
Data integration and predictive modeling methods for multi-omics datasets†
Minseung
Kim
 ab and
Ilias
Tagkopoulos
*ab
ab and
Ilias
Tagkopoulos
*ab
aDepartment of Computer Science, University of California, Davis, California 95616, USA
bGenome Center, University of California, Davis, California 95616, USA. E-mail: itagkopoulos@ucdavis.edu
First published on 20th December 2017
Abstract
Translating data to knowledge and actionable insights is the Holy Grail for many scientific fields, including biology. The unprecedented massive and heterogeneous data have created as many challenges to store, process and analyze as the opportunities and promises they hold. Here, we provide an overview of these opportunities and challenges in multi-omics predictive analytics.
Introduction
Machine learning and multi-omics technologies revolutionize the way we acquire and process data. At their core, machine learning (ML) algorithms dissect the data to learn their structure and associations within, often without the need of specific knowledge on processes and models that generated them.1 The strength of ML techniques is proportional to the size and quality of the data amassed. At the same time, sequencing and molecular technologies can generate a vast amount of high quality data in an inexpensive, reproducible way and hence they allow an unprecedented system-level view of any organism.2 These datasets, which can come from a variety of sources, equipment and experimental settings, are in their majority not ready to serve as training sets to computational models and machine learning methods, as they have not been created with that function in mind. As such, there is a clear need for methods that process, normalize, integrate and transform the plethora of heterogeneous multi-omics data to cohesive compendia that can be used as a training grounds for further analysis and learning.3,4Here, we review the current methods for preprocessing and analysis of heterogeneous omics data for various problems in computational biology. In line with previous reviews on similar topics in personalized medicine,5 genetics,6 and bio-imaging analysis,7 we extend these efforts to the description of multiple omics-types and to the characterization of the practical aspects of high-throughput technologies to profile such omics-types. We summarize the data universe for the most data-rich organisms across the five kingdoms and provide an overview of processing procedures for genome-wide raw data profiled from major high-throughput technologies. We then explore methods for integrating heterogeneous omics data and general principles and applications of quality assessment (QA) and quality control (QC) of genome-wide data, as well as the application of machine learning methods to these datasets across a wide spectrum of applications.
The general workflow of multi-omics integration and analysis consists of three major steps (Fig. 1). First, omics data are collected and processed to interrogate genome-wide molecular measurements from isolates. Then, the processed omics data are combined at different levels of depth (prior knowledge and degrees of coverage) and widths (across heterogeneous omics-types) after the quality assurance procedure is performed. On the integrated compendia, machine-learning analytics are applied to learn complex patterns, finally guiding new experimentation based on the model results. This high-level abstraction of the analytic pipeline for predictive biology is applicable to diverse domains including biomedicine,5,8 biotechnology,9 agriculture,10,11 and nutritional science.12,13 In the sections below, we review the types of data, preprocessing pipelines, predictive models and applications of omics data.
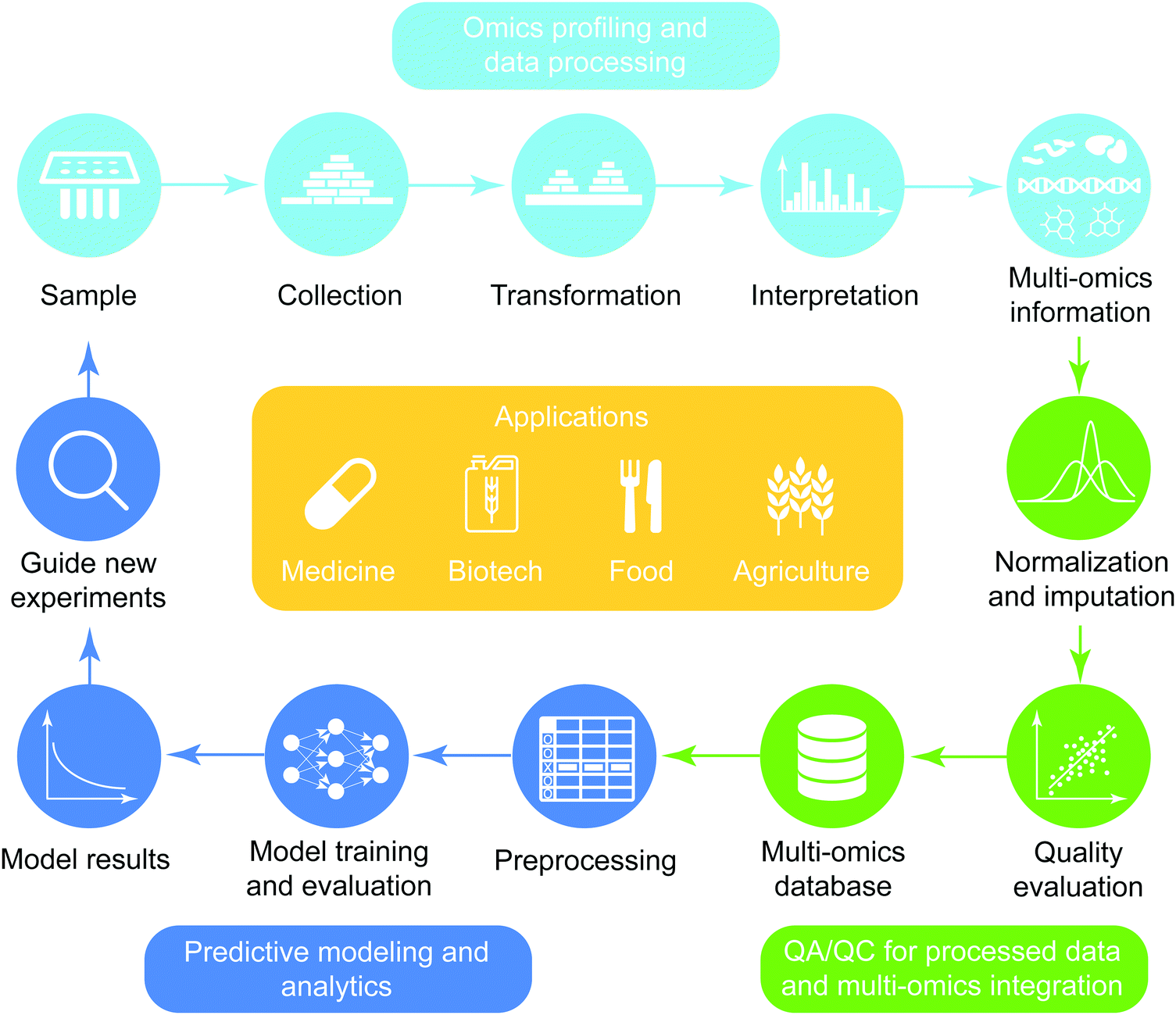 | ||
| Fig. 1 An end-to-end pipeline for multi-omics data. The three major steps involved are data acquisition, multi-omics integration and predictive modeling. For omics profiling and data processing (cyan icons), samples are collected and profiled using high-throughput technologies. The raw data are then processed, interpreted and translated to knowledge. For QA/QC and multi-omics integration (green icons), the quality of integrated data is ensured by performing normalization, imputation, and quality evaluation. Then, the genome-wide data across different omics-types are integrated into one or more databases. Finally, during predictive modeling and analysis (blue icons), analytics are applied after data have been transformed (pre-processed) to be suitable for training models. Trained models are then evaluated and interrogated to guide further experimentation, either as validation or as hypothesis generation steps for the next iteration of the omics cycle. This pipeline is applied with small variations in a variety of industries, including agriculture, food and nutrition, biotechnology and medicine. | ||
Omics data types
Overview
There are four main omics-types (genome, transcriptome, proteome, and metabolome) where each represents all molecules of a specific type (DNA, RNA, protein, and metabolite, respectively) within a cell or a group of cells. Here we describe each of the four omics-types and characterize many practical aspects of high-throughput technologies to interrogate such information at the genome scale (Table 1).| Omics-types | Platform | Utility | Cost | Quantity (%) | Quality review | Resources |
|---|---|---|---|---|---|---|
| Genome | DNA-Seq187 |
– Genome assembly
– Genetic variant identification |
$250–$650 | ∼95188 | 14 |
GEO16
SRA15 dbGaP17 |
| Transcriptome | RNA-Seq19 |
– Transcriptome profiling
– Novel transcript discovery – Novel splicing event18 |
$175–$450 | 80–9919 | 20 |
GEO16
ArrayExpress21 SRA15 |
| Proteome | MS189 |
– Proteome profiling
– Quantification of PTMs – Novel protein discovery22,23 |
$100–$171 | 55–9224,25 | 26 |
PRIDE27
ProteomeXchange28 ProteomicsDB24 |
| Metabolome | MS46 |
– Metabolome profiling
– Novel metabolite discovery29 |
$69–$90 | <2030 | 31 | MetaboLights32 |
| Interactome |
ChIP-Seq
ChIP-exo36–38 |
– Genome-wide mapping of protein–DNA interactions
+ Gene-regulatory network + Histone modification maps + Nucleosome maps |
$395–$415 | ∼95188 | 38 |
GEO16
SRA15 hmChIP190 |
| Y2H33 | – Protein–protein interaction | — | 34–5035 | 191 |
STRING39
BioGRID40 PPI database review41 |
Genome
A genome is the complete information of the DNA of an organism. A primary technique to interrogate such information is whole-genome sequencing (DNA-Seq), which can be used for novel assembly and for the discovery of genetic variants for a re-sequenced organism. The quantity and quality of the outcome depends on the read depth (i.e. how many reads on average are mapped on the reference genome at a single base position) and it has been extensively reviewed in the past.14 Two major databases collecting publicly available genomic data are the Sequence Read Archive (SRA), which stores raw sequence data,15 and the Gene Expression Omnibus (GEO), which stores processed genomic data with characterization metadata.16 The NCBI dbGaP (The database of Genotypes and Phenotypes) is a public repository for individual-level genotype, sequence data, and phenotype with controlled access.17Transcriptome
The transcriptome is the set of all messenger RNA molecules in a cell or a population of cells. The most common high-throughput techniques for transcriptional profiling are microarrays and more recently RNA-Seq. Raw data can be used for quantification of mRNAs, novel transcript identification as well as discovery of novel splicing sites.18 The coverage of genes that can be profiled by RNA-Seq experiments varies from 80% to 99% of the total count depending on the experimental setup.19 Past reviews have summarized the quality of RNA-Seq data.20 Most publicly available transcriptional profiling datasets can be found in the GEO database,16 ArrayExpress21 and SRA.15Proteome
The proteome is the entire universe of proteins that can be expressed by a cell. Mass-spectrometry (MS) is the main platform used for large-scale proteomic profiling. The output processed from mass-spectrometry can be used for quantification of proteins and PTMs (post-translational modifications), as well as for identifying novel proteins.22,23 Due to technological limitations, not all proteins can be detected. The coverage of detectable proteins ranges from 55% to 94% of the total proteome, depending on the specific organism.24,25 The quality of MS-produced proteome data has been reviewed in ref. 26. The three major repositories that store proteome profiling results are PRIDE,27 ProteomeXchange28 and ProteomicsDB.24Metabolome
The metabolome is the complete set of small-molecules present within an organism. The typical mass of metabolites in a cell spans from 50 to 1500 daltons (Da). Like the proteome, mass-spectrometry (MS) is a major class of technologies to interrogate genome-wide quantification of metabolites or to discover novel metabolites.29 Detection coverage is still limited to below 20% because of various technological limitations.30 A critical review about quality of MS-produced metabolome data can be found in ref. 31. Compared to other omics-types, the available databases collecting metabolome experiments are scarce. MetaboLights32 is a notable resource, although still with limited data (189 studies so far).Interactome
The interactome is a map of molecular dependencies in a cell. That is, the interactome can be considered as a collection of genome-wide interplays across genome, transcriptome, proteome, and metabolome. There are many different types of interactions depending on the type of interacting molecules, with protein–protein interaction (PPI) being one of the main ones. PPIs are usually identified by yeast-two hybrid screening33 and more recently by sequencing technology,34 although the coverage is believed to be limited to around 34–50%.35 Another interaction type is between a protein and DNA, which is typically profiled by ChIP-Seq, and more recently, by ChIP-Exo.36–38 The resulting information can be used for revealing the gene regulatory or histone modification maps. The public repositories curating molecular interactions include STRING39 and BioGRID.40 More extensive review on the resources is in ref. 41.Multi-omics data availability
The estimated availability of multi-omics data across five different kingdoms is shown in Table 2. As expected, genomic information is the most abundant of all omics-types (a total of 891k profiles for the 15 most popular organisms), followed by transcriptional profiling. Interestingly, the metabolome layer is more quantitatively explored than the proteome layer, which might reflect the lower profiling cost as reported in Table 1. As expected, Homo sapiens was the organism explored with the largest number of profiles across all omics-types except the fluxome layer, which was second ranked followed by E. coli. The number of available flux profiles is estimated to be more abundant in single cell organisms, due to their importance in biotechnology and metabolic engineering.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 950 search results as of 09/05/17 and 3823 profiles were found. We multiply the ratio (3823/44950 = 0.085) to other organisms. For proteome, in google scholar, we searched the keywords “[organism_name] proteome profiling mass-spectrometry”. This gives 49200 search results as of 09/05/17 and 137 profiles were found. We multiply the ratio (137/49200 = 0.00273) to other organisms. For metabolome, in google scholar, we searched the keywords “[organism_name] metabolome profiling mass-spectrometry”. This gives 16220 search results as of 09/05/17 and 696 profiles were found. We multiply the ratio (696/16220 = 0.042) to other organisms. For fluxome, in google scholar, we searched the keywords “[organism_name] 13C fluxome profiling”. This gives 1590 search results as of 09/05/17. In-depth investigation shows that there are 43 profiles in the results. We multiply the ratio of true number of profiles to number of google search results (43/1590 = 0.027) to other organisms
950 search results as of 09/05/17 and 3823 profiles were found. We multiply the ratio (3823/44950 = 0.085) to other organisms. For proteome, in google scholar, we searched the keywords “[organism_name] proteome profiling mass-spectrometry”. This gives 49200 search results as of 09/05/17 and 137 profiles were found. We multiply the ratio (137/49200 = 0.00273) to other organisms. For metabolome, in google scholar, we searched the keywords “[organism_name] metabolome profiling mass-spectrometry”. This gives 16220 search results as of 09/05/17 and 696 profiles were found. We multiply the ratio (696/16220 = 0.042) to other organisms. For fluxome, in google scholar, we searched the keywords “[organism_name] 13C fluxome profiling”. This gives 1590 search results as of 09/05/17. In-depth investigation shows that there are 43 profiles in the results. We multiply the ratio of true number of profiles to number of google search results (43/1590 = 0.027) to other organisms
| Kingdom | Species | Layer | ||||
|---|---|---|---|---|---|---|
| Genome | Transcriptome | Proteome | Metabolome | Fluxome | ||
| Monera | Escherichia coli | 35492 |
3579 | 137 | 696 | 43 |
| Bacillus subtilis | 445 | 967 | 56 | 180 | 13 | |
| Salmonella enterica | 67945 |
459 | 18 | 40 | 4 | |
| Protista | Chlamydomonas reinhardtii | 749 | 392 | 16 | 68 | 2 |
| Emiliania huxleyi | 38 | 45 | 2 | 8 | 0 | |
| Thalassiosira pseudonana | 14 | 83 | 3 | 15 | 0 | |
| Fungi | Saccharomyces cerevisiae | 39381 |
24392 |
97 | 381 | 20 |
| Chlamydomonas reinhardtii | 744 | 373 | 15 | 75 | 2 | |
| Schizosaccharomyces pombe | 2182 | 593 | 25 | 52 | 2 | |
| Plantae | Arabidopsis thaliana | 13501 |
30911 |
65 | 518 | 9 |
| Maize | 1998 | 10882 |
69 | 381 | 6 | |
| Oryza sativa (Rice) | 40910 |
10472 |
25 | 132 | 1 | |
| Animalia | Homo sapiens (Human) | 668718 |
210933 |
691 | 1519 | 45 |
| Caenorhabditis elegans | 8291 | 11198 |
64 | 184 | 5 | |
| Drosophila melanogaster | 12692 |
10918 |
52 | 13 | 2 | |
| Total | 893100 |
316197 |
1335 | 4262 | 154 | |
Omics profiling and data processing
In this section, we provide a high-level overview of procedures to process high-throughput raw-data produced from different platforms (Fig. 2). We focus on three major groups of high-throughput technologies (microarray, next generation sequencing, and mass-spectrometry). For more information about each platform, refer to ref. 42 and 43 for microarray, ref. 44 for sequencing technology, and ref. 45 and 46 for mass-spectrometry. Every processing platform can be divided into three phases, each with its own quality control: (i) early stage that directly handles raw data, (ii) middle stage that performs major data processing, and (iii) late stage that executes post-processing to finalize the molecular quantification.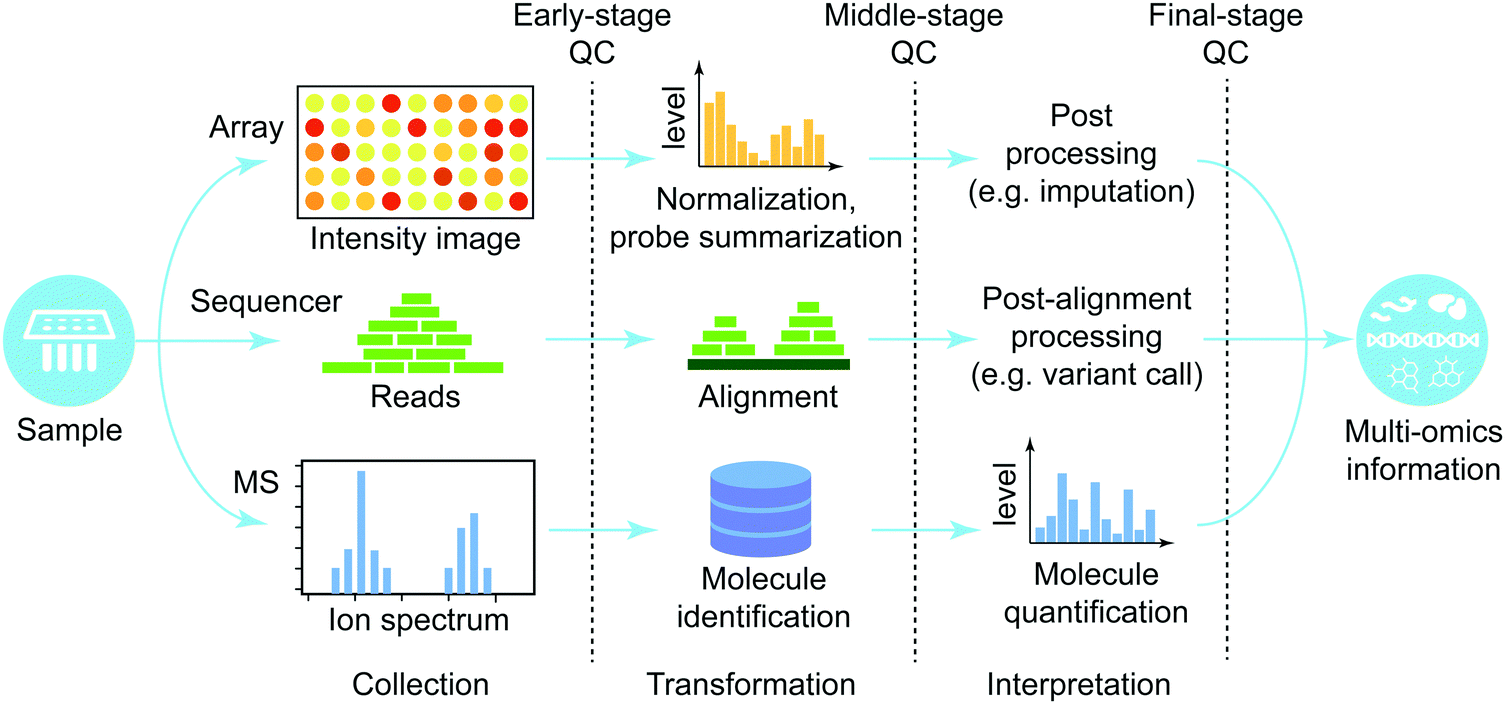 | ||
| Fig. 2 Omics data processing pipeline. The processing pipelines for three major high-throughput technologies are shown. It is comprised of three distinctive steps: (A) collection step, where the samples are processed and raw data are generated; (B) transformation step, where data are processed, reads mapped and molecules identified; (C) interpretation step, where data are interpreted based on existing knowledge of the corresponding organism. In all cases, quality control (QC) is applied at the end of each stage to ensure high data quality. | ||
Microarray
Microarray is a technique to probe massive amounts of molecules on a tiny slide based on hybridization principles.42,43 This general principle allows profiling of many different aspects of molecules ranging from genetic variants (DNA microarray) to quantification of transcripts (e.g. cDNA microarray). The raw output is an intensity image, which quantifies information about abundance of hybridized molecules. Higher intensity of molecular hybridization is regarded as that the specific molecule is present in higher abundance. The intensity image is in turn processed in a series of steps (e.g. background noise removal, normalization, and probe-set summarization). An example of the output is expression levels of molecules in cDNA microarray. Since the introduction of the technology nearly two decades ago, methods to process raw microarray data have been extensively developed and matured. A few suggested reviews on the microarray data processing methods are ref. 47–49 (more information is in Table 3).| High-throughput technologies | Type | Reviews on processing methods |
|---|---|---|
| Microarray | General | 43–45 |
| Gene-expression microarray | 194 | |
| DNA methylation microarray | 195 | |
| Sequencing | RNA-Seq | 196 |
| DNA-Seq for genotyping | 197 | |
| DNA-Seq for de novo assembly | 198 | |
| ChIP-Seq | 199 | |
| Mass-spectrometry | Protein mass-spectrometry | 200 |
| Metabolite mass-spectrometry | 201 | |
Whole-genome sequencing
Whole-genome sequencing technology is a method to interrogate complete information of DNA/RNA of an organism at a single time. The recent advance in this field is the so called next-generation sequencing which has ushered in a new era of genomics by reducing the cost and time to interrogate whole-genome information by profiling short sequence reads in a massively parallel way.44 The raw data of short reads from the sequencer are typically aligned on the reference genome to localize short reads. Then the post-alignment step finalizes the output and diverse omics information can be interrogated from the variants of this technology including profiling of protein–DNA binding events (e.g. ChIP-Seq and ChIP-exo). Processing methods of sequencing data have been extensively reviewed elsewhere (Table 3).Mass-spectrometry (MS)
Mass-spectrometry (MS) is a technique where ionization of chemical species is used to sort them based on the mass-to-charge ratio. This technology has been widely used for interrogating quantification of proteins and metabolites, and modification of the sample preparation step (e.g.13C labelling) allows profiling of metabolic fluxes.50 Unlike metabolites, proteins are usually first digested with a protease (e.g. trypsin) into short peptides to lower the mass to be detectable by the instrument. MS produces an ion spectrum which is then used to determine its molecular identify by matching to theoretical spectra measured from the existing databases.45,46 In the case of peptides, this step determines the sequences of peptides. Then the next step is to quantify the target molecules based on the amount of identified small molecules. Data processing methods for mass-spectrometry have been extensively reviewed and the suggested articles are in Table 3.Multi-omics integration
Methods
Omics data integration is not new, with the first review of the field appearing more than a decade ago,2,3 in both humans51 and plants.52 Methods for multi-omics integration can be mapped onto a discrete two-dimensional space (Fig. 3). One dimension represents whether integration is a single or multiple omics-type (breadth). The second dimension captures the depth of integration between data and data, data and knowledge or knowledge and knowledge. As such, multi-omics data integration can be categorized as follows.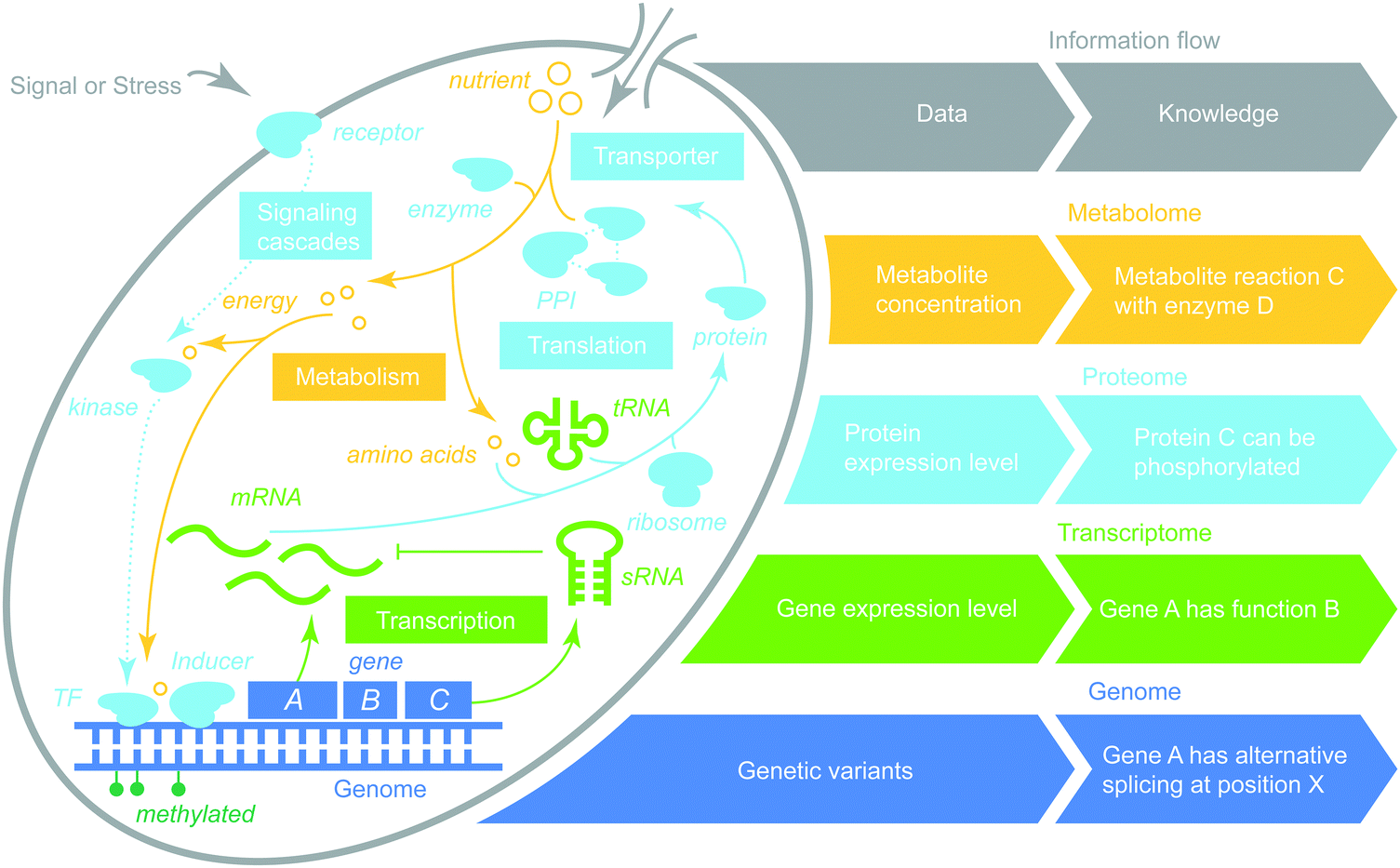 | ||
| Fig. 3 Multi-omics organization in a cell. Omics data can be integrated within a layer or across multiple layers. Depending on the information and data types involved, integration can be homogeneous (data to data) or heterogeneous (data to knowledge). | ||
Data-to-data. The integration of data within a layer typically refers to the combination of genome-wide data for the same omics type for a particular organism across different batches, studies and platforms. Most studies along this direction have been focused on the genomic layer and transcriptional layer, as they are the most profiled. SEEK is a transcriptome compendium for human, which provides 150k experiments with platform-adjusted gene correlation measures.53 COLOMBOS is a transcriptome compendium for 19 bacteria where all data are formatted in contrast of two profiles between a test condition and a corresponding control.54 Integration of expression profiles across different sources requires special attention in normalization as many artefacts due to lab-to-lab variation may arise.20 For more information, we refer readers to the review on normalization methods.55 Integration within the genome layer has been primarily performed across different types of genetic variations to augment the feature set including between SNPs and copy number variations56 and between common variants and rare variants.57
Data-to-knowledge. Integration of genome-wide omics data and other information about an organism. One notable method in this area is ANNOVAR,58 which performs functional annotation of genetic variants including gene annotation (e.g. splice site variant, non-synonymous SNP). CEGMA59 identifies the exon–intron structure from a novel genome sequence, which is useful for annotating the genome sequence of an unexplored organism. In addition, transcriptome profiles can be functionally annotated to identify novel transcriptional active regions and to reveal alternative splicing patterns.60 Interpretation of proteome data can be facilitated by STRAP, which automatically annotates and visualizes user's proteome data.61 Annotation of metabolomic data is relatively new, compared to genomics and transcriptomics and the tools to facilitate functional interpretation of metabolomic experiments are recently reviewed in ref. 62. A notable tool is MetaboAnalyst, which provides comprehensive characterization of large numbers of metabolites online.63
Knowledge-to-knowledge. Integration of facts about a single omics type of a specific organism that have been compiled and curated by separate groups and projects. Many of the existing biological databases belong to this category, where the primary goal is to curate functional molecular characteristics and their interactions from multiple sources. Molecular characterization is a resource-rich area, where a plethora of gene annotations exist for different organisms including EcoCyc for E. coli,64 TAIR for A. thaliana,65 SGD for S. cerevisiae,66 and NCBI OMIM for human disease genes.67 Proteome knowledge has been extensively curated in UniProtKB,68 which combines SWISS-PROT that is manually annotated and reviewed as well as TrEMNBL that is automatically annotated and not reviewed.69 Another example is the HAMAP project, which combines automated curation and manual curation of the microbial proteome database to facilitate the speed of the curation process while preserving the accuracy of the curated knowledge.70 For the metabolome, species-specific databases are available ranging from ECMDB for E. coli and YMDB for Yeast. For a more comprehensive list, refer to ref. 71. In addition, specialized collections based on the type of interaction exist, including protein–protein,41 gene-regulatory,72 and metabolic interactions.73
Data-to-data. Integration of genome-wide data for multiple omics types for a particular organism across different batches, studies and platforms. Co-analyses of genomic data with expression profiles from either the transcriptome, proteome, or methylome fall under this category. The main goal of these analyses is to identify the quantitative trait locus, and eQTL, pQTL or mQTL are some techniques that are used for this purpose.74 The integration of transcriptome and proteome data has also led to the discovery of post-translational activities and correlation between two omics-types under identical conditions.75 Proteogenome is an emerging field that employs proteomic data to annotate genome sequences.76 There has been a growing list of individual studies employing multi-omics data and recent reviews concerning this subject exist. Their focus ranges from grapevines77 to microbes,78 and single-cell technologies.79 Furthermore, there have been recent constructions of large-scale multi-omics compendia. For example, MOPED is a multi-omics compendium of four model organisms of human, mouse, worm and yeast where it collects publicly available transcriptome profiles and proteome profiles.80 Ecomics and MyMpn are multi-omics compendia for E. coli4 and M. pneumonia, respectively.81
Data-to-knowledge. Integration of genome-wide data for a multiple omics type for a particular organism and relevant facts about the integrated omics type of the organism that have been accumulated by a group of people through time. Many of the studies belonging to this category integrate transcriptome signatures with the protein–protein interaction network. The underlying assumption here is that transcriptional expression is a proxy of protein expression levels although its validity is arguable.82 For example, ref. 83 reveals topological features of cancer genes by combining the transcriptome and the interactome. More recently, the genome, transcriptome, and interactome were merged together to process mass-spectrometry data84 and plant regulatory networks were inferred by integrating known regulatory bindings, transcriptome, proteome, and metabolome data.85
Knowledge-to-knowledge. Integration of facts about a multiple omics type of a specific organism that have been compiled and curated by separate groups and projects. Integration of heterogeneous networks is the focus of the studies in this category. For example, ref. 86 and 87 integrate metabolic, transcriptional regulatory and signal transduction networks for E. coli for metabolic flux predictions. Ref. 88 identifies network patterns in the combined network of protein–protein interactions and transcription regulation for S. cerevisiae. Gene-to-phenotype associations in the context of biological networks have also been studied extensively. For example, CIPHER89 employed human disease genes and protein–protein interaction map to infer novel biomarkers. Moreover, the power to identify phenotype-associated genes can be improved by integrating findings from genetic association studies and biological networks and pathways.90
Furthermore, biased exploration often subsists in the experimental space of an organism,4 which limits our understanding and ability to predictively model an organism. For instance, among the top 5 strains and the top 5 media used for experimenting with E. coli, only 6 combinations out of 25 have been explored.4 This partial sampling generates knowledge gaps, which increase uncertainty. Computational methods, such as active learning93 and optimal experiment design,94 can guide experimentation to lower this uncertainty by selecting the experimental space that we need to explore to inform predictive models and gain a holistic view of an organism's physiological behavior.
Furthermore, the lack of widely adopted standards in metadata characterization prevents the efficient integration of such information across different studies. Often this process requires extensive manual labor to curate literature. There have been suggestions on standardizing the way of describing experimental metadata.95,96 Still we are in need of a more structured approach if we aspire to use the resulting datasets for training machine learning algorithms and predictive models. For example, a specific minimal medium called M9 used for growing bacteria can be created with different concentrations of each nutrient, while the meta-data information may not mention it and reading the corresponding publication may be necessary. Similarly, the growth stage in which cultured bacteria are profiled is not mentioned anywhere despite its significance.
Quality assurance and quality control (QA/QC) for processed data
Overview
We provide an overview of the QA/QC procedure at the final stage of omics data processing (Fig. 4). We focus on this stage than the two previous stages because the final stage of QC has a lot more commonalities than earlier QCs, which are heavily dependent on instruments and processing methods that are used at each step. We can classify the final-stage QA/QC methods based on the availability of control data into supervised and unsupervised.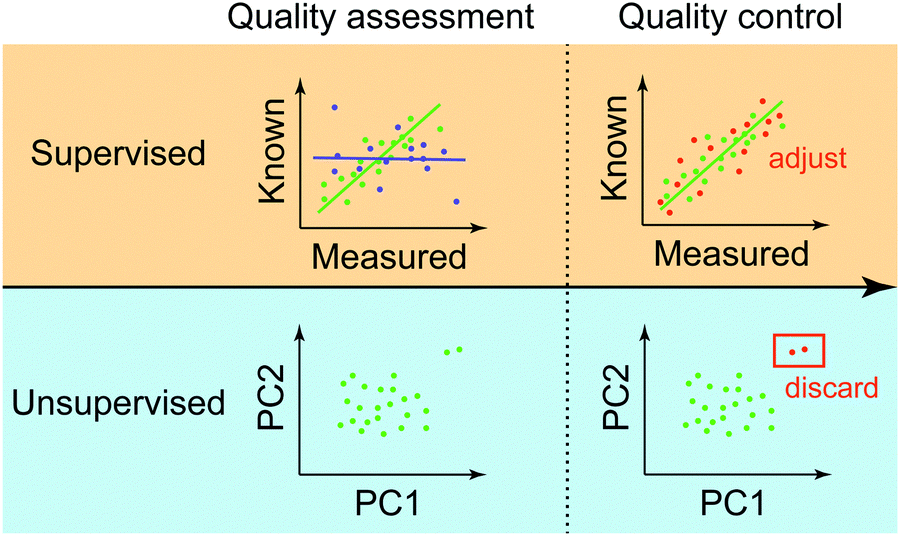 | ||
| Fig. 4 Quality assessment and quality control. For ensuring data quality, there exist supervised and unsupervised approaches. Supervised approaches rely on control data that can be considered as high-quality measurements (e.g. qPCR). In this way, the quality of data is determined based on how well high-throughput and low-throughput measurements are correlated. Highly correlated data (green points) are further used for bias correction in cases where the corresponding control measurements are missing. In the case of the unsupervised approach, measurements of molecules across multiple profiles are first clustered, as in the case of principal component analysis (PCA). Clustering and dimensionality reduction techniques can reveal outliers (like the two outliers in the figure), i.e. points that are distinctively far away from the clusters, possibly due to low-quality samples. In the quality control step, the outliers can be discarded to avoid any artefactual results arising from the outliers. The PC on the x-axis refers to the corresponding principal component. | ||
Supervised QA methods
Supervised approaches rely on control data with known and accurate molecular measurements. In this way, the quality of data is determined based on the goodness of fit between the processed and known molecular abundances in the control data. As shown in Fig. 4, data exhibiting high correlation (green points) may be used as anchors to adjust measurements of other molecules that don’t have corresponding control measurements based on the measured correlation. Typically, two major types of control data are used to assess and to adjust genome-wide measurements. One is called “spike-in control”, where known concentrations of selected molecules are profiled together with high-throughput experiments. This approach has been extensively used in different platforms to profile transcripts,97 proteins,98–101 and metabolites.102 In the case of genotypes such as SNPs, control data can be samples having known genotypes, for example, of individuals precisely studied from a large consortium (e.g. HapMap or 1000 Genomes Project). Another approach is to generate high-quality measurements of selected molecular species from the same isolate in an independent setting (e.g. qPCR as a control for RNA-Seq datasets).Unsupervised approach
In the unsupervised approach, the quality of molecular measurements is compared based on “relative” criteria. That is, parameters (e.g. expression levels of genes) consisting of samples are compared with each other to evaluate the data quality. The most popular method in this category is the clustering of multiple genome-wide experiments. This way, the clustering results can show how normal your experiment is compared to other experiments if all experiments arise from the same condition (a combination of environment, genotype, and phenotype). For example, the two outliers in Fig. 4 are distinctively far away from a major group, which “might” be an indication that those are of low-quality. During the quality control step, these outliers can be discarded to avoid any artefactual results. The unsupervised way of administering the quality of data has been widely used in a variety of omics data ranging from genome (e.g.ref. 103 based on PCA), transcriptome (e.g.ref. 104 based on clustering), and the integration of multi-omics data with knowledge.105 One limitation of the unsupervised approach, however, is its sensitivity to noise and bias due to data paucity and process variation.Predictive modeling and analytics
Overview
Machine learning analytics has been applied in biology to deal with the intrinsic complexity in omics data with a long history and its integration in recent years. The high-level overview of the machine-learning analytic pipeline for integrated multi-omics data is shown in Fig. 5 and consists of data preprocessing, modeling, and active learning. In this review, rather than extensively exploring all steps in the pipeline of predictive analytics, which has been studied elsewhere, we focus on surveying recent applications of machine-learning methods over integrated omics data (Table 4) and organize them based on characteristics of problems in the context of omics integration.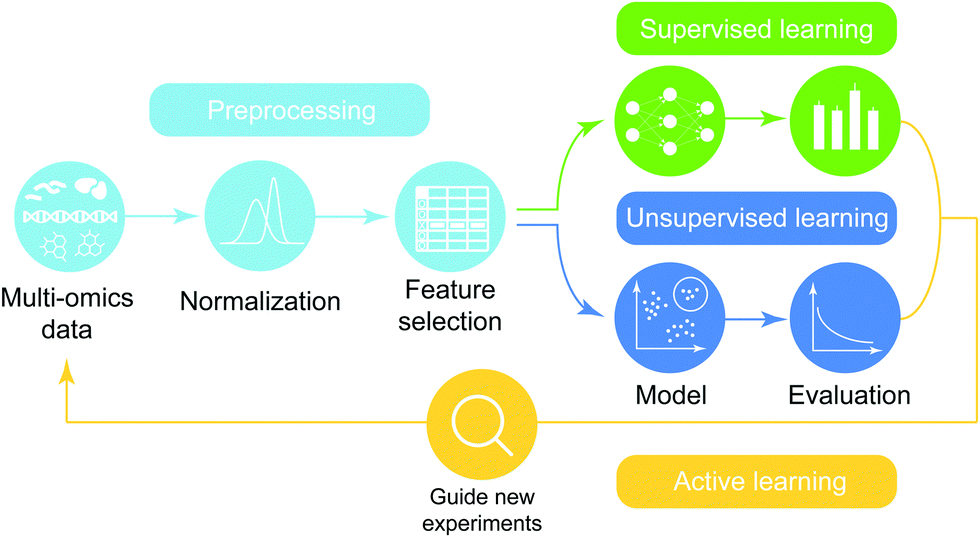 | ||
| Fig. 5 Iterative framework of predictive analytics over multi-omics data. In the preprocessing step, normalization and feature selection are performed for multi-omics data before model training and evaluation to make models more generalizable, predictive and less vulnerable to noise. Then supervised/unsupervised learning is performed to build a model from training data and then the performance of the model is evaluated by various criteria. The final model is then used to guide new experiments, thereby adding new multi-omics to the original dataset. | ||
| Output layer | Problems | Category | Methods and References | Input omics-types | |||||
|---|---|---|---|---|---|---|---|---|---|
| G | T | P | M | E | I | ||||
| Genome | – SNP imputation | Supervised | Shallow learning202 | O | |||||
| – Gene structure annotation | Supervised | 108 | O | ||||||
| – Alternative splicing and splice site | Supervised | 109 | O | ||||||
| – Promoter binding site | Supervised | 110 | O | ||||||
| – Essential genes | Supervised | 111 | O | O | |||||
| Transcriptome | – RNA structure | Supervised | 112 | O | |||||
| – eQTL | Supervised | 113 and 114 | O | ||||||
| – Pre-mRNA splicing | Supervised | 115 | O | ||||||
| – Gene expression | Supervised | 116 | O | O | |||||
| Proteome | – Protein function | Supervised | 118 | O | |||||
| 117 | O | ||||||||
| 119 | O | ||||||||
| – Secondary structure | Supervised | 120 | O | ||||||
| – Metal binding site | Supervised | 121 | O | ||||||
| – Glycosylation site | Supervised | 122 | O | ||||||
| – Subcellular organization | Supervised | 123 | O | ||||||
| – Post-translational modification | Supervised | 124 and 125 | O | ||||||
| Metabolome | – Substructure | Supervised | 126 | O | |||||
| – Metabolite type | Supervised | 127 | O | ||||||
| Epigenome | – Chromatin state | Supervised | Deep learning128,129 | O | |||||
| Shallow learning130 | O | ||||||||
| Unsupervised | 162 and 163 | O | |||||||
| – Methylated CpG | Supervised | 131 | O | ||||||
| Interactome | – Gene–gene interaction | Supervised | 132 | O | |||||
| – Protein/DNA-binding | Supervised | Deep learning134 | O | ||||||
| Shallow learning133 | O | ||||||||
| Unsupervised | 36 | O | |||||||
| – Gene-regulatory network | Supervised | 135 | O | ||||||
| Unsupervised | 160 | O | |||||||
| – Protein–protein interaction | Supervised | Active learning136 | O | ||||||
| 137–139 | O | ||||||||
| Unsupervised | 159 | O | |||||||
| – Protein/RNA-binding | Supervised | Deep learning134 | O | ||||||
| Shallow learning203 | O | ||||||||
| – Signaling network | Supervised | 140 | O | ||||||
| – Metabolic pathway | Supervised | 141 | O | O | |||||
| Phenome | – Microbial phenotype prediction | Supervised | Deep learning204 | O | |||||
| Ensemble8 | O | ||||||||
| Shallow142 | O | ||||||||
| Ensemble205 | O | ||||||||
| – Plant phenotype prediction | Supervised | 10 | O | ||||||
| Ensemble143 | O | O | O | ||||||
| – Human phenotype prediction | Supervised | Ensemble206 | O | ||||||
| Active learning167 | O | ||||||||
| 144 | O | ||||||||
| 145 | O | ||||||||
| 146 and 147 | O | ||||||||
| 148 | O | ||||||||
| 149 | O | O | |||||||
| – Biomarker | Supervised | 150 | O | ||||||
| Unsupervised | 158 | O | |||||||
| – Novel sub-phenotype identification | Unsupervised | 154 | O | ||||||
| 155 | O | ||||||||
| 156 | O | ||||||||
| 157 | O | ||||||||
| 207 | O | O | |||||||
| – Phylogenetic relationships | Supervised | Bayesian151 | O | ||||||
| Unsupervised | 161 | O | |||||||
Data preprocessing
First, the multi-omics data are normalized to ensure that the downstream analysis handles data effectively. For example, scaled data are prone to convergence when gradient descent is used. Feature normalization is another important topic and it is covered extensively in ref. 106. The next step is feature selection, which is to decide the subset of features that are useful for modeling. In supervised settings, relevant methods use similarity measures such as mutual information and Pearson correlation coefficient, while unsupervised approaches such as principal component analysis (PCA) are popular. Regularization techniques can supplement these methods107 and a relevant review on this is ref. 6.Modeling
(1) Genome, transcriptome, proteome, metabolome, and epigenome: the problems in predicting a genomic layer include imputation of SNPs, and annotation of a variety characteristics in genome including gene structure,108 splice site,109 and promoter binding site.110 Typical input of such problems is either of genome sequences or of genetic variants. Furthermore, essential genes of bacteria are predicted with support-vector machine using sequence characteristics and the co-expression pattern in transcriptome profiles.111 With regard to transcriptome output, the problems in predicting the transcriptome layer include prediction of RNA structure112 and prediction of eQTL113,114 given genome data. In addition to this, pre-mRNA splicing events were predicted with an ensemble of machine learning methods from genome data.115 Expression levels of transcripts were predicted from genetic and epigenetic signatures using a deep neural network.116 The problems in predicting the proteome layer include prediction of different characteristics of proteins. For example, protein function has been predicted using omics data from different layers,117–119 and other examples include secondary structure,120 metal binding site,121 glycosylation site,122 subcellular localization,123 and post-translational modification124,125 given proteome sequence data. For metabolome prediction, the primary goal is the prediction of metabolite substructure and functional type given metabolome information.126,127 For epigenome prediction, inferring chromatin state from non-coding variants has been of great interest as characterization of the functional effect remains a challenge. This has been investigated with different methods including deep learning methods128,129 and support vector machine.130 Furthermore, predicting methylated CpG from genome sequences has been studied with a long history.131
(2) Interactome: prediction of gene–gene interactions has been studied using many different machine-learning approaches (please see the review in Ref. 132 for more information). Moreover, prediction of protein/DNA binding events given genome sequences has been studied using a kernel-based method133 and a convolutional neural network.134 Another network type is a map of transcriptional regulation, which has been inferred based on transcriptome data using ensemble learning.135 Protein–protein interaction (PPI) networks have been predicted using random forest trained over previously identified PPIs136 and transcriptome dataset.137–139 The signaling network and metabolic pathways are predicted using decision tree over transcriptome dataset140 and a variety of machine learning methods over genome dataset,141 respectively.
(3) Phenome output: phenotype prediction is perhaps one of the most heavily studied subjects among others in this category as understanding genotype–phenotype relationships is a fundamental goal in biology. This can be sub-classified into bacterial phenotype prediction (e.g. growth rate prediction from transcriptome142), plant phenotype (e.g. stress prediction from three omics-types using ensemble learning143), and human phenotype (e.g. prediction of disease outcome and drug response from different omics-types144–148 and from integration149). Moreover, biomarker prediction has been studied using support vector machine trained with proteomic data.150 Finally, phylogenetic relationships between different organisms have been largely investigated based on genome sequences, for example, by the Bayesian approach.151
To investigate how well methods can use different omics data to predict phenotypic characteristics in novel environments, we curated the prediction performance of AUC reported in the literature in recent years and the results (Fig. 6) show that the reported predictability largely fluctuates depending on the types of problems (AUC: 0.81 ± 0.13). The prediction performance of phenotype more dramatically changes with the specific type of phenotype, compared to prediction of other types of omics data. The most challenging problem is the prediction of Parkinson's disease (AUC: 0.56) and pancreatic cancer (AUC: 0.58) from genomic signatures among the others we compared. Gene–gene interaction (AUC: 0.75) and protein function are some of the hardest problems among the others we curated. Furthermore, several machine learning methods have been used across different prediction problems. The list includes regression-based methods, Naive Bayes, Support Vector Machine, KNN, Ensemble method, and Neural network. The Ensemble method was mostly used among others. This is expected as the prediction based on multiple models is known to outperform one that relies on a single model. Interestingly, the regression-based methods (e.g. logistic regression, LASSO) were heavily used in the prediction of phenotype. Recent studies combine complex models for the same prediction problems although without much success, possibly due to the challenges involved in integration of two or more omics data sources.152
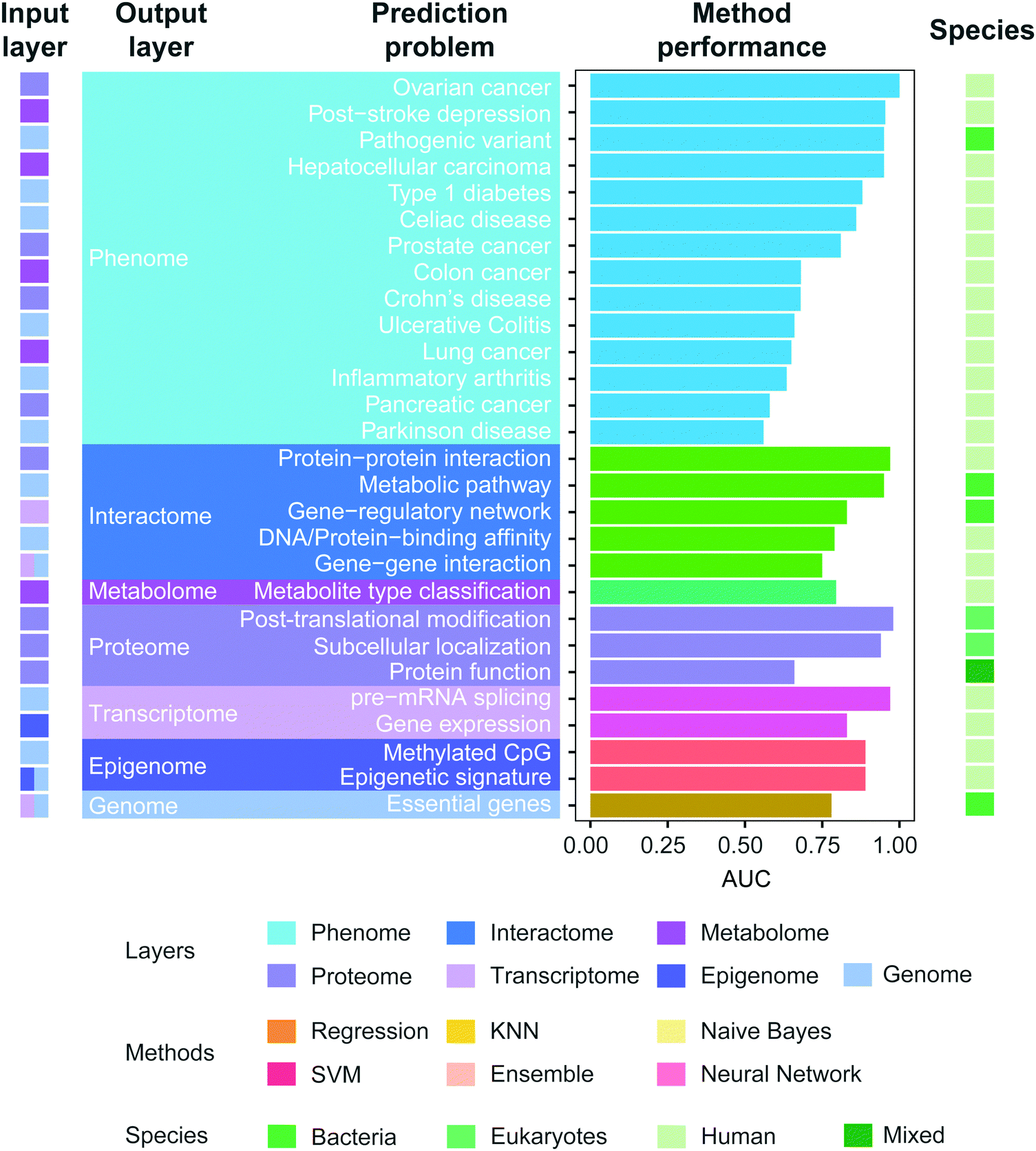 | ||
| Fig. 6 Prediction performance of multi-omics models. We curated the literature of multi-omics models published between 2007 and 2017 (Table S1, ESI†). We collected the reported AUC values and validated that the reported performance is indeed the highest for that specific problem, by also curating any articles citing the referenced publication. We only investigated the articles providing AUC of their methods. In the case where the authors reported multiple performance results in various settings, the highest AUC was included, while for different sub-tasks of the same prediction problem, we included their average performance. References of the listed prediction problems: colon cancer,208 hepatocellular carcinoma,209 post-stroke depression,210 lung cancer,211 ovarian cancer,212 prostate cancer,212 pancreatic cancer,212 celiac disease,213 Crohn's disease,213 ulcerative colitis,213 type 1 diabetes,214 Parkinson's disease,215 inflammatory arthritis,216 pathogenic variant,204 epigenetic signature,217 methylated CpG,131 DNA/protein-binding affinity,218 protein–protein interaction,219 gene–gene interaction,220 gene-regulatory network,221 metabolic pathway,141 post-translational modification (glycosylation site),122 protein function,222 subcellular localization,223 metabolite type classification,127 essential genes,111 pre-mRNA splicing,115 gene expression.116 | ||
Active learning
Once a model is constructed and evaluated, active learning guides what experiments to perform next to minimize uncertainty in the model.93 It was first actively studied in supervised setting, and more methods have been developed in unsupervised setting in recent years.165,166 Active learning is particularly a significant problem in the experimental design of genome-wide profiling because of high cost in data generation (Table 1). For example, the human protein–protein interaction network was actively learned based on random forest.136 Another example includes ref. 167, which argues that cancer classification can be improved with active profiling of transcriptome signatures based on ML methods such as Support Vector Machines. A recent review covers the topics of active learning on experimental design for uncovering molecular interactions.168Other classes of machine learning methods
Challenges and limitations
There are many caveats in machine learning analytics that must be carefully administered. A wide range of issues have been addressed in past reviews including overfitting, imbalanced class size, and the curse of dimensionality.6 To be brief, the overfitting problem arises when too complex a model (i.e. with a large number of parameters) is trained over a few data points and the trained model doesn’t behave well with unseen objects. This problem is related to the curse of dimensionality because most of the overfitting issues arise from too many parameters to fit compared to given data points, which can be overcome by feature selection before training a model. Moreover, imbalanced class size is a widespread problem in many applications of machine learning to omics data. This refers to the phenomenon that a trained model preferably assigns a specific class label due to highly skewed distribution of class labels. Many computational remedies have been devised to cure such biased prediction including weighting more cost in incorrect predictions to a minor class.178Applications
Machine-learning analytics over integrated multi-omics data has the capacity to make far-reaching impacts across multiple industries. In medical applications, finding therapeutic targets and biomarkers is one of the major issues in human health,5 and such efforts are being more and more translated into the real world (e.g. BERG, Eagle Genomics). Antibiotic resistance is of paramount importance as it is considered a global threat and machine learning methods can be applied for predicting antibiotic resistance from the molecular signature of clinical isolates to select effective antibiotics.8 Biotechnological applications include optimization of genetic and regulatory processes to produce maximum yield of a certain substance, which can be enabled by a prediction model trained over omics data.9 In agriculture, identifying stress response genes is of great significance in crop management and machine learning can accelerate such discovery.10,11 Finally, in food and nutrition science, optimizing nutrition treatment for individuals is enabled by machine learning over personal omics data accompanied with dietary information.12 Furthermore, machine learning and multi-omics analytics can be used in food engineering for producing the best quality of fermentation foods with desired flavors13 once genome-wide profiles collected over the course of fermentation process are available.Next wave and future directions
The ability to generate high-throughput omics data and to build intelligent systems based on large-scale data and convoluted knowledge has revolutionized the way we conduct biology. Most genome-wide technologies provide averages across population of cells, which ignores variability at individual cells.79 High-resolution understanding of molecular signatures in a cell is enabled with single-cell assays,179 which are being expanded to interrogate the multi-omics landscape of a cell.79 Furthermore, advances in community-level profiling of molecules (e.g. metagenome sequencing) facilitate the investigation of biodiversity that is directly collected from the environment, which is not possible with conventional cultivation-based technologies, and the type of molecular species that can be profiled by such advancement is becoming more diverse.180–183Accompanied with the explosion of available data, rapid advances in the development of cognitive systems facilitated by artificial intelligence (AI) are revolutionizing many industries. For example, IBM Watson that was first developed for human-like question-answering is expanding for supporting decision of experts in different domains ranging from healthcare to finance.184 We believe that biology is not an exception to this ongoing paradigm shifting. That is, we envision building a cognitive system for every single organism that has the ability to process data, transfer information, bring new knowledge, represent a knowledge map of the organism in a structured way and suggest new experiments based on machine-produced hypotheses.185,186 We firmly believe that such systems can be a powerful assistant that can empower, rather than replace, humans in their pursuit of scientific knowledge.
Conflicts of interest
There are no conflicts to declare.References
- P. Simon, Too Big to Ignore: The Business Case for Big Data, John Wiley & Sons, 2013, vol. 72 Search PubMed.
- A. R. Joyce and B. O. Palsson, The model organism as a system: integrating ‘omics’ data sets, Nat. Rev. Mol. Cell Biol., 2006, 7(3), 198–210 CrossRef CAS PubMed.
- M. Bersanelli, et al., Methods for the integration of multi-omics data: mathematical aspects, BMC Bioinf., 2016, 17(suppl 2), 15 CrossRef PubMed.
- M. Kim, et al., Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli, Nat. Commun., 2016, 7, 13090 CrossRef CAS PubMed.
- A. Ahmad and H. Fröhlich, Integrating Heterogeneous omics Data via Statistical Inference and Learning Techniques, Genomics and Computational Biology, 2016, 2(1), e32 CrossRef.
- M. W. Libbrecht and W. S. Noble, Machine learning applications in genetics and genomics, Nat. Rev. Genet., 2015, 16(6), 321–332 CrossRef CAS PubMed.
- C. Angermueller, et al., Deep learning for computational biology, Mol. Syst. Biol., 2016, 12(7), 878 CrossRef PubMed.
- J. J. Davis, et al., Antimicrobial Resistance Prediction in PATRIC and RAST, Sci. Rep., 2016, 6, 27930 CrossRef CAS PubMed.
- L. J. Sweetlove, R. L. Last and A. R. Fernie, Predictive metabolic engineering: a goal for systems biology, Plant Physiol., 2003, 132(2), 420–425 CrossRef CAS PubMed.
- R. Shaik and W. Ramakrishna, Machine learning approaches distinguish multiple stress conditions using stress-responsive genes and identify candidate genes for broad resistance in rice, Plant Physiol., 2014, 164(1), 481–495 CrossRef CAS PubMed.
- C. Ma, H. H. Zhang and X. Wang, Machine learning for Big Data analytics in plants, Trends Plant Sci., 2014, 19(12), 798–808 CrossRef CAS PubMed.
- D. Zeevi, et al., Personalized Nutrition by Prediction of Glycemic Responses, Cell, 2015, 163(5), 1079–1094 CrossRef CAS PubMed.
- R. F. Schwan and A. E. Wheals, The microbiology of cocoa fermentation and its role in chocolate quality, Crit. Rev. Food Sci. Nutr., 2004, 44(4), 205–221 CrossRef CAS PubMed.
- N. J. Loman, et al., Performance comparison of benchtop high-throughput sequencing platforms, Nat. Biotechnol., 2012, 30(5), 434–439 CrossRef CAS PubMed.
- Y. Kodama, et al., The Sequence Read Archive: explosive growth of sequencing data, Nucleic Acids Res., 2012, 40(Database issue), D54–D56 CrossRef CAS PubMed.
- E. Clough and T. Barrett, The Gene Expression Omnibus Database, Methods Mol. Biol., 2016, 1418, 93–110 Search PubMed.
- M. D. Mailman, et al., The NCBI dbGaP database of genotypes and phenotypes, Nat. Genet., 2007, 39(10), 1181–1186 CrossRef CAS PubMed.
- T. Weirick, et al., The identification and characterization of novel transcripts from RNA-seq data, Briefings Bioinf., 2016, 17(4), 678–685 CrossRef PubMed.
- Z. Wang, M. Gerstein and M. Snyder, RNA-Seq: a revolutionary tool for transcriptomics, Nat. Rev. Genet., 2009, 10(1), 57–63 CrossRef CAS PubMed.
- Seqc/Maqc-Iii Consortium, A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium, Nat. Biotechnol., 2014, 32(9), 903–914 CrossRef PubMed.
- N. Kolesnikov, et al., ArrayExpress update-simplifying data submissions, Nucleic Acids Res., 2015, 43(Database issue), D1113–D1116 CrossRef CAS PubMed.
- E. S. Witze, et al., Mapping protein post-translational modifications with mass spectrometry, Nat. Methods, 2007, 4(10), 798–806 CrossRef CAS PubMed.
- M. Brosch, et al., Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome, Genome Res., 2011, 21(5), 756–767 CrossRef CAS PubMed.
- M. Wilhelm, et al., Mass-spectrometry-based draft of the human proteome, Nature, 2014, 509(7502), 582–587 CrossRef CAS PubMed.
- A. Schmidt, et al., The quantitative and condition-dependent Escherichia coli proteome, Nat. Biotechnol., 2016, 34(1), 104–110 CrossRef CAS PubMed.
- J. E. Elias, et al., Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations, Nat. Methods, 2005, 2(9), 667–675 CrossRef CAS PubMed.
- P. Jones, et al., PRIDE: a public repository of protein and peptide identifications for the proteomics community, Nucleic Acids Res., 2006, 34(Database issue), D659–D663 CrossRef CAS PubMed.
- J. A. Vizcaino, et al., ProteomeXchange provides globally coordinated proteomics data submission and dissemination, Nat. Biotechnol., 2014, 32(3), 223–226 CrossRef CAS PubMed.
- E. J. Want, B. F. Cravatt and G. Siuzdak, The expanding role of mass spectrometry in metabolite profiling and characterization, ChemBioChem, 2005, 6(11), 1941–1951 CrossRef CAS PubMed.
- Z. Lei, D. V. Huhman and L. W. Sumner, Mass spectrometry strategies in metabolomics, J. Biol. Chem., 2011, 286(29), 25435–25442 CrossRef CAS PubMed.
- J. M. Buscher, et al., Cross-platform comparison of methods for quantitative metabolomics of primary metabolism, Anal. Chem., 2009, 81(6), 2135–2143 CrossRef CAS PubMed.
- N. S. Kale, et al., MetaboLights: An Open-Access Database Repository for Metabolomics Data, Curr. Protoc. Bioinformatics, 2016, 53, 14 Search PubMed.
- M. Baker, Proteomics: the interaction map, Nature, 2012, 484(7393), 271–275 CrossRef CAS PubMed.
- B. Suter, et al., Next-Generation Sequencing for Binary Protein–Protein Interactions, Front. Genet., 2015, 6, 346 Search PubMed.
- J. De Las Rivas and C. Fontanillo, Protein–protein interactions essentials: key concepts to building and analyzing interactome networks, PLoS Comput. Biol., 2010, 6(6), e1000807 Search PubMed.
- T. S. Furey, ChIP-seq and beyond: new and improved methodologies to detect and characterize protein–DNA interactions, Nat. Rev. Genet., 2012, 13(12), 840–852 CrossRef CAS PubMed.
- D. S. Johnson, et al., Genome-wide mapping of in vivo protein–DNA interactions, Science, 2007, 316(5830), 1497–1502 CrossRef CAS PubMed.
- H. S. Rhee and B. F. Pugh, Comprehensive genome-wide protein–DNA interactions detected at single-nucleotide resolution, Cell, 2011, 147(6), 1408–1419 CrossRef CAS PubMed.
- D. Szklarczyk, et al., STRINGv10: protein–protein interaction networks, integrated over the tree of life, Nucleic Acids Res., 2015, 43(Database issue), D447–D452 CrossRef CAS PubMed.
- A. Chatr-aryamontri, et al., The BioGRID interaction database: 2017 update, Nucleic Acids Res., 2017, 45(D1), D369–D379 CrossRef PubMed.
- D. Szklarczyk and L. J. Jensen, Protein–protein interaction databases, Methods Mol. Biol., 2015, 1278, 39–56 CAS.
- M. J. Heller, DNA microarray technology: devices, systems, and applications, Annu. Rev. Biomed. Eng., 2002, 4, 129–153 CrossRef CAS PubMed.
- Y. F. Leung and D. Cavalieri, Fundamentals of cDNA microarray data analysis, Trends Genet., 2003, 19(11), 649–659 CrossRef CAS PubMed.
- M. L. Metzker, Sequencing technologies – the next generation, Nat. Rev. Genet., 2010, 11(1), 31–46 CrossRef CAS PubMed.
- X. Han, A. Aslanian and J. R. Yates, 3rd, Mass spectrometry for proteomics, Curr. Opin. Chem. Biol., 2008, 12(5), 483–490 CrossRef CAS PubMed.
- K. Dettmer, P. A. Aronov and B. D. Hammock, Mass spectrometry-based metabolomics, Mass Spectrom. Rev., 2007, 26(1), 51–78 CrossRef CAS PubMed.
- J. Quackenbush, Microarray data normalization and transformation, Nat. Genet., 2002, 32(suppl), 496–501 CrossRef CAS PubMed.
- H. J. Ruskin, Computational Modeling and Analysis of Microarray Data: New Horizons, Microarrays, 2016, 5, 4 CrossRef PubMed.
- D. B. Allison, et al., Microarray data analysis: from disarray to consolidation and consensus, Nat. Rev. Genet., 2006, 7(1), 55–65 CrossRef CAS PubMed.
- T. H. Yang, 13C-based metabolic flux analysis: fundamentals and practice, Methods Mol. Biol., 2013, 985, 297–334 CAS.
- M. D. Ritchie, et al., Methods of integrating data to uncover genotype–phenotype interactions, Nat. Rev. Genet., 2015, 16(2), 85–97 CrossRef CAS PubMed.
- D. Rajasundaram and J. Selbig, More effort – more results: recent advances in integrative ‘omics’ data analysis, Curr. Opin. Plant Biol., 2016, 30, 57–61 CrossRef CAS PubMed.
- Q. Zhu, et al., Targeted exploration and analysis of large cross-platform human transcriptomic compendia, Nat. Methods, 2015, 12(3), 211–214 CrossRef CAS PubMed.
- M. Moretto, et al., COLOMBOSv3.0: leveraging gene expression compendia for cross-species analyses, Nucleic Acids Res., 2016, 44(D1), D620–D623 CrossRef CAS PubMed.
- J. Rudy and F. Valafar, Empirical comparison of cross-platform normalization methods for gene expression data, BMC Bioinf., 2011, 12, 467 CrossRef PubMed.
- S. A. McCarroll, et al., Integrated detection and population-genetic analysis of SNPs and copy number variation, Nat. Genet., 2008, 40(10), 1166–1174 CrossRef CAS PubMed.
- International HapMap 3 Consortium, Integrating common and rare genetic variation in diverse human populations, Nature, 2010, 467(7311), 52–58 CrossRef PubMed.
- H. Yang and K. Wang, Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR, Nat. Protoc., 2015, 10(10), 1556–1566 CrossRef CAS PubMed.
- G. Parra, K. Bradnam and I. Korf, CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes, Bioinformatics, 2007, 23(9), 1061–1067 CrossRef CAS PubMed.
- T. Lu, et al., Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq, Genome Res., 2010, 20(9), 1238–1249 CrossRef CAS PubMed.
- V. N. Bhatia, et al., Software tool for researching annotations of proteins: open-source protein annotation software with data visualization, Anal. Chem., 2009, 81(23), 9819–9823 CrossRef CAS PubMed.
- M. Chagoyen and F. Pazos, Tools for the functional interpretation of metabolomic experiments, Briefings Bioinf., 2013, 14(6), 737–744 CrossRef PubMed.
- J. Xia, et al., MetaboAnalyst 3.0-making metabolomics more meaningful, Nucleic Acids Res., 2015, 43(W1), W251–W257 CrossRef CAS PubMed.
- P. D. Karp, et al., The EcoCyc Database, EcoSal Plus, 2014, 6, 1 CrossRef CAS PubMed.
- D. Swarbreck, et al., The Arabidopsis Information Resource (TAIR): gene structure and function annotation, Nucleic Acids Res., 2008, 36(Database issue), D1009–D1014 CAS.
- J. M. Cherry, et al., Saccharomyces Genome Database: the genomics resource of budding yeast, Nucleic Acids Res., 2012, 40(Database issue), D700–D705 CrossRef CAS PubMed.
- A. Hamosh, et al., Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders, Nucleic Acids Res., 2005, 33(Database issue), D514–D517 CrossRef CAS PubMed.
- E. Boutet, et al., UniProtKB/Swiss-Prot, Methods Mol. Biol., 2007, 406, 89–112 CAS.
- C. O'Donovan, et al., High-quality protein knowledge resource: SWISS-PROT and TrEMBL, Briefings Bioinf., 2002, 3(3), 275–284 CrossRef.
- A. Gattiker, et al., Automated annotation of microbial proteomes in SWISS-PROT, Comput. Biol. Chem., 2003, 27(1), 49–58 CrossRef CAS PubMed.
- M. R. Viant, et al., How close are we to complete annotation of metabolomes?, Curr. Opin. Chem. Biol., 2017, 36, 64–69 CrossRef CAS PubMed.
- S. A. Teichmann and M. M. Babu, Gene regulatory network growth by duplication, Nat. Genet., 2004, 36(5), 492–496 CrossRef CAS PubMed.
- J. Schellenberger, et al., BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions, BMC Bioinf., 2010, 11, 213 CrossRef PubMed.
- C. Liu, QTL Mapping of Molecular Traits for Studies of Human Complex Diseases, Applied Computational Genomics, Springer, 2012, pp. 61–82 Search PubMed.
- D. Kumar, et al., Integrating transcriptome and proteome profiling: strategies and applications, Proteomics, 2016, 16(19), 2533–2544 CrossRef CAS PubMed.
- A. I. Nesvizhskii, Proteogenomics: concepts, applications and computational strategies, Nat. Methods, 2014, 11(11), 1114–1125 CrossRef CAS PubMed.
- P. Jullian Fabres, et al., A concise review on multi-omics data integration for terroir analysis in Vitis vinifera, Front. Recent Dev. Plant Sci., 2017, 8, 1065 CrossRef PubMed.
- D. J. Beale, A. V. Karpe and W. Ahmed, Beyond Metabolomics: A Review of Multi-Omics-Based Approaches, Microbial Metabolomics, Springer, 2016, pp. 289–312 Search PubMed.
- C. Bock, M. Farlik and N. C. Sheffield, Multi-Omics of Single Cells: Strategies and Applications, Trends Biotechnol., 2016, 34(8), 605–608 CrossRef CAS PubMed.
- E. Montague, et al., Beyond protein expression, MOPED goes multi-omics, Nucleic Acids Res., 2015, 43(Database issue), D1145–D1151 CrossRef CAS PubMed.
- W. H. Chen, et al., Integration of multi-omics data of a genome-reduced bacterium: Prevalence of post-transcriptional regulation and its correlation with protein abundances, Nucleic Acids Res., 2016, 44(3), 1192–1202 CrossRef CAS PubMed.
- C. Vogel and E. M. Marcotte, Insights into the regulation of protein abundance from proteomic and transcriptomic analyses, Nat. Rev. Genet., 2012, 13(4), 227–232 CAS.
- S. Wachi, K. Yoneda and R. Wu, Interactome–transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues, Bioinformatics, 2005, 21(23), 4205–4208 CrossRef CAS PubMed.
- X. Wang and B. Zhang, Integrating genomic, transcriptomic, and interactome data to improve Peptide and protein identification in shotgun proteomics, J. Proteome Res., 2014, 13(6), 2715–2723 CrossRef CAS PubMed.
- M. A. Moreno-Risueno, W. Busch and P. N. Benfey, Omics meet networks-using systems approaches to infer regulatory networks in plants, Curr. Opin. Plant Biol., 2010, 13(2), 126–131 CrossRef PubMed.
- M. W. Covert, et al., Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli, Bioinformatics, 2008, 24(18), 2044–2050 CrossRef CAS PubMed.
- J. M. Lee, et al., Dynamic analysis of integrated signaling, metabolic, and regulatory networks, PLoS Comput. Biol., 2008, 4(5), e1000086 Search PubMed.
- E. Yeger-Lotem, et al., Network motifs in integrated cellular networks of transcription–regulation and protein–protein interaction, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(16), 5934–5939 CrossRef CAS PubMed.
- X. Wu, et al., Network-based global inference of human disease genes, Mol. Syst. Biol., 2008, 4, 189 CrossRef PubMed.
- Y. V. Sun, Integration of biological networks and pathways with genetic association studies, Hum. Genet., 2012, 131(10), 1677–1686 CrossRef PubMed.
- C. J. Mitchell, et al., A multi-omic analysis of human naive CD4 + T cells, BMC Syst. Biol., 2015, 9, 75 CrossRef PubMed.
- J. N. Weinstein, et al., The Cancer Genome Atlas Pan-Cancer analysis project, Nat. Genet., 2013, 45(10), 1113–1120 CrossRef PubMed.
- B. Settles, Active learning literature survey, University of Wisconsin, Madison, 2010, vol. 52(55–66), p. 11 Search PubMed.
- M. Alipoor, et al., Optimal Experiment Design for Monoexponential Model Fitting: Application to Apparent Diffusion Coefficient Imaging, BioMed Res. Int., 2015, 2015, 138060 Search PubMed.
- L. N. Soldatova and R. D. King, An ontology of scientific experiments, J. R. Soc., Interface, 2006, 3(11), 795–803 CrossRef PubMed.
- A. Brazma, Minimum information about a microarray experiment (MIAME)-successes, failures, challenges, Sci. World J., 2009, 9, 420–423 CrossRef CAS PubMed.
- J. Loven, et al., Revisiting global gene expression analysis, Cell, 2012, 151(3), 476–482 CrossRef CAS PubMed.
- B. Hoekman, et al., msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies, Mol. Cell. Proteomics, 2012, 11(6), M111 015974 Search PubMed.
- C. C. Tsou, et al., IDEAL-Q, an automated tool for label-free quantitation analysis using an efficient peptide alignment approach and spectral data validation, Mol. Cell. Proteomics, 2010, 9(1), 131–144 CAS.
- B. Valot, et al., MassChroQ: a versatile tool for mass spectrometry quantification, Proteomics, 2011, 11(17), 3572–3577 CrossRef CAS PubMed.
- H. P. Benton, et al., XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization, Anal. Chem., 2008, 80(16), 6382–6389 CrossRef CAS PubMed.
- P. Franceschi, et al., A benchmark spike-in data set for biomarker identification in metabolomics, J. Chemom., 2012, 26(1–2), 16–24 CrossRef CAS.
- C. A. Anderson, et al., Data quality control in genetic case-control association studies, Nat. Protoc., 2010, 5(9), 1564–1573 CrossRef CAS PubMed.
- T. Raman, et al., Quality control in microarray assessment of gene expression in human airway epithelium, BMC Genomics, 2009, 10, 493 CrossRef PubMed.
- S. Yoo, et al., MODMatcher: multi-omics data matcher for integrative genomic analysis, PLoS Comput. Biol., 2014, 10(8), e1003790 Search PubMed.
- S. Aksoy and R. M. Haralick, Feature normalization and likelihood-based similarity measures for image retrieval, Pattern Recognit. Lett., 2001, 22(5), 563–582 CrossRef.
- H. Zou and T. Hastie, Regularization and variable selection via the elastic net, J. R. Stat. Soc. Series B, Stat. Methodol., 2005, 67(2), 301–320 CrossRef.
- G. Ratsch, et al., Improving the Caenorhabditis elegans genome annotation using machine learning, PLoS Comput. Biol., 2007, 3(2), e20 Search PubMed.
- S. Sonnenburg, et al., Accurate splice site prediction using support vector machines, BMC Bioinf., 2007, 8(suppl 10), S7 CrossRef PubMed.
- F. Anwar, et al., Pol II promoter prediction using characteristic 4-mer motifs: a machine learning approach, BMC Bioinf., 2008, 9, 414 CrossRef PubMed.
- K. Plaimas, R. Eils and R. König, Identifying essential genes in bacterial metabolic networks with machine learning methods, BMC Syst. Biol., 2010, 4(1), 56 CrossRef PubMed.
- B. A. Shapiro, et al., Bridging the gap in RNA structure prediction, Curr. Opin. Struct. Biol., 2007, 17(2), 157–165 CrossRef CAS PubMed.
- M. Ackermann, et al., Teamwork: improved eQTL mapping using combinations of machine learning methods, PLoS One, 2012, 7(7), e40916 CAS.
- T. Huang and Y. D. Cai, An information-theoretic machine learning approach to expression QTL analysis, PLoS One, 2013, 8(6), e67899 CAS.
- X. Jian, E. Boerwinkle and X. Liu, In silico prediction of splice-altering single nucleotide variants in the human genome, Nucleic Acids Res., 2014, 42(22), 13534–13544 CrossRef CAS PubMed.
- J. Li, et al., Using epigenomics data to predict gene expression in lung cancer, BMC Bioinf., 2015, 16(5), S10 CrossRef PubMed.
- L. Han, et al., Recent progresses in the application of machine learning approach for predicting protein functional class independent of sequence similarity, Proteomics, 2006, 6(14), 4023–4037 CrossRef CAS PubMed.
- V. G. Krishnan and D. R. Westhead, A comparative study of machine-learning methods to predict the effects of single nucleotide polymorphisms on protein function, Bioinformatics, 2003, 19(17), 2199–2209 CrossRef CAS PubMed.
- R. Sharan, I. Ulitsky and R. Shamir, Network-based prediction of protein function, Mol. Syst. Biol., 2007, 3, 88 CrossRef PubMed.
- M. Agathocleous, et al., Protein Secondary Structure Prediction with Bidirectional Recurrent Neural Nets: Can Weight Updating for Each Residue Enhance Performance? in Artificial Intelligence Applications and Innovations: 6th IFIP WG 12.5 International Conference, AIAI 2010, Larnaca, Cyprus, October 6–7, 2010. Proceedings, ed. H. Papadopoulos, A. S. Andreou, and M. Bramer, 2010, Springer Berlin Heidelberg, Berlin, Heidelberg, pp. 128–137.
- M. Brylinski and J. Skolnick, FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level, Proteins, 2011, 79(3), 735–751 CrossRef CAS PubMed.
- C. Caragea, et al., Glycosylation site prediction using ensembles of Support Vector Machine classifiers, BMC Bioinf., 2007, 8(1), 438 CrossRef PubMed.
- Z. Lu, et al., Predicting subcellular localization of proteins using machine-learned classifiers, Bioinformatics, 2004, 20(4), 547–556 CrossRef CAS PubMed.
- S. Li, et al., Predicting O-glycosylation sites in mammalian proteins by using SVMs, Comput. Biol. Chem., 2006, 30(3), 203–208 CrossRef CAS PubMed.
- G. Bologna, et al., N-Terminal myristoylation predictions by ensembles of neural networks, Proteomics, 2004, 4(6), 1626–1632 CrossRef CAS PubMed.
- J. Hummel, et al., Decision tree supported substructure prediction of metabolites from GC-MS profiles, Metabolomics, 2010, 6(2), 322–333 CrossRef CAS PubMed.
- M. J. Embrechts and S. Ekins, Classification of metabolites with kernel-partial least squares (K-PLS), Drug Metab. Dispos., 2007, 35(3), 325–327 CrossRef CAS PubMed.
- J. Zhou and O. G. Troyanskaya, Predicting effects of noncoding variants with deep learning-based sequence model, Nat. Methods, 2015, 12(10), 931–934 CrossRef CAS PubMed.
- D. R. Kelley, J. Snoek and J. L. Rinn, Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks, Genome Res., 2016, 26(7), 990–999 CrossRef CAS PubMed.
- M. Ghandi, et al., Enhanced regulatory sequence prediction using gapped k-mer features, PLoS Comput. Biol., 2014, 10(7), e1003711 Search PubMed.
- M. Bhasin, et al., Prediction of methylated CpGs in DNA sequences using a support vector machine, FEBS Lett., 2005, 579(20), 4302–4308 CrossRef CAS PubMed.
- B. A. McKinney, et al., Machine learning for detecting gene–gene interactions: a review, Appl. Bioinf., 2006, 5(2), 77–88 CrossRef CAS.
- N. Bhardwaj, et al., Kernel-based machine learning protocol for predicting DNA-binding proteins, Nucleic Acids Res., 2005, 33(20), 6486–6493 CrossRef CAS PubMed.
- B. Alipanahi, et al., Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning, Nat. Biotechnol., 2015, 33, 831–838 CrossRef CAS PubMed.
- D. Marbach, et al., Wisdom of crowds for robust gene network inference, Nat. Methods, 2012, 9(8), 796–804 CrossRef CAS PubMed.
- T. P. Mohamed, J. G. Carbonell and M. K. Ganapathiraju, Active learning for human protein–protein interaction prediction, BMC Bioinf., 2010, 11(suppl 1), S57 CrossRef PubMed.
- J. D. Han, et al., Evidence for dynamically organized modularity in the yeast protein–protein interaction network, Nature, 2004, 430(6995), 88–93 CrossRef CAS PubMed.
- N. Bhardwaj and H. Lu, Correlation between gene expression profiles and protein–protein interactions within and across genomes, Bioinformatics, 2005, 21(11), 2730–2738 CrossRef CAS PubMed.
- R. Jansen, et al., A Bayesian networks approach for predicting protein-protein interactions from genomic data, Science, 2003, 302(5644), 449–453 CrossRef CAS PubMed.
- S. Hautaniemi, et al., Modeling of signal-response cascades using decision tree analysis, Bioinformatics, 2005, 21(9), 2027–2035 CrossRef CAS PubMed.
- J. M. Dale, L. Popescu and P. D. Karp, Machine learning methods for metabolic pathway prediction, BMC Bioinf., 2010, 11, 15 CrossRef PubMed.
- E. M. Airoldi, et al., Predicting cellular growth from gene expression signatures, PLoS Comput. Biol., 2009, 5(1), e1000257 Search PubMed.
- A. Acharjee, et al., Integration of multi-omics data for prediction of phenotypic traits using random forest, BMC Bioinf., 2016, 17(suppl 5), 180 CrossRef PubMed.
- M. Xu, et al., Automated multidimensional phenotypic profiling using large public microarray repositories, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(30), 12323–12328 CrossRef CAS PubMed.
- H. W. Ressom, et al., Classification algorithms for phenotype prediction in genomics and proteomics, Front. Biosci., 2008, 13, 691–708 CrossRef CAS.
- L. C. Kenny, et al., Novel biomarkers for pre-eclampsia detected using metabolomics and machine learning, Metabolomics, 2005, 1(3), 227–234 CrossRef.
- S. Mahadevan, et al., Analysis of metabolomic data using support vector machines, Anal. Chem., 2008, 80(19), 7562–7570 CrossRef CAS PubMed.
- M. P. Menden, et al., Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties, PLoS One, 2013, 8(4), e61318 CAS.
- L. C. Stetson, et al., Computational identification of multi-omic correlates of anticancer therapeutic response, BMC Genomics, 2014, 15(suppl 7), S2 CrossRef PubMed.
- M. Wagner, et al., Computational protein biomarker prediction: a case study for prostate cancer, BMC Bioinf., 2004, 5, 26 CrossRef PubMed.
- G. McGuire, M. C. Denham and D. J. Balding, MAC5: Bayesian inference of phylogenetic trees from DNA sequences incorporating gaps, Bioinformatics, 2001, 17(5), 479–480 CrossRef CAS PubMed.
- T. T. Wu, et al., Genome-wide association analysis by lasso penalized logistic regression, Bioinformatics, 2009, 25(6), 714–721 CrossRef CAS PubMed.
- H. B. Barlow, Unsupervised learning, Neural Comput., 1989, 1(3), 295–311 CrossRef.
- J. Lapointe, et al., Gene expression profiling identifies clinically relevant subtypes of prostate cancer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(3), 811–816 CrossRef CAS PubMed.
- S. J. Deeb, et al., Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles, Mol. Cell. Proteomics, 2012, 11(5), 77–89 CAS.
- P. Chinnaiyan, et al., The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism, Cancer Res., 2012, 72(22), 5878–5888 CrossRef CAS PubMed.
- M. E. Figueroa, et al., DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia, Cancer Cell, 2010, 17(1), 13–27 CrossRef CAS PubMed.
- M. Lauten, et al., Unsupervised proteome analysis of human leukaemia cells identifies the Valosin-containing protein as a putative marker for glucocorticoid resistance, Leukemia, 2006, 20(5), 820–826 CrossRef CAS PubMed.
- C. C. Friedel, J. Krumsiek and R. Zimmer, Bootstrapping the Interactome: Unsupervised Identification of Protein Complexes in Yeast, in Research in Computational Molecular Biology: 12th Annual International Conference, RECOMB 2008, Singapore, March 30 – April 2, 2008. Proceedings, ed. M. Vingron and L. Wong, 2008, Springer Berlin Heidelberg: Berlin, Heidelberg, pp. 3–16.
- T. Schaffter, D. Marbach and D. Floreano, GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods, Bioinformatics, 2011, 27(16), 2263–2270 CrossRef CAS PubMed.
- N. Zamani, et al., Unsupervised genome-wide recognition of local relationship patterns, BMC Genomics, 2013, 14, 347 CrossRef CAS PubMed.
- M. M. Hoffman, et al., Unsupervised pattern discovery in human chromatin structure through genomic segmentation, Nat. Methods, 2012, 9(5), 473–476 CrossRef CAS PubMed.
- J. Ernst and M. Kellis, Discovery and characterization of chromatin states for systematic annotation of the human genome, Nat. Biotechnol., 2010, 28(8), 817–825 CrossRef CAS PubMed.
- M. Halkidi, Y. Batistakis and M. Vazirgiannis, On clustering validation techniques, J. Intell. Inf. Syst., 2001, 17(2–3), 107–145 CrossRef.
- S. Berardo, E. Favero and N. Neto, Active Learning with Clustering and Unsupervised Feature Learning, Canadian Conference on Artificial Intelligence, Springer, Cham, 2015 Search PubMed.
- H. Steck and T. S. Jaakkola, Unsupervised active learning in large domains, Proceedings of the Eighteenth conference on Uncertainty in artificial intelligence, Morgan Kaufmann Publishers Inc, 2002 Search PubMed.
- Y. Liu, Active learning with support vector machine applied to gene expression data for cancer classification, J. Chem. Inf. Comput. Sci., 2004, 44(6), 1936–1941 CrossRef CAS PubMed.
- Y. Sverchkov and M. Craven, A review of active learning approaches to experimental design for uncovering biological networks, PLoS Comput. Biol., 2017, 13(6), e1005466 Search PubMed.
- T. P. Nguyen and T. B. Ho, Detecting disease genes based on semi-supervised learning and protein-protein interaction networks, Artif. Intell. Med., 2012, 54(1), 63–71 CrossRef PubMed.
- N. Zhao, et al., Determining effects of non-synonymous SNPs on protein–protein interactions using supervised and semi-supervised learning, PLoS Comput. Biol., 2014, 10(5), e1003592 Search PubMed.
- D. Kim, et al., Knowledge boosting: a graph-based integration approach with multi-omics data and genomic knowledge for cancer clinical outcome prediction, J. Am. Med. Inform. Assoc., 2015, 22(1), 109–120 Search PubMed.
- L. P. Kaelbling, M. L. Littman and A. W. Moore, Reinforcement learning: a survey, J. Intell. Inf. Syst., 1996, 4, 237–285 Search PubMed.
- A. Tsoukalas, T. Albertson and I. Tagkopoulos, From data to optimal decision making: a data-driven, probabilistic machine learning approach to decision support for patients with sepsis, JMIR Med. Inform., 2015, 3(1), e11 CrossRef PubMed.
- Y. LeCun, Y. Bengio and G. Hinton, Deep learning, Nature, 2015, 521(7553), 436–444 CrossRef CAS PubMed.
- P. Mamoshina, et al., Applications of Deep Learning in Biomedicine, Mol. Pharmaceutics, 2016, 13(5), 1445–1454 CrossRef CAS PubMed.
- S. Min, B. Lee and S. Yoon, Deep learning in bioinformatics, Briefings Bioinf., 2017, 18(5), 851–869 Search PubMed.
- T. Ching, et al., Opportunities And Obstacles For Deep Learning In Biology And Medicine, bioRxiv, 2017, p. 142760.
- W. Liu and S. Chawla, Class confidence weighted knn algorithms for imbalanced data sets, Advances in Knowledge Discovery and Data Mining, Springer, 2011, pp. 345–356 Search PubMed.
- D. Wang and S. Bodovitz, Single cell analysis: the new frontier in ‘omics’, Trends Biotechnol., 2010, 28(6), 281–290 CrossRef CAS PubMed.
- E. A. Rebollar, et al., Using “Omics” and Integrated Multi-Omics Approaches to Guide Probiotic Selection to Mitigate Chytridiomycosis and Other Emerging Infectious Diseases, Front. Microbiol., 2016, 7, 68 Search PubMed.
- J. Hultman, et al., Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes, Nature, 2015, 521(7551), 208–212 CrossRef CAS PubMed.
- A. Heintz-Buschart, et al., Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes, Nat. Microbiol., 2016, 2, 16180 CrossRef CAS PubMed.
- E. A. Franzosa, et al., Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling, Nat. Rev. Microbiol., 2015, 13(6), 360–372 CrossRef CAS PubMed.
- Y. Chen, J. D. Elenee Argentinis and G. Weber, IBM Watson: How Cognitive Computing Can Be Applied to Big Data Challenges in Life Sciences Research, Clin. Ther., 2016, 38(4), 688–701 CrossRef PubMed.
- R. D. King, et al., Functional genomic hypothesis generation and experimentation by a robot scientist, Nature, 2004, 427(6971), 247–252 CrossRef CAS PubMed.
- R. D. King, et al., The automation of science, Science, 2009, 324(5923), 85–89 CrossRef CAS PubMed.
- J. Shendure and H. Ji, Next-generation DNA sequencing, Nat. Biotechnol., 2008, 26(10), 1135–1145 CrossRef CAS PubMed.
- D. Sims, et al., Sequencing depth and coverage: key considerations in genomic analyses, Nat. Rev. Genet., 2014, 15(2), 121–132 CrossRef CAS PubMed.
- M. Bantscheff, et al., Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present, Anal. Bioanal. Chem., 2012, 404(4), 939–965 CrossRef CAS PubMed.
- L. Chen, G. Wu and H. Ji, hmChIP: a database and web server for exploring publicly available human and mouse ChIP-seq and ChIP-chip data, Bioinformatics, 2011, 27(10), 1447–1448 CrossRef CAS PubMed.
- M. E. Cusick, et al., Literature-curated protein interaction datasets, Nat. Methods, 2009, 6(1), 39–46 CrossRef CAS PubMed.
- R. H. Davis, The age of model organisms, Nat. Rev. Genet., 2004, 5(1), 69–76 CrossRef CAS PubMed.
- S. B. Hedges, The origin and evolution of model organisms, Nat. Rev. Genet., 2002, 3(11), 838–849 CrossRef CAS PubMed.
- G. Parmigiani, et al., The analysis of gene expression data: an overview of methods and software, The analysis of gene expression data, Springer, 2003, pp. 1–45 Search PubMed.
- C. S. Wilhelm-Benartzi, et al., Review of processing and analysis methods for DNA methylation array data, Br. J. Cancer, 2013, 109(6), 1394–1402 CrossRef CAS PubMed.
- M. Garber, et al., Computational methods for transcriptome annotation and quantification using RNA-seq, Nat. Methods, 2011, 8(6), 469–477 CrossRef CAS PubMed.
- R. Nielsen, et al., Genotype and SNP calling from next-generation sequencing data, Nat. Rev. Genet., 2011, 12(6), 443–451 CrossRef CAS PubMed.
- J. R. Miller, S. Koren and G. Sutton, Assembly algorithms for next-generation sequencing data, Genomics, 2010, 95(6), 315–327 CrossRef CAS PubMed.
- H. Kim, et al., A short survey of computational analysis methods in analysing ChIP-seq data, Hum. Genomics, 2011, 5(2), 117–123 CrossRef CAS PubMed.
- A. I. Nesvizhskii, O. Vitek and R. Aebersold, Analysis and validation of proteomic data generated by tandem mass spectrometry, Nat. Methods, 2007, 4(10), 787–797 CrossRef CAS PubMed.
- M. Katajamaa and M. Oresic, Data processing for mass spectrometry-based metabolomics, J. Chromatogr. A, 2007, 1158(1–2), 318–328 CrossRef CAS PubMed.
- E. Halperin and D. A. Stephan, SNP imputation in association studies, Nat. Biotechnol., 2009, 27(4), 349–351 CrossRef CAS PubMed.
- Y. D. Cai and S. L. Lin, Support vector machines for predicting rRNA-, RNA-, and DNA-binding proteins from amino acid sequence, Biochim. Biophys. Acta, 2003, 1648(1–2), 127–133 CrossRef CAS.
- D. Quang, Y. Chen and X. Xie, DANN: a deep learning approach for annotating the pathogenicity of genetic variants, Bioinformatics, 2015, 31(5), 761–763 CrossRef CAS PubMed.
- M. Kim, V. Zorraquino and I. Tagkopoulos, Microbial forensics: predicting phenotypic characteristics and environmental conditions from large-scale gene expression profiles, PLoS Comput. Biol., 2015, 11(3), e1004127 Search PubMed.
- M. Kim and S. H. Kim, Empirical prediction of genomic susceptibilities for multiple cancer classes, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(5), 1921–1926 CrossRef CAS PubMed.
- C. Curtis, et al., The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups, Nature, 2012, 486(7403), 346–352 CAS.
- L. Deng, et al., Development and Validation of a High-Throughput Mass Spectrometry Based Urine Metabolomic Test for the Detection of Colonic Adenomatous Polyps, Metabolites, 2017, 7(3), 32 CrossRef PubMed.
- R. Gao, et al., Serum metabolomics to identify the liver disease-specific biomarkers for the progression of hepatitis to hepatocellular carcinoma, Sci. Rep., 2015, 5, 18175 CrossRef CAS PubMed.
- J. Xiao, et al., Discriminating poststroke depression from stroke by nuclear magnetic resonance spectroscopy-based metabonomic analysis, Neuropsychiatr. Dis. Treat., 2016, 12, 1919 CrossRef PubMed.
- T. Ligor, Ł. Pater and B. Buszewski, Application of an artificial neural network model for selection of potential lung cancer biomarkers, J. Breath Res., 2015, 9(2), 027106 CrossRef PubMed.
- T. Nguyen, et al., Mass spectrometry cancer data classification using wavelets and genetic algorithm, FEBS Lett., 2015, 589(24), 3879–3886 CrossRef CAS PubMed.
- D. Speed and D. J. Balding, MultiBLUP: improved SNP-based prediction for complex traits, Genome Res., 2014, 24(9), 1550–1557 CrossRef CAS PubMed.
- C. Kooperberg, M. LeBlanc and V. Obenchain, Risk prediction using genome-wide association studies, Genet. Epidemiol., 2010, 34(7), 643–652 CrossRef PubMed.
- F. Mittag, et al., Use of support vector machines for disease risk prediction in genome-wide association studies: concerns and opportunities, Hum. Mutat., 2012, 33(12), 1708–1718 CrossRef CAS PubMed.
- S. J. Schrodi, et al., Genetic-based prediction of disease traits: prediction is very difficult, especially about the future, Front. Genet., 2014, 5, 162 Search PubMed.
- J. Zhou and O. G. Troyanskaya, Predicting effects of noncoding variants with deep learning-based sequence model, Nat. Methods, 2015, 12(10), 931–934 CrossRef CAS PubMed.
- B. Alipanahi, et al., Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning, Nat. Biotechnol., 2015, 33(8), 831–838 CrossRef CAS PubMed.
- Y.-A. Huang, et al., Sequence-based prediction of protein–protein interactions using weighted sparse representation model combined with global encoding, BMC Bioinf., 2016, 17(1), 184 CrossRef PubMed.
- X. Lu, et al., Predicting human genetic interactions from cancer genome evolution, PLoS One, 2015, 10(5), e0125795 Search PubMed.
- S. R. Maetschke, et al., Supervised, semi-supervised and unsupervised inference of gene regulatory networks, Briefings Bioinf., 2013, 15(2), 195–211 CrossRef PubMed.
- P. Radivojac, et al., A large-scale evaluation of computational protein function prediction, Nat. Methods, 2013, 10(3), 221–227 CrossRef CAS PubMed.
- K. Lee, et al., Protein networks markedly improve prediction of subcellular localization in multiple eukaryotic species, Nucleic Acids Res., 2008, 36(20), e136 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mo00051k |
| This journal is © The Royal Society of Chemistry 2018 |