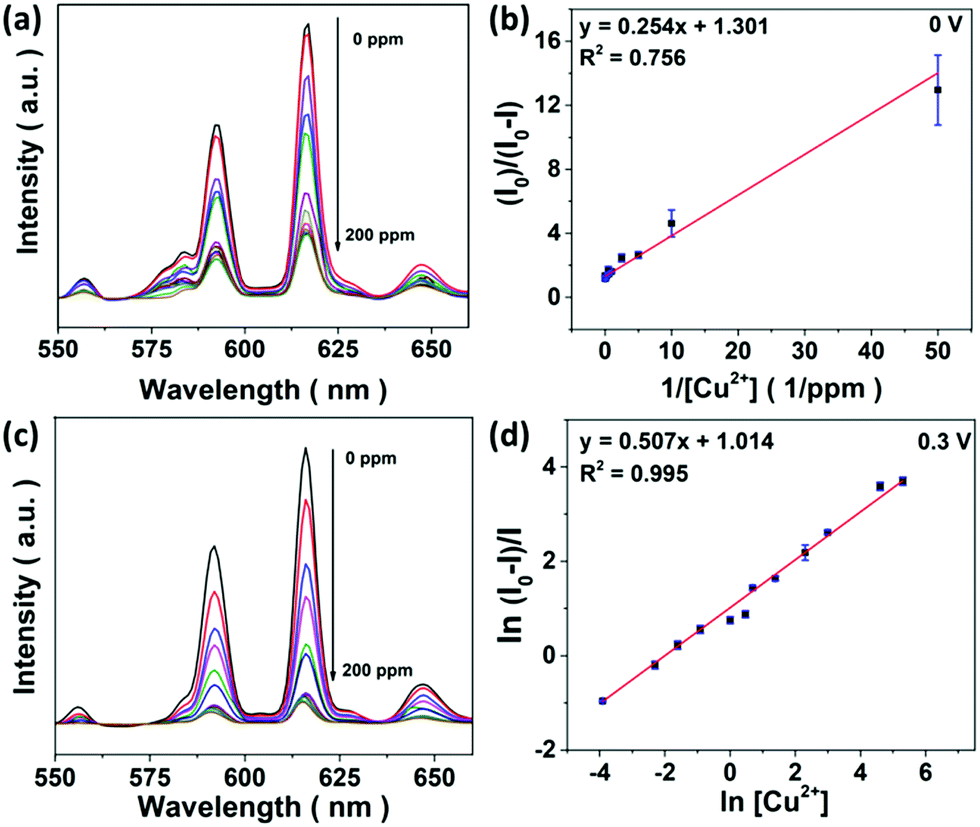 | ||
| Fig. 3 (a) UCL spectra of the CFC-UCNP probe for sensing 0–200 ppm Cu2+ ions at 0 V and (b) the corresponding linear relationship at 615 nm emission based on the modified SV equation. (c) UCL spectra of the CFC-UCNP probe for sensing 0–200 ppm Cu2+ ions at 0.3 V and (d) the corresponding linear relationship at 615 nm emission based on the double natural logarithm SV equation. Io and I refer to the UCL intensity before and after adding Cu2+ ions, respectively. | ||
The UCL quenching of the CFC-UCNP probe was evaluated using the Stern–Volmer (SV) relationship:19–21
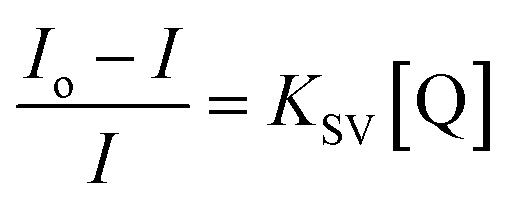 | (1) |
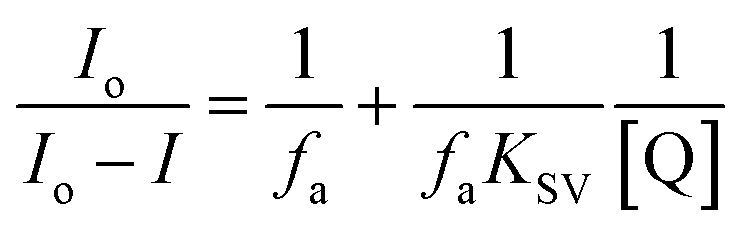 | (2) |
E-chem performance and voltage effects on the CFC-UCNP probe
The E-chem performance of the CFC-UCNP probe was studied in order to evaluate if the proposed E-chem-assisted sensing approach is feasible. Fig. 4a illustrates the working principle of a typical E-chem sensor utilizing the composite probe as a working electrode. It provides a platform to support redox reactions of analyte ions at given voltages and produce various