
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous photo(electro)catalysis with eumelanin thin films†
Ludovico
Migliaccio
a,
Maciej
Gryszel
bc,
Vedran
Đerek
bcd,
Alessandro
Pezzella
 aef and
Eric Daniel
Głowacki
*bc
aef and
Eric Daniel
Głowacki
*bc
aDepartment of Chemical Sciences, University of Naples “Federico II”, Naples, Italy
bLaboratory of Organic Electronics, ITN Norrköping, Linköping University, Norrköping, Sweden
cWallenberg Centre for Molecular Medicine, Linköping University, Linköping, Sweden. E-mail: eric.glowacki@liu.se
dCenter of Excellence for Advanced Materials and Sensing Devices, Ruđer Bošković Institute, Zagreb, Croatia
eInstitute for Polymers, Composites and Biomaterials (IPCB), CNR, Pozzuoli (Na), Italy
fNational Interuniversity Consortium of Materials Science and Technology (INSTM), Florence, Italy
First published on 14th August 2018
Abstract
We report that eumelanin, the ubiquitous natural pigment found in most living organisms, is a photocatalytic material. Though the photoconductivity of eumelanin and its photochemical reactions with oxygen have been known for some time, eumelanins have not been regarded as photofaradaic materials. We find that eumelanin shows photocathodic behavior for both the oxygen reduction reaction and the hydrogen evolution reaction. Eumelanin films irradiated in aqueous solutions at pH 2 or 7 with simulated solar light photochemically reduce oxygen to hydrogen peroxide with accompanying oxidation of sacrificial oxalate, formate, or phenol. Autooxidation of the eumelanin competes with the oxidation of donors. Deposition of thin films on electrodes yields photoelectrodes with higher photocatalytic stability compared with the case of pure photocatalysis, implicating the successful extraction of positive charges from the eumelanin layer. These results open up new potential applications for eumelanin as a photocatalytically-active biomaterial, and inform the growing fundamental body of knowledge about the physical chemistry of eumelanins.
Conceptual insightsMelanins are biopigments ubiquitous in plants and animals. Thier unusual physical properties have made them the most-studied biomacromolecules in the context of (opto)electronic behavior. In recent years, the physical understanding of these complex materials has converged and melanins are playing an increasing role in emerging bioelectronic devices. In this article we present a completely different and previously unexplored aspect of melanin – its photocatalytic properties. We show detailed evidence that melanin performs as a classic semiconductor photocatalyst, participating in a true photocatalytic redox cycle of oxygen reduction and oxidation of various donors. While previous research has elaborated the possibility of photochemical generation of peroxide, and more recently the photoconductivity of melanin has been explained in detail, the discovery of true photocatalytic cycles being possible in melanin, including those featuring extraction of holes, is a novel conceptual insight. Our work represents a milestone in understanding the biomacromolecule melanin, and shows the wholly-new application of photo(electro)catalysts. This represents a major finding about melanins and opens up a new research direction of using biomolecules as potential photocatalysts for solar-to-chemical energy conversion, or harnessing this photochemistry for biomedical purposes. |
Introduction
The electrical properties of biomolecules such as proteins,1–3 DNA,4 polysaccharides,5,6 and biopigments7–9 have attracted a growing amount of scientific interest in recent years. This attention is motivated by expanding fundamental understanding of the roles electronic transport may play in biology, as well as the possibility of using bio-origin materials as active components in bioelectronics devices.10,11 The long history of research into eumelanins, the most common organic pigmentary materials found in nature, is as complex as the chemistry and physics of eumelanins themselves. From the biophysical perspective, the physiological roles played by eumelanins have been the topic of the most scrutiny.12 Agreement exists that the primary purpose of eumelanin is as a photoprotectant, as it is ideally suited to absorbing potentially damaging high energy photons.13,14 Eumelanins have however been implicated in other important biochemical processes, and are also crucial for mediating light management in the mammalian eye.15 Since the seminal work of McGinness16 on electrical transport in eumelanin in 1974, many have endeavored to exploit eumelanin as a natural active material for bioelectronics.10,17–19 Understanding the nature of eumelanin conductivity is complicated by its heterogeneous nature, and two models for its structure exist: the extended heteromacromolecule model versus the stacked oligomer model, where planar molecular units assemble via π–π stacking. The nature of the conductivity of eumelanin remains debated, though the recent consensus is that observations of macroscopic conductivity in eumelanin are primarily protonic in nature. Meredith et al. and Santato et al. have elaborated a charge transport model in a series of papers20–22 showing the relationship between protons as the major charge carrier and the paramagnetic species observed in eumelanin, with the phenomenon of photoconductivity in eumelanin also being accounted for in ref. 23. There are two distinct radical species in eumelanin, a carbon-centered and semiquinone one, with the latter being responsible for protonic photoconductivity and implicated in potential photochemical reactivity.24 Several devices exploiting protonic transport in eumelanin thin films have been demonstrated,22,25 though there is also evidence that there may be electronic charge carrier mobility in eumelanin in the form of hopping negative charges stored in the semiquinone structure.26 Eumelanin thin films can be electrochemically-addressed and intercalated with ions,27 thus they have been used in sodium ion batteries.18It has been recently demonstrated that some organic semiconducting small molecules can act as aqueous photocatalysts28,29 or photoelectrocatalysts30,31 for O2 reduction to H2O2. A starting off question for this work is: can eumelanin be a bio-origin material with intrinsic photocatalytic activity? The photochemical properties of eumelanins have been studied in the biological context. Sarna et al. discovered the photochemical production of hydrogen peroxide via oxygen reduction during eumelanin autooxidation,32,33 inspiring detailed investigation into the role of eumelanin interactions with reactive oxygen species (ROS). In his 1990 paper, Sarna noted that the molar quantity of H2O2 produced during the photochemical degradation of eumelanin was 20% higher than the molar equivalent of the starting monomers. This foreshadows the possibility of a photochemical redox cycle where photoexcited electrons in eumelanin reduce oxygen to hydrogen peroxide, while the photogenerated positive species oxidize some other donor. It follows that these holes can be, in principle, thanks to the (photo)redox properties of the pigment, neutralized by electrons originating from an electrode to complete the cycle. In this work, we provide evidence that it is indeed the case that eumelanin behaves in a way analogous to a semiconductor photocatalyst, and is suitable for photoelectrodes as well. Eumelanin can behave as a photocathode in both oxygenated and deoxygenated conditions, demonstrating higher catalytic stability than in the case of pure photocatalytic conditions. This shows that eumelanin can be electrochemically addressed by an underlying electrode with an impressive effectiveness.
Results and discussion
As a commonly-used standard eumelanin, we prepared synthetic eumelanin thin films by spin coating a 30 mg mL−1 5,6-dihydroxyindole (DHI) methanolic solution onto PET foil substrates followed by an ammonia-induced solid-state polymerization19 procedure (Fig. 1a). For photochemical experiments, eumelanin-coated foil circles were placed in solutions at two different pH values (pH = 2, pH = 7) in the presence and in the absence of different sacrificial electron donors (phenol, formate, and oxalate). The “blank” condition, referring to electrolyte alone, indicates that oxygen reduction to hydrogen peroxide, or proton reduction to H2, can be accompanied by the autooxidation of eumelanin itself. If a photocatalytic redox cycle can be supported, more peroxide should be produced in conditions with added donors, and the eumelanin must show higher stability. The hypothesized photo-redox processes with eumelanin as a photocatalyst are illustrated in Fig. 1b. The two pH values were chosen since neutral pH is relevant to biological applications of melanin and also corresponds to the majority of existing experimental data on melanin photochemistry, and low pH was chosen to see if the proton-enhanced reaction of peroxide production (or hydrogen evolution) proceeded more efficiently. High pH conditions cannot be tested for photochemical experiments, as the eumelanin degrades in alkaline conditions (pH > 9).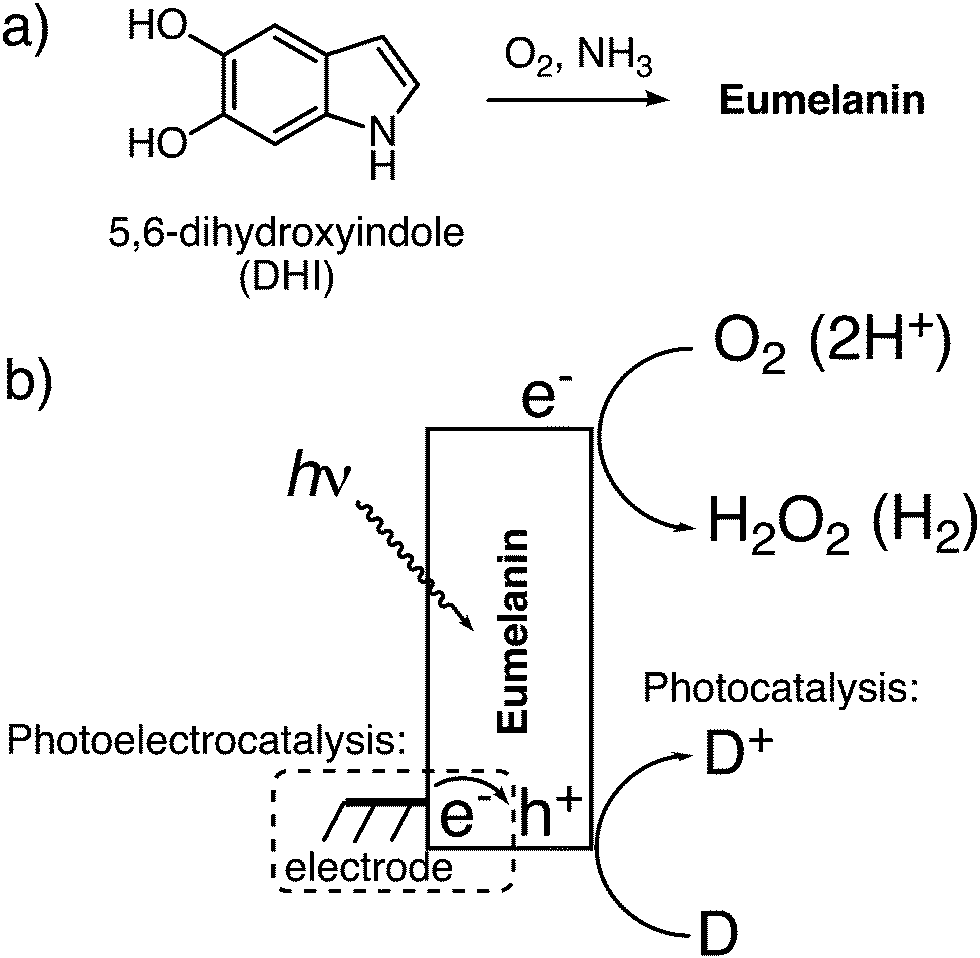 | ||
| Fig. 1 Eumelanin as a photocatalyst. (a) Eumelanin thin films obtained via oxidation of 5,6-dihydroxyindole (DHI) were used in this study as a model for natural eumelanin. (b) Schematic of eumelanin photocatalysis, where photogenerated electrons and holes participate in redox reactions. Reduction of oxygen to hydrogen peroxide proceeds in oxygenated conditions, or reduction of protons to hydrogen in deoxygenated conditions. Holes can oxidize suitable electron donors, or be neutralized by electrons injected by an electrode in the case of photoelectrocatalysis. If the hole is not quenched by an electron, it will lead to eumelanin autooxidation. | ||
We first conducted photolysis experiments by irradiation of eumelanin films in water under a pure O2 atmosphere, using a solar simulator light source with an irradiance of 1.05 sun. (Fig. 2a). Aliquots of solution were periodically removed to test for the evolution of hydrogen peroxide using the tetramethyl benzidine/horseradish peroxidase (TMB/HRP) assay, well known from biological applications (see Experimental methods section and Fig. S1, ESI†).34 The results for hydrogen peroxide evolution over 12 hours are shown in Fig. 2b. Samples with added electron donors yield about twice as much peroxide as “blank” samples. To quantify the difference between the contribution of eumelanin autooxidation and the oxidation of donors, we calculated the catalytic turnover number (TON) expressed as nmol of H2O2 generated/nmol DHI (starting indole equivalent) regarding a simplified picture in which DHI acts as monomer and eumelanin as its polymer, although the picture would be more complex.12 The quantity of degraded DHI is calculated from measurement of the optical absorbance of the eumelanin in the film before and after the entire process and comparing to the absorbance of standard curves, as detailed in the Experimental methods section. The TONs, along with max final [H2O2] values are given in Table 1. The presence of donors increases the TON in all cases, giving values 2–4 times higher than eumelanin alone (autooxidation). This result confirms the presence of a true photo-redox cycle in eumelanin (Fig. 1b).
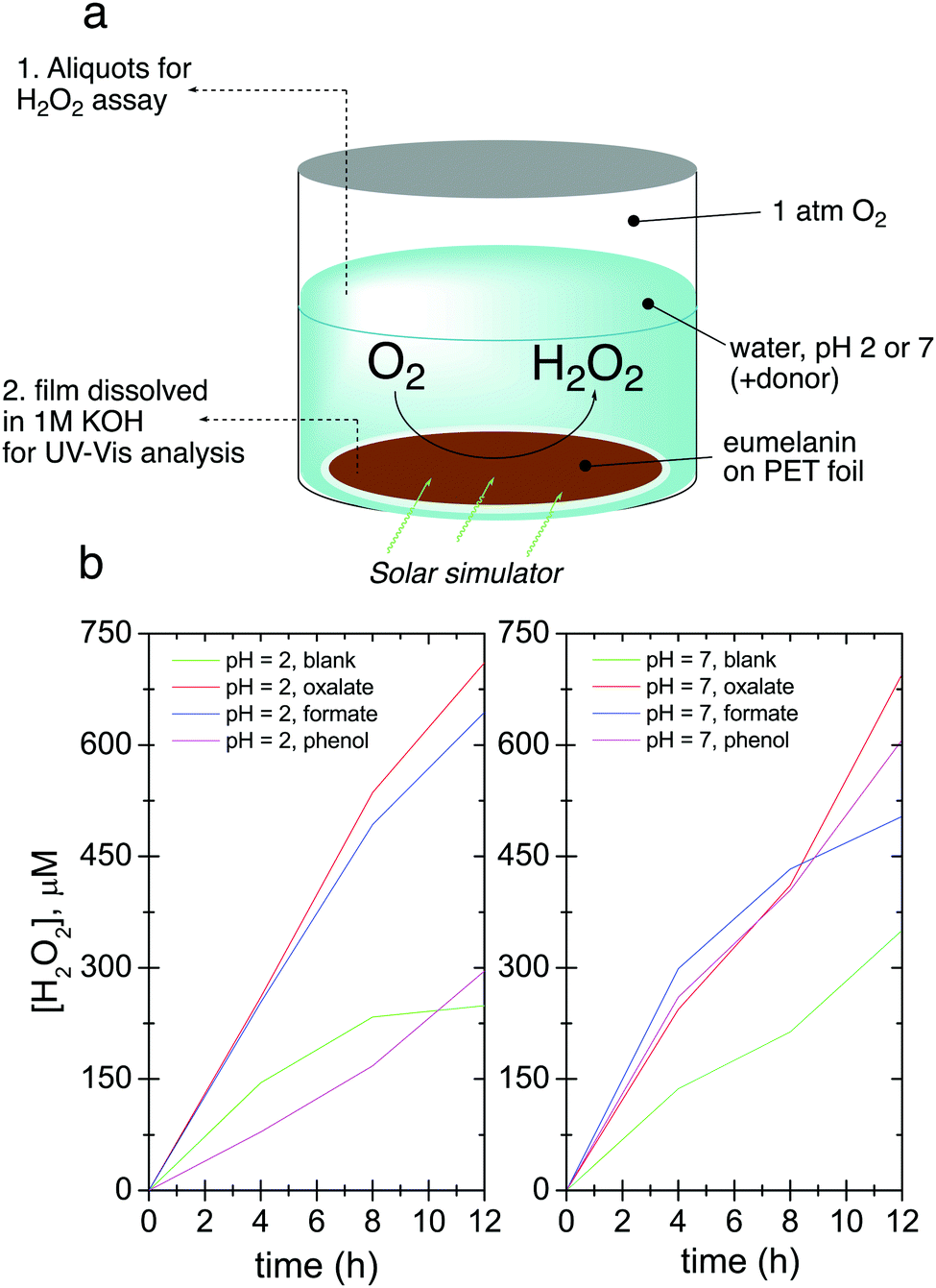 | ||
| Fig. 2 Photochemical evolution of hydrogen peroxide by eumelanin. (a) The experiment involves irradiating eumelanin films in an oxygenated aqueous solution with a broad-spectrum light source and removing aliquots to determine H2O2 concentration. At the end of the experiment, the film is removed and dissolved to determine the amount of degraded eumelanin by measuring optical absorption. (b) The amount of evolved H2O2 over time for conditions of pH 2 and pH 7, without donor (blank case) and with readily-oxidizable donors oxalate, formate, and phenol. | ||
| pH | Donor | [H2O2], μM | Catalytic TON [nH2O2/nDHI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) consumed] consumed] |
|---|---|---|---|
| 2 | Blank | 248.8 | 1.0 |
| