
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a hydroxyapatite/poly(methyl methacrylate) nanocomposite using dolomite
W. P. S. L.
Wijesinghe
 *ab,
M. M. M. G. P. G.
Mantilaka
ab,
T. S. E. F.
Karunarathne
b and
R. M. G.
Rajapakse
a
*ab,
M. M. M. G. P. G.
Mantilaka
ab,
T. S. E. F.
Karunarathne
b and
R. M. G.
Rajapakse
a
aDepartment of Chemistry, Faculty of Science, University of Peradeniya, Sri Lanka. E-mail: slwijesinghe@gmail.com
bSri Lanka Institute of Nanotechnology, Pitipana, Homagama, Sri Lanka
First published on 21st September 2018
Abstract
Hydroxyapatite/poly(methyl methacrylate) (HA–PMMA) nanocomposites are extensively used in biomedical fields. Therefore, the design and development of low-cost and industrially viable novel methods are essential to synthesize HA–PMMA nanoparticles. In this letter, we report such an economical, simple and industrially applicable novel method to synthesize nanosized HA–PMMA composite particles using extensively distributed dolomite.
Hydroxyapatite [HA, Ca10(PO4)6(OH)2], also known as a bioceramic, is the major component of human bones and teeth (hard tissues) and is chemically compatible with synthetic HA nanoparticles.1 Hence, synthetic HA nanoparticles are extensively employed in biomedical fields due to their excellent biocompatibility.2,3 In the preparation of artificial bones or bone cement, it is important to maintain the mechanical properties of the materials in order to mimic natural bones.4 However, HA alone cannot perform all the requirements of clinical applications due to the brittleness and stiffness of HA nanoparticles.5,6 Therefore, many researchers have developed HA/polymer nanocomposites to enhance the bioactive properties and mechanical properties of the artificial bone or bone cement. Poly(methyl methacrylate) (PMMA) has become one of the attractive and frequently used polymers in the synthesis of bone cements. Poly(methyl methacrylate) (PMMA) is the first synthetic polymer used in biomedical applications in 1937.7–9 HA can be combined with PMMA in order to synthesize HA/PMMA nanocomposites. When PMMA is associated with HA, it increases the biocompatibility, osteoconductivity, and mechanical properties of the nanocomposites. Therefore, PMMA–HA nanocomposites have good ability to fill dental cavities and also to generate a strong bond between the bone and the prosthesis.10,11 Numerous methods have been developed to synthesize HA–PMMA composites.10–12 For instance, Teodora et al. (2009) described the biocompatibility of HA–PMMA composites which indicates the better cell adhesion on the synthesized HA–PMMA composites.13 Andrei et al. (2016) synthesized HA–PMMA composites for application in dentistry with a better biocompatibility14 and Nieto et al. (2012) used the HA–PMMA composite as a coating material for polythene substrates for use in biomedical applications while increasing mechanical properties.15 Therefore, HA–PMMA composites can be used in different biomedical applications such as bone filler materials, bone cements, bone implants and dental implants. However, simplicity and economic factors are timely needed requirements for the industrial scale manufacture of HA–PMMA. Therefore, in this manuscript, we report a novel, economical and potential industrially applicable method to synthesize HA–PMMA composites using readily available and extensively distributed dolomite as a calcium source. This method is industrially important due to the usage of readily available cheap naturally occurring dolomite as a raw material rather than using expensive chemicals and due to the involvement of a one pot in situ synthesis route.16,17
Herein, sucrose, potassium persulphate (K2S2O8), ammonium dihydrogen orthophosphate ((NH4)H2PO4) and methyl methacrylate (MMA) were purchased from Sigma-Aldrich to use as raw materials for the synthesis. First, a calcium sucrate solution was prepared by adding 5.00 g of calcined dolomite (CaO·MgO) into 0.5 M sucrose (100 mL) while stirring and by continuing the stirring for 6 h. The mixture was filtered under suction and the solution was collected.18 Then, 1.00 g of potassium persulfate was dissolved in 100 mL of 0.5 M prepared calcium sucrate solution in a three-neck rounded bottom flask and heated to 80 °C. 10 mL of freshly distilled MMA and 100 mL of 0.3 M ammonium dihydrogen orthophosphate were added (until Ca/P ratio of 1.67) to the reaction mixture using two dropping funnels while stirring. The mixture was further stirred for 12 h and filtered under suction to obtain a precipitate. The precipitate was washed with distilled water 3 times and allowed to dry under ambient conditions. The yield of the obtained HA was 84%.
X-ray diffraction (XRD) patterns of the synthesized products were obtained using a Siemens D5000 powder diffractometer. Fourier Transform Infrared (FT-IR) spectra of the products were recorded on a Shimadzu IR Prestige 21 instrument with the KBr pellet method. Raman spectra of both HA and HA–PMMA samples were recorded on a Bruker Senterra 2 instrument. The morphology of the products was examined using Transmission Electron Microscopy (TEM) with the help of a JEOL JEM-2000FX Electron Microscope. Differential scanning calorimetry (DSC) was performed using an STA S-1500 instrument.
Preparation of HA–PMMA nanocomposites through calcium sucrate without using CaO directly is a better solution to prevent the inclusion of impurities such as MgO, and unreacted CaO and CaCO3 in the final product.18 Hence, dolomite is employed as a raw material for the proposed method.18,19 A white precipitate is formed towards the end of stirring (Section 2.2). The synthesized HA–PMMA composite is a white colored crystalline powder with fine particles. The XRD pattern of HA/PMMA given in Fig. 1A(b) has peaks at 2-theta values of 25.78°, 31.74°, 32.88°, 39.68°, 46.62°, 49.43°, and 53.02° which represent hydroxyapatite (JCPDS card no. 72-1243). The XRD pattern of the synthesized HA–PMMA is similar to the XRD pattern of bare HA (Fig. 1A(a)). Also, the synthesized product only contains pure HA as the crystalline material and hence, it does not contain any other crystalline impurities such as unreacted reactants. The estimated average particle size of HA in the composite from the Debye–Scherrer formula is 11 nm. XRD peaks of PMMA are not found in the XRD spectrum of the composite because it is amorphous (Fig. 1A(c)).
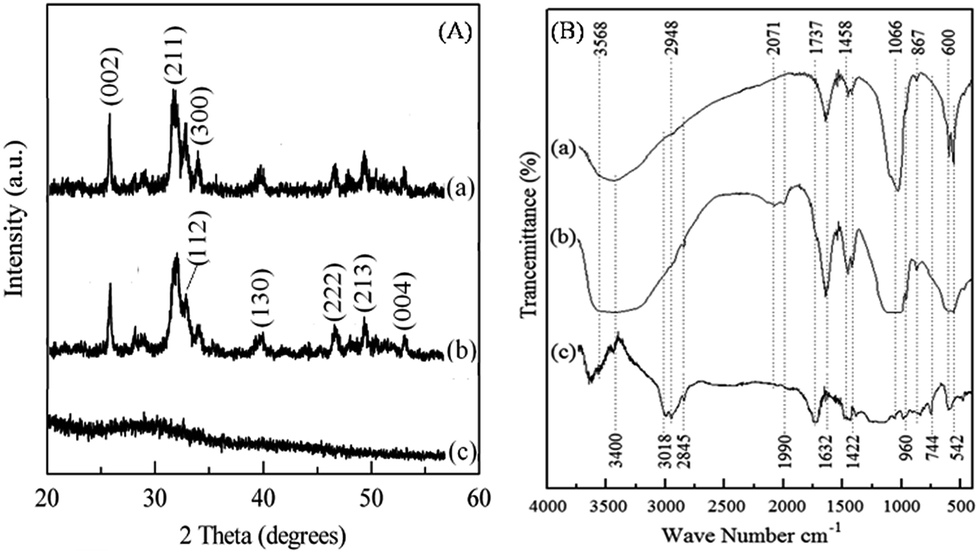 | ||
| Fig. 1 (A) XRD pattern of (a) HA, (b) the HA–PMMA composite and (c) PMMA. (B) FTIR spectrum of (a) HA, (b) the HA–PMMA composite and (c) PMMA. | ||
In Fig. 1B(b), the characteristic absorption bands of the FT-IR spectrum of HA at 542 cm−1, 600 cm−1, 960 cm−1 and 1066 cm−1 reveal the presence of HA in the HA–PMMA composite.20,21 FT-IR spectra of PMMA and HA are recorded to compare with the spectrum of the composite. It is clearly identified that absorption bands at 744 cm−1 and 2848 cm−1 are common for both PMMA and the composite. These bands are not found in HA synthesized in the absence of PMMA. This confirms the presence of PMMA in the composite.19,20 The bands at 867 cm−1, 1428 cm−1 and 1458 cm−1 are attributed to the C–O bonds of PMMA which are hydrogen bonded with HA.21 The band at 2845 cm−1 is related to the C–H bonds which are present in PMMA groups. A new band appears at around 2000 cm−1 which can be assigned to the carbonyl group.20,21 This band indicates the formation of chelate bonds between the carbonyl group and Ca2+ in HA. This may be due to the breakage of the ester bond between –CO and –OCH3, in order to make bonds between HA and carbonyl carbon. Also, some of the bands of PMMA have been overlapped by bands of HA in the composite. This may be due to the absorption of the –COOCH3 groups of PMMA for the growth of HA particles during the nucleation process.18,20
Fig. 2 shows the Raman spectra of both HA and HA–PMMA samples. The Raman spectrum in Fig. 2a shows the characteristic bands of HA at 440 cm−1, 600 cm−1, 960 cm−1 and 1050 cm−1 which reveal the presence of HA in the HA–PMMA composite.22 Raman spectra of PMMA and HA are recorded to compare with the spectrum of the composite. It is clearly identified that absorption bands at 440 cm−1, 600 cm−1 and 960 cm−1 are common for both HA and the composite.22,23 Raman shifts of the major bands of PMMA those appeared in our HA–PMMA spectrum (Fig. 2b) are comparable with those reported by Thomas et al. (2008).24 The bands around 1400 cm−1 are attributed to the C–H, O–CH3 and –CH3 groups of PMMA in the HA–PMMA composite. The intense band at around 1260 cm−1 is attributed to the C–O and C–COO functional groups which are present in the PMMA component of the composite.24 The bands around 1100 cm−1 are related to the symmetric stretching mode (ν3) of the PO4 groups of HA. The bands 999 cm−1 (O–CH3) and 1081 cm−1 (C–C skeletal mode) are masking each other in the prepared HA–PMMA composite due to the hydrogen bonding and coordination bonding. These bands are not found in HA synthesized in the absence of PMMA. These results are also comparable with FTIR spectra and further confirm the presence of PMMA in the composites.
 | ||
| Fig. 2 Raman spectrum of (a) HA and (b) the HA–PMMA composite. | ||
The TEM images of the prepared HA–PMMA are shown in Fig. 3. The calculated average particle size of the HA–PMMA nanocomposite, using TEM images, is approximately 30 nm. Also, the synthesized composite presents a spherical morphology. Thermal characterization results confirm the presence of PMMA in the synthesized HA–PMMA nanocomposite. The DSC curve of HA–PMMA (Fig. 4) shows an exothermic peak in the temperature range 445–479 °C and the TGA curve of HA–PMMA (Fig. 4) shows a mass loss which reveal the combustion of PMMA. The DSC exothermic peak further confirms the presence of PMMA in the composite. Similarly, CaCO3–PMMA nanocomposites have been synthesized and characterized by Mantilaka et al. 2013 and the PMMA decomposition range in CaCO3–PMMA was 435–453 °C in their study.18 This indicates the increment of thermal properties of PMMA during the formation of PMMA composites. Also, several studies have been carried out to demonstrate the increment of decomposition temperature with the formation of PMMA composites.11,25,26
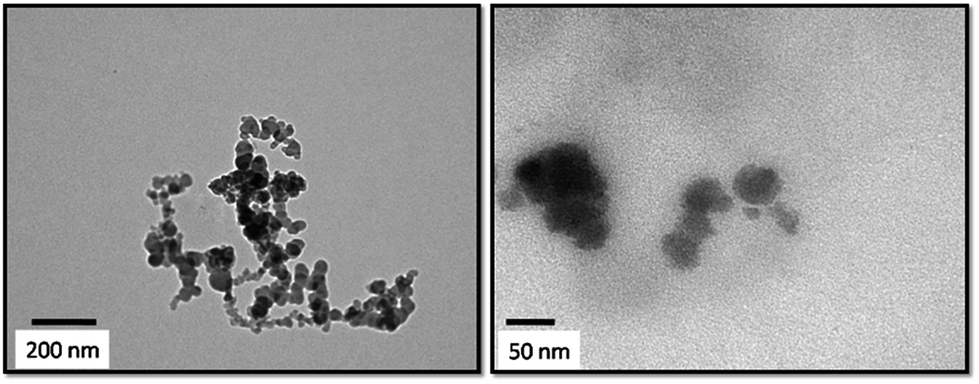 | ||
| Fig. 3 TEM images of the HA–PMMA composite at two different magnifications. | ||
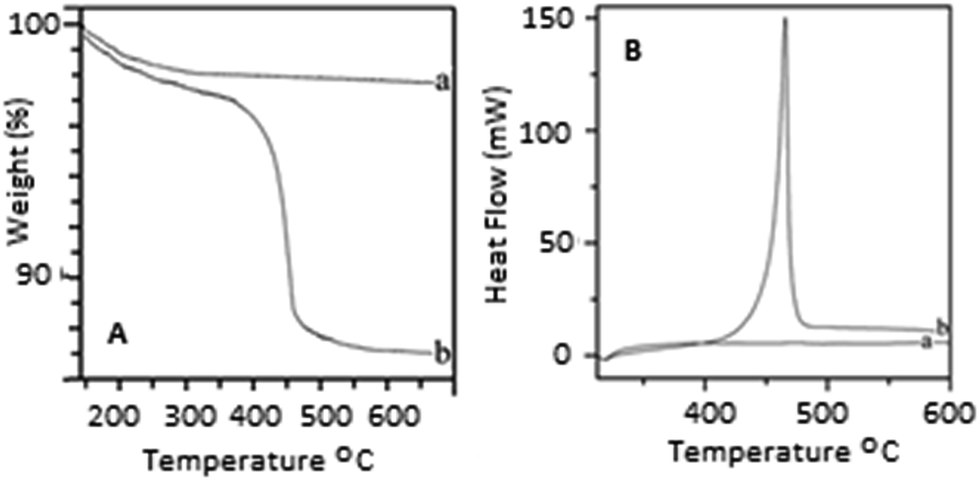 | ||
| Fig. 4 [A] TGA curves of (a) HA and (b) the PMMA–HA nanocomposite [B] DSC curves of (a) HA and (b) the PMMA–HA nanocomposite. | ||
Conclusions
A nanosized HA–PMMA composite was prepared by a novel, simple and industrially applicable method using dolomite. The synthesized HA–PMMA nanocomposite was 30 nm in size and had a spherical morphology with 84% yield.Conflicts of interest
There is no conflicts of interest.Acknowledgements
The research was funded by the National Research Council, Sri Lanka (Grant no. NRC/11-46).Notes and references
- Y. Tseng, C. Kuo, Y. Li and C. Huang, Mater. Sci. Eng., C, 2009, 29, 819 CrossRef CAS.
- Y. Han, S. Li, X. Wang, I. Bauer and M. Yin, Ultrason. Sonochem., 2007, 14, 286 CrossRef CAS PubMed.
- B. Cengiz, Y. Gokce, N. Yildiz, Z. Aktas and A. Calimli, Colloids Surf., A, 2008, 322, 29 CrossRef CAS.
- K. E. Tanner, Proc. Inst. Mech. Eng., Part H, 2010, 224, 1359 CrossRef CAS PubMed.
- Z. Dong, Y. Li and Q. Zou, Appl. Surf. Sci., 2009, 255, 6087 CrossRef CAS.
- Y. Fujishiro, H. Yabuki, K. Kawamura, T. Sato and A. Okuwaki, J. Chem. Technol. Biotechnol., 1993, 57, 349 CrossRef CAS PubMed.
- R. Murugan and S. Ramakrishna, Compos. Sci. Technol., 2005, 65, 2385 CrossRef CAS.
- M. K. Singh, P. A. Marques, A. C. Sousa, J. Gracio, V. Silva, P. Goncalves and S. Olhero, Int. J. Nano Biomater., 2009, 2, 442 CrossRef CAS.
- D. Gnanasekaran, K. Madhavan and B. S. R. Reddy, J. Sci. Ind. Res., 2009, 68, 437 CAS.
- A. S. Hamizah, M. Mariatti, R. Othman, M. Kawashita and A. R. N. Hayati, J. Appl. Polym. Sci., 2012, 125, 661 CrossRef.
- Y. Wang, Y. Xiao, X. Huang and M. Lang, J. Colloid Interface Sci., 2011, 360, 415 CrossRef CAS PubMed.
- S. Zebarjad, S. Sajjadi, T. Sdrabadi, S. Sajjadi, A. Yaghmaei and B. Naderi, Engineering, 2011, 3, 795 CrossRef CAS.
- T. G. Tihan, M. D. Ionita, R. G. Popescu and D. Iordachescu, Mater. Chem. Phys., 2009, 118, 165 CrossRef.
- A. T. Cucuruza, E. Andronescu, A. Ficai, A. Ilie and F. Iordache, Int. J. Pharm., 2016, 510, 516 CrossRef PubMed.
- V. M. Nieto, C. H. Navarroa, K. J. Morenoa, A. A. Morquecho, A. C. Valdez, S. G. Miranda and J. F. L. Hernández, Prog. Org. Coat., 2013, 76, 204 CrossRef.
- Y. R. Somarathna, M. M. M. G. P. G. Mantilaka, D. G. G. P. Karunaratne, R. M. G. Rajapakse, H. M. T. G. A. Pitawala and K. G. U. Wijayantha, Cryst. Res. Technol., 2016, 51, 207 CrossRef CAS.
- M. M. M. G. P. G. Mantilaka, W. P. S. L. Wijesinghe, H. M. T. G. A. Pitawala, R. M. G. Rajapakse and D. G. G. P. Karunaratne, J. Natl. Sci. Found. Sri Lanka, 2014, 42, 221 Search PubMed.
- M. M. M. G. P. G. Mantilaka, D. G. G. P. Karunaratne, R. M. G. Rajapakse and H. M. T. G. A. Pitawala, Powder Technol., 2013, 235, 628 CrossRef CAS.
- W. P. S. L. Wijesinghe, M. M. M. G. P. G. Mantilaka, E. V. A. Premalal, H. M. T. U. Herath, S. Mahalingam, M. Edirisinghe, R. P. V. J. Rajapakse and R. M. G. Rajapakse, Mater. Sci. Eng., C, 2014, 42, 83 CrossRef CAS PubMed.
- C. H. Navarro, K. J. Moreno, A. C. Valdez, F. L. Hernández, J. S. G. Miranda, R. Lesso and A. A. Morquecho, Wear, 2012, 282, 76 CrossRef.
- A. Angelopoulou, E. K. Efthimiadou, N. Boukos and G. Kordas, Colloids Surf., B, 2014, 117, 322 CrossRef CAS PubMed.
- S. Koutsopoulosy, Inc. J Biomed Mater Res, 2002, 62, 600 CrossRef PubMed.
- C. A. Zamperini, R. S. André, V. M. Longo, E. G. Mima, C. E. Vergani, A. L. Machado, J. A. Varela and E. Longo, J. Nanomater., 2013, 174398 Search PubMed.
- K. J. Thomas, M. Sheeba, V. P. N. Nampoori, C. P. G. Vallabhan and P. Radhakrishnan, J. Opt. A: Pure Appl. Opt., 2008, 10, 055303 CrossRef.
- A. Singhal, A. Dubey, Y. K. Bhardwaj, D. Jain, S. Choudhury and A. K. Tyagi, RSC Adv., 2013, 3, 20913 RSC.
- N. W. Elshereksi, S. H. Mohamed, A. Arifin and Z. A. M. Ishak, J. Phys. Sci., 2014, 25, 15 CAS.
| This journal is © The Royal Society of Chemistry 2019 |