
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Short oligopeptides with three cysteine residues as models of sulphur-rich Cu(I)- and Hg(II)-binding sites in proteins†
Edit
Mesterházy
ab,
Colette
Lebrun
b,
Serge
Crouzy
c,
Attila
Jancsó
 *b and
Pascale
Delangle
*a
*b and
Pascale
Delangle
*a
aINAC/SYMMES/Université Grenoble Alpes, CEA, CNRS, 38000 Grenoble, France. E-mail: pascale.delangle@cea.fr
bDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, Szeged H-6720, Hungary. E-mail: jancso@chem.u-szeged.hu
cBIG/LCBM/Université Grenoble Alpes, CEA, CNRS, (UMR 5249), 38000 Grenoble, France
First published on 16th July 2018
Abstract
The essential Cu(I) and the toxic Hg(II) ions possess similar coordination properties, and therefore, similar cysteine rich proteins participate in the control of their intracellular concentration. In this work we present the metal binding properties of linear and cyclic model peptides incorporating the three-cysteine motifs, CxCxxC or CxCxC, found in metallothioneins. Cu(I) binding to the series of peptides at physiological pH revealed to be rather complicated, with the formation of mixtures of polymetallic species. In contrast, the Hg(II) complexes display well-defined structures with spectroscopic features characteristic for a HgS2 and HgS3 coordination mode at pH = 2.0 and 7.4, respectively. Stability data reflect a ca. 20 orders of magnitude larger affinity of the peptides for Hg(II) (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) βpH7.4HgP ≈ 41) than for Cu(I) (logβpH7.4CuP ≈ 18). The different behaviour with the two metal ions demonstrates that the use of Hg(II) as a probe for Cu(I), coordinated by thiolate ligands in water, may not always be fully appropriate.
βpH7.4HgP ≈ 41) than for Cu(I) (logβpH7.4CuP ≈ 18). The different behaviour with the two metal ions demonstrates that the use of Hg(II) as a probe for Cu(I), coordinated by thiolate ligands in water, may not always be fully appropriate.
Significance to metallomicsMercury(II) and copper(I) detoxification in cells involves similar sulphur-rich proteins, including metallothioneins. Short model peptides are used here to study the molecular interactions of these two soft cations with sequences containing three cysteine moieties found in these proteins. We demonstrate that the binding of Hg(II) and Cu(I) with these representative cysteine-rich sequences involves different molecular mechanisms, with the formation of various coordination complexes. Hence, the use of Hg(II) as a probe for Cu(I) coordination with sulphur-rich peptides or proteins in physiological conditions is demonstrated here to be not fully appropriate. |
Introduction
Many metal ions, including Fe(II/III), Cu(I/II) or Zn(II), are essential micronutrients. Since both their deficiency and excess are harmful for the cells, the optimal concentration of these ions is controlled by regulation systems involving lots of proteins. Other metal ions, like Hg(II) and Cd(II), are toxic without any advantageous effects. The high affinity of these soft metal ions for soft sulphur bases, found in proteins and small ligands, such as glutathione, is one of the main factors leading to their toxicity. Indeed, interactions between these toxic metal ions and thiol-containing molecules induce misfolded protein structures or perturbation in the redox balance of the cells.As a consequence of the similar coordination properties of the essential Cu(I) and the toxic Hg(II) ions, the cellular regulation and protection systems for controlling or reducing the concentration of these ions involve proteins with sulphur-rich metal coordination sites, usually dominated by Cys-thiolate donor groups. A varying number of Cys residues may allow the binding of Cu(I) or Hg(II) in isolated monometallic coordination sites or in polymetallic clusters. The strong preference of Hg(II) for a bis-thiolate coordination environment is reflected by the published structures of several elements of the bacterial mercury resistance system, MerP,1 MerT,2 the N-terminal domain of MerA3 or the Hg(II)-bound form of the copper chaperone Atx1.4 Nevertheless, there are also examples of structures with more Hg(II)–thiolate bonds, e.g. the distorted tetrahedral tetrathiolate Hg(II)-centre of rubredoxin,5 or the tri- or tetra-coordinated metal ion environments in Hg(II)-bridged protein dimers, like those observed in the MerR metalloregulatory protein6 or in the human copper chaperone HAH1.7,8 Cu(I) can also accommodate a linear CuS2-type coordination mode, as indicated by structural data on the copper efflux regulator CueR9 or the metal binding domains of the copper transporter protein ATP7B.10 Trigonal CuS3 centres were proposed in the binuclear Cu(I)–thiolate core of the repressor protein CopY11 and in one of the suggested metal-bridged dimeric forms of the copper chaperone CopZ,12 whereas the Cu(I)-bridged HAH1 dimer7 was shown also to include a fourth, weakly bound, thiolate in a pseudotetrahedral environment.
Di-, tri- or tetrathiolate coordination modes are also typical in polymetallic clusters. Metallothioneins (MTs) are small, cysteine-rich proteins, found in all kingdoms of life from prokaryotes to mammals. Several isoforms of MTs evolved with high diversity of amino acid sequences and, pursuant to this, with different metal ion preferences, 3D structures and proposed biological functions.13,14 A novel approach for the classification of such a big family of proteins was developed by the groups of Capdevila and Atrian based on the metal binding features of MTs, i.e. on the criterion whether the MT binds Zn(II) either in homo or in heterometallic form, or only Cu(I) under physiological conditions.15,16 Trigonally coordinated Cu(I) ions strongly dominate in Cu(I)-loaded MTs displaying also less abundant Cu(I)S2 centres.17,18 The only published Cu(I)-loaded MT crystal structure, a truncated form of the Cu(I)–MT of yeast, bears a Cu(I)8S10 core with six tri- and two di-coordinated metal ions where all Cys residues bridge two or three Cu(I) centres.19 It was also suggested that a Cu(I)4S6 cluster could form in yeast MTs under low Cu(I) availability.20 Indeed, the latter core contains only tri-coordinated Cu(I) and is widely used by nature in many other types of metalloproteins,21e.g. in the C-terminal domain of the membrane copper transporter Ctr122 or in various Cu(I)-activated transcription factors.23,24 In contrast, Hg(II)–thiolate clusters in MTs were found to display a complicated mixture of di-, tri- and tetra-coordinated Hg(II) centres, with strong dependence on the applied conditions (pH, temperature, Hg(II) concentration, counter ions, etc.).25,26
An interesting strategy to better understand the individual contributions of specific short sequences to the overall affinity of proteins for metal ions is to design and study model peptides. This approach could eventually lead to the identification of relevant peptide-based motifs for heavy metal chelation. Some of the above described metal-binding sites have been probed by using oligopeptides encompassing the relevant metal binding sequences of the modelled proteins. The metal-binding features of the metalloregulator CueR and the metallochaperone Atx1, both encompassing 2 cysteines in their metal-binding domain, with different sequences between the Cys residues, were investigated via oligopeptide models of the relevant metal-binding loops.27–33 A general finding was that the linear unstructured ligands, while displaying high affinity and interesting metal ion selectivity for Hg(II) or Cu(I) chelation, could not fully reproduce the efficiency of the modelled proteins.28,31,33 Cyclisation of the linear Atx1 model, resulting in a more rigid skeleton with preoriented Cys-sidechains, was an important step forward leading to notably increased metal binding affinities.27,28 The more challenging imitation of the HgS3 type metal coordination sites was successfully achieved by different approaches. Pecoraro et al. successfully applied single oligopeptide chain three-helix bundles34 and three-stranded coiled coils35 to settle three Cys residues into optimal position for accommodating the Hg(II) ion in a tri-coordinate fashion under slightly alkaline conditions. These tris-thiolate bound species were in a pH-dependent equilibrium with HgS2 type complexes, characterized by apparent pKa values ≥7.34,35 Using a different strategy, tripodal pseudopeptide ligands were designed with cysteine or D-penicillamine (D-Pen) moieties grafted onto a nitrilotriacetic acid scaffold.36,37 These constructs were able to stabilize the HgS3 coordination mode in a broad pH-range, starting even at pH ∼ 5.5 with one of the D-Pen ligands.36 Interestingly, the behaviour of the same ligands in binding Cu(I) was less straightforward. Depending on the bulkiness and hydrophobicity of the arms attached to the tripodal template, the formation of mononuclear and (Cu2Lig)x type complexes with (Cu2S3)x cores was observed with high stabilities.38,39 In a recently published article, the Hg(II) binding of the cyclic peptide P3C incorporating a three Cys variant of the metal binding loop of Atx1 was discussed.40 This cyclic solvent accessible peptide was considered as a predisposed structure that could pre-orient the Cys-sidechains for an easy accommodation of metal ions in a tri-coordinate fashion. The results confirmed the formation of a mononuclear Hg(II) complex with properties indicative of an HgS3 coordination.
Peptide scaffolds similar to P3C and comprising two typical metal binding motifs found in metallothioneins, namely the CxCxxC and CxCxC sequences,41–43 are investigated in the present paper. Their binding properties for the two soft metal ions Cu(I) and Hg(II), exhibiting the largest affinities for metallothioneins, are studied and discussed in relation to their binding by detoxification systems.26,44–46
Experimental section
Materials
Materials and solvents were purchased from Sigma Aldrich, Fluka, Acros Organics and used without further purification. Amino acid building blocks, resins and coupling agents were purchased from NovaBiochem. For aqueous solutions, ultrapure laboratory grade MilliQ water was used (resistivity 18 MΩ cm).Abbreviations
Ac2O: acetic anhydride; AcN: acetonitrile; BCS: bathocuproinedisulfonate; DCM: dichloromethane; DIEA: N,N-diisopropylethylamine; DTNB: 5,5′-dithio-bis-(2-nitrobenzoic acid); EDT: ethanedithiol; Et2O: diethyl ether; Fmoc: 9-fluoroenylmethoxycarbonyl; HBTU: 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOBt: hydroxybenzotriazole; MeOH: methanol; NMP: N-methyl-2-pyrrolidone; TFA: trifluoro acetic acid; TIS: triisopropylsilane.Peptide synthesis and purification
The crude products were purified by reversed phase HPLC on a Shimadzu LC-20 instrument equipped with a Phenomenex Synergi 4 μm fusion-RP 80 Å semi-preparative column. H2O and acetonitrile with 0.1% TFA were used as eluents. The purity was checked by analytical HPLC and ESI-MS. Analytical RP-HPLC was performed using an analytical column (Chromolith® performance RP-18e 100-4.6 mm) at 1 mL min−1 flow rate (gradient 0–60% of AcN in 30 min). The synthesis of P3C has been published earlier.40 For the other peptides analytical HPLC and ESI-MS characterization is available in Table S1 and Fig. S1 in the ESI.†
Sample preparation for physicochemical studies
The cysteine residues of the peptides and Cu(I) are sensitive to oxidation; therefore all sample preparations and manipulations were performed in a glove box, under an argon atmosphere. Fresh solution was made for each experiment in the appropriate buffer. 10% acetonitrile in volume was added to the Cu(I) containing samples to overcome Cu(I) disproportion in water.48 The final peptide concentration was determined by Ellman's procedure, where DTNB reacts with the free thiols forming TNB− (2-nitro-5-thiobenzoate) with ε412 = 14150 M−1 cm−1.49,50 Cu(I) solution was made from Cu(CH3CN)4PF6 salt in acetonitrile. The final concentration was determined by using BCS forming a stable Cu(BCS)2 complex with ε483nm = 13300 M−1 cm−1.51 A precise weight of high purity HgCl2 was used to prepare Hg(II) stock solution in water (∼3 mM).
UV-Vis absorption and circular dichroism (CD) measurements
UV-Vis spectra were recorded on a Varian Cary 50 spectrophotometer equipped with optical fibers connected to an external cell holder in the glovebox. CD spectra were acquired with an Applied Photophysics Chirascan photometer. (1S)-(+)-10-Camphorsulfonic acid served as the calibration compound for the instrument. Spectra were recorded in the 320–190 nm wavelength range with 1 nm bandwidth and 2 s dwell time per point. For each sample 3 parallel spectra were recorded and the average of these spectra was smoothed by the Savitzky–Golay method with a “window size” 7. CD spectra are reported in molar ellipticity ([Θ] in units of deg cm2 dmol−1). [Θ] = θobs/(10lc), where θobs is the observed ellipticity in millidegrees, l is the optical path length in cm and c is the peptide concentration in mol dm−3.With Cu(I), titrations were performed in individually prepared samples because of the slow complex formation. 2.5 mL volumes of peptide solution (20–30 μM) in phosphate buffer (20 mM, pH = 7.4) were transferred into UV cells (1.0 cm path length) and then different (0.0–3.0) equivalents of Cu(I) were added to these samples. The samples were equilibrated for at least 2 hours. To ensure the thermodynamic equilibrium was reached, the absorbance was measured regularly. In the case of Hg(II) titrations, 2.5 mL of peptide solution (20–30 μM) in phosphate buffer (20 mM, pH = 7.4) was transferred into a UV cell (1.0 cm path length) and then aliquots of the Hg(II) ion solution (∼3 mM) were gradually added from 0.0 to 3.0 equivalents. pH titrations of the Hg(II) complexes were performed in pure water in the pH range of 2–11 by adding aliquots of 0.1 M KOH. The pH was measured using a Metrohm 702 SM Titrino equipped with a Mettler Toledo InLab® Micro electrode. pKa values for the observed deprotonation processes were obtained by fitting the data by using the SPECFIT computer program.52–55
ESI-MS experiments
Mass spectra were acquired on a LXQ-linear ion trap (THERMO Scientific) instrument equipped with an electrospray ion source. Electrospray full scan spectra in the range of m/z = 50–2000 amu were obtained by infusion through a fused silica tubing at a flow rate of 2–10 μL min−1. The solutions were analysed in negative and positive ion modes. The LXQ calibration (m/z = 50–2000) was achieved according to the standard calibration procedure from the manufacturer (mixture of caffeine, MRFA and Ultramark 1621). The temperature of the heated capillary for the LXQ was set in the range of 200–250 °C, the ion-spray voltage was in the range of 3–6 kV and the injection time was 5–200 ms. The ligand solution (100 μM) was prepared in ammonium acetate buffer (20 mM, pH = 6.9) and aliquots of the appropriate metal ion were then added.Determination of apparent stabilities of the metal ion complexes
The apparent stability constants at a given pH were determined by UV-Vis titration in the presence of a competitor. The Cu(I) complexes were prepared by adding 0.9 equivalents of Cu(I) to three samples of each peptide (50 μM) in phosphate buffer (20 mM, pH = 7.4)/acetonitrile (9/1 V/V). Then 4, 8 and 12 equivalents (with respect to Cu(I) concentration) of BCS were added, respectively. The samples were equilibrated until the absorbance stabilized. Stability constants were calculated considering the formation of the CuP mononuclear complexes at the beginning of the titration according to the following equation:where CuP and P represent the complexed and free peptide at the pH of the studies, independent of the protonation state of the ligand, and Cu stands for the free Cu(I) ions.
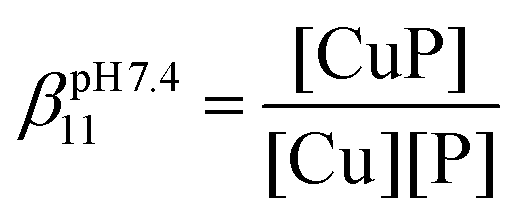
The stabilities of the Hg(II)–peptide complex were determined at pH = 2.0 by using iodide (I−) as a competitor. Stability constants of the HgIn (n = 1–4) complexes were taken from the literature56 and recalculated for the conditions of the experiments (pH = 2.0, I = 0.1 M NaClO4) by applying the SIT model.57,58 The molar absorption spectra of the four Hg(II)–I− complexes were determined by titrating a Hg(II) solution by the continuous addition of aliquots of I− solutions (0.01, 0.1 and 0.5 M KI). The recorded spectra were fitted with SPECFIT52–55 by fixing the stability constants of the HgIn complexes. The peptide solutions were prepared in water (30 μM, I = 0.1 M NaClO4) and the pH was adjusted by a 1.0 M HCl solution. Samples for the competition experiments were prepared by adding 1.0 equivalent of Hg(II) to the peptide solutions. They were then titrated by the I− solutions by the same procedure and under the same conditions as described above. The pH of samples was under control throughout the experiments. SPECFIT52–55 was used for the evaluation of spectra. The molar absorption spectra and the stabilities of the HgIn complexes were fixed in the fitting procedure, allowing one to calculate the apparent stability of the HgP binary and HgPI ternary complexes (see the equations below), as well as their molar absorption spectra.
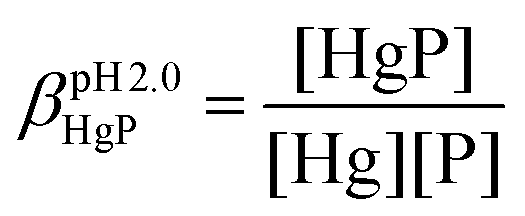
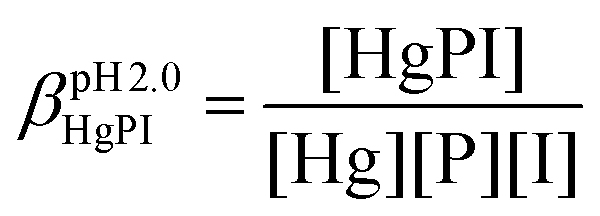
In the equations above, HgP (or HgPI) and P represent the complexed and free peptide at the pH of the studies, independent of the protonation state of the ligand, and Hg and I stand for the free Hg(II) and I− ions, respectively.
Determination of acid dissociation constants (pKa)
Deprotonation processes of one of the linear peptide ligands, 1L, were followed by pH-potentiometric titrations in aqueous solution (T = 298.0 ± 0.1 K, cligand = 1.0 × 10−3 M, I = 0.1 M NaClO4) following a protocol described earlier.29 An automatic titration set, including a PC-controlled Dosimat 665 (Metrohm) autoburette and an Orion 710A precision digital pH-meter equipped with an Orion 8103BNUWP Ross Ultra semi micro pH electrode, was used to carry out the experiments. Argon atmosphere in the titration cell was applied in order to prevent the oxidation of the ligand. The data obtained in two parallel titrations were fitted using the PSEQUAD computer program59 based on the following general equilibrium process with the related equilibrium formation constants allowing the calculation of the pKa values for the individual deprotonation steps:| qH + rL ⇌ HqLr |
where L denotes the non-protonated peptide and H the protons (charges are omitted for simplicity). Consequently, the composition of the neutral fully protonated peptide is H3L. These pKa values were used in the calculation of the thermodynamic stability constants for the HgL and HgHL complexes from the apparent stabilities obtained at pH = 2.0. Details of these calculations are found in the ESI.† Note that ‘L’ for the ligands is used in the text where the actual protonation states of the ligands/complexes are taken into account in calculations or in the description of species, whereas ‘P’ denotes the peptides in general, independent of their protonation states, as in the calculation of the apparent stabilities.
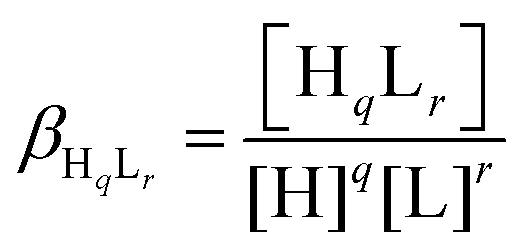
Modelling of the trithiolate mercury complexes
The six peptides were modelled in the apo and Hg(II)-bound form with a HgS3 trigonal coordination. In the absence of structures for all the peptides but P3C, initial coordinates were built with the CHARMM program60 from standard values for the internal bonds, angles and dihedrals of amino acids in proteins. For peptide P3C, initial coordinates were either generated as for other peptides or taken from the solution NMR structure of the HgP3C complex.40Results and discussion
Peptide design and synthesis
The cyclic peptide P3C with three cysteine sidechains preoriented for Hg(II) coordination was recently reported by our group.40 All spectroscopic data obtained for the HgP3C mononuclear complex point to a HgS3 coordination mode, which is stable over a large pH range (pH = 5–9).Five new peptides (Scheme 1) were designed in this work to monitor the effects of introducing a CxCxC motif instead of the CxCxxC sequence found in P3C on the Cu(I) and Hg(II) complexation features. The positions of the three cysteines in the sequence of the newly designed linear and cyclic peptides were varied. An xPGx β turn inducing motif was introduced in the sequences of the cyclic peptides to rigidify the scaffold containing the metal-binding fragment CxCxC next to the turn in 1C or one amino acid apart in 2C.28,67–691L and 2L linear analogues of the cyclic decapeptides 1C and 2C were synthesized to determine the impact of the cyclisation and flexibility of the backbone on the coordination properties. Finally, an additional amino acid, namely alanine, was inserted in peptide 3C with the idea of promoting larger flexibility in the cysteine sidechain orientation.
 | ||
| Scheme 1 Sequences of the studied peptides. Bonds in the turn motif are indicated in bold and the coordinating cysteine residues are highlighted in red. | ||
Cu(I) complexes
One of the six investigated peptides, P3C, was previously demonstrated to stabilize the HgS3 coordination in a mononuclear complex.40 This raised the question whether a CuS3 geometry could also be observed with the latter peptide and with the new 3-Cys containing peptides presented in this paper. Cu(I) complexation of the ligands was studied by UV-Vis and CD spectroscopy and ESI-MS. All the six peptides display a rather similar behaviour; therefore studies with peptide 1C are presented here to illustrate the Cu(I)-binding features of the whole series. The titration of the peptides with Cu(I) at pH 7.4 was followed by UV-Vis spectroscopy. The absorbance stabilizes surprisingly slowly with the equilibrium reached only after ca. 2 hours. Such long equilibration times were not observed before in similar systems with thiolate containing peptides or pseudopeptides, even when Cu(I) clusters were formed from the very beginning of the titration.28,38,39 Titrations using individual samples were therefore conducted with Cu(I) to peptide concentration ratios from 0.0 to 3.0 in 0.25–0.5 steps to allow for long equilibration times and ensure that the thermodynamic equilibrium is reached. Fig. 1 shows the results of the UV titration of peptide 1C with Cu(I) at equilibrium. An intense band, characteristic for the S− to Cu(I) charge transfer transition (LMCT),70 emerges at λ = 263 nm with increasing Cu(I) concentration. The absorbance increases linearly up to 2.0 equiv. of Cu(I). The breakpoint observed for 2 equiv. of Cu(I) per peptide is indicative of the formation of polymetallic species with (Cu2P)n overall stoichiometry as previously observed with 3-Cys-containing tripodal peptide-like ligands.38,39 As seen before, the intensity of the S− to Cu(I) LMCT is not sensitive to the composition of the complex and provides information only about the number of Cu(I)–thiolate bonds.38,39,71,72 The extinction coefficients ε = 6000–7000 M−1 cm−1 per bound Cu(I) reported in Table 1 are in good agreement with data reported with other thiolate–Cu(I) complexes.71,73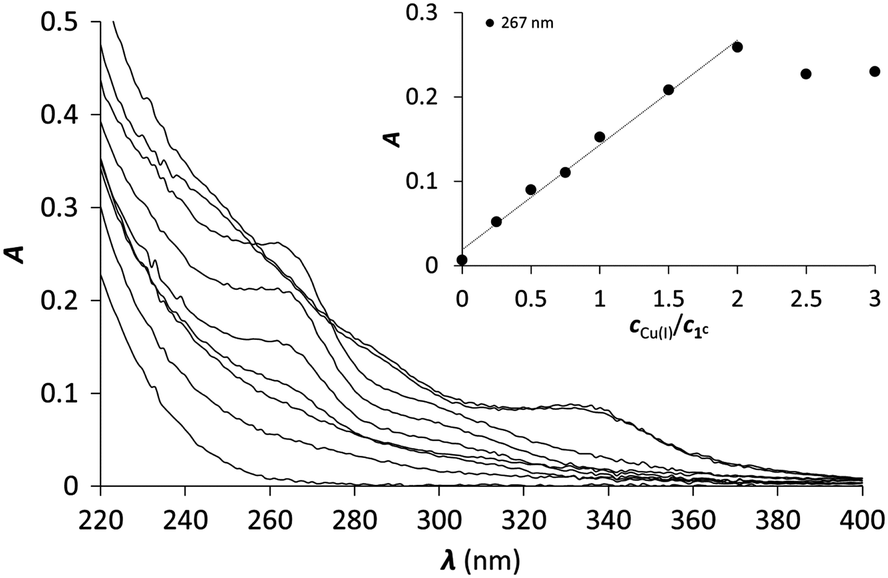 | ||
| Fig. 1 UV spectra of 1C titrated with Cu(I) (equilibration time = 2 hours). The inset shows the increase of the absorbance at 263 nm as a function of cCu(I)/cpeptide ratio (cpeptide = 30 μM in phosphate buffer, 20 mM, pH = 7.4). | ||
| CuP | HgP | |||
|---|---|---|---|---|
| λ (nm) | ε (M−1 cm−1) | λ (nm) | ε (M−1 cm−1) | |
| P3C | 259 | 7500 | 240 | 1750040 |
| 270 | 10200 |
|||
| 1C | 267 | 6700 | 240 | 13800 |
| 280 | 7300 | |||
| 1L | 267 | 6100 | 240 | 14900 |
| 280 | 9400 | |||
| 2C | 267 | 6200 | 240 | 15000 |
| 280 | 7700 | |||
| 2L | 262 | 6500 | 240 | 14600 |
| 280 | 7700 | |||
| 3C | 261 | 6400 | 240 | 14500 |
| 280 | 7800 | |||
Cu(I) complex formation was also followed by recording CD spectra of the individually prepared samples. As a result of Cu(I) additions, the spectra significantly change compared to the free ligand and signals appear in the S− to Cu(I) LMCT region (260–320 nm). However, the molar ellipticities are relatively low ([Θ] < 1 × 104 deg cm2 dmol−1) compared to those obtained with other species involving 3 coordinated Cys ([Θ] = 4 × 104 – 8 × 104 deg cm2 dmol−1).38,39,71 Moreover, no clear tendency can be observed in the evolution of the spectra upon Cu(I) addition up to 2.0 equivalents, as shown in Fig. S2 (ESI†). The mostly affected region falls below 250 nm, where the negative CD band is attributed to π → π* and possibly overlapping n → π* transitions, both belonging to the amide bonds of the peptide backbone,74,75 which suggests a conformational rearrangement upon Cu(I) addition. Since CD spectroscopy is more sensitive to the structure around Cu(I) than UV, these results indicate that the free peptides transform into more than one Cu(I) species. This assumption is confirmed by ESI-MS measurements in ammonium acetate buffer at pH 6.9 (Fig. S3, ESI†). Spectra recorded for samples with the peptides and 0.9 equivalents of Cu(I) show the formation of mononuclear complexes, CuP, and several polynuclear species, like Cu4P2 and Cu4P3. With the increase of the Cu(I) concentration, further species of higher nuclearity (Cu8P4, Cu7P3, Cu9P3) are detected. Therefore, the CD and ESI-MS experiments demonstrate that the apparently simple evolution of the LMCT bands in the UV titration is in fact due to the formation of a mixture of many polynuclear thiolate complexes.
Copper binding affinities were determined in the presence of BCS as a competitor. BCS forms a well-characterised Cu(BCS)2 complex with Cu(I) according to eqn (1).
| Cu+ + 2BCS2− ⇌ [Cu(BCS)2]3− logβ = 19.8 | (1) |
300 M−1 cm−1.51 Solutions of the Cu(I)–peptide complexes (Cu(I):P ratio = 0.9:1) were titrated with BCS and the amount of Cu(I) displaced from the peptide by BCS was quantified based on the known absorption of the Cu(BCS)2 complex. Since several Cu(I) complexes are formed with the six peptides, the fit of the spectroscopic competition results could not be perfectly implemented with a given complex stoichiometry. Hence, the apparent stability constants were calculated considering the formation of a CuP complex. The logβpH7.4CuP values are presented in Table 2. All peptides display high affinity towards Cu(I) in the range typical of Cu(I) chaperone proteins, like Atx1.22
| logβpH7.4CuPa |
logβpH2.0HgP |
logβpH7.4HgPb |
pKHgLHgHLc |
logβHgHLb |
logβHgLb |
|
|---|---|---|---|---|---|---|
| a The apparent stabilities were calculated by assuming the presence of only mononuclear complexes. b Apparent stability constants for pH 7.4 and formation constants of the HgHL and HgL species were estimated from the apparent stabilities obtained at pH = 2.0, as described in the ESI. c The pK values characterize the HgHL ⇌ HgL + H+ process. | ||||||
| P3C | 18.1(1) | 27.2(1) | 40.9 | 4.3(1)40 | 48.7 | 44.4 |
| 1C | 17.3(3) | 27.3(1) | 40.5 | 4.8(1) | 48.8 | 44.0 |
| 1L | 17.4(2) | 27.1(1) | 40.0 | 5.1(1) | 48.6 | 43.5 |
| 2C | 17.8(4) | 27.2(1) | 40.7 | 4.5(1) | 48.7 | 44.2 |
| 2L | 17.8(2) | 27.0(1) | 40.0 | 5.0(1) | 48.5 | 43.5 |
| 3C | 17.9(4) | 27.5(1) | 41.0 | 4.5(1) | 49.0 | 44.5 |
Hence, Cu(I) binding to the investigated 3-Cys containing peptides is characterized by a rather complicated speciation at pH = 7.4. Indeed, although the UV profile appears simple with a single endpoint at 2 equivalents of Cu(I), ESI-MS and CD clearly evidence several clusters, even with low Cu(I) concentration. This complicated mixture of species observed for the whole series of peptides highly contrasts with previous results obtained with tripodal pseudopeptides also incorporating three cysteine moieties, which form Cu(I) complexes, resembling those formed in metallothioneins.39,71,76 Indeed, the pre-orientation of the three thiolate groups in the tripodal pseudopeptides induces a well-defined metal binding cavity, stabilized by a network of hydrogen bonds.39 This structure perfectly controls the speciation of the Cu(I) complexes with only two identified species, namely the mononuclear complex and the cluster Cu6S9 both with Cu(I) ions in trithiolate environments.39,76 More recently, the highly constrained tetrapeptide Ac-Cys-D-Pro-Pro-Cys-NH2 with a strong turn was shown to form exclusively a Cu4S6 core.77 The larger flexibility of the peptides described here could be responsible for the lack of control of the speciation of the Cu(I) complexes and ultimately for the formation of a mixture of polymetallic species, with quite large stability. The determined apparent stability constants are similar in the whole peptide series, within the range of experimental errors, indicating that the structural differences have only a minor effect, if any, on the stability of the Cu(I) complexes.
Hg(II) complexes
Hg(II) is a metal ion with soft character according to Pearson's theory,78 and as such, an often used probe of the oxygen and water sensitive Cu(I). The cyclic peptide P3C has been recently demonstrated to form a complex with Hg(II) in a HgS3 coordination environment, which is stable over a large pH range. Besides, the protonation of the mononuclear complex HgP3C happens at a relatively low pH (pKa value of 4.3) to produce a species with HgS2 geometry.40 Hg(II) binding of the new series of peptides was therefore studied to reveal whether the behaviour of P3C with Hg(II) is specific to the structure of this cyclic peptide or common for peptide sequences encompassing three Cys residues in a CxCxxC or CxCxC arrangement.:1 Hg(II)–peptide ratios, which reflects the formation of a single mononuclear complex, where Hg(II) is very likely coordinated by the three cysteine thiolates of the peptides. In the presence of Hg(II) in excess, a decrease of the absorbances at the selected wavelength values can be observed, indicating that the mononuclear complex transforms into polymetallic species with Hg(II) ions coordinated by only two cysteine thiolate residues.28,80,81 Besides, the second endpoint observed at 1.5 equiv. of Hg(II) strongly suggests the formation of Hg3P2 complexes.
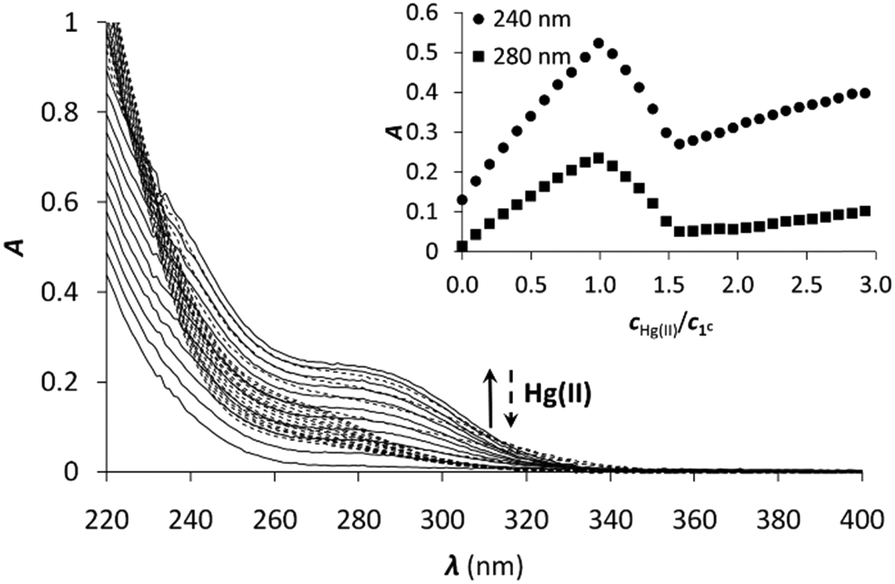 | ||
| Fig. 2 UV titration of 1C with Hg(II) in phosphate buffer (20 mM, pH = 7.4). The inset shows the increase of the absorbance at 240 and 280 nm as a function of cHg(II)/cpeptide ratio (cpeptide = 30 μM). | ||
ESI-MS experiments performed in ammonium acetate buffer at pH 6.9 support well the UV spectroscopic results (Fig. 3). The spectrum of 1C with 1.0 equivalent of Hg(II) clearly shows the presence of the mononuclear HgP complex in the forms of a [Hg1C]2+ and a [Hg1C–H]+ ion with m/z = 576.8 and 1152.3, respectively. The spectrum recorded with 1.5 equivalents of Hg(II) indicates the formation of new species. The major detected complex is Hg3P2 in the forms of [Hg3(1C)2–4H + Na]3+ and [Hg3(1C)2–4H]2+ with m/z = 841.7 and 1250.8, respectively. This species is compatible with the HgS2 coordination environment of the metal ions. When 2.0 equivalents of Hg(II) are added, Hg3P2 and Hg2P ([Hg2(1C)–2H]2+; m/z = 675.8) can be detected in similar amounts. This mixture of complexes may explain the slight increase of the absorbance above 1.5 equivalents of Hg(II) per peptide.
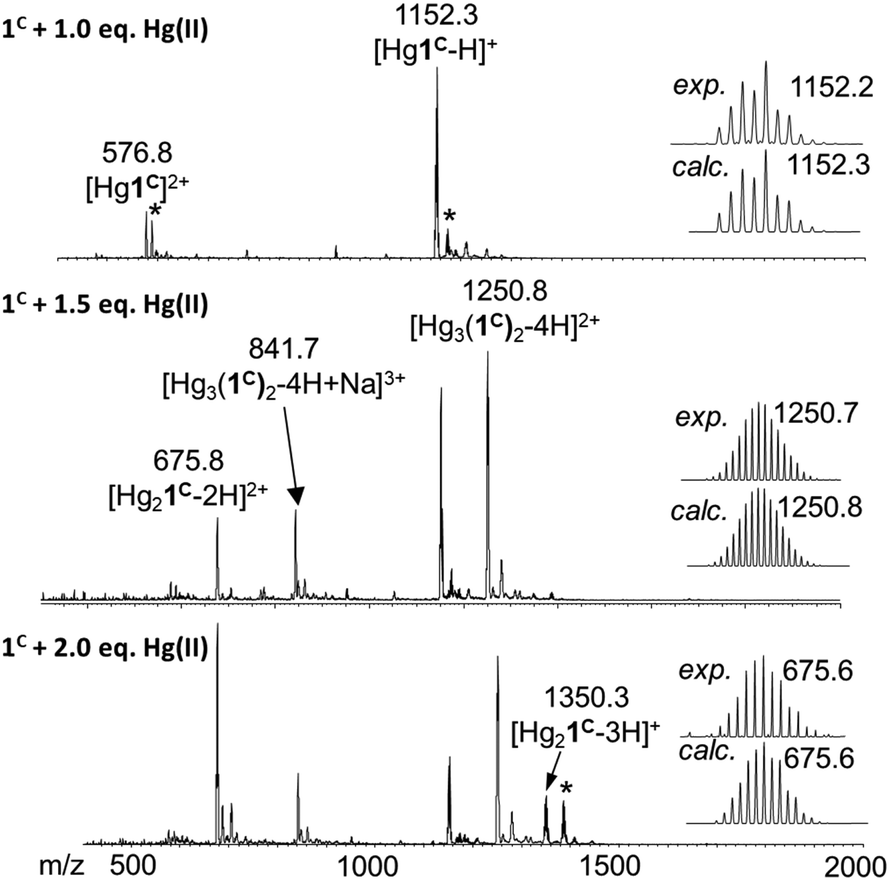 | ||
| Fig. 3 (+)ESI-MS spectra registered for 1C with different equivalents of Hg(II) in ammonium acetate buffer (20 mM, pH = 6.9). The comparison of the experimental and calculated isotopic envelop of the detected species is also presented. Asterisks mark the sodium adduct of the corresponding species. The notation 1C refers here to the neutral free peptide. | ||
All the peptides showed the formation of Hg(II) complexes with a simple speciation as described recently with the P3C cyclic decapeptide.40 The mononuclear complexes with features characterizing a trithiolate coordination are formed at physiological pH, as also extensively reported for the Hg(II) complexes of triple coiled-coil peptides.34,35 Contrary to the latter systems, which define a highly protected hydrophobic metal-binding pocket, the flexible structure of the cyclic and linear peptides makes the transformation of the trithiolate coordination into the preferred dithiolate coordination possible in excess of Hg(II) in Hg3P2 complexes.
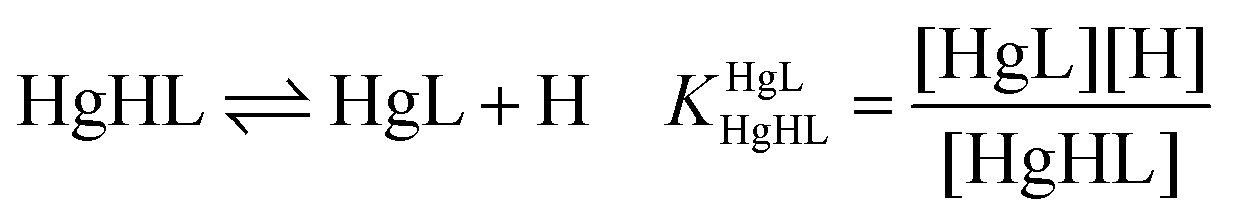 | (2) |
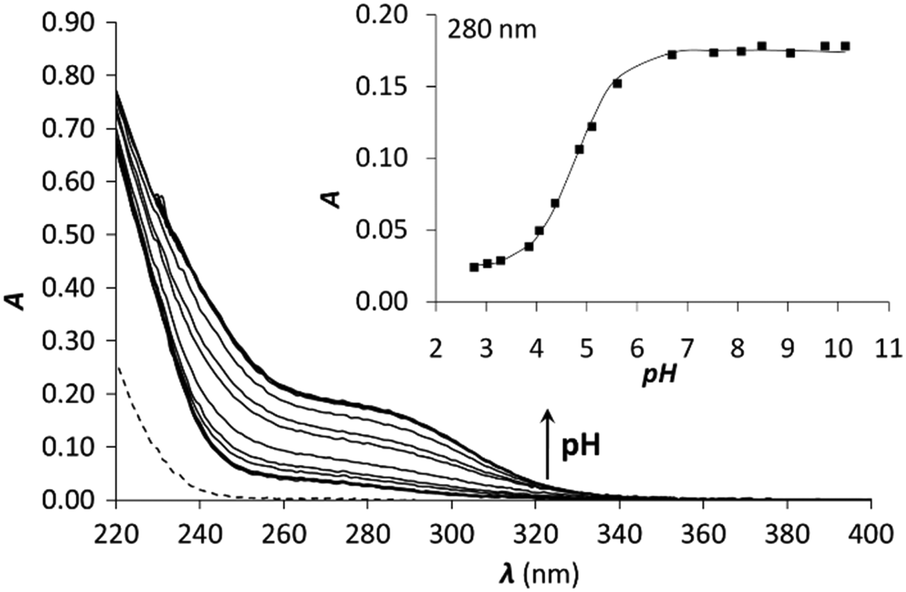 | ||
| Fig. 4 pH titration of 1C with 1.0 equivalent of Hg(II). The dashed line represents the spectrum of free 1C at pH = 2. The inset shows the evolution of the absorbance as a function of pH at 280 nm. Symbols represent the experimental points and the line is the fitted absorbance obtained by SPECFIT. | ||
The pKa of the Hg(II)–peptide complexes (see Table 2) follow the order of P3C < 3C ≈ 2C < 1C < 2L ≈ 1L. However, the differences observed between the pKa values in the series of peptides (maximum difference = 0.8) are quite small, which reflects the weak influence of the pattern of the three Cys residues (CxCxC or CxCxxC) and the separation of the metal binding fragment from the turn motif. P3C incorporating the CxCxxC sequence forms the HgS3 coordination mode at a slightly lower pH than the other cyclic peptides. A larger separation from the PG-turn also seems to be favourable for the formation of the tris-thiolate complex in 3C. The highest pKa values are seen for the two linear peptides, which may be a consequence of their larger flexibility and a more significant reorganization necessary for the coordination of the third Cys sidechain. It is rather interesting that even the latter data (Table 2) are 1.7–1.8log units lower than the pKa observed for the Hg(II)-complex of the tris-cysteine functionalized tripodal pseudopeptide ligand with amidated carboxyl groups,37 or of the three-stranded coiled coils (pKa values of 8.6 and 7.6 for sites d and a, respectively).34 This might be related to the presumably very different water-accessibility of the thiol groups, as hinted recently.40
Therefore, the published stabilities of the iodo complexes were recalculated by applying the SIT model to the conditions of the experiments,57,58 leading to the following formation constants: logβ[HgI]+ = 13.05, logβ[HgI2] = 24.09, logβ[HgI3]− = 27.84, logβ[HgI4]2− = 29.91. The result of the titration of Hg(II) with KI and the obtained molar spectra of the forming Hg(II)–I− complexes can be seen in Fig. 5A and Fig. S4 (ESI†), respectively.
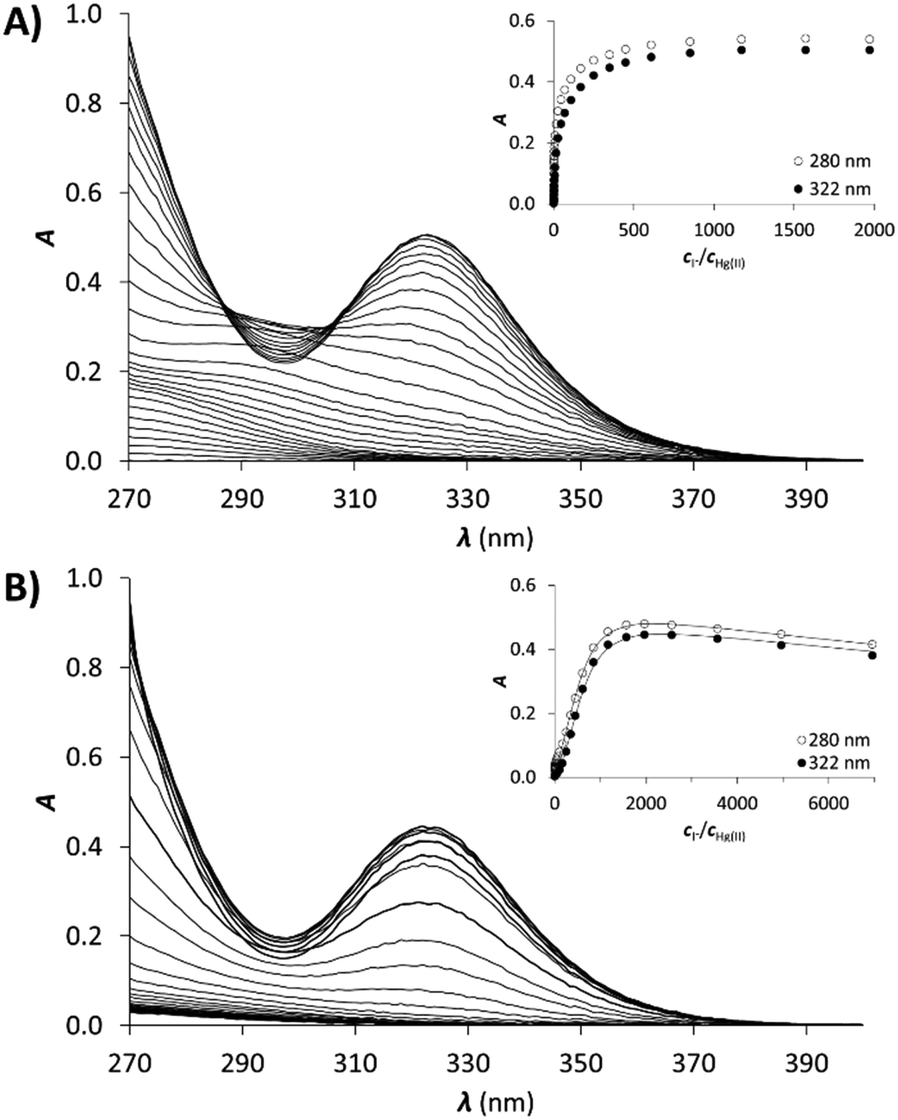 | ||
| Fig. 5 UV spectra recorded in the titration of (A) Hg(II) and (B) Hg1C with I−. pH = 2.0, cHg(II) = 30 μM (A), cHg(II) = cpeptide = 30 μM (B). The insets show the evolution of the absorbance at 322 nm (●) and 280 nm (○). Symbols represent the experimental data, and solid lines represent the absorbances calculated by SPECFIT. | ||
Samples of the peptides containing 1.0 equivalent of Hg(II) were titrated with I− and the recorded spectra are presented in Fig. 5B. The first phase of the titrations, i.e. up to the presence of 10 equivalents of I−, does not reflect considerable changes in the recorded spectra. Further addition of I− ions results in the appearance of new bands characteristic for the [HgI3]− and [HgI4]2− species (compare to the spectra in Fig. S4, ESI†). A complete displacement of Hg(II) from the peptide complexes is achieved at ca. 2000 equiv. of I−.
The obtained spectra were fitted by SPECFIT by fixing the logβ values and the molar spectra of the Hg(II)–I− complexes. The best fits were obtained when the formation of a mixed ligand complex, HgPI, was also included in the models, besides the HgP species (inset in Fig. 5B). The appearance of such species is probably a consequence of the flexibility of the peptide structures. The HgPI complexes are present only in the beginning part of the titrations in a ca. 20% relative proportion, except for HgP3C where a somewhat larger fraction of HgPI could be observed. The formation of the mixed ligand complexes, according to the HgP + I ⇌ HgPI equation, is characterized by stabilities falling in the range of logK ∼ 1.5–2.5. The apparent stability constants determined for the HgP complexes (Table 2) indicate rather similar affinities of Hg(II) to all peptides. Considering that at pH = 2.0 Hg(II) is coordinated only by two thiolate units, it is a plausible assumption that the preorientation of the donor groups has only a modest influence on the stabilities and the high thiophilicity of Hg(II) easily governs the formation of the favoured HgS2 structures. Formation constants for the different forms of the HgP species, i.e. for HgHL and HgL (where L denotes the fully deprotonated peptide), and apparent stabilities for pH = 7.4, were also estimated from the relevant conditional stability constants (logβpH2.0HgP) by applying the pKa values of the HgHL ⇌ HgL + H processes (Table 2) and the pKa values obtained for one of the free ligands, pKHLa = 9.26(1); 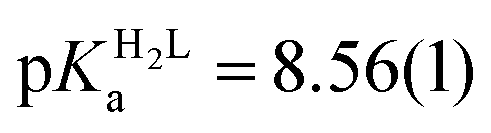 ;
; 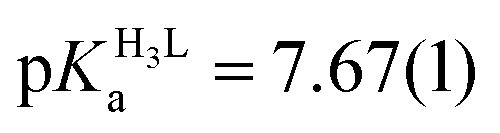 (see the Experimental part and the ESI†). The stability constants estimated for the Hg(II)–trithiolate HgL complexes span over a small range (maximum difference, ΔlogβHgP = 1) demonstrating a weak influence of the Cys-sidechain orientations in the chosen sequences (Table 2). Nevertheless, the two linear peptides display a slightly weaker affinity suggesting the need for a more pronounced rearrangement of the Cys sidechains. It is noteworthy to compare these data to the stability constants of Hg(II)–bisthiolate complexes of highly constrained bis-thiol ligands. Our peptides, indeed, display very similar Hg(II)-binding affinities to that of the well-known soft metal ion chelator 2,3-dimercaptopropan-1-ol (BAL) (logβHgL = 44.8),82 which forms a highly stable 5-membered chelate ring around the metal ion. Comparison of our data to the stability of the HgL species of the tetrapeptide CDPPC (logβHgL = 40.0)81 clearly indicates that the structure of our peptides is prone to easily rearrange to a suitable form for the tridentate coordination of Hg(II) and thus the larger number of Hg(II)–thiolate bonds is revealed by higher affinities.
(see the Experimental part and the ESI†). The stability constants estimated for the Hg(II)–trithiolate HgL complexes span over a small range (maximum difference, ΔlogβHgP = 1) demonstrating a weak influence of the Cys-sidechain orientations in the chosen sequences (Table 2). Nevertheless, the two linear peptides display a slightly weaker affinity suggesting the need for a more pronounced rearrangement of the Cys sidechains. It is noteworthy to compare these data to the stability constants of Hg(II)–bisthiolate complexes of highly constrained bis-thiol ligands. Our peptides, indeed, display very similar Hg(II)-binding affinities to that of the well-known soft metal ion chelator 2,3-dimercaptopropan-1-ol (BAL) (logβHgL = 44.8),82 which forms a highly stable 5-membered chelate ring around the metal ion. Comparison of our data to the stability of the HgL species of the tetrapeptide CDPPC (logβHgL = 40.0)81 clearly indicates that the structure of our peptides is prone to easily rearrange to a suitable form for the tridentate coordination of Hg(II) and thus the larger number of Hg(II)–thiolate bonds is revealed by higher affinities.
Modelling of the trithiolate HgL complexes
The six peptides were modelled in the apo and Hg(II)-bound form with a HgS3 trigonal coordination (see Experimental section for methods). Several independent simulations were run: first, for the Hg(II)-bound peptides three different simulations were run varying the orders of the cysteine sulfur atoms defining the Hg–S–S–S improper angle; then, for each best energy conformation in each system, new simulations for the related systems were run starting from there. The energy values reported in Table S2 (ESI†) refer to the simulation leading to the minimum average total energy for each peptide (over all simulations). Binding of Hg(II) to linear or cyclic peptides always results in stabilization meaning that the peptide structure organizes the 3 cysteine residues in an environment appropriate for HgS3 coordination. The stabilization expressed as ΔE = E(HgP) − E(P) is remarkably similar for all four cyclic peptides (between −11 and −12.8 kcal mol−1 from Table S2, ESI†) with a relative error (ΔE/E) less than 1%. This stabilization varies from −13.1 to −9.5 kcal mol−1 for the two linear peptides; however, this slightly larger difference may be due to an incomplete conformational search of the highly flexible linear apo peptides. Hence, the calculated stabilization energies, ΔE, are very similar in the series of peptides, which is in agreement with the comparable values of the stability constants of the HgL complexes determined experimentally (Table 2). Models of energy minimized structures of the Hg(II)-bound peptides, starting from the frame with the lowest potential energy during the dynamics simulation leading to minimum average total energy, are shown in Fig. 6 and Fig. S5 (ESI†) for cyclic and linear peptides, respectively. The three cysteine binding chains are well-disposed to afford the trithiolate coordination of the mercury ion in the six structures. In all the complexes, the positively charged sidechain of arginine (charge +1) is capping the HgS3, negatively charged binding site (charge −1), providing stabilizing electrostatic interactions.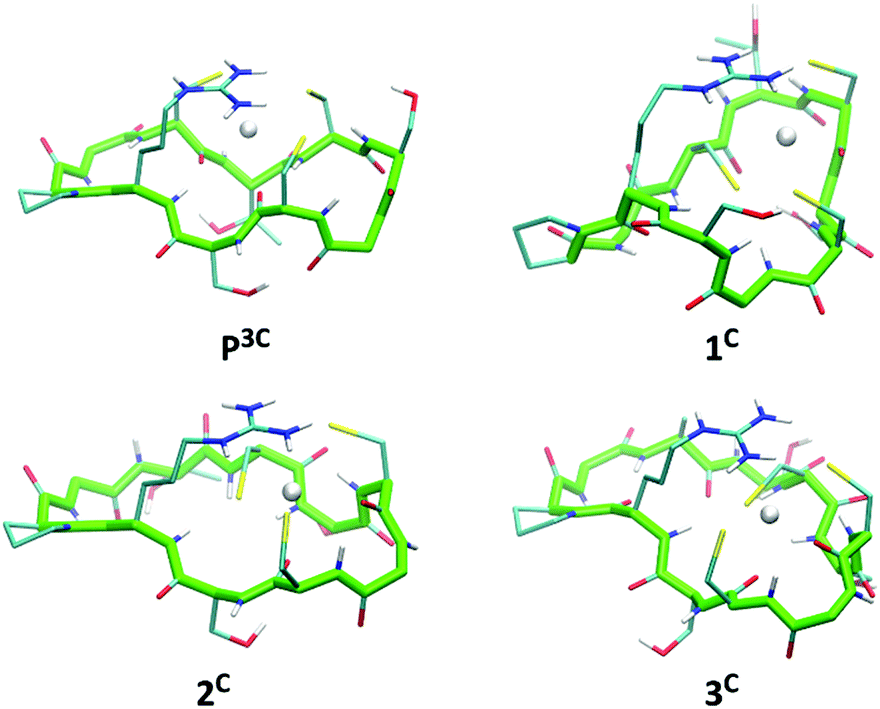 | ||
| Fig. 6 Energy minimized structures of the 4 cyclic peptides in their Hg(II)-bound form (oriented with respect to the position of backbone atom coordinates of residues 1 to 10). | ||
Conclusions
Model peptides containing cysteine-rich sequences found in metallothioneins were studied for their metal-binding properties in relation to metal detoxification mechanisms. The two soft ions Cu(I) and Hg(II) were selected since they exhibit the largest affinities for these small detoxification proteins among endogenous and toxic metal ions, respectively. Three cysteine residues were introduced in CxCxxC and CxCxC motifs in different positions within the sequence, in linear and cyclic derivatives. Overall, the six peptides display rather similar behaviour, which evidences minor contributions of the position of the three cysteine residues or cyclisation to the formation and stability of the Hg(II) and Cu(I) complexes.Cu(I) binding to the series of peptides at physiological pH revealed to be rather complicated, with the formation of a mixture of polymetallic species. In contrast, cysteine-rich highly structured peptides77 or peptide-like ligands39,76 are able to control the formation of well-defined Cu(I) complexes. Consequently, the complicated Cu(I)-complex speciation of the series of peptides, reported in this paper, has been assigned to their significantly larger flexibility. However, despite the formation of many polymetallic species, large affinity is achieved for the soft Cu(I) cation at physiological pH (1017–1018).
The binding of Hg(II), another soft metal ion often used as a probe for the oxygen and water sensitive Cu(I), demonstrates that the complexity of the Cu(I) speciation is due to the peculiar behaviour of Cu(I)–thiolate complexes in water and not to the cysteine-rich sequences chosen for the peptides. Interestingly, the HgS3 coordination mode is stable over a large pH-range for all studied peptide complexes. Indeed, the protonation of the complex to give the HgS2 linear coordination is observed with pKa values ranging from 4.3 to 5.1, making the trithiolate coordination the major binding mode at physiological pH whatever the peptide sequence. The stabilities of the Hg(II) complexes (1040–1041 at pH 7.4) are of the same order of magnitude as those reported for high affinity sulphur chelating agents such as BAL.82 The large stability constants together with the low pKa values and simulated structures clearly indicate that all the peptide sequences studied in this paper are adapted for an efficient trithiolate coordination of the thiophilic cation Hg(II).
Importantly, the striking differences observed in the coordination of Hg(II) and Cu(I) with the series of peptides indicate different molecular mechanisms involved in their binding to detoxification proteins. The sulphur-rich peptides studied here show more than 20 orders of magnitude larger affinity at pH 7.4 for Hg(II) (logβpH7.4HgP ≈ 41) than for Cu(I) (logβpH7.4CuP ≈ 18), due to the significantly softer character of Hg(II). Most importantly, Hg(II) forms well-defined complexes, whereas Cu(I)-coordination leads to mixtures of polymetallic species. This demonstrates the peculiar behaviour of Cu(I) thiolate complexes in water. Only highly constrained peptide sequences are able to promote the formation of well-defined Cu(I) complexes. The peptides studied here are probably too flexible to achieve such a control for Cu(I). Hence, the use of Hg(II) as a probe for Cu(I) coordination with sulphur-rich peptides or proteins in physiological conditions is demonstrated here to be not fully appropriate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Labex ARCANE (Grant ANR-11-LABX-0003-01), the “Fondation pour la Recherche Médicale” (grant DCM20111223043), the Hungarian National Research, Development and Innovation Office-NKFIH through project GINOP-2.3.2-15-2016-00038 and grant no. K_16/120130. Edit Mesterházy kindly acknowledges the financial support of Campus France.References
- R. A. Steele and S. J. Opella, Structures of the Reduced and Mercury-Bound Forms of MerP, the Periplasmic Protein from the Bacterial Mercury Detoxification System, Biochemistry, 1997, 36, 6885–6895 CrossRef PubMed.
- E. Rossy, O. Sénèque, D. Lascoux, D. Lemaire, S. Crouzy, P. Delangle and J. Covès, Is the cytoplasmic loop of MerT, the mercuric ion transport protein, involved in mercury transfer to the mercuric reductase?, FEBS Lett., 2004, 575, 86–90 CrossRef PubMed.
- R. Ledwidge, B. Patel, A. Dong, D. Fiedler, M. Falkowski, J. Zelikova, A. O. Summers, E. F. Pai and S. M. Miller, NmerA, the Metal Binding Domain of Mercuric Ion Reductase, Removes Hg2+ from Proteins, Delivers It to the Catalytic Core, and Protects Cells under Glutathione-Depleted Conditions, Biochemistry, 2005, 44, 11402–11416 CrossRef PubMed.
- A. C. Rosenzweig, D. L. Huffman, M. Y. Hou, A. K. Wernimont, R. A. Pufahl and T. V. O’Halloran, Crystal structure of the Atx1 metallochaperone protein at 1.02 Å resolution, Structure, 1999, 7, 605–617 CrossRef PubMed.
- P. Faller, B. Ctortecka, W. Tröger, T. Butz and M. Vašák, Optical and TDPAC spectroscopy of Hg(II)-rubredoxin: model for a mononuclear tetrahedral [Hg(CysS)4]2− center, J. Biol. Inorg. Chem., 2000, 5, 393–401 CrossRef PubMed.
- C.-C. Chang, L.-Y. Lin, X.-W. Zou, C.-C. Huang and N.-L. Chan, Structural basis of the mercury(II)-mediated conformational switching of the dual-function transcriptional regulator MerR, Nucleic Acids Res., 2015, 43, 7612–7623 CrossRef PubMed.
- A. K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. O'Halloran and A. C. Rosenzweig, Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins, Nat. Struct. Biol., 2000, 7, 766 CrossRef PubMed.
- M. Łuczkowski, B. A. Zeider, A. V. H. Hinz, M. Stachura, S. Chakraborty, L. Hemmingsen, D. L. Huffman and V. L. Pecoraro, Probing the Coordination Environment of the Human Copper Chaperone HAH1: Characterization of HgII-Bridged Homodimeric Species in Solution, Chem. – Eur. J., 2013, 19, 9042–9049 CrossRef PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. Halloran and A. Mondragón, Molecular Basis of Metal-Ion Selectivity and Zeptomolar Sensitivity by CueR, Science, 2003, 301, 1383 CrossRef PubMed.
- C. Ariöz, Y. Li and P. Wittung-Stafshede, The six metal binding domains in human copper transporter, ATP7B: molecular biophysics and disease-causing mutations, Biometals, 2017, 30, 823–840 CrossRef PubMed.
- P. A. Cobine, G. N. George, C. E. Jones, W. A. Wickramasinghe, M. Solioz and C. T. Dameron, Copper Transfer from the Cu(I) Chaperone, CopZ, to the Repressor, Zn(II)CopY: Metal Coordination Environments and Protein Interactions, Biochemistry, 2002, 41, 5822–5829 CrossRef PubMed.
- M. A. Kihlken, A. P. Leech and N. E. L. E. Brun, Copper-mediated dimerization of CopZ, a predicted copper chaperone from Bacillus subtilis, Biochem. J., 2002, 368, 729 CrossRef PubMed.
- C. A. Blindauer and O. I. Leszczyszyn, Metallothioneins: unparalleled diversity in structures and functions for metal ion homeostasis and more, Nat. Prod. Rep., 2010, 27, 720–741 RSC.
- M. Capdevila, R. Bofill, Ò. Palacios and S. Atrian, State-of-the-art of metallothioneins at the beginning of the 21st century, Coord. Chem. Rev., 2012, 256, 46–62 CrossRef.
- R. Bofill, M. Capdevila and S. Atrian, Independent metal-binding features of recombinant metallothioneins convergently draw a step gradation between Zn- and Cu-thioneins, Metallomics, 2009, 1, 229–234 RSC.
- M. Valls, R. Bofill, R. González-Duarte, P. González-Duarte, M. Capdevila and S. l. Atrian, A New Insight into Metallothionein (MT) Classification and Evolution: The in vivo and in vitro Metal Binding Features of Homarus Americanus Recombinant MT, J. Biol. Chem., 2001, 276, 32835–32843 CrossRef PubMed.
- M. J. Stillman, Metallothioneins, Coord. Chem. Rev., 1995, 144, 461–511 CrossRef.
- G. Henkel and B. Krebs, Metallothioneins: Zinc, Cadmium, Mercury, and Copper Thiolates and Selenolates Mimicking Protein Active Site Features – Structural Aspects and Biological Implications, Chem. Rev., 2004, 104, 801–824 CrossRef PubMed.
- V. Calderone, B. Dolderer, H.-J. Hartmann, H. Echner, C. Luchinat, C. Del Bianco, S. Mangani and U. Weser, The crystal structure of yeast copper thionein: The solution of a long-lasting enigma, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 51–56 CrossRef PubMed.
- L. Zhang, J. Pickering Ingrid, R. Winge Dennis and N. George Graham, X-Ray Absorption Spectroscopy of Cuprous-Thiolate Clusters in Saccharomyces cerevisiae Metallothionein, Chem. Biodiversity, 2008, 5, 2042–2049 CrossRef PubMed.
- M. J. Pushie, L. Zhang, I. J. Pickering and G. N. George, The fictile coordination chemistry of cuprous-thiolate sites in copper chaperones, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 938–947 CrossRef PubMed.
- Z. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd, C-Terminal Domain of the Membrane Copper Transporter Ctr1 from Saccharomyces cerevisiae Binds Four Cu(I) Ions as a Cuprous-Thiolate Polynuclear Cluster: Sub-femtomolar Cu(I) Affinity of Three Proteins Involved in Copper Trafficking, J. Am. Chem. Soc., 2004, 126, 3081–3090 CrossRef PubMed.
- J. A. Graden, M. C. Posewitz, J. R. Simon, G. N. George, I. J. Pickering and D. R. Winge, Presence of a Copper(I)–Thiolate Regulatory Domain in the Copper-Activated Transcription Factor Amt1, Biochemistry, 1996, 35, 14583–14589 CrossRef PubMed.
- X. Chen, H. Hua, K. Balamurugan, X. Kong, L. Zhang, G. N. George, O. Georgiev, W. Schaffner and D. P. Giedroc, Copper sensing function of Drosophila metal-responsive transcription factor-1 is mediated by a tetranuclear Cu(I) cluster, Nucleic Acids Res., 2008, 36, 3128–3138 CrossRef PubMed.
- W. Lu, A. J. Zelazowski and M. J. Stillman, Mercury binding to metallothioneins: formation of the Hg18-MT species, Inorg. Chem., 1993, 32, 919–926 CrossRef.
- À. Leiva-Presa, M. Capdevila and P. Gonzàlez-Duarte, Mercury(II) binding to metallothioneins, Eur. J. Biochem., 2004, 271, 4872–4880 CrossRef PubMed.
- O. Seneque, S. Crouzy, D. Boturyn, P. Dumy, M. Ferrand and P. Delangle, Novel model peptide for Atx1-like metallochaperones, Chem. Commun., 2004, 770–771 RSC.
- P. Rousselot-Pailley, O. Sénèque, C. Lebrun, S. Crouzy, D. Boturyn, P. Dumy, M. Ferrand and P. Delangle, Model Peptides Based on the Binding Loop of the Copper Metallochaperone Atx1: Selectivity of the Consensus Sequence MxCxxC for Metal Ions Hg(II), Cu(I), Cd(II), Pb(II), and Zn(II), Inorg. Chem., 2006, 45, 5510–5520 CrossRef PubMed.
- A. Jancsó, B. Gyurcsik, E. Mesterházy and R. Berkecz, Competition of zinc(II) with cadmium(II) or mercury(II) in binding to a 12-mer peptide, J. Inorg. Biochem., 2013, 126, 96–103 CrossRef PubMed.
- D. Szunyogh, B. Gyurcsik, F. H. Larsen, M. Stachura, P. W. Thulstrup, L. Hemmingsen and A. Jancsó, ZnII and HgII binding to a designed peptide that accommodates different coordination geometries, Dalton Trans., 2015, 44, 12576–12588 RSC.
- E. Mesterházy, B. Boff, C. Lebrun, P. Delangle and A. Jancsó, Oligopeptide models of the metal binding loop of the bacterial copper efflux regulator protein CueR as potential Cu(I) chelators, Inorg. Chim. Acta, 2018, 472, 192–198 CrossRef.
- A. Jancsó, D. Szunyogh, F. H. Larsen, P. W. Thulstrup, N. J. Christensen, B. Gyurcsik and L. Hemmingsen, Towards the role of metal ions in the structural variability of proteins: CdII speciation of a metal ion binding loop motif, Metallomics, 2011, 3, 1331–1339 RSC.
- D. Szunyogh, H. Szokolai, P. W. Thulstrup, F. H. Larsen, B. Gyurcsik, N. J. Christensen, M. Stachura, L. Hemmingsen and A. Jancsó, Specificity of the Metalloregulator CueR for Monovalent Metal Ions: Possible Functional Role of a Coordinated Thiol?, Angew. Chem., Int. Ed., 2015, 54, 15756–15761 CrossRef PubMed.
- S. Chakraborty, J. Yudenfreund Kravitz, P. W. Thulstrup, L. Hemmingsen, W. F. DeGrado and V. L. Pecoraro, Design of a Three-Helix Bundle Capable of Binding Heavy Metals in a Triscysteine Environment, Angew. Chem., Int. Ed., 2011, 50, 2049–2053 CrossRef PubMed.
- O. Iranzo, P. W. Thulstrup, S.-b. Ryu, L. Hemmingsen and V. L. Pecoraro, The Application of 199Hg NMR and 199mHg Perturbed Angular Correlation (PAC) Spectroscopy to Define the Biological Chemistry of HgII: A Case Study with Designed Two- and Three-Stranded Coiled Coils, Chem. – Eur. J., 2007, 13, 9178–9190 CrossRef PubMed.
- A.-S. Jullien, C. Gateau, C. Lebrun and P. Delangle, Mercury Complexes with Tripodal Pseudopeptides Derived from d-Penicillamine Favour a HgS3 Coordination, Eur. J. Inorg. Chem., 2015, 3674–3680 CrossRef.
- A. M. Pujol, C. Lebrun, C. Gateau, A. Manceau and P. Delangle, Mercury-Sequestering Pseudopeptides with a Tris(cysteine) Environment in Water, Eur. J. Inorg. Chem., 2012, 3835–3843 CrossRef.
- A.-S. Jullien, C. Gateau, C. Lebrun, I. Kieffer, D. Testemale and P. Delangle, D-Penicillamine Tripodal Derivatives as Efficient Copper(I) Chelators, Inorg. Chem., 2014, 53, 5229–5239 CrossRef PubMed.
- A. M. Pujol, C. Gateau, C. Lebrun and P. Delangle, A Series of Tripodal Cysteine Derivatives as Water-Soluble Chelators that are Highly Selective for Copper(I), Chem. – Eur. J., 2011, 17, 4418–4428 CrossRef PubMed.
- O. Sénèque, P. Rousselot-Pailley, A. Pujol, D. Boturyn, S. Crouzy, O. Proux, A. Manceau, C. Lebrun and P. Delangle, Mercury Trithiolate Binding (HgS3) to a de Novo Designed Cyclic Decapeptide with Three Preoriented Cysteine Side Chains, Inorg. Chem., 2018, 57, 2705–2713 CrossRef PubMed.
- M. Imagawa, T. Onozawa, K. Okumura, S. Osada, T. Nishihara and M. Kondo, Characterization of metallothionein cDNAs induced by cadmium in the nematode Caenorhabditis elegans, Biochem. J., 1990, 268, 237 CrossRef PubMed.
- S. Perez-Rafael, A. Kurz, M. Guirola, M. Capdevila, O. Palacios and S. Atrian, Is MtnE, the fifth Drosophila metallothionein, functionally distinct from the other members of this polymorphic protein family?, Metallomics, 2012, 4, 342–349 RSC.
- D. R. Winge, K. B. Nielson, W. R. Gray and D. H. Hamer, Yeast metallothionein. Sequence and metal-binding properties, J. Biol. Chem., 1985, 260, 14464–14470 Search PubMed.
- M. Vasak, J. H. R. Kaegi and H. A. O. Hill, Zinc(II), cadmium(II), and mercury(II) thiolate transitions in metallothionein, Biochemistry, 1981, 20, 2852–2856 CrossRef PubMed.
- P. Faller, Neuronal growth-inhibitory factor (metallothionein-3): reactivity and structure of metal–thiolate clusters, FEBS J., 2010, 277, 2921–2930 CrossRef PubMed.
- A. Presta, A. R. Green, A. Zelazowski and M. J. Stillman, Copper Binding to Rabbit Liver Metallothionein, Eur. J. Biochem., 1995, 227, 226–240 CrossRef PubMed.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides, Anal. Biochem., 1970, 34, 595–598 CrossRef PubMed.
- P. Kamau and R. B. Jordan, Complex Formation Constants for the Aqueous Copper(I)–Acetonitrile System by a Simple General Method, Inorg. Chem., 2001, 40, 3879–3883 CrossRef PubMed.
- G. L. Ellman, Tissue sulfhydryl groups, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef PubMed.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Methods Enzymol., 1983, 91, 49–60 Search PubMed.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, Unification of the Copper(I) Binding Affinities of the Metallo-chaperones Atx1, Atox1, and Related Proteins: Detection Probes and Affinity Standards, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Calculation of equilibrium constants from multiwavelength spectroscopic data—I, Talanta, 1985, 32, 95–101 CrossRef PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Calculation of equilibrium constants from multiwavelength spectroscopic data—II, Talanta, 1985, 32, 257–264 CrossRef PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Calculation of equilibrium constants from multiwavelength spectroscopic data—III, Talanta, 1985, 32, 1133–1139 CrossRef PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Calculation of equilibrium constants from multiwavelength spectroscopic data—IV, Talanta, 1986, 33, 943–951 CrossRef PubMed.
- L. G. Sillen, Electrometric Investigation of Equilibria between Mercury and Halogen Ions. Vlll. Survey and Conclusions, Acta Chem. Scand., 1949, 3, 539–553 CrossRef.
- I. Grenthe, A. V. Plyasunov and K. Spahiu, Estimations of Medium Effects on Thermodynamic Data, in Modelling in aquatic chemistry, ed. I. Grenthe and I. Puigdomenech, OECD Publications, 1997, ch. IX, pp. 325–426 Search PubMed.
- J. Powell Kipton, L. Brown Paul, H. Byrne Robert, T. Gajda, G. Hefter, S. Sjöberg and H. Wanner, Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+, Cl−, OH−, CO32−, SO42−, and PO43− aqueous systems, Pure Appl. Chem., 2005, 77, 739 Search PubMed.
- L. Zékány, I. Nagypál and G. Peintler, PSEQUAD for chemical equilibria, Technical Software Distributors, Baltimore, MD, 1991 Search PubMed.
- R. Brooks Bernard, E. Bruccoleri Robert, D. Olafson Barry, J. States David, S. Swaminathan and M. Karplus, CHARMM: A program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4, 187–217 CrossRef.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, N. Rega, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, revision A.11.3, Gaussian, Inc., Pittsburgh, PA, 2002 Search PubMed; (b) M. J. Frisch, et al., Gaussian03 Search PubMed.
- F. Šebesta, V. Sláma, J. Melcr, Z. Futera and J. V. Burda, Estimation of Transition-Metal Empirical Parameters for Molecular Mechanical Force Fields, J. Chem. Theory Comput., 2016, 12, 3681–3688 CrossRef PubMed.
- L. Themis and K. Martin, Effective energy function for proteins in solution, Proteins: Struct., Funct., Bioinf., 1999, 35, 133–152 CrossRef.
- P. Dumy, I. M. Eggleston, G. Esposito, S. Nicula and M. Mutter, Solution structure of regioselectively addressable functionalized templates: An NMR and restrained molecular dynamics investigation, Biopolymers, 1996, 39, 297–308 CrossRef PubMed.
- A. M. Pujol, M. Cuillel, O. Renaudet, C. Lebrun, P. Charbonnier, D. Cassio, C. Gateau, P. Dumy, E. Mintz and P. Delangle, Hepatocyte Targeting and Intracellular Copper Chelation by a Thiol-Containing Glycocyclopeptide, J. Am. Chem. Soc., 2011, 133, 286–296 CrossRef PubMed.
- C. S. Bonnet, P. H. Fries, S. Crouzy, O. Sénèque, F. Cisnetti, D. Boturyn, P. Dumy and P. Delangle, A Gadolinium-Binding Cyclodecapeptide with a Large High-Field Relaxivity Involving Second-Sphere Water, Chem. – Eur. J., 2009, 15, 7083–7093 CrossRef PubMed.
- M. Beltramini and K. Lerch, Spectroscopic studies on Neurospora copper metallothionein, Biochemistry, 1983, 22, 2043–2048 CrossRef PubMed.
- A. M. Pujol, C. Gateau, C. Lebrun and P. Delangle, A Cysteine-Based Tripodal Chelator with a High Affinity and Selectivity for Copper(I), J. Am. Chem. Soc., 2009, 131, 6928–6929 CrossRef PubMed.
- C. T. Dameron, D. R. Winge, G. N. George, M. Sansone, S. Hu and D. Hamer, A copper-thiolate polynuclear cluster in the ACE1 transcription factor, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6127–6131 CrossRef.
- D. L. Pountney, I. Schauwecker, J. Zarn and M. Vasak, Formation of Mammalian Cu8-Metallothionein in vitro: Evidence for the Existence of Two Cu(I)4-Thiolate Clusters, Biochemistry, 1994, 33, 9699–9705 CrossRef PubMed.
- S. M. Kelly and N. C. Price, The Use of Circular Dichroism in the Investigation of Protein Structure and Function, Curr. Protein Pept. Sci., 2000, 1, 349–384 CrossRef PubMed.
- Y. J. Li and U. Weser, Circular dichroism, luminescence, and electronic absorption of copper binding sites in metallothionein and its chemically synthesized alpha. and beta domains, Inorg. Chem., 1992, 31, 5526–5533 CrossRef.
- A.-S. Jullien, C. Gateau, I. Kieffer, D. Testemale and P. Delangle, X-ray Absorption Spectroscopy Proves the Trigonal-Planar Sulfur-Only Coordination of Copper (I) with High-Affinity Tripodal Pseudopeptides, Inorg. Chem., 2013, 52, 9954–9961 CrossRef PubMed.
- E. Mesterházy, C. Lebrun, A. Jancsó and P. Delangle, A Constrained Tetrapeptide as a Model of Cu(I) Binding Sites Involving Cu4S6 Clusters in Proteins, Inorg. Chem., 2018, 57, 5723–5731 CrossRef PubMed.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef.
- M. Łuczkowski, M. Stachura, V. Schirf, B. Demeler, L. Hemmingsen and V. L. Pecoraro, Design of Thiolate Rich Metal Binding Sites within a Peptidic Framework, Inorg. Chem., 2008, 47, 10875–10888 CrossRef PubMed.
- G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. O'Halloran, J. E. Penner-Hahn, W. F. DeGrado and V. L. Pecoraro, De Novo Design of Mercury-Binding Two- and Three-Helical Bundles, J. Am. Chem. Soc., 1997, 119, 6195–6196 CrossRef.
- S. Pires, J. Habjanič, M. Sezer, C. M. Soares, L. Hemmingsen and O. Iranzo, Design of a Peptidic Turn with High Affinity for HgII, Inorg. Chem., 2012, 51, 11339–11348 CrossRef PubMed.
- J. S. Casas and M. M. Jones, Mercury(II) complexes with sulfhydryl containing chelating agents: Stability constant inconsistencies and their resolution, J. Inorg. Nucl. Chem., 1980, 42, 99–102 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mt00113h |
| This journal is © The Royal Society of Chemistry 2018 |