currents. These processes can be simply presented as Cu+ (ads.) ⇌ Cu2+ (ads.) + e− for oxidation and reversed for reduction. Cyclic voltammetry (CV) is a traditional and reliable technique that can be used to detect these current signals by applying a range of voltages cyclically to the system. Hence, the current signals across the CFC-UCNP probe were measured in the potential window of 0–0.5 V at a scan rate of 100 mV s−1. The nearly identical CV curves of the CFC and CFC-UCNP electrodes (Fig. S10†) revealed the electrochemically inert nature of the nanophosphors at low potentials, which is beneficial for obtaining E-chem responses only from the analyte ions. After mixing different aliquots of CuSO4 (aq.) with the electrolyte, redox peaks appeared at around 0.3 V (forward bias) and 0.2 V (reverse bias) (Fig. 4b). They are ascribed to the oxidation of Cu+ ions and reduction of Cu2+ ions, respectively.38 Positive linear relationships between the current density and [Cu2+] were individually obtained from 0.3 V (Fig. 4c) and 0.2 V (Fig. S11†). Based on the 3σ/slope rule,39 their LODs were estimated at 125 ppb and 132 ppb, respectively.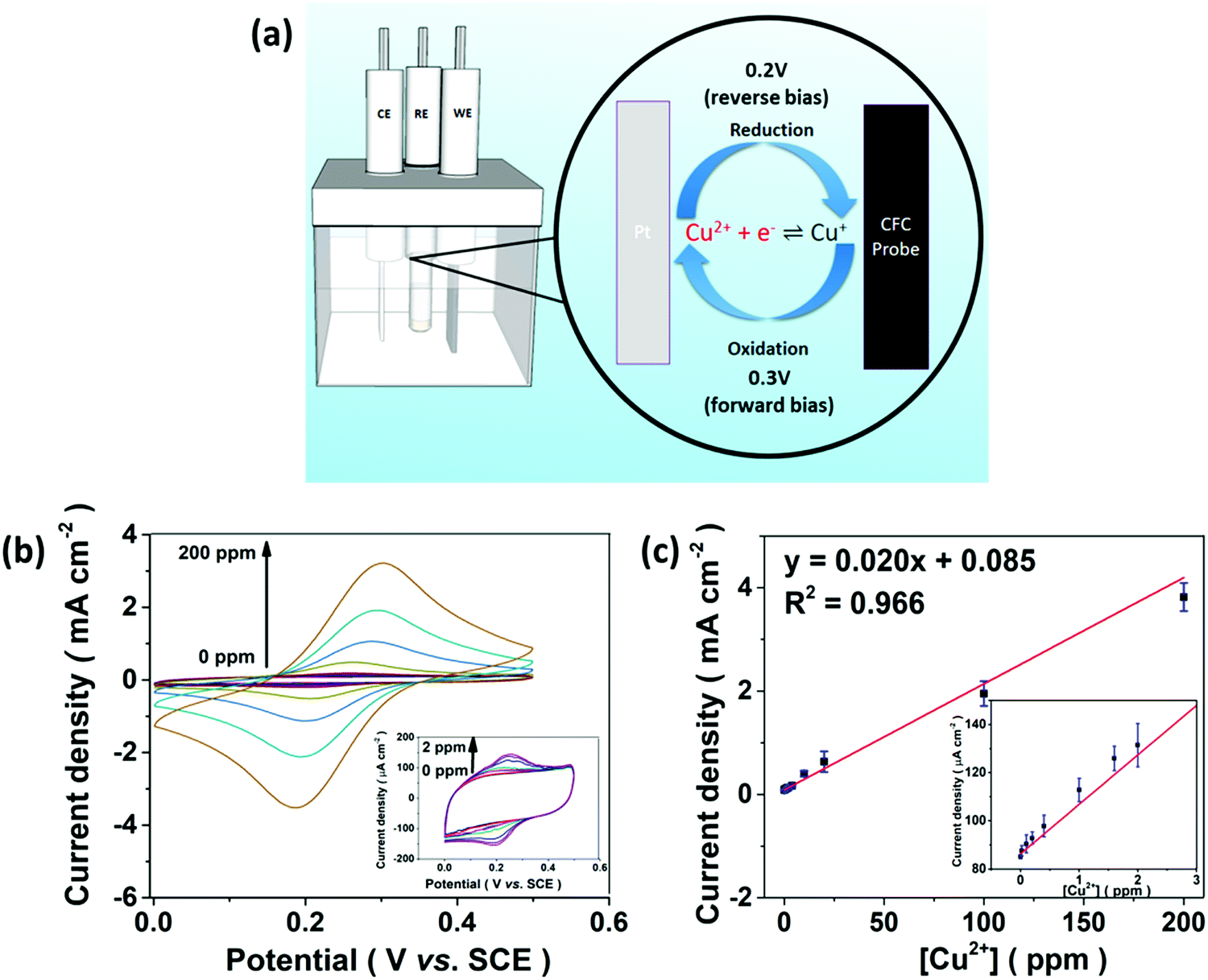 | ||
| Fig. 4 (a) A diagram illustrating the E-chem processes on the CFC-UCNP probe. (b) CV curves of the CFC-UCNP probe in response to 0–200 ppm Cu2+ ions recorded within the potential window of 0–0.5 V at 100 mV s−1. (c) The linear relationship between the E-chem signals and [Cu2+] obtained at 0.3 V. The insets are the enlarged plots for 0–2 ppm Cu2+ ions. | ||
Encouraged by the effective oxidation of Cu+ ions on the cloth, voltage effects on UCL at 615 nm of the composite probe were examined. It was found that low anodic potential could induce UCL deactivation (Fig. S12a†) along with a shortened lifetime from 5.07 to 4.88 ms (Fig. S12b†) because of high susceptibility of UCL to the applied electric-field.40 Notably, voltages beyond 0.2 V fostered a steady quenching effect on the red emission and 0.3 V triggered UCL fluctuation as slight as 0 V in the time-lapse experiments (Fig. S12c and d†). The nanophosphors are less likely to detach from the CFC in the electrolyte, so the low anodic potential at 0.3 V poses a small effect on the composite probe for the optical sensing.
UCL sensing under E-chem assistance
UCL sensing of Cu2+ ions with the CFC-UCNP probe was accessed by monitoring the 615 nm emission at 0.3 V (Fig. 3c). A good linearity was obtained from the double natural logarithm of the SV equation (Fig. 3d) while concave downward curves were observed in either the SV or the modified SV plots (Fig. S9b and d†). This may indicate the involvement of reduced heterogeneity or additional mechanisms in the assays. Based on the enhanced E-chem signals with increasing [Cu2+] (Fig. 4c), it is speculated that more analyte ions are adsorbed on the CFC-UCNP probe at 0.3 V and facilitate UCL deactivation. This E-chem-assisted accumulation effect can last for a period of time as revealed by the steady decrease in the UCL of the composite probe over time at fixed [Cu2+] (Fig. S13†). By comparing the relative emission intensity in the time-lapse experiments, higher [Cu2+] displayed greater quenching rate. This is ascribed to a larger [Cu+] gradient between the bulk solution and the CFC-UCNP probe, at which Cu+ ions are consumed via oxidation rapidly. As a result, such an improved diffusion behavior can lead to better accessibility and accumulation of metal ion quenchers to the core@shell UCNPs inside the cloth pores. This accounts for the reduced heterogeneity and more efficient UCL quenching compared to the detection assays at 0 V.The quenching mechanism was investigated by analyzing the decay behavior of the UCL at 615 nm. The lifetimes were gradually shortened from 4.90 to 4.13 ms for 0–50 ppm Cu2+ ions at 0.3 V; subsequently, it was slightly increased at high [Cu2+] (Fig. S14†). In contrast, the detection at 0 V could only result in a small lifetime shift of 0.13 ms at low [Cu2+] and a drastic recovery of the lifetime occurred at higher [Cu2+] (Fig. S15†). Lifetime shifts indicate that dynamic quenching processes are largely responsible for the detections, while the lifetime recoveries at high [Cu2+] are ascribed to the increasing significance of the static quenching factor.19,20 The findings are in agreement with the SV analysis as discussed. In view of a small spectral overlap of the metal ion absorbance and 615 nm UCL, radiative processes such as LRET and the inner filter effect less likely dominate the UCL deactivation. In other words, the greater the collisional probability of analyte ions with the core@shell UCNPs for electron transfer, the higher the participation of a dynamic quenching mechanism in the metal ion sensing. Therefore, the large lifetime change observed in E-chem-assisted sensing suggests that oxidation of Cu+ ions promotes the dynamic quenching factor, likely by improving the diffusion behavior and accumulation of the analyte ions onto composite probes. Such a shortened interaction distance with the core@shell UCNPs results in more efficient UCL deactivation. In addition, more concentrated [Cu2+] can reduce collisional events with the UCL probes, particularly in 0 V assays whose static quenching factor becomes important. It is noted that the complicated quenching mechanisms behind this E-chem-assisted optical sensing scheme are yet to be well understood and other possible routes should not be excluded. For instance, the voltage-triggered crystal field changes in Cu+, Cu2+ and Eu3+ ions may prompt the electron transfer and non-radiative relaxation processes to give greater optical responses.
This sensing strategy offered higher stability and a much more outstanding LOD at 82 ppb for 0–200 ppm Cu2+ ions, an about 300-fold enhancement in sensitivity compared to the detection at 0 V. It displayed comparable sensitivity and a wider working range compared to some reported UCNPs functionalized with Cu2+-chelating molecules.14–16 Hence, the E-chem technique can help establish a versatile, simple, rapid and sensitive UCL sensor. Apart from being developed as an optical sensor, the CFC-UCNP probe is also promising for UCL/E-chem dual modal sensing because the sensitivity of the E-chem sensing mode is comparable to the UCL sensing at 0.3 V.
Selectivity of the CFC-UCNP probe for Cu2+ ion sensing
Selectivity is known to be another figure of merit of a sensor; therefore UCL of the CFC-UCNP probe was examined for some non-specific cations including Zn2+, Ni2+, Pb2+ and so on. As shown in Fig. S16,† the optical responses were rather sensitive to most of the tested non-specific cations at 0 V. The inferior selectivity is attributed to the lack of Cu2+-responsive molecules on the nanophosphors and the efficient electron transfer to the transition metal ions leads to observable UCL quenching effects.20 In particular, Ni2+ and Cr3+ ions exhibited relatively better quenching capability among the hazardous cations which are at low concentrations. The observation is likely because of their spectral overlap with UCL from the probe as an additional quenching factor (Fig. S7b†). By contrast, the selectivity for Cu2+ ions was significantly enhanced at 0.3 V (Fig. 5a). The quenching effects of the non-specific cations were largely reduced at 0.3 V while the quenching efficiency was high due to Cu2+ ions present in the co-existing systems. This improvement can confirm the specific accumulation of analyte ions on the probe by the E-chem reaction. To further verify the specific oxidation of Cu+ ions on the CFC-UCNP probe, its selectivity was studied based on E-chem signals. The currents across the CFC-UCNP probe were drastically increased after adding CuSO4 (aq.) to any of the non-specific cations (Fig. 5b). Therefore, Cu+ ions are the most responsive to the applied voltage of 0.3 V and are specifically adsorbed onto the composite probe resulting in a remarkable improvement in its optical sensing performance.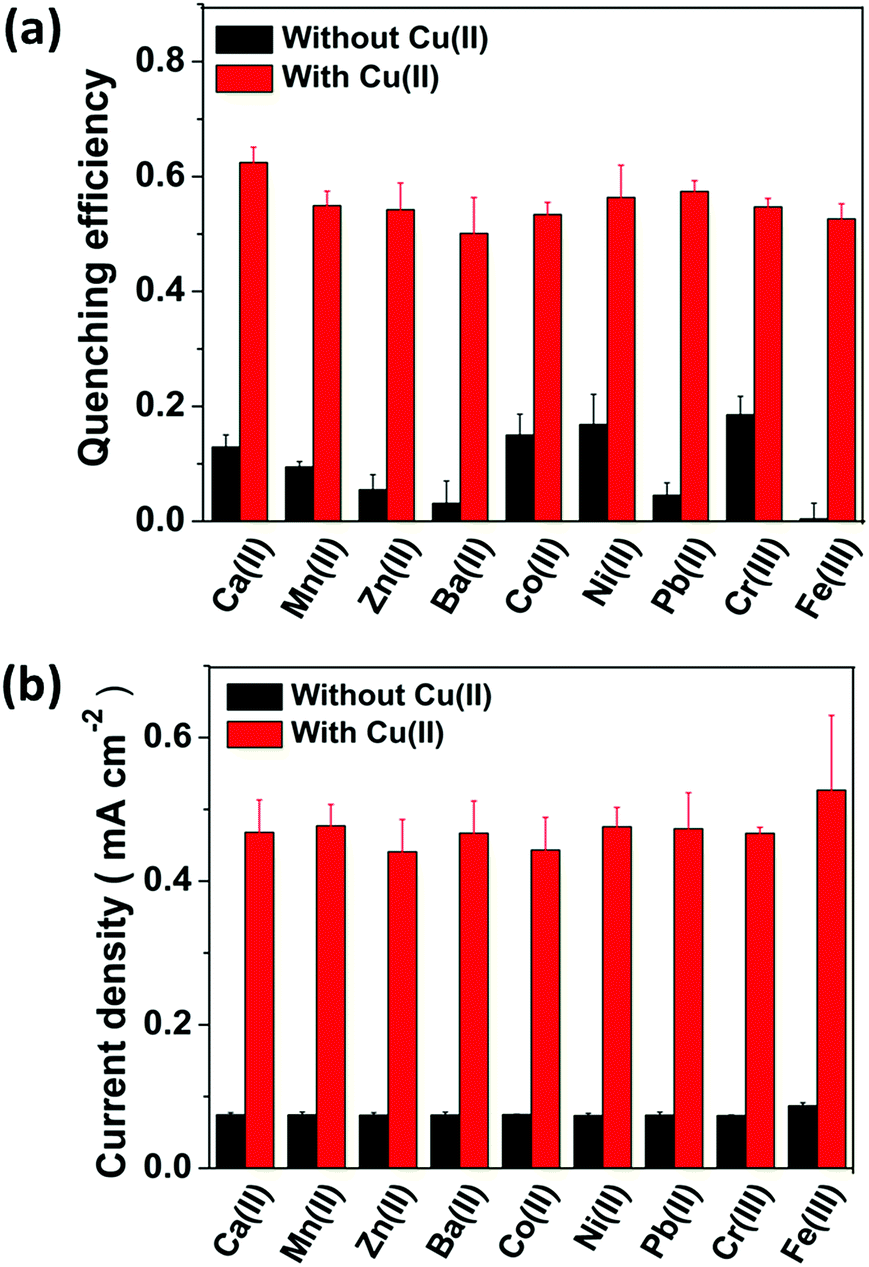 | ||
| Fig. 5 (a) The optical responses and (b) the E-chem responses of the CFC-UCNP probe at 0.3 V for sensing various non-specific cations. Black bars correspond to the single-ion system while red bars correspond to the co-existing non-specific cation/Cu2+ ion system. All error bars were obtained from three separated experiments ([Cu2+] = 315 μM, [Ba2+/Ni2+/Pb2+/Co2+/Cr3+] = 25 μM and [Ca2+/Mn2+/Zn2+/Fe3+] = 500 μM). | ||
Conclusions
In conclusion, a composite probe was constructed by the use of CFC incorporated with core@shell UCNPs for Cu2+ ion sensing. An E-chem technique was introduced for the first time for the UCL probe, which demonstrated a significant improvement in its sensitivity and selectivity due to specific accumulation of analyte ions on the probe. The LOD of the sensor was drastically improved from 25.8 ppm to 82 ppb under a small potential, which is lower than the tolerance levels suggested by WHO and USEPA. Our work suggests that the proposed flexible composite probe is a promising sensor for on-site monitoring of contaminated drinking water with high sensitivity and selectivity. The sensing scheme can be potentially extended to sensing other heavy metal ions by choosing suitable applied voltages and electrolytes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by grants from the Research Grants Council (RGC) of Hong Kong (RGC GRF No. PolyU 153036/14P) and PolyU Project (4-BCCA).Notes and references
- J. A. Cotruvo Jr, A. T. Aron, K. M. Ramos-Torres and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC.
- S. G. Kaler, Nat. Rev. Neurol., 2011, 7, 15–29 CrossRef CAS PubMed.
- X. Pu, Z. Jiang, B. Hu and H. Wang, J. Anal. At. Spectrom., 2004, 19, 984–989 RSC.
- X. Duan, R. L. McLaughlin, I. D. Brindle and A. Conn, J. Anal. At. Spectrom., 2002, 17, 227–231 RSC.
- K. C. Teo and J. Chen, Analyst, 2001, 126, 534–537 RSC.
- H. Dong, S. R. Du, X. Y. Zheng, G. M. Lyu, L. D. Sun, L. D. Li, P. Z. Zhang, C. Zhang and C. H. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS PubMed.
- P. Huang, W. Zheng, S. Zhou, D. Tu, Z. Chen, H. Zhu, R. Li, E. Ma, M. Huang and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 1252–1257 CrossRef CAS PubMed.
- J. Peng, W. Xu, C. L. Teoh, S. Han, B. Kim, A. Samanta, J. C. Er, L. Wang, L. Yuan, X. Liu and Y.-T. Chang, J. Am. Chem. Soc., 2015, 137, 2336–2342 CrossRef CAS PubMed.
- Y. Liu, D. Tu, W. Zheng, L. Lu, W. You, S. Zhou, P. Huang, R. Li and X. Chen, Nano Res., 2018, 11, 3164–3174 CrossRef CAS.
- M. K. Tsang, G. Bai and J. Hao, Chem. Soc. Rev., 2015, 44, 1585–1607 RSC.
- Z. Yi, X. Li, Z. Xue, X. Liang, W. Lu, H. Peng, H. Liu, S. Zeng and J. Hao, Adv. Funct. Mater., 2015, 25, 7119–7129 CrossRef CAS.
- M.-K. Tsang, Y.-T. Wong and J. Hao, Small Methods, 2018, 2, 1700265 CrossRef.
- X. Lei, R. Li, D. Tu, X. Shang, Y. Liu, W. You, C. Sun, F. Zhang and X. Chen, Chem. Sci., 2018, 9, 4682–4688 RSC.
- J. Zhang, B. Li, L. Zhang and H. Jiang, Chem. Commun., 2012, 48, 4860–4862 RSC.
- C. Li, J. Liu, S. Alonso, F. Li and Y. Zhang, Nanoscale, 2012, 4, 6065–6071 RSC.
- Z. Cui, W. Bu, W. Fan, J. Zhang, D. Ni, Y. Liu, J. Wang, J. Liu, Z. Yao and J. Shi, Biomaterials, 2016, 104, 158–167 CrossRef CAS PubMed.
- Q. Liu, J. Peng, L. Sun and F. Li, ACS Nano, 2011, 5, 8040–8048 CrossRef CAS PubMed.
- S. Xu, S. Xu, Y. Zhu, W. Xu, P. Zhou, C. Zhou, B. Dong and H. Song, Nanoscale, 2014, 6, 12573–12579 RSC.
- T. Liang, Z. Li, D. Song, L. Shen, Q. Zhuang and Z. Liu, Anal. Chem., 2016, 88, 9989–9995 CrossRef CAS PubMed.
- S. M. Saleh, R. Ali and O. S. Wolfbeis, Chem.–Eur. J., 2011, 17, 14611–14617 CrossRef CAS PubMed.
- B. C. Barja and P. F. Aramendia, Photochem. Photobiol. Sci., 2008, 7, 1391–1399 RSC.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- J. Qi, J. Chen, G. Li, S. Li, Y. Gao and Z. Tang, Energy Environ. Sci., 2012, 5, 8937–8941 RSC.
- L. He, Y. Liu, J. Liu, Y. Xiong, J. Zheng, Y. Liu and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 3741–3745 CrossRef CAS PubMed.
- M.-K. Tsang, W. Ye, G. Wang, J. Li, M. Yang and J. Hao, ACS Nano, 2016, 10, 598–605 CrossRef CAS PubMed.
- Y. Li, J. Tang, L. He, Y. Liu, Y. Liu, C. Chen and Z. Tang, Adv. Mater., 2015, 27, 4075–4080 CrossRef CAS PubMed.
- G. Bai, M.-K. Tsang and J. Hao, Adv. Funct. Mater., 2016, 26, 6330–6350 CrossRef CAS.
- W. Yantasee, Y. Lin, T. S. Zemanian and G. E. Fryxell, Analyst, 2003, 128, 467–472 RSC.
- Y. Liu, N. Fu, G. Zhang, M. Xu, W. Lu, L. Zhou and H. Huang, Adv. Funct. Mater., 2017, 27, 1605307 CrossRef.
- Z. Xu and C. Gao, Mater. Today, 2015, 18, 480–492 CrossRef CAS.
- WHO, Guidelines for drinking-water quality: fourth edition incorporating the first addendum, World Health Organization, Geneva, 2017 Search PubMed.
- USEPA, National Primary Drinking Water Regulations, United States Environmental Protection Agency, 2009 Search PubMed.
- F. Wang, R. Deng and X. Liu, Nat. Protoc., 2014, 9, 1634–1644 CrossRef CAS PubMed.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS PubMed.
- Z. Wu, C. U. Pittman and S. D. Gardner, Carbon, 1995, 33, 597–605 CrossRef CAS.
- W. Shao, G. Pattanaik and G. Zangari, J. Electrochem. Soc., 2007, 154, D201–D207 CrossRef CAS.
- S. Basak, P. S. Zacharias and K. Rajeshwar, J. Electroanal. Chem. Interfacial Electrochem., 1991, 319, 111–123 CrossRef CAS.
- J. Peng, J. Li, W. Xu, L. Wang, D. Su, C. L. Teoh and Y.-T. Chang, Anal. Chem., 2018, 90, 1628–1634 CrossRef CAS PubMed.
- D. Tu, Y. Liu, H. Zhu, R. Li, L. Liu and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 1128–1133 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00012c |
| ‡ Y.-T. Wong and S.-Y. Pang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |