
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined multivariate analysis and machine learning reveals a predictive module of metabolic stress response in Arabidopsis thaliana†
Lisa
Fürtauer‡
 a,
Alice
Pschenitschnigg‡
b,
Helene
Scharkosi‡
b,
Wolfram
Weckwerth
bc and
Thomas
Nägele
*a
a,
Alice
Pschenitschnigg‡
b,
Helene
Scharkosi‡
b,
Wolfram
Weckwerth
bc and
Thomas
Nägele
*a
aLudwig-Maximilians-Universität München, Department Biology I, Plant Evolutionary Cell Biology, Großhadernerstr. 2-4, D-82152 Planegg-Martinsried, Germany. E-mail: Thomas.Naegele@lmu.de; Fax: +49-89-2180-74661; Tel: +49-89-2180-74660
bDepartment of Ecogenomics and Systems Biology, University of Vienna, Vienna, Austria
cVienna Metabolomics Center, University of Vienna, Vienna, Austria
First published on 2nd November 2018
Abstract
Abiotic stress exposure of plants induces metabolic reprogramming which is tightly regulated by signalling cascades connecting transcriptional with translational and metabolic regulation. Complexity of such interconnected metabolic networks impedes the functional understanding of molecular plant stress response compromising the design of breeding strategies and biotechnological processes. Thus, defining a molecular network to enable the prediction of a plant's stress mode will improve the understanding of stress responsive biochemical regulation and will yield novel molecular targets for technological application. Arabidopsis wild type plants and two mutant lines with deficiency in sucrose or starch metabolism were grown under ambient and combined cold/high light stress conditions. Stress-induced dynamics of the primary metabolome and the proteome were quantified by mass spectrometry. Wild type data were used to train a machine learning algorithm to classify mutant lines under control and stress conditions. Multivariate analysis and classification identified a module consisting of 23 proteins enabling the reliable prediction of combined temperature/high light stress conditions. 18 of these 23 proteins displayed putative protein–protein interactions connecting transcriptional regulation with regulation of primary and secondary metabolism. The identified stress-responsive core module supports prediction of complex biochemical regulation under changing environmental conditions.
Introduction
The exposure of plants to abiotic stress conditions induces metabolic reprogramming to prevent chronic tissue damage and to stabilize growth and development. Plant stress tolerance and adaptation mechanisms represent multigenic and complex physiological traits.1 While significance of metabolic dynamics depends on stress type, intensity and duration, plant stress response can generally be subdivided into an early (alarm) phase, an acclimation phase and the maintenance phase of the newly acclimated metabolic homeostasis.2 Additionally, metabolic reprogramming affects various cellular processes on multiple molecular levels comprising regulation of transcriptional, translational and enzymatic processes.3 For example, the C-repeat binding factor (CBF) pathway of Arabidopsis plays a crucial role in freezing tolerance.4 Only minutes after transferring Arabidopsis plants from ambient to low temperature, CBF1, CBF2 and CBF3 genes are induced5 to alter the expression of more than hundred cold-regulated (COR) genes, also known as the CBF regulon.6 This results in an increase of freezing tolerance7 and recent work indicated extensive co-regulation of the CBF regulon by other transcription factors. This provides evidence for a complex and highly interconnected low-temperature regulatory network.8 Further, comparison of CBF regulon gene induction in natural Arabidopsis accessions with a differing freezing tolerance revealed a potential contribution of the CBF pathway to the adaptive evolution of those populations.9Stress-induced dynamics in transcriptional and translational processes significantly affect protein levels under stress conditions.10Vice versa, changes in protein concentrations, e.g. transcription factors, affect transcriptional and translational processes. Previous work has revealed stress responsive proteins, e.g. dehydrins11,12 and RNA-binding protein CP29 and GRP7,13–15 to strongly accumulate during exposure to low temperature. Stress-responsive proteins are distributed across various cell compartments involved in numerous signalling cascades, metabolic pathways of primary and secondary metabolism, protein folding, membrane stabilization, energy and redox regulation.16 Carbohydrates are primary photosynthetic products, thus playing a central role in energy metabolism, developmental processes and stress signalling. Starch and soluble sugars have been found to be tightly linked by the circadian clock ensuring a continuous carbohydrate availability.17,18 Further, sugars play a crucial role in entrainment of the circadian clock,19 and clock components have been found to be significantly influenced by abiotic stress conditions, e.g. low temperature.20 Previous work indicated an important role of starch degradation during initial response to cold stress augmenting hexose and raffinose accumulation.21 Yet, also in cold acclimated plants reprogramming of the starch degradation machinery was found to be a characteristic part of naturally occurring cold and freezing tolerance of Arabidopsis thaliana.22 Regulation of starch and sucrose metabolism affects carbon and energy metabolism on a whole plant level and, hence, its reprogramming is central to plant abiotic stress response. Starch biosynthesis is directly linked to the Calvin–Benson cycle by a sequential action of phosphoglucose isomerase (PGI), phosphoglucomutase (PGM1) and ADP-glucose pyrophosphorylase (AGPase) yielding ADP-glucose, the direct substrate for biosynthesis of the starch granule.23 Sucrose synthesis is regulated by sucrose–phosphate synthase (SPS) catalysing the cytosolic synthesis of sucrose-6-phosphate (S6P) from UDP-glucose and fructose-6-phosphate.24 Finally, sucrose phosphate phosphatase (SPP) releases inorganic phosphate from S6P, yielding sucrose (see Fig. S1, ESI†). Mutant lines, deficient in PGM1 (pgm1) or SPS (spsa1) activity, have previously been reported to be significantly affected not only in the central carbohydrate metabolism but also in metabolism of organic and amino acids.25 In starchless pgm1 plants, increased sugar concentrations were observed in root and shoot tissue and were accompanied by significantly reduced growth.26
Due to the pivotal role of sugars in plant stress response, analysing mutants with a deficiency in the central carbohydrate metabolism promises to unravel metabolic network components with a crucial role in stress tolerance. However, deriving patterns of stress-induced metabolic regulation from experimental data sets is challenging due to the vast amount of involved molecular and regulatory processes, affecting diverse transcripts, proteins and metabolites simultaneously. Particularly, due to the multidimensional output of high-throughput experiments, theoretical approaches are needed to support the detection of characteristic metabolic patterns and the generation of predictive models. Machine learning techniques are suitable to classify data based on patterns recognized in multivariate large-scale data sets.27 Typically, training data sets comprising predictor and response variables are used to generate a trained model which can be applied to predict structures and classes in a new and, hitherto, unknown data set. Recently, a machine learning method was developed to predict whether proteins localize to the apoplast independent of the presence of a signal peptide.28 Potentially, this method improves the accuracy of predicting whether plant pathogen-derived effectors localize still to the apoplast or entered already the plant cell. Other studies have applied machine learning techniques for stress phenotyping in plants from high-resolution images or the classification of proteomics data of field-grown potato cultivars.29,30 Conclusively, developing and applying machine learning techniques for classification, regression or clustering of biological systems represents a promising approach to derive conclusive metabolic patterns crucial to generate predictive models.
The present study aimed to identify central and predictive molecular components of plant metabolic stress response. Frequently, cold and high light occur simultaneously under natural conditions and extrapolation from each stress response to a combined one is not possible.31,32 Therefore, a combined abiotic stress treatment was chosen to induce stress response which potentially reflects a broad scenario of plant growth conditions. The interface between primary and secondary metabolism was expected to characteristically shape stress response across a wide range of metabolic states. Thus, stress-induced dynamics in protein and metabolite levels were recorded in wild type and mutant plants being affected in starch or sucrose biosynthesis. Multivariate statistics and machine learning techniques were combined to identify a molecular network enabling the prediction of metabolic stress response in metabolic mutants based on wild type data.
Materials and methods
Plant material
Plants of Arabidopsis thaliana, accession Columbia (Col-0), mutant line pgm1 (plastidial PGM, At5g51820, TAIRstock CS3092), and mutant line spsa1 (At5g20280, SALK line 148643C) were grown within a growth cabinet under controlled conditions (Conviron® Adaptis). Light intensity under control condition was 50 μmol m−2 s−1 in a 12/12 h day/night cycle. Relative air humidity was 60% and temperature was 22 °C during the day and 18 °C during the night. All plants were grown on soil which was composed of Einheitserde® ED63 and perlite. Plants were watered daily and fertilized once with NPK fertilization solution (WUXAL® Super; MANNA-Dünger, Ammerbuch). After 42 days, leaf rosettes were sampled after 3 hours in the light (= control samples). All other plants were transferred to 5 °C and 300 μmol m−2 s−1 for 72 hours. Like control samples, stress samples were collected after 3 hours in the light and immediately quenched in liquid nitrogen. Each sample consisted of a whole leaf rosette which was ground to a fine powder and lyophilized. Seven leaf rosettes were sampled for each genotype and condition.Chlorophyll fluorescence
To quantify the impact of stress exposure on the photosynthetic apparatus, parameters of chlorophyll fluorescence were recorded using a WALZ MINI-PAM II/B (Heinz Walz GmbH, Germany). Rapid light curves were recorded after 5 minutes of dark adaptation. Intensities of actinic illumination were increased as follows: 0, 24, 44, 64, 89, 124, 189, 284, 419, 630, 822, 1149, and 1498 μmol m−2 s−1.Starch quantification
Leaf starch content was quantified photometrically as described earlier.33 In brief, soluble sugars were extracted from ground plant material by adding 80% ethanol and heating to 80 °C for 30 minutes. The supernatant was discarded, and the pellet was used for starch quantification. Pellets were suspended in 0.5 M NaOH at 95 °C for 45 min. Following acidification with 1 M CH3COOH, the suspension was digested with amyloglucosidase for 2 h at 55 °C. Glucose content of the supernatant was quantified photometrically in a coupled glucose oxidase/peroxidase/dianisidine assay.SPS activity measurement
SPS activity was determined as described previously with slight modification.34 Leaf tissue was suspended in 50 mM Hepes/KOH (pH 7.5), 10 mM MgCl2, 1 mM EDTA, 2.5 mM dithiothreitol, 10% glycerol and 0.1% Triton X-100. Suspensions were centrifuged at 4 °C and SPS activity was assayed in the supernatant. The reaction buffer consisted of 50 mM Hepes/KOH (pH 7.5), 15 mM MgCl2, 2.5 mM dithiothreitol, 35 mM UDP-glucose, 35 mM fructose 6-phosphate and 140 mM glucose 6-phosphate, 30% KOH was added to the control of each assay. Reactions were incubated for 30 min at 25 °C, and then for 5 min at 95 °C. Anthrone 0.14% in H2SO4 was added, and the samples were incubated for 30 min at 40 °C. Extinction was determined photometrically at 620 nm.Metabolite analysis
Primary metabolites were semi-quantitatively analysed via gas chromatography coupled to time-of-flight mass spectrometry applying a previously published protocol with slight modifications.22,35 Metabolites were extracted twice with a methanol–chloroform–water mixture (MCW, 5/2/1, v/v/v) followed by an extraction step with 80% ethanol in which the samples were heated to 80 °C for 30 minutes. Addition of water to the MCW supernatant induced separation of polar and apolar phases. The polar phase was mixed with the ethanol extract and dried in a vacuum concentrator (ScanVac, LaboGene). The dried extracts were derivatized applying procedures of methoximation (methoxyamine hydrochloride in pyridine) and silylation (N-methyl-N-(trimethylsilyl)trifluoroacetamide). For methoximation, samples were incubated for 90 minutes at 30 °C. For silylation, samples were incubated for 30 minutes at 37 °C. Derivatized samples were transferred into glass vials and sealed with a crimp cap. GC-ToF-MS analysis was performed on an Agilent 6890 gas chromatograph (Agilent Technologies®, Santa Clara, USA) coupled to a LECO Pegasus® GCxGC-TOF mass spectrometer (LECO Corporation, St. Joseph, USA). Compounds were separated on an Agilent column HP5MS (length: 30 m, diameter: 0.25 mm, film: 0.25 μm). Deconvolution of the total ion chromatogram and peak integration was performed using the software LECO Chromatof®. Compounds were manually annotated using retention index information and mass spectrum comparison to the GMD database36 with a minimum match factor of 700. Retention index information was derived from even numbered analytical alkane standard solution dissolved in hexane (C10–C40; Sigma-Aldrich). Additionally, all compounds were confirmed by and normalized to the area of a pure standard calibration mix peak (Table SI, ESI†).Proteome analysis
Plant material was solubilized in 8 M urea, 50 mM Hepes/KOH (pH = 7.8) on ice. Samples were precipitated in acetone with 0.5% beta-mercaptoethanol. Pellets were washed 2 times with methanol and acetone, and afterwards again solubilized in 8 M urea, 50 mM Hepes/KOH (pH 7.8). Protein concentration was determined via Bio-Rad Bradford-assay using BSA as standard (Bio-Rad, USA). Equal amounts of protein (15 μg) were reduced with dithiothreitol (DTT) at a concentration of 5 mM for 45 minutes at 37 °C and alkylated at a concentration of 10 mM with iodacetamid (IAA) and dark incubated for 60 minutes at 23 °C. Alkylation was stopped by increasing DTT concentration to 10 mM and dark incubated for 15 minutes. Samples were diluted 2-fold by 20% acetonitrile (ACN) and 100 mM ammonium bicarbonate (AmBic), proteins were predigested with Lys-C (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 w:w; Sigma-Aldrich, USA) at 30 °C for 2.5 hours dark. Samples were diluted 2-fold to 2 M urea by 10% ACN, 25 mM AmBic, 10 mM CaCl2 and digested with sequencing grade modified trypsin (poroszyme, immobilized trypsin; 1:100 v:w) for 12 hours. Digested proteins were acidified with formic acid (pH ∼ 3.0) and desalted with C18 materials (Agilent, Bond Elut SPEC) and dried in a vacuum concentrator (ScanVac, LaboGene). Peptides were dissolved in 2% ACN, 0.1% formic acid and the same amount of total protein was loaded and separated on a PepMap RSLC 75 μm, 50 cm column (Thermo Fisher Scientific Inc., Waltham, USA). The flowrate was set to 300 nl min−1 with 2% to 40% in 120 minutes of mobile phase B (mobile phase A: 0.1% formic acid (FA) in water [v/v]; mobile phase B: 0.1% FA in 90% ACN [v/v]). The run ended with 60 minutes of equilibrium. Subsequently, mobile phase B was increased from 40% to 90% within 1 minute and held stable for 10 minutes, followed by decreased mobile phase B from 90% to 2%. MS analysis was performed with an Orbitrap Elite instrument (Thermo Fisher Scientific Inc., Waltham, USA) in positive mode and full scan in FT with a resolution of 60000 in profile mode. Precursor masses ranged between 360–1800 m/z. MS/MS analysis was done in the linear ion trap with CID fragmentation and rapid scan mode for the 20 most intense ions with a minimal signal threshold of 500 counts. Prediction of ion injection time was enabled (5 × 102 ions for up to 10 ms). Dynamic exclusion was enabled with repeat count 1 and repeat duration of 30 seconds. Exclusion list size was set to 500 and exclusion duration to 30 seconds. Excluded mass was set to ±10 ppm relative to reference mass, early expiration enabled with 1 count and the s/n threshold was 2.0.
:1000 w:w; Sigma-Aldrich, USA) at 30 °C for 2.5 hours dark. Samples were diluted 2-fold to 2 M urea by 10% ACN, 25 mM AmBic, 10 mM CaCl2 and digested with sequencing grade modified trypsin (poroszyme, immobilized trypsin; 1:100 v:w) for 12 hours. Digested proteins were acidified with formic acid (pH ∼ 3.0) and desalted with C18 materials (Agilent, Bond Elut SPEC) and dried in a vacuum concentrator (ScanVac, LaboGene). Peptides were dissolved in 2% ACN, 0.1% formic acid and the same amount of total protein was loaded and separated on a PepMap RSLC 75 μm, 50 cm column (Thermo Fisher Scientific Inc., Waltham, USA). The flowrate was set to 300 nl min−1 with 2% to 40% in 120 minutes of mobile phase B (mobile phase A: 0.1% formic acid (FA) in water [v/v]; mobile phase B: 0.1% FA in 90% ACN [v/v]). The run ended with 60 minutes of equilibrium. Subsequently, mobile phase B was increased from 40% to 90% within 1 minute and held stable for 10 minutes, followed by decreased mobile phase B from 90% to 2%. MS analysis was performed with an Orbitrap Elite instrument (Thermo Fisher Scientific Inc., Waltham, USA) in positive mode and full scan in FT with a resolution of 60000 in profile mode. Precursor masses ranged between 360–1800 m/z. MS/MS analysis was done in the linear ion trap with CID fragmentation and rapid scan mode for the 20 most intense ions with a minimal signal threshold of 500 counts. Prediction of ion injection time was enabled (5 × 102 ions for up to 10 ms). Dynamic exclusion was enabled with repeat count 1 and repeat duration of 30 seconds. Exclusion list size was set to 500 and exclusion duration to 30 seconds. Excluded mass was set to ±10 ppm relative to reference mass, early expiration enabled with 1 count and the s/n threshold was 2.0.
Peptide identification and protein quantification was performed with MaxQuant (http://www.maxquant.org) and implemented algorithms of version 1.5.5.137 against the TAIR10 (http://www.arabidopsis.org) protein database.38 A maximum of 2 missed cleavages was applied. Maximally 5 variable modifications per peptide were allowed for N-terminal acetylation and methionine oxidation. Carbamidomethylation was set as a fixed modification (due to previous methylation). For identification a minimum of 2 peptides and 2 minimum razor + unique peptides were requested.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE39 partner repository with the dataset identifier PXD010580.
Data analysis
Statistical analysis (Pearson correlation, ANOVA, PCA, Clustering) was performed in R (The R Project for Statistical Computing; http://www.r-project.org/).40 For statistical analysis, outliers were removed, and missing values were filled by mean values. Venn diagrams were created using the online tool Venny 2.1.41 Data classification by supervised machine learning was performed in MATLAB® (R2017a, 9.2.0.538062) (http://www.mathworks.com) applying the Classification Learner App. First, classification of a data set comprising all metabolite and protein levels of Col-0, spsa1 and pgm1 under both control and stress conditions was performed applying diverse classifiers: (i) decision tree classifier, (ii) support vector machines (SVMs) and (iii) nearest neighbour classifiers. To prevent overfitting, model training was performed with 25% holdout validation. False negative rates of decision tree classifiers were 13% for control samples while true positive rates were 100% for stress samples. SVMs with linear, quadratic, cubic and radial basis function kernels displayed 100% true positive rates for both control and stress samples. Applying nearest neighbour classifiers resulted in a false negative rate of 17% for stress samples and 100% true positive rates for control samples. Classification training was performed using the wild type (Col-0) data set as described in Table 2 (first column) for SVM, decision tree and nearest neighbour classification. The trained wild type model was exported into the MATLAB® workspace and applied to predict the classification of control and stress samples of pgm1 and spsa1 mutants.Results
Stress-induced effects on chlorophyll fluorescence parameters
The exposure of wild type and mutant plants to combined cold and high light stress resulted in a decreased effective photochemical quantum yield of PSII, ϕPSII (Fig. 1). This effect significantly differed between control and stress condition (ANOVA, p < 0.001) and was similar in all genotypes. ϕPSII measures the proportion of light absorbed by chlorophyll associated with PSII being used in photochemistry, thus being an indicator of overall photosynthesis.42 Together with significant condition-dependent effects on coefficients of photochemical (qP) and non-photochemical quenching (qN; Fig. S2, ESI†) these observations indicated a similar stress-induced effect on photobiochemistry in all genotypes. | ||
| Fig. 1 Effective photochemical quantum yield of PSII, ϕPSII, as a function of actinic illumination intensity. ϕPSII was measured in (A) Col-0, (B) pgm1, and (C) spsa1 under control (green) and cold/high light stress (blue) conditions with an increasing actinic light intensity (PAR; n = 4). Conditions significantly differed within all three genotypes (ANOVA, p < 0.001). | ||
Deficiency of phosphoglucomutase affects the capacity of sucrose biosynthesis
Under ambient conditions, maximum sucrose phosphate synthase activity was significantly elevated in the plastidial pgm1 mutant line compared to Col-0 (Fig. 2A). Under stress, SPS activity of the wild type slightly increased while it decreased significantly in pgm1 (ANOVA, p < 0.01). Compared to Col-0, stress exposure resulted in lower SPS activity in pgm1 while SPS activity in spsa1 was 10–20% of the wild type under both conditions (Fig. 2A; for comparison of SPSA1 protein levels see Fig. S3, ESI†).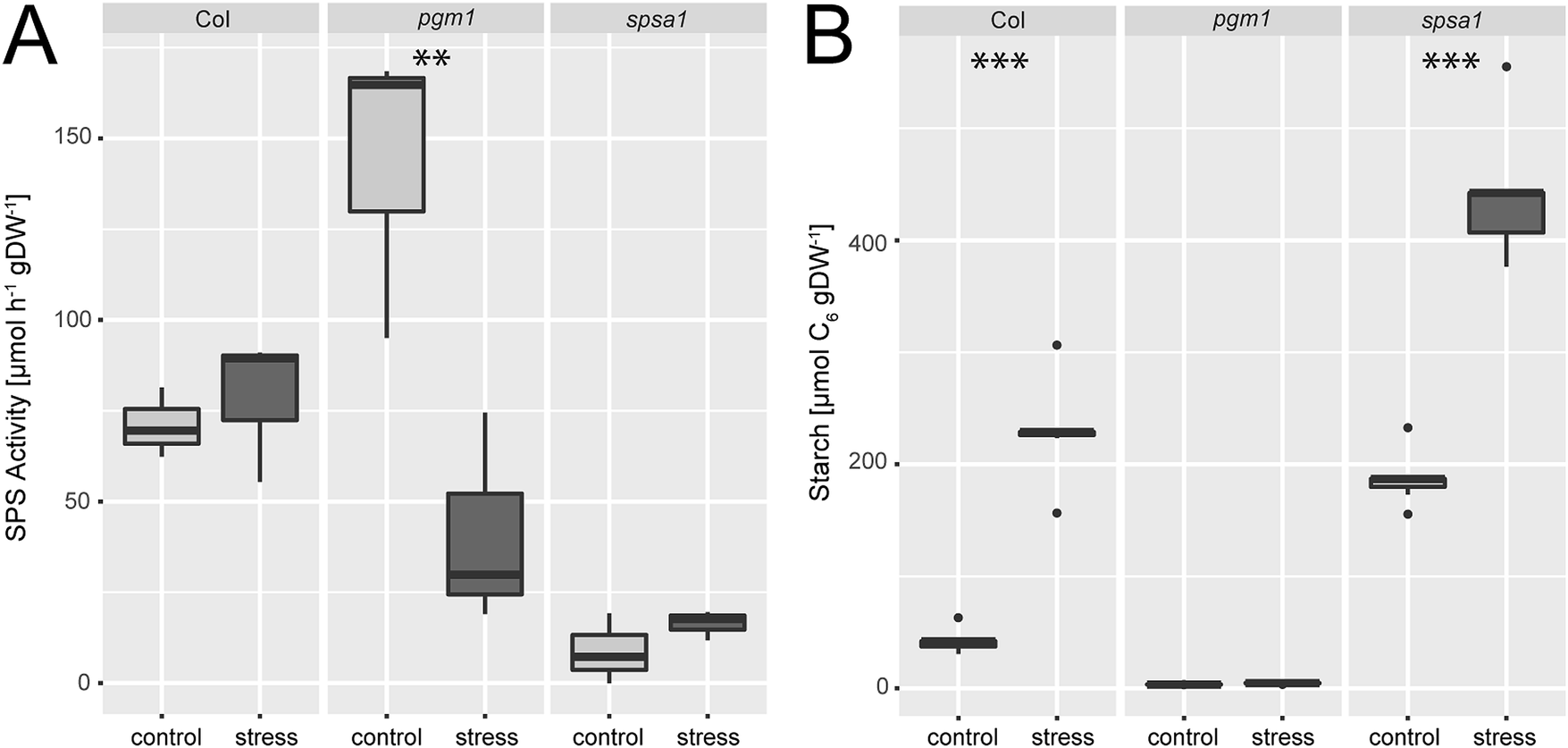 | ||
| Fig. 2 (A) SPS activity and (B) starch levels under control and combined cold/high light stress. Asterisks indicate significant differences within the genotypes revealed by an ANOVA. **p < 0.01; ***p < 0.001 (SPS activity: n = 3; starch: n = 5). | ||
Starch levels of pgm1 were less than 5% of Col-0 under both analysed conditions (Fig. 2B) while Col-0 and spsa1 displayed a significant stress-induced increase of starch levels (ANOVA, p < 0.001). Under both ambient and stress conditions starch levels were highest in spsa1. Starch levels of stressed spsa1 plants were almost twice as high as in Col-0 (p < 0.001).
Pearson correlation of SPS activity, starch levels and concentrations of primary metabolites across all genotypes and conditions revealed a significantly positive correlation of SPS activity with sucrose concentration (Fig. 3; p < 0.05). Additionally, SPS activity was negatively correlated to levels of starch, serine, malate and proline (Fig. 3). Further, starch was negatively correlated with sucrose concentration while it showed positive correlation with central compounds at the interface of carbon/nitrogen metabolism, e.g. glutamate, glutamine, glycine and serine. Strongest correlation of TCA cycle intermediates was observed between citrate, succinate and malate while fumarate was only weakly correlated with succinate (Fig. 3). Interestingly, raffinose was found to positively correlate with its metabolic precursor galactinol, yet not sucrose, pointing to a substrate limitation of raffinose biosynthesis by galactinol biosynthesis.43 Further, raffinose concentration was negatively correlated (p < 0.05) with pyruvate concentration which might indicate a trade-off between synthesis of stress protective substances and energy metabolism.
 | ||
| Fig. 3 Pearson correlation of primary metabolites, starch and SPS activity across all genotypes and conditions. Correlation coefficients are indicated by the colour (blue: positive correlation; red: negative correlation). Only significant correlations (p < 0.05) were colour-coded, blank fields indicate non-significant correlations. | ||
Stress-induced dynamics of primary metabolites and proteome
Principal component analysis (PCA) revealed significant stress-induced dynamics in primary metabolism (Fig. 4A). Principle component 1 (PC1), which explains most of the variance (here 44%), was found to separate control from stress samples in the genotypes Col-0 and spsa1 (Fig. 4A). This separation was significant for spsa1 (p < 0.05) while 95% confidence ellipses slightly overlapped in Col-0. Control and stress samples of pgm1 were significantly separated on a diagonal of PC1 and PC2 (Fig. 4A). Associated loadings, i.e. metabolites, showing a significant increase during stress in Col-0 and spsa1 but staying constant in pgm1 were citrate, succinate, threonine, glutamate and starch (Fig. 2B and 5A–D). Pyruvate was the only metabolite which decreased in its concentration during stress in Col-0 (p = 0.07) but was constant in both mutant lines (Fig. 5E). While sucrose decreased in pgm1, it slightly increased in Col-0 and spsa1 (Fig. 5F). Further, glucose and fructose concentrations increased significantly in Col-0 and spsa1 except for pgm1 where it decreased (Fig. 5G and H). Similar dynamics were observed for maltose and asparagine (Fig. 5K and M). Under stress, myo-inositol significantly decreased in pgm1 while it increased in spsa1 and did not change in Col-0 (Fig. 5N). | ||
| Fig. 4 Statistical analysis of the (A) primary metabolome and the (B) proteome. (A) Colours and symbols indicate genotypes and conditions as indicated in the legend. Significance is indicated by 95% confidence ellipses. (B) Euclidean distance clustering of protein mean values (n = 3). For clustering, mean values were scaled to zero mean and unit variance. | ||
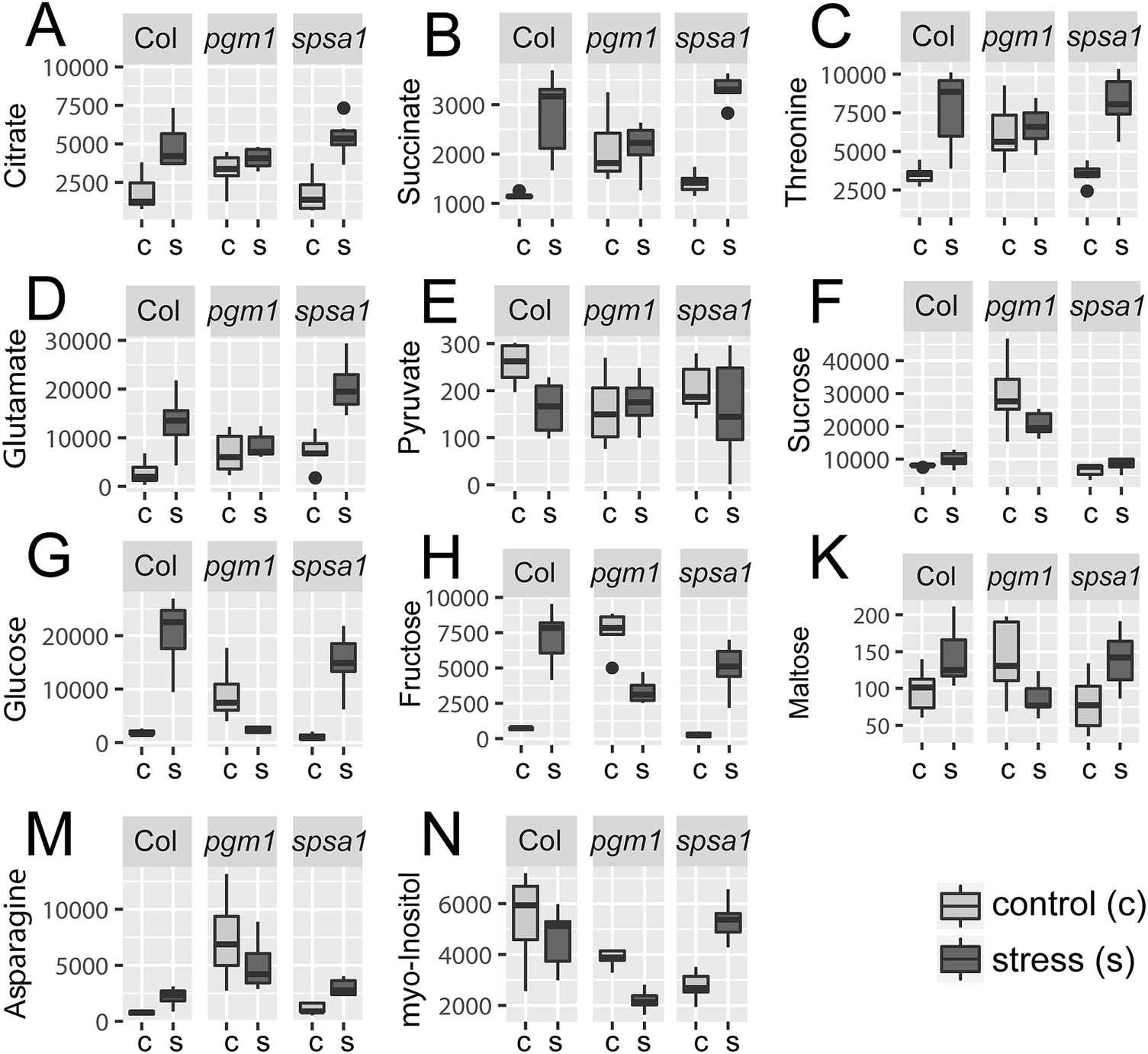 | ||
| Fig. 5 Stress-induced reprogramming of the primary metabolome. Concentration of metabolites under control (c) and stress (s) conditions are summarized genotype-wise as indicated on the top of each sub-figure. All levels are given in [a.u. gDW−1]. Control samples are shown in light grey, stress samples in dark grey (n = 7). Only a selection of metabolites is shown which increase significantly in all genotypes but stay constant in pgm1 (A–D), decrease only in Col-0 (E), increase in all genotypes but decrease in pgm1 (F–N). | ||
The Euclidean distance-based clustering of mean protein levels revealed a similar protein constitution in Col-0 and spsa1 under control conditions (Fig. 4B). In pgm1, mean protein levels under control condition clustered together with the pgm1 stress condition and was separated from the stress response in Col-0 and spsa1. Genotype-wise comparison of stress-induced significant changes in the proteome (p < 0.05) revealed a much higher number of changing proteins in Col-0 (199) and spsa1 (120) than in pgm1 (46) (Fig. 6). The number of genotype-specific and uniquely changing proteins was lowest in pgm1 (15) followed by spsa1 (58) and Col-0 (141). Further, 23 proteins (out of 1644 proteins) were observed to be significantly reprogrammed in all genotypes (see Fig. 6) indicating a central stress-responsive set of proteins which seemed to respond independently of the cellular carbohydrate status. Levels of all 23 proteins were significantly increased under stress condition in all genotypes while none of them decreased (ANOVA p < 0.05; see Fig. S4, ESI†).
 | ||
| Fig. 6 Stress-induced reprogramming of the proteome. The Venn diagram comprises only significantly changed proteins due to stress treatment (ANOVA, p < 0.05). Col-0: blue; pgm1: yellow; spsa1: green. | ||
To analyse whether this set of proteins potentially constituted a stress-induced interaction and/or signalling network, information about potential protein–protein interactions were derived from the STRING database (https://string-db.org/; Minimum interaction score: 0.4 (medium confidence);44 (Table 1)). Interaction analysis revealed a protein–protein interaction network comprising 18 out of the 23 selected proteins. The interaction network comprised well-known cold-induced proteins like COR15b and COR78 being part of the CBF regulon. Interestingly, both COR proteins showed potential interaction with an unknown transmembrane protein predicted to be located in the ER or in the extracellular region (AT1G16850). Further, both COR proteins showed interaction with the ABA and cold-inducible protein KIN1, a potential anti-freeze protein. Beyond molecular chaperones in mitochondria (mtHsc70-1; AT4G37910) and plastids (CLPB3; AT5G15450), also delta 1-pyrroline-5-carboxylate synthase 2 (P5CS2), involved in proline biosynthesis, was part of the identified stress-responsive network with potential interaction with COR78. Further proteins of the network were a central part of carbohydrate metabolism (RHM1; SUS1 and MIPS1) or belonged to the interface of primary and secondary metabolism, e.g. phenylalanine ammonia-lyase 1 and 2, flavanone 3-hydroxylase and chalcone synthase/isomerase family proteins (PAL1 and PAL2, F3H, TT4, TT5; see Table 2). Finally, two plastidial RNA-binding proteins, CP29 and AT2G37220, were identified as potential protein–protein interaction partners. Proteins which were part of the genotype-independent stress-responsive proteome but did not show any potential interaction within this set were CCR2 (AT2G21660), fibrillin precursor protein (FIB1A, AT4G04020), ferritin 1 (FER1, AT5G01600), glycine-rich RNA-binding protein 4 (GRP4, AT3G23830) and SVR3 (AT5G13650) which is involved in the elongation process during protein biosynthesis.
| AGI | Synonym | Function | Subcellular localization | (Potential) interaction with | Processes involved in |
|---|---|---|---|---|---|
| AT5G52310 | COR78 | Low-temperature-induced 78 | Nucleus | KIN1 | Cold stress response/has cis-acting regulatory elements that can impart cold-regulated gene expression |
| LTI78 | COR15B | ||||
| LTI140 | AT1G16850 | ||||
| RD29A | P5CS2 | ||||
| ADH1 | |||||
| AT2G42530 | COR15B | Cold regulated 15b | Chloroplast | COR78 | Cold stress response/protects chloroplast membranes during freezing |
| KIN1 | |||||
| AT1G16850 | |||||
| AT5G15960 | KIN1 | Stress-induced protein (KIN1) | Cytosol | COR78 | Hormone metabolism/ABA |
| COR15B | Cold and ABA inducible protein kin1/possibly functions as an anti-freeze protein. Transcript level of this gene is induced by cold, ABA, dehydration and osmoticum (mannitol). | ||||
| AT1G16850 | x | Unknown protein | Endoplasmic reticulum/extracellular region | COR15B | Not assigned/response to salt stress |
| Transmembrane protein | COR78 | ||||
| AT3G55610 | P5CS2 | Delta 1-pyrroline-5-carboxylate synthase 2 | Cytosol/chloroplast/mitochondria/plasmodesma | COR78 | Amino acid metabolism/gene expression is induced by dehydration, high salt and ABA |
| mtHsc70-1 | |||||
| CLPB3 | |||||
| PAL2 | |||||
| ADH1 | |||||
| PAL1 | |||||
| AT4G37910 | mtHsc70-1 | Mitochondrial heat shock protein 70-1 | Mitochondrion/cell wall/vacuolar membrane | CLPB3 | Heat stress response/mitochondrial heat shock protein |
| P5CS2 | |||||
| AT5G15450 | CLPB3 | Casein lytic proteinase B3 | Chloroplast (stroma)/cytoplasm | mtHsc70-1 | Heat stress response/functions as a molecular chaperone/involved in plastid differentiation mediating internal thylakoid membrane formation/conferring thermotolerance to chloroplasts during heat stress |
| CLPB-P | P5CS2 | ||||
| APG6 | |||||
| ATCLPB3 | |||||
| AT1G77120 | ADH1 | Alcohol dehydrogenase 1 | Cytosol/nucleus/plasma membrane | F3H | Cellular respiration/oxidation–reduction process/positive regulation of cellular response to hypoxia/response to abscisic acid |
| ATADH | COR78 | ||||
| P5CS2 | |||||
| SUS1 | |||||
| TT4 | |||||
| TT5 | |||||
| AT2G37040 | PAL1 | Phenylalanine ammonia-lyase 1 | Cytosol | F3H | L-Phenylalanine catabolic process/cinnamic acid biosynthetic process/defense response, drought recovery/lignin catabolic process |
| ATPAL1 | P5CS2 | ||||
| PHE ammonia lyase 1 | PAL2 | ||||
| TT4 | |||||
| TT5 | |||||
| AT3G53260 | PAL2 | Phenylalanine ammonia-lyase 2 | Cytosol | P5CS2 | Secondary metabolism/phenylpropanoids/lignin synthesis |
| ATPAL2 | |||||
| F3H | |||||
| TT4 | |||||
| PAL1 | |||||
| AT3G51240 | F3H | Flavanone 3-hydroxylase | Cytosol | TT4 | Secondary metabolism/flavonoids/dihydroflavonols/regulates flavonoid biosynthesis |
| TT6 | TT5 | ||||
| PAL2 | |||||
| RHM1 | |||||
| ADH1 | |||||
| PAL1 | |||||
| AT5G13930 | TT4 | Chalcone and stilbene synthase family protein | Endoplasmic reticulum/nucleus/cytoplasm | F3H | Secondary metabolism/flavonoids/chalcones, encodes chalcone synthase (CHS), a key enzyme involved in the biosynthesis of flavonoids/required for the accumulation of purple anthocyanins in leaves and stems. |
| CHS | PAL2 | ||||
| ATCHS | TT5 | ||||
| RHM1 | |||||
| ADH1 | |||||
| PAL1 | |||||
| AT3G55120 | TT5 | Chalcone–flavanone isomerase family protein | Endoplasmic reticulum/nucleus/chloroplast | TT4 | Secondary metabolism/flavonoids/chalcones, catalyzes the conversion of chalcones into flavanones/required for the accumulation of purple anthocyanins in leaves and stems. Co-expressed with CHS. |
| A11 | F3H | ||||
| CFI | ADH1 | ||||
| CHI | PAL1 | ||||
| ATCHI | |||||
| AT1G78570 | RHM1 | Rhamnose biosynthesis 1 | Cytosol/chloroplast/plasmodesma | F3H | Cell wall/precursor synthesis/UDP-glucose 4,6-dehydratase/encodes a UDP-L-rhamnose synthase involved in the biosynthesis of rhamnose, a major monosaccharide component of pectin. |
| ROL1 | TT4 | ||||
| ATRHM1 | SUS1 | ||||
| AT5G20830 | SUS1 | Sucrose synthase 1 | Cytosol | RHM1 | Major CHO metabolism/UDP-glycosyltransferase activity/sucrose synthase activity |
| ASUS1 | MIPS1 | ||||
| ATSUS1 | ADH1 | ||||
| AT4G39800 | MIPS1 | Myo-inositol-1-phosphate synthase 1 | Cytosol | SUS1 | Minor CHO metabolism/myo-inositol/InsP synthases |
| ATMIPS1 | |||||
| ATIPS1 | Myo-inositol-3-phosphate synthase 1 | ||||
| MI-1-P SYNTHASE | |||||
| AT3G53460 | CP29 | Chloroplast RNA-binding protein 29 | Chloroplast | AT2G37220 | Regulation of transcription/RNA binding/encodes a nuclear gene with a consensus RNA-binding domain that is localized to the chloroplast |
| AT2G37220 | x | RNA-binding (RRM/RBD/RNP motifs) family protein | Chloroplast | CP29 | RNA binding/encodes a chloroplast RNA binding protein. |
| AT3G23830 | GRP4 | Glycine-rich RNA-binding protein 4 | Mitochondria | — | Response to cold/response to osmotic stress/response to salt stress/response to water deprivation |
| RBGA4 | |||||
| AT2G21660 | CCR2 | Cold, circadian rhythm, and RNA binding 2 | Nucleus/cytosol/peroxisome/chloroplast | — | RNA binding/encodes a small glycine-rich RNA binding protein that is part of a negative-feedback loop through which AtGRP7 regulates the circadian oscillations of its own transcript/gene expression is induced by cold. |
| ATGRP7 | |||||
| GR-RBP7 | |||||
| GRP7 | |||||
| RBGA3 | |||||
| AT4G04020 | FIB1A | Fibrillin precursor protein | Chloroplast/stroma | — | Cell organisation/fibrillin precursor protein. The fibrillin preprotein, but not the mature protein, interacts with ABI2/regulated by ABA response regulators/involved in ABA-mediated photoprotection. |
| FIB | |||||
| PGL35 | |||||
| AT5G01600 | FER1 | Ferritin 1 | Chloroplast | — | Metal handling, binding/chelation/storage ferric iron/iron binding. |
| ATFER1 | Encodes a ferritin protein that is targeted to the chloroplast. | ||||
| AT5G13650 | SVR3 | Elongation factor family protein/suppressor of variegation | Chloroplast/plasma membrane | — | Protein synthesis/elongation/encodes SVR3, a putative chloroplast TypA translation elongation GTPase. |
| Exp. training data | pgm1 | spsa1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lSVM | qSVM | cSVM | Decision tree | Nearest neighbour | lSVM | qSVM | cSVM | Decision tree | Nearest neighbour | |
| (A) Col-0, full metabolome | 67 | 87 | 73 | 87 | 60 | 100 | 100 | 100 | 100 | 100 |
| (B) Col-0, metabolome, starch excluded | 73 | 67 | 67 | 87 | 60 | 100 | 100 | 100 | 100 | 100 |
| (C) Col-0, metabolome, Suc excluded | 67 | 80 | 73 | 87 | 73 | 100 | 100 | 100 | 100 | 100 |
| (D) Col-0, metabolome, Hex excluded | 80 | 87 | 87 | 87 | 60 | 100 | 100 | 100 | 100 | 100 |
| (E) Col-0, metabolome, maltose excluded | 87 | 87 | 67 | 87 | 60 | 100 | 100 | 100 | 100 | 100 |
| (F) Col-0, metabolome, Suc/Hex excluded | 87 | 87 | 87 | 87 | 87 | 100 | 100 | 100 | 100 | 100 |
| (G) Col-0, full proteome (1644 proteins) | 67 | 100 | 100 | 50 | 100 | 100 | 100 | 100 | 83 | 83 |
| (H) Col-0, stress-responsive core proteome (23 proteins) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| (I) Col-0, stress-responsive core proteome (23 proteins) + full metabolome | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
Predicting stress response in metabolic mutants
Classification of metabolic stress response was analysed using decision tree classifiers, support vector machines (SVMs) and nearest neighbour classifiers. Models were trained using experimental metabolite and protein data of Col-0. Subsequently, trained models were applied to experimental data of pgm1 and spsa1 to predict whether sample data belong to the class “control” or “stress” (Table 2).Using experimental training data of Col-0 allowed for the accurate prediction of control and stress samples in spsa1 mutants. This was particularly true for SVM classification. None of the classifiers predicted stress state of pgm1 correctly if metabolite concentrations were applied (Table 2, A–F). Prediction of condition in pgm1 based on the primary metabolome including starch resulted in maximally 87% accuracy. To test the effect of starch dynamics on prediction accuracy in pgm1, training and prediction was performed excluding starch levels (Table 2, B). However, prediction accuracy of pgm1 samples could not be rescued to 100% as it was observed for spsa1. Next, sucrose, hexoses and maltose were excluded separately (Table 2, C–E) and in combination with each other from training and prediction due to their significantly inverse stress dynamics in pgm1 compared to all other genotypes (see Fig. 5). Excluding both sucrose and hexoses from the data sets most efficiently raised the prediction accuracy from 67% to 87% across all applied SVM kernel functions (Table 2, F).
Applying SVM classification of the full proteome information, prediction accuracy was similar in pgm1 and spsa1 (Table 2, G–I). Except for the linear SVM kernel function, where prediction accuracy of pgm1 was still at 67%, i.e. 2 out of 3 samples were predicted correctly (Table 2, G lSVM), all samples were classified correctly by the quadratic and cubic SVM kernel function. In contrast, decision tree and nearest neighbour classification of the proteome was less accurate and yielded results in the range of 50–100% for pgm1 and spsa1. Using only the set of 23 genotype-independent stress responsive proteins a predictive model with 100% accuracy was curated across all classifiers, kernel functions and genotypes (Table 2, H). Finally, using this set of proteins together with the full metabolome information increased the predictability of pgm1 samples to 100% (Table 2, I).
Discussion
Capacity of sucrose biosynthesis is affected by plastidial phosphoglucomutase during the initial light phase
Starch and sucrose represent two major products of photosynthetic carbon assimilation. Regulation of their metabolism crucially affects carbon partitioning and carbon balance on a whole-plant level.45 Thus, perturbing biosynthetic pathways of both carbohydrates, i.e. by T-DNA insertion, results in dramatic alteration of photosynthesis, metabolite concentrations and stress tolerance.21,46 In the present study, metabolomic and proteomic consequences of altered sucrose and starch metabolism were analysed during the early day phase, i.e. after 3 hours in the light. Previous studies have shown strong dynamics of carbohydrate metabolism during this day period (see e.g.ref. 26 and 47). Hence, analysis at such an early point of time in the day promises to reveal the role of sucrose and starch metabolism in establishing a daily equilibrium. While the applied stress combination of increased light intensity and decreased temperature cannot resolve the individual stress response, it provides insight into the synergistic stress response which might not be predictable by single stress application.48 Chlorophyll fluorescence parameters revealed a significant stress effect (ANOVA, p < 0.001) on the effective photochemical quantum yield as well as on photochemical and non-photochemical quenching, yet no significant genotype-effect was detected (Fig. 1). This implies a similar constitution of photosystems in all genotypes and allows for an interpretation of observed differences in metabolism independent of (potential) photosystem damage.Deficiency of plastidial phosphoglucomutase, resulting in starch-deficient plants (<5% of wild type, Fig. 2B), has been shown in many previous studies to enhance diurnal and stress-induced dynamics of soluble carbohydrates due to a loss of buffering capacity for carbon acquisition rates affecting both source and sink tissue.26 However, maximum capacity of sucrose biosynthesis (vmax,SPS) in leaf tissue of Arabidopsis was not found to correlate with increased sucrose concentrations, neither before nor after cold acclimation.25 In contrast, SPS activity correlated weakly, yet significantly, positive with sucrose concentrations in the present study (Fig. 3). Already under control conditions a significantly increased vmax of SPS was observed in pgm1 plants (Fig. 2A). Similarly, previous analyses indicated slightly increased SPS activity in pgm1 compared to Col-0 under 12/12 day/night growth conditions.47 However, in the present study the difference to Col-0 was more pronounced which might be due to a different light regime. Here, plants were grown at PAR 50 μmol m−2 s−1 while Gibon and co-workers applied a light regime which was almost threefold higher (140 μmol m−2 s−1). Also, on the protein level, a significant increase of SPSA1 protein was observed in pgm1 mutants under control conditions (Fig. S3, ESI†), indicating that activity reflected the protein level of the SPS enzyme. Interestingly, activity under combined stress decreased in the pgm1 mutant while protein levels increased, pointing to an inactivation by phosphorylation.49–51 While the exact mechanism of SPS inactivation remains speculative in the present study, it seems probable that the interplay of cytosolic protein kinase/phosphatase activity and SPS is differentially regulated in pgm1 compared to Col-0.
A central core proteome is sufficient for accurate predictions of plant growth conditions using machine learning techniques
Identification of patterns in experimental data sets suitable for prediction of growth conditions, developmental stages or stress tolerance is a central aim of quantitative plant biology. Such patterns potentially reveal conserved molecular mechanisms and molecular interaction networks fundamental for complex cellular organisation. Promisingly, machine learning techniques enable such pattern recognition in large, heterogeneous and multidimensional data sets, hence being suitable for biological data integration.52 Using support vector machines trained by a Col-0 data set, control and stress conditions of spsa1 plants were predictable with high accuracy. For pgm1, predictability of metabolome data from the Col-0 model was weak (∼67% accuracy) due to the strong deviation from patterns observed in Col-0 (see Fig. 4 and 5). Yet, when combining the metabolome with a statistically selected subset of the proteome which was observed to be stress-induced independent of the genotype (see Table 1), conditions were predicted correctly by all classifiers. Beyond, prediction accuracy was independent of the metabolome information if the selected proteome subset, described in Table 1, was applied for training. This indicated that the proteome subset dominantly affected the prediction accuracy (see Table 2, H and I). Predictions based solely on the proteome resulted in much higher accuracy of predictions than predictions based on metabolomics data. Together with multivariate analysis of metabolomics and proteomics data this indicates that stress-induced reprogramming of the pgm1 proteome occurs in higher similarity to Col-0 and spsa1 than reprogramming of the metabolome. Previous findings on cold response of pgm1 revealed a similar multivariate pattern of integrated metabolite/protein data after 3 days at 4 °C.53 In this study, the authors detected metabolic processes which distinguish genotype- and temperature-specific effects.Conclusively, these findings suggest proteomics data can help enable high-accuracy predictions of growth conditions with a varying light intensity and temperature regime. In particular, the identified stress core proteome, resulting from the overlap of all significantly changed proteins across all genotypes (see Fig. 6), strongly contributed to these predictions and was sufficient to increase prediction accuracy of the metabolome from 67% to 100%. The resulting putative protein–protein interaction network comprised several well-described stress responsive proteins, e.g. COR15b, COR78 or molecular chaperones like CLPB3.54–56 Proteins with most interactions were COR78 (n = 5), P5CS2 (delta 1-pyrroline-5-carboxylate synthase 2, n = 6), F3H (n = 6) and TT4 (n = 6), PAL1 (n = 5), ADH1 (n = 6). P5CS2 is involved in biosynthesis of proline which is well-known to accumulate during stress response in many plant species playing diverse roles in signalling, cryoprotection and redox balance.57 F3H, a flavanone 3-hydroxylase, hydroxylates naringenin to form dihydrokaempferol which can further be hydroxylated to form dihydroquercetin, the substrates for flavonol and anthocyanin biosynthesis.58 TT4, a chalcone and stilbene synthase family protein, and TT5, a chalcone–flavanone isomerase family protein, catalyse the biosynthesis of naringenin and, hence, provide the substrate for F3H enzymes.58 Previously, mathematical modelling of metabolomics data identified biochemical reactions being strongly involved in metabolic reprogramming during simultaneous application of cold and light stress.59 One of the most significantly reprogrammed reactions in this study was the entry point of flavonoid and anthocyanin biosynthesis being directly related to PAL1/PAL2 abundance and activity.59,60 In the present study, several additional proteins which are involved in this pathway were identified indicating the central role of its regulation during abiotic stress response. Furthermore, a predicted potential interaction between PAL1, PAL2 and P5CS2 establishes an interface between primary and secondary metabolism and indicates how redox balance might affect stress-induced branches of secondary metabolism.
An additional branch of the genotype-independent stress-responsive protein network comprised steps of central carbohydrate metabolism (MIPS1, SUS1) and cell wall synthesis (RHM1). MIPS1, a myo-inositol-1-phosphate synthase, catalyses the limiting step of inositol biosynthesis and in response to stress, the transcription of MIPS1 is induced promoting the biosynthesis of inositol and derivatives. Previously, the light signalling protein FAR-RED ELONGATED HYPOCOTYL3 (FHY3) and its homolog FAR-RED IMPAIRED RESPONSE1 (FAR1) were shown to regulate light-induced inositol biosynthesis and oxidative stress responses by directly binding to the promoter of MIPS1 and activating its expression.61 Hence, findings of the present study suggest a central role of MIPS1 under combined cold/high light stress conditions where oxidative stress response might be even more pronounced than under single stress conditions. Together with previous reports about the potential involvement of sucrose synthase (SUS1) in starch and cellulose biosynthesis,62 the finding that RHM1 protein levels are significantly increased suggests reprogramming of carbon allocation towards cell wall biosynthesis. Another central core protein was ADH1, which reduces acetaldehyde to ethanol to regenerate NAD+ to maintain energy-generating glycolysis. ADH1 is known to accumulate during various stresses like hypoxia, salt, dehydration and cold.63–65 The interaction of ADH1 with the cold response genes (COR78), amino acid metabolism (P5CS2), secondary metabolites (F3H, TT4, TT5) and sucrose synthase (SUS1) integrates all main targets of metabolism found in the core set. However, earlier findings indicated that although ADH mRNA accumulated during cold exposure, its activity was not required for cold acclimation.66 Although this prevents the direct interpretation of ADH1 function from the present study, it still suggests a potential role in response to combined stress which might be relevant during the early and intermediate rather than the late acclimation phase.
Six further proteins involved in transcriptional and circadian regulation, cell organisation and protein biosynthesis were part of the identified stress responsive core proteome. All of them shared the chloroplast as a subcellular localization. FIB1A, a fibrillin precursor protein, has earlier been shown to play a role in abscisic acid (ABA)-mediated photoprotection.67 These authors reported enhanced tolerance of PSII towards light-stress induced photoinhibition due to ABA treatment and fibrillin accumulation. In the present study, no significant genotype effect was detected for chlorophyll fluorescence parameters. Although the contribution of FIB1A to photosystem stabilization remains to be elucidated in the background of pgm1 and spsa1, the significant increase of FIB1A is likely to contribute to the observed similar photosystem constitution in all genotypes.
In summary, the stress-responsive core proteome identified in this study interconnects cellular processes across various subcellular compartments and biological functions. Due to their consistent stress-response across different metabolic constitutions, it is likely that the identified 23 protein candidates are dominantly involved in general high light and low temperature response. Finally, resolving the evolutionary conservation and the ecological role of this core proteome will potentially provide novel insights into complex stress tolerance mechanisms in plants.
Author contribution
LF, AP and HS performed experiments, data evaluation and wrote the manuscript. WW revised the manuscript. TN designed the study, performed experiments, data evaluation and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the whole MoSys Team at the University of Vienna for support and fruitful discussion. Particularly, we would like to thank Jakob Weiszmann and Martin Brenner for critical discussion and technical assistance. Finally, we thank the gardeners, Thomas Joch and Andreas Schröfl, for excellent plant cultivation. This work was supported by the Austrian Science Fund, FWF (Project Number I2071).References
- B. Vinocur and A. Altman, Curr. Opin. Biotechnol., 2005, 16, 123–132 CrossRef CAS PubMed.
- K. Kosova, P. Vitamvas, I. T. Prasil and J. Renaut, J. Proteomics, 2011, 74, 1301–1322 CrossRef CAS PubMed.
- D. C. Haak, T. Fukao, R. Grene, Z. Hua, R. Ivanov, G. Perrella and S. Li, Front. Plant Sci., 2017, 8, 1564 CrossRef PubMed.
- M. F. Thomashow, Plant Physiol., 2010, 154, 571–577 CrossRef CAS PubMed.
- S. J. Gilmour, D. G. Zarka, E. J. Stockinger, M. P. Salazar, J. M. Houghton and M. F. Thomashow, Plant J., 1998, 16, 433–442 CrossRef CAS PubMed.
- S. Fowler and M. F. Thomashow, Plant Cell, 2002, 14, 1675–1690 CrossRef CAS.
- K. R. Jaglo-Ottosen, S. J. Gilmour, D. K. Zarka, O. Schabenberger and M. F. Thomashow, Science, 1998, 280, 104–106 CrossRef CAS PubMed.
- S. Park, C. M. Lee, C. J. Doherty, S. J. Gilmour, Y. Kim and M. F. Thomashow, Plant J., 2015, 82, 193–207 CrossRef CAS PubMed.
- M. A. Gehan, S. Park, S. J. Gilmour, C. An, C. M. Lee and M. F. Thomashow, Plant J., 2015, 84, 682–693 CrossRef CAS PubMed.
- K. Nakaminami, A. Matsui, H. Nakagami, A. Minami, Y. Nomura, M. Tanaka, T. Morosawa, J. Ishida, S. Takahashi, M. Uemura, K. Shirasu and M. Seki, Mol. Cell. Proteomics, 2014, 13, 3602–3611 CrossRef CAS PubMed.
- S. A. Campbell and T. J. Close, New Phytol., 1997, 137, 61–74 CrossRef CAS.
- S. Amme, A. Matros, B. Schlesier and H.-P. Mock, J. Exp. Bot., 2006, 57, 1537–1546 CrossRef CAS PubMed.
- K. Kosová, P. Vítámvás and I. T. Prášil, Front. Plant Sci., 2014, 5, 711 Search PubMed.
- J. S. Kim, H. J. Jung, H. J. Lee, K. Kim, C. H. Goh, Y. Woo, S. H. Oh, Y. S. Han and H. Kang, Plant J., 2008, 55, 455–466 CrossRef CAS PubMed.
- C. Kupsch, H. Ruwe, S. Gusewski, M. Tillich, I. Small and C. Schmitz-Linneweber, Plant Cell, 2012, 24, 4266–4280 CrossRef CAS PubMed.
- K. Kosová, P. Vítámvás, M. O. Urban, I. T. Prášil and J. Renaut, Front. Plant Sci., 2018, 9, 122 CrossRef PubMed.
- A. Graf, A. Schlereth, M. Stitt and A. M. Smith, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9458–9463 CrossRef CAS PubMed.
- B. Pommerrenig, F. Ludewig, J. Cvetkovic, O. Trentmann, P. A. W. Klemens and H. E. Neuhaus, Plant Cell Physiol., 2018, 59, 1290–1299 Search PubMed.
- M. J. Haydon, O. Mielczarek, F. C. Robertson, K. E. Hubbard and A. A. Webb, Nature, 2013, 502, 689 CrossRef CAS PubMed.
- K. Miura and T. Furumoto, Int. J. Mol. Sci., 2013, 14, 5312–5337 CrossRef CAS PubMed.
- R. Sicher, Plant Sci., 2011, 181, 167–176 CrossRef CAS PubMed.
- M. Nagler, E. Nukarinen, W. Weckwerth and T. Nägele, BMC Plant Biol., 2015, 15, 284 CrossRef PubMed.
- M. Stitt and S. C. Zeeman, Curr. Opin. Plant Biol., 2012, 15, 282–292 CrossRef CAS PubMed.
- Y. L. Ruan, Annu. Rev. Plant Biol., 2014, 65, 33–67 CrossRef CAS PubMed.
- I. I. Hoermiller, T. Naegele, H. Augustin, S. Stutz, W. Weckwerth and A. G. Heyer, Plant, Cell Environ., 2017, 40, 602–610 CrossRef CAS PubMed.
- K. Brauner, I. Hörmiller, T. Nägele and A. G. Heyer, Plant J., 2014, 79, 82–91 CrossRef CAS PubMed.
- C. Ma, H. H. Zhang and X. Wang, Trends Plant Sci., 2014, 19, 798–808 CrossRef CAS PubMed.
- J. Sperschneider, P. N. Dodds, K. B. Singh and J. M. Taylor, New Phytol., 2018, 217, 1764–1778 CrossRef CAS PubMed.
- A. Singh, B. Ganapathysubramanian, A. K. Singh and S. Sarkar, Trends Plant Sci., 2016, 21, 110–124 CrossRef CAS PubMed.
- W. Hoehenwarter, A. Larhlimi, J. Hummel, V. Egelhofer, J. Selbig, J. T. van Dongen, S. Wienkoop and W. Weckwerth, J. Proteome Res., 2011, 10, 2979–2991 CrossRef CAS PubMed.
- R. Mittler, Trends Plant Sci., 2006, 11, 15–19 CrossRef CAS PubMed.
- C. M. Prasch and U. Sonnewald, Environ. Exp. Bot., 2015, 114, 4–14 CrossRef CAS.
- T. Nägele, S. Stutz, I. Hörmiller and A. G. Heyer, Plant J., 2012, 72, 102–114 CrossRef PubMed.
- T. Nägele, B. A. Kandel, S. Frana, M. Meissner and A. G. Heyer, FEBS J., 2011, 278, 506–518 CrossRef PubMed.
- W. Weckwerth, K. Wenzel and O. Fiehn, Proteomics, 2004, 4, 78–83 CrossRef CAS PubMed.
- J. Kopka, N. Schauer, S. Krueger, C. Birkemeyer, B. Usadel, E. Bergmuller, P. Dormann, W. Weckwerth, Y. Gibon, M. Stitt, L. Willmitzer, A. R. Fernie and D. Steinhauser, Bioinformatics, 2005, 21, 1635–1638 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367 CrossRef CAS PubMed.
- P. Lamesch, T. Z. Berardini, D. Li, D. Swarbreck, C. Wilks, R. Sasidharan, R. Muller, K. Dreher, D. L. Alexander, M. Garcia-Hernandez, A. S. Karthikeyan, C. H. Lee, W. D. Nelson, L. Ploetz, S. Singh, A. Wensel and E. Huala, Nucleic Acids Res., 2012, 40, D1202–D1210 CrossRef CAS PubMed.
- J. A. Vizcaino, A. Csordas, N. del-Toro, J. A. Dianes, J. Griss, I. Lavidas, G. Mayer, Y. Perez-Riverol, F. Reisinger, T. Ternent, Q. W. Xu, R. Wang and H. Hermjakob, Nucleic Acids Res., 2016, 44, D447–D456 CrossRef CAS PubMed.
- R Core Team, R Foundation for Statistical Computing, Vienna, Austria, 2008, ISBN 3-900051-07-0.
- J. C. Oliveros, Venny, An interactive tool for comparing lists with Venn's diagrams, 2007–2015, http://bioinfogp.cnb.csic.es/tools/venny/index.html.
- K. Maxwell and G. N. Johnson, J. Exp. Bot., 2000, 51, 659–668 CrossRef CAS PubMed.
- F. Keller and D. M. Pharr, Photoassimilate distribution in plants and crops: source-sink relationships, Marcel Dekker, New York, 1996, pp. 157–183 Search PubMed.
- C. von Mering, L. J. Jensen, B. Snel, S. D. Hooper, M. Krupp, M. Foglierini, N. Jouffre, M. A. Huynen and P. Bork, Nucleic Acids Res., 2005, 33, D433–D437 CrossRef CAS PubMed.
- A. M. Smith and M. Stitt, Plant, Cell Environ., 2007, 30, 1126–1149 CrossRef CAS PubMed.
- J. Sun, J. Zhang, C. T. Larue and S. C. Huber, Plant, Cell Environ., 2011, 34, 592–604 CrossRef CAS PubMed.
- Y. Gibon, O. E. Bläsing, N. Palacios-Rojas, D. Pankovic, J. H. Hendriks, J. Fisahn, M. Höhne, M. Günther and M. Stitt, Plant J., 2004, 39, 847–862 CrossRef CAS PubMed.
- C. M. Prasch and U. Sonnewald, Environ. Exp. Bot., 2015, 114, 4–14 CrossRef CAS.
- R. W. McMichael, R. R. Klein, M. E. Salvucci and S. C. Huber, Arch. Biochem. Biophys., 1993, 307, 248–252 CrossRef CAS PubMed.
- S. C. Huber and J. L. Huber, Plant Physiol., 1992, 99, 1275–1278 CrossRef CAS PubMed.
- W. M. Kaiser and S. C. Huber, J. Exp. Bot., 2001, 52, 1981–1989 CrossRef CAS PubMed.
- Y. Li, F. X. Wu and A. Ngom, Briefings Bioinf., 2018, 19, 325–340 Search PubMed.
- S. Wienkoop, K. Morgenthal, F. Wolschin, M. Scholz, J. Selbig and W. Weckwerth, Mol. Cell. Proteomics, 2008, 7, 1725–1736 CrossRef CAS PubMed.
- D. P. Horvath, B. K. McLarney and M. F. Thomashow, Plant Physiol., 1993, 103, 1047–1053 CrossRef CAS PubMed.
- A. Thalhammer, M. Hundertmark, A. V. Popova, R. Seckler and D. K. Hincha, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1812–1820 CrossRef CAS PubMed.
- J. Larkindale and E. Vierling, Plant Physiol., 2008, 146, 748–761 CrossRef CAS PubMed.
- L. Szabados and A. Savouré, Trends Plant Sci., 2010, 15, 89–97 CrossRef CAS PubMed.
- W. Jiang, Q. Yin, R. Wu, G. Zheng, J. Liu, R. A. Dixon and Y. Pang, J. Exp. Bot., 2015, 66, 7165–7179 CrossRef CAS PubMed.
- H. Doerfler, D. Lyon, T. Nägele, X. Sun, L. Fragner, F. Hadacek, V. Egelhofer and W. Weckwerth, Metabolomics, 2013, 9, 564–574 CrossRef CAS PubMed.
- A. Rohde, K. Morreel, J. Ralph, G. Goeminne, V. Hostyn, R. De Rycke, S. Kushnir, J. Van Doorsselaere, J.-P. Joseleau and M. Vuylsteke, Plant Cell, 2004, 16, 2749–2771 CrossRef CAS PubMed.
- L. Ma, T. Tian, R. Lin, X. W. Deng, H. Wang and G. Li, Mol. Plant, 2016, 9, 541–557 CrossRef CAS PubMed.
- E. Baroja-Fernandez, F. J. Munoz, J. Li, A. Bahaji, G. Almagro, M. Montero, E. Etxeberria, M. Hidalgo, M. T. Sesma and J. Pozueta-Romero, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 321–326 CrossRef CAS PubMed.
- R. Dolferus, G. De Bruxelles, E. S. Dennis and W. J. Peacock, Ann. Bot., 1994, 74, 301–308 CrossRef CAS.
- J. A. Jarillo, A. Leyva, J. Salinas and J. M. Martinez-Zapater, Plant Physiol., 1993, 101, 833–837 CrossRef CAS PubMed.
- H. Shi, W. Liu, Y. Yao, Y. Wei and Z. Chan, Plant Sci., 2017, 262, 24–31 CrossRef CAS PubMed.
- J. A. Jarillo, A. Leyva, J. Salinas and J. M. Martínez-Zapater, Plant Physiol., 1993, 101, 833–837 CrossRef CAS PubMed.
- Y. Yang, R. Sulpice, A. Himmelbach, M. Meinhard, A. Christmann and E. Grill, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6061–6066 CrossRef CAS PubMed.
- O. Thimm, O. Bläsing, Y. Gibon, A. Nagel, S. Meyer, P. Krüger, J. Selbig, L. A. Müller, S. Y. Rhee and M. Stitt, Plant J., 2004, 37, 914–939 CrossRef CAS PubMed.
- C. M. Hooper, S. K. Tanz, I. R. Castleden, M. A. Vacher, I. D. Small and A. H. Millar, Bioinformatics, 2014, 30, 3356–3364 CrossRef CAS PubMed.
- L. J. Jensen, M. Kuhn, M. Stark, S. Chaffron, C. Creevey, J. Muller, T. Doerks, P. Julien, A. Roth and M. Simonovic, Nucleic Acids Res., 2008, 37, D412–D416 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mo00095f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |