
Anatomical redistribution of endogenous copper in embryonic mice overexpressing SOD1
K.
Kysenius
 *ab,
J. B.
Hilton
a,
B.
Paul
bc,
D. J.
Hare
bde and
P. J.
Crouch
ab
*ab,
J. B.
Hilton
a,
B.
Paul
bc,
D. J.
Hare
bde and
P. J.
Crouch
ab
aDepartment of Pharmacology and Therapeutics, the University of Melbourne, Victoria 3052, Australia. E-mail: kai.kysenius@unimelb.edu.au
bFlorey Institute of Neuroscience and Mental Health, the University of Melbourne, Victoria 3052, Australia
cSchool of Earth Sciences, the University of Melbourne, Parkville, Victoria 3052, Australia
dMelbourne Dementia Research Centre at the Florey Institute of Neuroscience and Mental Health, Parkville, Victoria 3052, Australia
eElemental Bio-imaging Facility, University of Technology Sydney, Broadway, New South Wales 2007, Australia
First published on 26th September 2018
Abstract
Mutations in the copper (Cu)- and zinc (Zn)-binding metalloenzyme Cu/Zn-superoxide dismutase (SOD1) cause familial forms of amyotrophic lateral sclerosis (ALS), a fatal adult-onset neurodegenerative disorder of the central nervous system (CNS). Transgenic over-expression of mutant SOD1 produces a robust ALS-like phenotype in mice. Despite being ubiquitously expressed from the moment of conception, the mechanisms underlying the CNS-selective phenotype of mutant SOD1 expression remain poorly understood. We have previously shown that the physiological requirement for copper in SOD1 is unsatiated in the CNS of adult mice overexpressing mutant SOD1 and that suboptimal delivery of Cu to SOD1 in these mice progressively worsens with age. An age-related impediment to Cu availability may therefore contribute to the adult onset of disease in cases of ALS caused by mutant SOD1. Here, we have extended the age-related investigation of Cu in SOD1 overexpressing transgenic mice to the embryonic stage of development. We used the quantitative in situ elemental imaging method, laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS), to assess the endogenous distribution of Cu, Zn and other endogenous elements (carbon, phosphorus, sulphur, magnesium, manganese and iron) in the embryonic day 14 (E14) embryos of transgenic mice overexpressing wild-type human SOD1 (hSOD1Wt) or mutant human SOD1 (hSOD1G37R). We show that in contrast to adult mice, SOD1 overexpression (both wild-type and mutant) is associated with an overt redistribution of Cu from the liver to the CNS during embryonic development. Also in contrast to adult mice, Zn redistribution to the CNS in response to SOD1 over-expression is relatively modest in embryonic mice, being limited to the brainstem. No other elemental changes between genotypes were observed. Our application of quantitative LA-ICP-MS in situ imaging details the first anatomical mapping of endogenous elements in embryonic mice. The observed redistribution of Cu from the liver to the CNS in response to SOD1 overexpression during embryogenesis indicates that the impediment of Cu delivery to SOD1, which is evident in adult mutant SOD1 overexpressing mice, only occurs at a later stage in life.
 K. Kysenius | Kai Kysenius is a Postdoctoral Research Fellow at the University of Melbourne, Australia. After receiving his PhD in Physiology and Neuroscience from the University of Helsinki in 2015, the Finnish native was awarded the Sigrid Juselius Fellowship and relocated to Melbourne, Australia, to research therapies for motor neurone disease. Kai's current work incorporates the use of metal imaging to further our understanding of metal biology and its role in disease pathogenesis. |
Significance to metallomicsMetals are required by nearly half of all known enzymes and disruptions to the natural metal distribution within the body are intimately involved in both ageing and the development of neurodegenerative diseases. Ageing-related disruptions in copper are seen in mouse models of amyotrophic lateral sclerosis (ALS), and are amenable to therapeutic restoration leading to symptomatic improvement. In this study we explore the anatomical distribution of metals, including copper, during the embryonic developmental stages of a mouse model of ALS using advanced anatomical elemental mapping to provide insight into disease-related metal redistribution in ALS. |
Introduction
Metals are essential to the survival of organisms and are intimately involved in the functionality of almost half of all known enzymes.1 These metalloenzymes account for significant proportion of each of the six different enzyme classes and the metals can be involved in either a direct catalytic capacity or provide structural integrity to the protein.1 Considering their importance in enzymatic function, essential metals, which include iron, zinc, magnesium, manganese and copper, are held under strict homeostatic control within the cell via a system of dedicated chaperones and transporters (reviewed by Martinez-Finley et al.2). Genetic defects affecting these chaperones and transporters can have serious implications for human health. For example, mutations affecting the copper transporters ATP7B and ATP7A lead to systemic copper dyshomeostasis and subsequent CNS pathology, and are the molecular basis of the neurodegenerative disorders Wilson's disease and Menkes disease, respectively.3 Moreover, if left unchecked, redox metals are capable of mediating widespread oxidative damage.4Substitution mutations in the ubiquitously expressed antioxidant enzyme Cu/Zn-superoxide dismutase (SOD1) were the first discovered genetic cause of familial cases of amyotrophic lateral sclerosis (ALS),5 a degenerative condition that selectively affects motor neurones in the central nervous system (CNS). Despite ubiquitous expression of mutant SOD1 throughout life, the initial symptoms of ALS caused by mutant SOD1 (limb weakness, for example) only manifest in the mid to late stages of life. After disease onset, symptoms progress rapidly, culminating in paralysis and the inability to move, breathe, speak and swallow. Most people with ALS will not survive more than three years after diagnosis. This poor prognosis is due to the current absence of effective treatment options, which in turn is due to an incomplete understanding of the biochemical causes of the disease.
Transgenic animals overexpressing mutant SOD1 are a robust model of ALS pathology and have been used extensively since their initial development.6,7 Despite overexpressing mutant SOD1 to levels greatly exceeding those seen in the human condition, these models allow for the interrogation of the toxic gain-of-function mechanisms through which SOD1 mutations induce motor neurone degeneration. Physiologically, the functional form of SOD1 exists as a dimer with each subunit binding one catalytic copper atom and one structural zinc atom.8 Mutations to the SOD1 gene associated with ALS have been established to adversely affect the stability of translated protein giving rise to misfolding and subsequent aggregation implicated in the disease.9,10
In vitro assessments of mutant SOD1 using protein folding kinetic approaches have demonstrated that ALS-causing mutations drive the formation of copper-deficient misfolded species and that resultant metal-deficient aggregates formed in cells contained significantly diminished levels of copper.11,12 Furthermore, interrogation of mutant SOD1 in vivo has demonstrated that approximately 50% of SOD1 in the spinal cords of SOD1G37R and SOD1G93A mice is copper-deficient,12,13 and since the copper is required for SOD1 activity,14 SOD1 activity in the CNS of mutant SOD1-expressing mice is not commensurate with the large increase in SOD1 protein levels.10,15
However, these observations fail to explain why motor neurones in the CNS are selectively affected despite mutant SOD1 being expressed ubiquitously. One possible explanation may lie in the disproportional rates of bioavailable copper delivery across different tissues, with the adult CNS showing a substantially slower rate of copper turnover compared to peripheral tissues.16 In the context of SOD1G37R mice this was found to translate into a selective inability of the CNS to satiate the elevated copper requirements commanded by mutant SOD1 overexpression. In peripheral tissues of the SOD1G37R mice, this increased mutant SOD1 expression led to a commensurate increase in levels of both zinc and copper which bind to SOD1. Contrarily, whilst zinc levels increased commensurately in the SOD1G37R mouse CNS, levels of copper were either unchanged or only marginally increased.17 Taken together, these outcomes indicate that a bottleneck in the CNS is present for copper, leaving limited recourse under circumstances that require elevated levels of bioavailable copper.
More recently, it has been shown that the CNS-specific bottleneck that affects copper delivery to SOD1 also affects additional cuproenzymes in the CNS of SOD1G37R mice, with activity of the copper-dependent ferroxidase ceruloplasmin also diminished in the adult SOD1G37R mouse spinal cord.15 Moreover, it was shown that while the impact of the copper bottleneck on these two cuproenzymes in the CNS worsens with age, it can be overcome by genetically increasing copper levels in the CNS; concomitant overexpression of the copper uptake transporter CTR1 in SOD1G37R mice improves the animals’ ALS-like phenotype10 and restores SOD1 and ceruloplasmin activity.15
Although these studies provide evidence for endogenous copper availability playing a role in forms of ALS involving mutant SOD1, an understanding of why SOD1 mutations expressed from the time of conception only cause ALS later in life remains elusive. The present study was therefore undertaken to examine whether the inability for the CNS to satiate the elevated requirement for copper in mutant SOD1 overexpressing mice is a phenomenon that is restricted to the adult CNS. To achieve this, we examined the anatomical distribution of copper across various tissues in embryonic mice that overexpress mutant SOD1 (SOD1G37R) or wild-type SOD1 (SOD1Wt) using an in situ laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) approach. This approach enables simultaneous collection of quantitative data for multiple elements within biological samples combined with the capacity to retain spatial information at a near cellular level.18 By investigating the early developmental stages of the SOD1G37R mice we aimed to better understand the elemental cues underpinning the late-stage development of ALS.
Experimental methods
Animal breeding
All experimentation involving mice was approved by an Animal Experimentation Ethics Committee (University of Melbourne Approval #1513772). All procedures were conducted in accordance with National Health and Medical Research Council guidelines.Male transgenic mice expressing human SOD1 harbouring the G37R substitution mutation (SOD1G37R)19 or wild-type human SOD1 (SOD1Wt) on the congenic C57BL/6 background were mated with non-transgenic C57BL/6 females. Timed matings were performed and at day 14 of embryonic development (E14), pregnant dams were euthanised by CO2 asphyxiation followed by cervical dislocation. Stomach surface was sprayed with 80% ethanol and a sagittal cut made using scissors to open the stomach cavity to expose the two embryo-containing uterine horns. Each uterine horn was removed and placed into ice-cold tris-buffered saline (TBS) for 30 min. Embryos were dissected from their individual embryonic sacs, rinsed in TBS, dabbed dry on a moist paper towel, weighed on a scale and then a 1 mm section of the tail removed for genotyping as previously described.10 Whole embryos were placed into 15 × 15 × 5 mm Biopsy Cryomolds® (Tissue-Tek®) and covered with approximately 1 mL of Tissue-Tek® Optimal Cutting Temperature (OCT) compound and then placed directly onto dry ice. Once frozen, samples were stored at −80 °C until used for cryosectioning and analysis. Embryos from 6 pregnant dams (3 × SOD1Wt, 3 × SOD1G37R) were used for this study.
Cryosectioning and histological staining
Embryos frozen in OCT were oriented on the cryostat stage to enable sectioning through the sagittal plane. The cryostat (Leica CM1850) chamber and stage were maintained at −20 °C with the samples temperature-equilibrated in the chamber for at least 1 hour before cutting. Block faces were first trimmed then sectioned at 40 μm to progress through to the anatomical mid-sagittal plane. Sequential midline sections cut at 25 μm were mounted onto SuperFrost® Plus (Thermo Scientific) microscope slides. Embryo sections adjacent to the ablated sections were stained with haematoxylin and eosin stain (H&E stain). Whole slide scanning was used to obtain microscopic images of the H&E stained sections (APN Slide Scanning Service, Department of Anatomy, University of Melbourne).Quantitative in situ elemental imaging
LA-ICP-MS analyses were performed using sections mounted onto microscope slides as described recently.20,21 Briefly, unstained 25 μm thick sections on slides were air-dried overnight and placed in the ablation cell together with matrix-matched elemental standards for quantitative imaging.22 The sections were ablated using a 40 μm square laser spot size at a scanning speed of 160 μm s−1 on an Elemental Scientific Lasers NWR213 ablation system (Kennelec Scientific, Mitcham, Victoria, Australia). Ablated material was transferred into the Agilent 8800 QQQ-ICP-MS (Mulgrave, Victoria, Australia) using an argon gas flow at 1.2 L min−1. Analysis time for each embryo section was roughly 6–8 hours, with a maximum of four sections run on the same slide. Ablated samples were analysed for the following isotopes: 13C, 24Mg, 31P, 33S, 55Mn, 56Fe, 63Cu and 66Zn, accounting for between 100% (Mn, P) and 0.75% (S) of the total pool of their stable element levels (Table 1).23 The measured isotopes for each element were selected to optimise signal intensity and signal/noise ratios (Table 1) calculated as the ratio of average signal intensities from the tissue and slide surface. Data were analysed using the Iolite analysis software (School of Earth Sciences, University of Melbourne) operating under the Igor Pro suite.| Isotope | Relative stable isotope abundance (%) | Signal/noise ratio |
|---|---|---|
| Average isotope-specific signal/noise ratios as calculated from three different imaging experiments analysed at 40 μm resolution. | ||
| 13C | 1.1 | 38 |
| 24Mg | 24.0 | 332 |
| 31P | 100 | 946 |
| 33S | 0.75 | 18 |
| 55Mn | 100 | 9 |
| 56Fe | 91.8 | 200 |
| 63Cu | 69.2 | 366 |
| 66Zn | 27.7 | 418 |
Statistical analyses
All statistical analyses were performed using GraphPad Prism 7.0. All data were tested for normality (Shapiro Wilk test), outliers (Grubbs’ test with 0.05 stringency) and significance for normally distributed data between three groups were calculated using one-way ANOVA with Fisher's LSD test. Non-parametric Kruskal–Wallis test without Dunn's correction was used for data failing the normality test. P values <0.05 are considered statistically significant. All data are presented as box plots (Tukey) where the box shows median value ±95% C.I. and the whiskers show minimum and maximum values not excluded by the outlier test.Results
Tissue identification using H&E staining and LA-ICP-MS imaging
To orientate our elemental maps and to verify tissue identity for quantitative measurements, we used consecutive midline sagittal sections of each mouse embryo for H&E staining (Fig. 1A). The embryo sections were then correlated with available online embryo atlases to confirm their identity and draw masks for each tissue region (Fig. 1B). Given the organs identifiable at the sagittal midline and given the imaging resolution used in this study (40 μm pixel size) we drew masks for the peripheral organs heart (H), sternum (St) and liver (L) for further quantitation. | ||
| Fig. 1 Tissue identification using H&E and LA-ICP-MS imaging. Identification of different tissue types using H&E staining (A) to create masks (B) for quantitative analysis. Endogenous distribution of copper at 40 μm resolution (C) identifies distinct Cu rich regions within the embryonic brain (black arrows), corresponding to the lining of ventricles. Abbreviations used: SC = spinal cord; St = sternum; H = heart; L = liver; BrSt = brainstem; Mb = midbrain; Dce = diencephalon; Cx = cerebral cortex; VZ = ventricular lining of the lateral ventricle. Scalebars = 2 mm (B), 1 mm (C). | ||
As evident from the masks superimposed on the LA-ICP-MS Cu map (Fig. 1C), the brain exhibits regional differences in elemental abundance. Most prominently, regions surrounding the ventricles and Aqueduct of Sylvius (indicated by black arrows) show higher levels of Cu. To sufficiently capture these regional differences, CNS tissue analysis was divided between the spinal cord (SC) and the brain regions brainstem (BrSt), midbrain (Mb), diencephalon (Dce), cerebral cortex (Cx) and the ventricular lining surrounding the lateral ventricle (VZ) (Fig. 1C). These ventricular regions, as well as the choroid plexus, are known to have high Cu buffering capacity and function to modulate CNS Cu levels.24 This image demonstrates that the resolution used in this study was sufficient to identify sub-regions of the brain.
Anatomical mapping and tissue quantitation of carbon, phosphorus, sulphur and magnesium in E14 embryos
Based on the anatomical mapping of C, P, S and Mg (Fig. 2) we report no significant differences in the sub-regional analysis between genotypes (Fig. 3). Carbon, P, S and Mg are among the most abundant elements in the human body contributing 18% (C), 1% (P), 0.3% (S) and 0.1% (Mg) of all elements as principal components of lipids, proteins and DNA.25 The concentrations presented for C and S are in arbitrary units due to the lack of quantitative information on the standards used in this study. Phosphorus and magnesium concentrations are at around 6000 μg g−1 (Fig. 3C and D) and 550 μg g−1 wet tissue weight (Fig. 3G and H), respectively, consistent with their reported relative abundance ratio of ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.25 The tissue specific analysis for C and S show no significant differences between tissue types (Fig. 3). The lowest concentration of P is seen in the heart, at around 500 μg g tissue−1, and highest magnesium concentrations are seen in the heart and the ventricular lining in the brain at around 600 μg g−1.
:1.25 The tissue specific analysis for C and S show no significant differences between tissue types (Fig. 3). The lowest concentration of P is seen in the heart, at around 500 μg g tissue−1, and highest magnesium concentrations are seen in the heart and the ventricular lining in the brain at around 600 μg g−1.
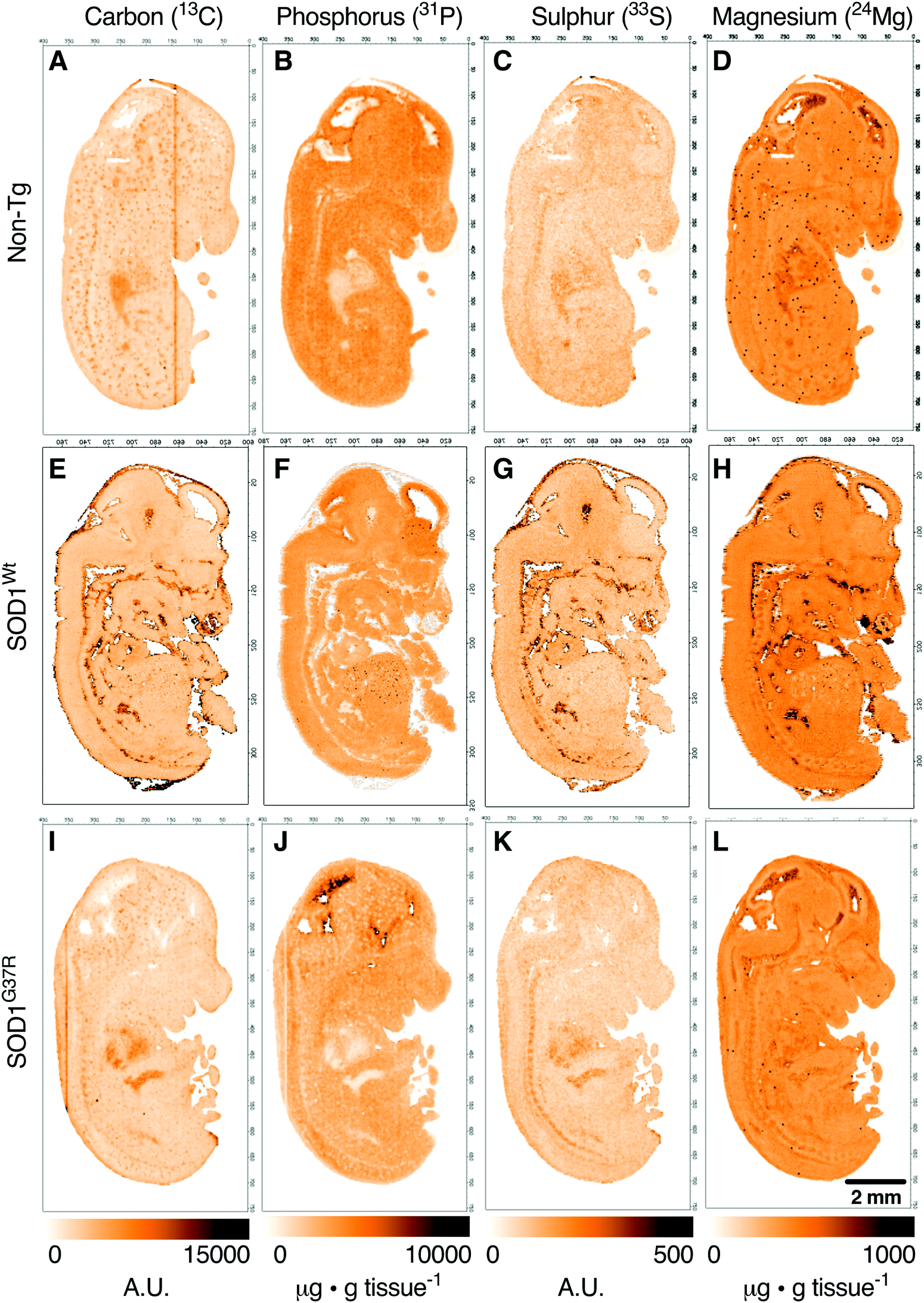 | ||
| Fig. 2 LA-ICP-MS imaging of endogenous distribution of carbon, phosphorus, sulphur and magnesium in E14 embryos. Representative elemental maps of non-transgenic (non-Tg) (A–D), transgenic wild-type SOD1 (SOD1Wt) (E–H) and mutant SOD1 (SOD1G37R) (I–L) embryos analysed using LA-ICP-MS. Signal intensities for 13C and 33S are presented in arbitrary units (A.U.) and in μg g tissue−1 for 31P and 24Mg showing relatively even distribution throughout the embryo. All images normalised to 31P, except for 31P (normalised to 13C). Scalebar = 2 mm. | ||
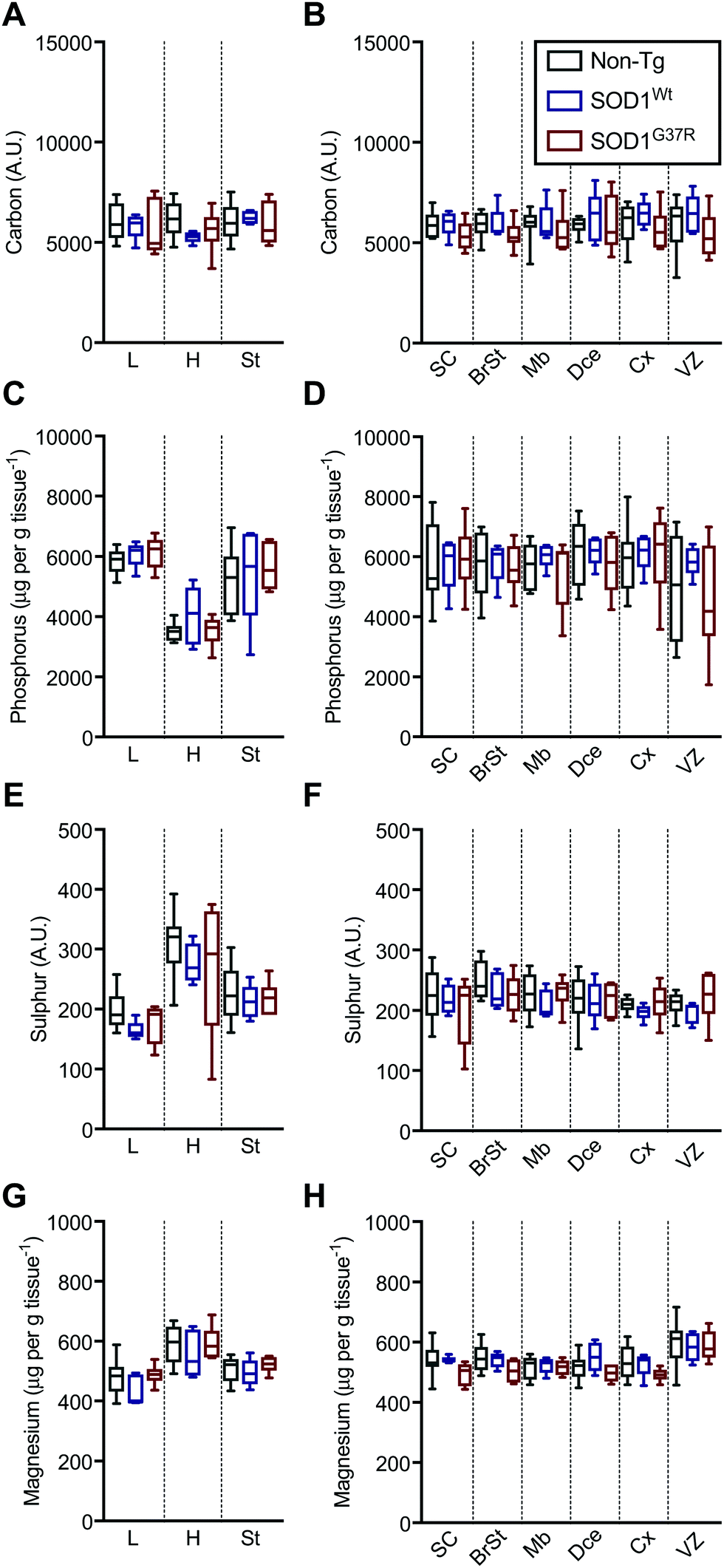 | ||
| Fig. 3 Sub-region analysis of C, P, S and Mg. Quantitation of mean concentrations of C (A and B), P (C and D), S (E and F) and Mg (G and H) from LA-ICP-MS elemental maps (Fig. 2) in peripheral organs and CNS sub-regions identified using H&E's (Fig. 1) showing no significant differences between SOD1-overexpressing embryos and their non-Tg littermates. All data are presented as box blots (Tukey) where the box shows median value ±95% C.I. and the whiskers show minimum and maximum values, with grey for non-Tg, blue for SOD1Wt and red for SOD1G37R. All data analysed using one-way ANOVA with Fisher's LSD multiple comparisons post hoc analysis. Abbreviations used: SC = spinal cord; St = sternum; H = heart; L = liver; BrSt = brainstem; Mb = midbrain; Dce = diencephalon; Cx = cerebral cortex; VZ = ventricular lining of the lateral ventricle. Non-Tg n = 8, SOD1Wtn = 5, SOD1G37Rn = 6. | ||
Anatomical mapping and quantitation of the trace element metals manganese, iron, copper and zinc in E14 embryos
Based on the anatomical mapping of the trace element metals Mn, Fe, Cu and Zn (Fig. 4), we report that as per S and Mg, the overexpression of neither SOD1Wt nor SOD1G37R has an impact on the abundance of Mn or Fe in the various embryonic tissues (Fig. 5). Manganese shows the highest levels of 0.4 μg g tissue−1 in the spinal cord and brainstem (Fig. 5B), consistent with reports of accumulation in the CNS around ventricular regions.26 The rest of the embryo contains lower Mn levels, with concentrations averaging around 0.15 μg g tissue−1. | ||
| Fig. 4 Quantitative LA-ICP-MS imaging of endogenous distribution of the metals manganese, iron, copper and zinc in E14 embryos. Representative elemental maps of non-transgenic (non-Tg) (A–D), transgenic wild-type SOD1 (SOD1Wt) (E–H) and mutant SOD1 (SOD1G37R) (I–L) embryos analysed using LA-ICP-MS at 40 μm resolution. Signal intensities for all elements are presented as μg g tissue−1 on the ‘SeaLandandFire’ colorscale with black indicating values above the highest value. Signal intensity for Fe is presented in log-scale due to the high 20-fold difference of concentration between liver and other tissues. The metals show distinct patterns of distribution and concentration levels, with the liver showing highest concentrations for Fe, Cu and Zn, although Zn concentration is more evenly distributed. On the contrary, Mn levels are the highest in the spinal cord and brainstem region. Scalebar = 2 mm. | ||
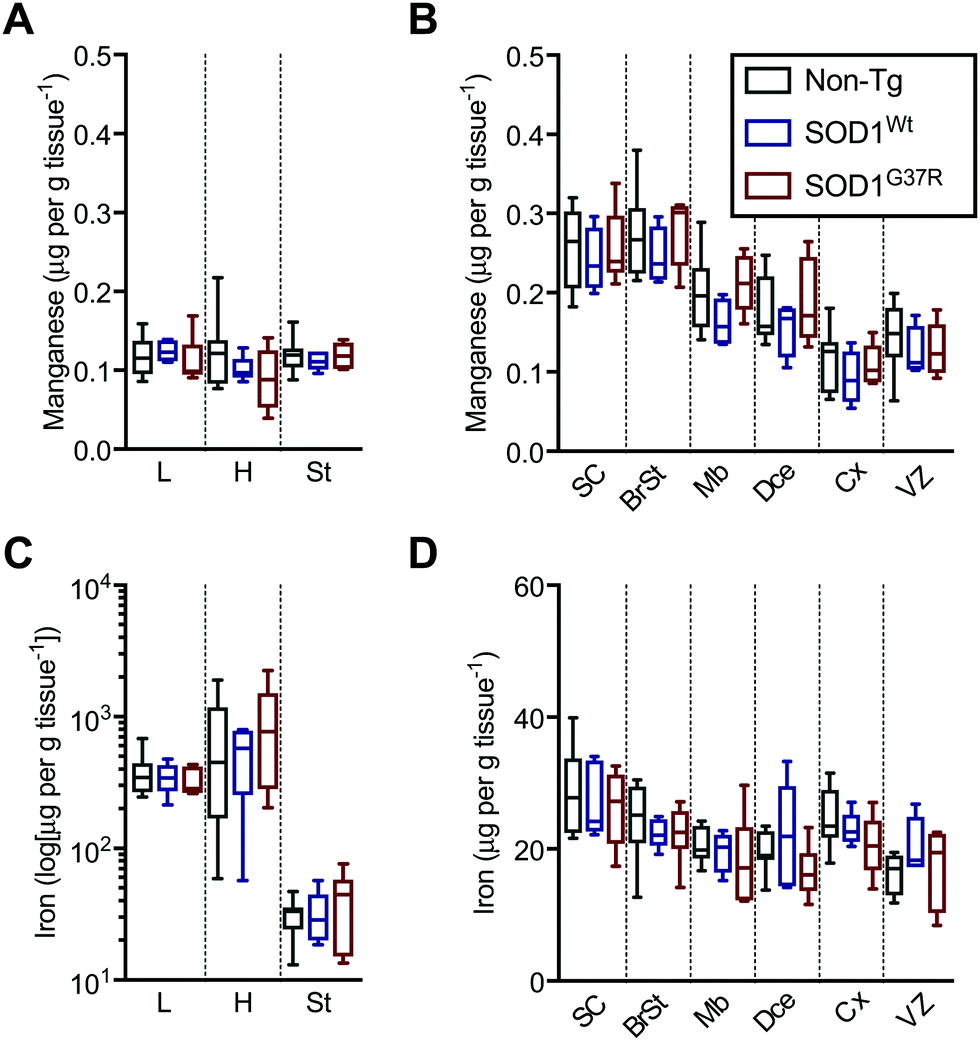 | ||
| Fig. 5 Sub-region analysis of Mn and Fe. Quantitation of mean concentrations of Mn (A and B) and Fe (C and D) from LA-ICP-MS elemental maps (Fig. 4) in peripheral organs and CNS sub-regions identified using H&E's (Fig. 1) showing no significant differences between SOD1-overexpressing mice embryos and their non-Tg littermates. Fe concentrations in the peripheral organs (C) are presented in log-scale due to the high levels detected in the heart and liver. All data are presented as box blots (Tukey) where the box shows median value ±95% C.I. and the whiskers show minimum and maximum values, with grey for non-Tg, blue for SOD1Wt and red for SOD1G37R. All data analysed using one-way ANOVA with Fisher's LSD multiple comparisons post hoc analysis. Abbreviations used: SC = spinal cord; St = sternum; H = heart; L = liver; BrSt = brainstem; Mb = midbrain; Dce = diencephalon; Cx = cerebral cortex; VZ = ventricular lining of the lateral ventricle. Non-Tg n = 8, SOD1Wtn = 5, SOD1G37Rn = 6. | ||
The liver and heart present with the highest concentrations for Fe (Fig. 5C), likely due to haemoglobin-bound Fe in the blood, with decreasing concentration of 30 μg g tissue−1 Fe from the rostral (spinal cord; SC) to 20 μg g tissue−1 Fe in the caudal (cerebral cortex; Cx) areas of the CNS similar to Mn distribution.
In contrast to Fe and Mn, the overexpression of SOD1Wt or SOD1G37R impacts the distribution of Cu and Zn in the embryonic mouse tissues (Fig. 4). This is most evident for Cu levels where a substantial 30% decrease of Cu in the liver is accompanied by an increase in Cu levels of all other tissues analysed, with the highest increases of 83–98% seen in the midbrain and diencephalon (Fig. 6A and B). These increases are more pronounced when all values are normalised to the total amount of liver copper measured for each individual embryo (Fig. 6C and D).
 | ||
| Fig. 6 SOD1 overexpression results in redistribution of Cu and Zn in the mouse embryo. Quantitation of mean concentrations of Cu (A and B) and Zn (E and F) from LA-ICP-MS elemental maps (Fig. 4) in peripheral organs and CNS sub-regions identified using H&E's (Fig. 1) showing an overt Cu redistribution from the liver to the CNS and peripheral organs in SOD1-overexpressing mice embryos. When expressed as the percentage of mean liver concentrations, significant increases are seen throughout the embryo for Cu (C and D) and more subtle increases for Zn in the heart and the brainstem region (G and H). All data are presented as box blots (Tukey) where the box shows median value ±95% C.I. and the whiskers show minimum and maximum values, with grey for non-Tg, blue for SOD1Wt and red for SOD1G37R. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA with Fisher's LSD multiple comparisons post hoc analysis. Abbreviations used: SC = spinal cord; St = sternum; H = heart; L = liver; BrSt = brainstem; Mb = midbrain; Dce = diencephalon; Cx = cerebral cortex; VZ = ventricular lining of the lateral ventricle. Non-Tg n = 8, SOD1Wtn = 5, SOD1G37Rn = 6. | ||
Although holo-SOD1 binds equimolar amounts of Cu and Zn, increased expression of SOD1 in the SOD1G37R and SOD1Wt embryos did not have a detectable impact on total Zn levels in the tissues analysed (Fig. 6E and F). However, expressing these data relative to liver Zn levels for each embryo did indicate some relatively moderate Zn elevations in the heart and the brainstem (Fig. 6G and H). The inability to detect overt changes to Zn in the SOD1 overexpressing embryos may be related to the naturally higher levels of Zn in all tissues masking a small change in Zn caused by SOD1 overexpression; Zn levels in all tissues examined (except the liver) are at least an order of magnitude higher than Cu (Fig. 6A, B, E, and F).
Discussion
Approximately 90% of ALS cases are sporadic and occur without a defined genetic cause, yet compelling, direct evidence for Cu-related mechanisms being involved in sporadic forms of ALS is yet to be published. In contrast, previous outcomes from cell culture studies and transgenic mice provide support for the involvement of Cu-related mechanisms in forms of ALS caused by mutant SOD1. Endogenous bio-availability of Cu to SOD1 is a limiting factor in the maturation of SOD1 in CNS tissues of adult mice overexpressing mutant SOD1, whereas Cu availability to SOD1 in non-CNS tissues is sufficient to ensure complete SOD1 maturation.17 Moreover, Zn availability to SOD1 is sufficient in all adult tissues, including the CNS.17 In other words, the adult CNS fails to satiate the increased requirement for Cu driven by over-expression of the mutant SOD1.In this study we show that at embryonic development stage E14, ubiquitous overexpression of SOD1Wt and SOD1G37R results in an increase of Cu in the CNS relative to non-transgenic controls. By contrast, Cu levels are decreased in the liver of the SOD1G37R and SOD1WT embryos. These data therefore suggest that unlike the adult CNS, the elevated requirement for Cu in the embryonic CNS due to SOD1 overexpression can be satiated via redistribution from the liver. This potential ability for the liver to act as a repository for the redistribution of bioavailable copper could be explained by the bulk liver copper levels seen at early developmental stages. In contrast to adult non-transgenic mice,17 the liver tissue from embryonic non-transgenic mice contains approximately four-fold more copper – an observation supported by findings showing that hepatic copper progressively accumulates in embryonic and postnatal rats before decreasing into adulthood.27 As such, early developmental stage liver tissue replete with copper may facilitate the satiation of elevated copper requirements driven by SOD1 overexpression.
The mechanistic basis for these differences between Cu bio-availability to SOD1 in the embryonic CNS and the adult CNS are not yet clear. Differences in permeability between the developing and adult blood-brain-barrier would appear to be the most accessible explanation, however, stable levels of Zn in the CNS of both embryonic and adult SOD1G37R mice17 suggest a more Cu-specific mechanism. Thus, an alternate explanation might be that pre-existing pools of Zn in the adult CNS may be relatively more labile than pre-existing pools of Cu in the CNS, thereby making the pool of Zn in the adult CNS more accessible and able to accommodate the increased requirement for Zn driven by SOD1 over-expression.
The course of metal distribution changes during postnatal development of SOD1-overexpressing mice remains largely unexplored. A recent study investigating the levels of Cu, Zn and Fe concentrations in spinal cord, brain, blood, muscle and feces of mutant SOD1G93A expressing mice at pre-symptomatic and post-symptomatic stages, describes a selective increase of Cu concentration in the muscle tissue.28 However, the reported metal values display high spread between tissues and ages (potentially due to residual blood contamination and/or incomplete cardiac perfusion) masking potential subtle changes in metal distribution. The full anatomical mapping of relevant tissues including the liver and blood–brain-barrier systems in future studies is likely to further our understanding of Cu metabolism in the context of SOD1-overexpression.
Specific differences in the cellular pathways that regulate Cu bioavailability, such as the expression, and cellular and tissue distribution of chaperones and transporters, likely contribute to the age-related changes in Cu bio-availability to the CNS.27 Significant differences in mRNA transcripts relating to copper chaperones and transporters have been reported between new-born and adult rats.27 Expression of membrane-bound copper transporter Ctr1 has also been shown to be lower at early embryonic stages with enhanced expression in particular tissues, such as the liver and CNS, only being observed after E16.5 embryonic stage.24 By contrast, expression of the copper transporter ATP7A steadily decreases in the postnatal mouse CNS during neurodevelopment until adulthood.29 Whilst ATP7A levels were found to be highest in the cortical layer V pyramidal neurons associated with disease-affected regions in ALS, global ATP7A expression was diminished. Given the importance of ATP7A in the copper-loading mechanism for vital cuproenzymes, this observation may imply an attenuated ability for the CNS to satisfy elevated copper requirements later in life.
Attributing such a mechanism to the adult-onset of ALS-like symptoms in SOD1 overexpressing mice, however, would need to encapsulate some salient differences between mutant and wild-type SOD1; while overexpression of mutant SOD1 causes a clear ALS-like phenotype in transgenic mice, the overexpression of wild-type SOD1 at equivalent levels does not.5 It is only when wild-type SOD1 is overexpressed at much higher levels that the transgenic mice develop phenotypic features reminiscent of ALS.30 Thus, it appears that the inability for the adult CNS to satiate the requirement for Cu in overexpressed mutant SOD1 cannot be explained by a simple disconnect between supply and demand. Instead, it appears plausible that the relative recalcitrance of the adult CNS to respond to the changed requirement for Cu due to SOD1 overexpression is exacerbated by the presence of an ALS-causing SOD1 mutation.
Notwithstanding, here we have presented data which show that unlike the CNS of adult mice, the CNS of embryonic mice displays large increase in copper in response to mutant SOD1 overexpression. If the pathogenesis of ALS in mutant SOD1 cases of the disease involves CNS-specific perturbations to copper availability, these data indicate that the ability of the embryonic CNS to seemingly more readily obtain Cu may explain why the symptoms of the disease do not occur at an earlier developmental stage.
Conclusions
SOD1 mutations are an unequivocal cause of familial cases of ALS and transgenic mice over-expressing mutant SOD1 are a robust animal model of the disease. Our application of quantitative in situ imaging reveals an early developmental redistribution of Cu in SOD1-transgenic mice that is distinct from the distribution observed in adults. This study highlights the need for further studies to dissect the age-related changes in Cu bio-availability to the CNS, which contribute to the age-related development of ALS.Conflicts of interest
BP receives salary support from sales of the University of Melbourne's commercialised iolite software package. DJH receives research and materials support from Agilent Technologies via Australian Government industrial partnership grants listed in the Acknowledgements. Other authors declare no conflicts of interest.Acknowledgements
This research was supported by funds from an Australian National Health and Medical Research Council (NHMRC; Project Grant GNT1061550 – PJC), the University of Melbourne, the Motor Neurone Disease Research Institute of Australia (Betty Laidlaw MND Research Grant DJH, PJC; Jenny Simko Research Grant – DJH, PJC), and Parkinson's Victoria (Argyrou Family Fellowship – DJH). K. K. was recipient of the Sigrid Juselius Postdoctoral Fellowship. J. B. H. was recipient of the Australian Postgraduate Award and the Nancy Frances Curry Scholarship. P. J. C. is recipient of the NHMRC R. D. Wright Biomedical Research Fellowship (CDF2, 1084927). DJH is supported by an NHMRC Career Development Fellowship – Industry (GNT1122981) in partnership with Agilent Technologies. This study utilised the Australian Phenomics Network Histopathology and Organ Pathology Service, University of Melbourne.Notes and references
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Metalloproteins and metal sensing, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- E. J. Martinez-Finley, S. Chakraborty, S. J. B. Fretham and M. Aschner, Cellular transport and homeostasis of essential and nonessential metals, Metallomics, 2012, 4, 593–605 RSC.
- R. Linz and S. Lutsenko, Copper-transporting ATPases ATP7A and ATP7B: Cousins, not twins, J. Bioenerg. Biomembr., 2007, 39, 403–407 CrossRef CAS PubMed.
- A. J. Nappi and E. Vass, Iron, metalloenzymes and cytotoxic reactions, Cell. Mol. Biol., 2000, 46, 637–647 CAS.
- M. E. Gurney, H. Pu, A. Y. Chiu, M. C. Dal Canto, C. Y. Polchow, D. D. Alexander, J. Caliendo, A. Hentati, Y. W. Kwon and H. X. Deng, et al., Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation, Science, 1994, 264, 1772–1775 CrossRef CAS PubMed.
- D. R. Rosen, T. Siddique, D. Patterson, D. A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J. P. O'Regan and H. X. Deng, et al., Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis, Nature, 1993, 362, 59–62 CrossRef CAS PubMed.
- J. B. Hilton, A. R. White and P. J. Crouch, Metal-deficient SOD1 in amyotrophic lateral sclerosis, J. Mol. Med., 2015, 93, 481–487 CrossRef CAS.
- E. Tokuda and Y. Furukawa, Copper Homeostasis as a Therapeutic Target in Amyotrophic Lateral Sclerosis with SOD1 Mutations, Int. J. Mol. Sci., 2016, 17 Search PubMed.
- C. P. Soon, P. S. Donnelly, B. J. Turner, L. W. Hung, P. J. Crouch, N. A. Sherratt, J. L. Tan, N. K. Lim, L. Lam, L. Bica, S. Lim, J. L. Hickey, J. Morizzi, A. Powell, D. I. Finkelstein, J. G. Culvenor, C. L. Masters, J. Duce, A. R. White, K. J. Barnham and Q. X. Li, Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model, J. Biol. Chem., 2011, 286, 44035–44044 CrossRef CAS PubMed.
- B. R. Roberts, N. K. Lim, E. J. McAllum, P. S. Donnelly, D. J. Hare, P. A. Doble, B. J. Turner, K. A. Price, S. C. Lim, B. M. Paterson, J. L. Hickey, T. W. Rhoads, J. R. Williams, K. M. Kanninen, L. W. Hung, J. R. Liddell, A. Grubman, J. F. Monty, R. M. Llanos, D. R. Kramer, J. F. Mercer, A. I. Bush, C. L. Masters, J. A. Duce, Q. X. Li, J. S. Beckman, K. J. Barnham, A. R. White and P. J. Crouch, Oral treatment with CuII(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis, J. Neurosci., 2014, 34, 8021–8031 CrossRef CAS.
- E. Tokuda, S. Watanabe, E. Okawa and S. Ono, Regulation of Intracellular Copper by Induction of Endogenous Metallothioneins Improves the Disease Course in a Mouse Model of Amyotrophic Lateral Sclerosis, Neurotherapeutics, 2015, 12, 461–476 CrossRef CAS PubMed.
- J. R. Williams, E. Trias, P. R. Beilby, N. I. Lopez, E. M. Labut, C. S. Bradford, B. R. Roberts, E. J. McAllum, P. J. Crouch, T. W. Rhoads, C. Pereira, M. Son, J. L. Elliott, M. C. Franco, A. G. Estevez, L. Barbeito and J. S. Beckman, Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD mice co-expressing the Copper-Chaperone-for-SOD, Neurobiol. Dis., 2016, 89, 1–9 CrossRef CAS PubMed.
- B. R. Roberts, N. K. Lim, E. J. McAllum, P. S. Donnelly, D. J. Hare, P. A. Doble, B. J. Turner, K. A. Price, S. C. Lim, B. M. Paterson, J. L. Hickey, T. W. Rhoads, J. R. Williams, K. M. Kanninen, L. W. Hung, J. R. Liddell, A. Grubman, J. F. Monty, R. M. Llanos, D. R. Kramer, J. F. Mercer, A. I. Bush, C. L. Masters, J. A. Duce, Q. X. Li, J. S. Beckman, K. J. Barnham, A. R. White and P. J. Crouch, Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis, J. Neurosci., 2014, 34, 8021–8031 CrossRef CAS PubMed.
- H. J. Forman and I. Fridovich, On the stability of bovine superoxide dismutase. The effects of metals, J. Biol. Chem., 1973, 248, 2645–2649 CAS.
- J. B. Hilton, K. Kysenius, A. R. White and P. J. Crouch, The accumulation of enzymatically inactive cuproenzymes is a CNS-specific phenomenon of the SOD1(G37R) mouse model of ALS and can be restored by overexpressing the human copper transporter hCTR1, Exp. Neurol., 2018, 307, 118–128 CrossRef CAS PubMed.
- C. W. Levenson and M. Janghorbani, Long-Term Measurement of Organ Copper Turnover in Rats by Continuous Feeding of a Stable-Isotope, Anal. Biochem., 1994, 221, 243–249 CrossRef CAS PubMed.
- J. B. Hilton, A. R. White and P. J. Crouch, Endogenous Cu in the central nervous system fails to satiate the elevated requirement for Cu in a mutant SOD1 mouse model of ALS, Metallomics, 2016, 8, 1002–1011 RSC.
- D. J. Hare, E. J. New, M. D. de Jonge and G. McColl, Imaging metals in biology: balancing sensitivity, selectivity and spatial resolution, Chem. Soc. Rev., 2015, 44, 5941–5958 RSC.
- P. C. Wong, C. A. Pardo, D. R. Borchelt, M. K. Lee, N. G. Copeland, N. A. Jenkins, S. S. Sisodia, D. W. Cleveland and D. L. Price, An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria, Neuron, 1995, 14, 1105–1116 CrossRef CAS.
- D. J. Hare, K. Kysenius, B. Paul, B. Knauer, R. W. Hutchinson, C. O'Connor, F. Fryer, T. P. Hennessey, A. I. Bush, P. J. Crouch and P. A. Doble, Imaging Metals in Brain Tissue by Laser Ablation – Inductively Coupled Plasma – Mass Spectrometry (LA-ICP-MS), J. Visualized Exp., 2017, 119 DOI:10.3791/55042.
- B. Paul, D. J. Hare, D. P. Bishop, C. Paton, V. T. Nguyen, N. Cole, M. M. Niedzwiecki, E. Andreozzi, A. Vais, J. L. Billings, L. Bray, A. I. Bush, G. McColl, B. R. Roberts, P. A. Adlard, D. I. Finkelstein, J. Hellstrom, J. M. Herg, J. D. Woodhead and P. A. Doble, Visualising mouse neuroanatomy and function by metal distribution using laser ablation-inductively coupled plasma-mass spectrometry imaging, Chem. Sci., 2015, 6, 5383–5393 RSC.
- D. J. Hare, J. Lear, D. P. Bishop, A. B. Beavis and P. A. Doble, Protocol for production of matrix-matched brain tissue standards for imaging by laser ablation-inductively coupled plasma-mass spectrometry, Anal. Methods, 2013, 5, 1915–1921 RSC.
- T. W. May and R. H. Wiedmeyer, A table of polyatomic interferences in ICP-MS, At. Spectrosc., 1998, 19, 150–155 CAS.
- Y. M. Kuo, B. Zhou, D. Cosco and J. Gitschier, The copper transporter CTR1 provides an essential function in mammalian embryonic development, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6836–6841 CrossRef CAS PubMed.
- J. J. R. Dasilva and R. J. P. Williams, The Biological Chemistry of the Elements - the Inorganic-Chemistry of Life, Nature, 1991, 354, 367 CrossRef.
- N. A. Bock, F. F. Paiva, G. C. Nascimento, J. D. Newman and A. C. Silva, Cerebrospinal fluid to brain transport of manganese in a non-human primate revealed by MRI, Brain Res., 2008, 1198, 160–170 CrossRef CAS PubMed.
- Y. A. Zatulovskaia, E. Y. Ilyechova and L. V. Puchkova, The Features of Copper Metabolism in the Rat Liver during Development, PLoS One, 2015, 10 Search PubMed.
- T. G. Enge, H. Ecroyd, D. F. Jolley, J. J. Yerbury and A. Dosseto, Longitudinal assessment of metal concentrations and copper isotope ratios in the G93A SOD1 mouse model of amyotrophic lateral sclerosis, Metallomics, 2017, 9, 161–174 RSC.
- M. J. Niciu, X. M. Ma, R. El Meskini, G. V. Ronnett, R. E. Mains and B. A. Eipper, Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment, Neuroscience, 2006, 139, 947–964 CrossRef CAS PubMed.
- K. S. Graffmo, K. Forsberg, J. Bergh, A. Birve, P. Zetterstrom, P. M. Andersen, S. L. Marklund and T. Brannstrom, Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis, Hum. Mol. Genet., 2013, 22, 51–60 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |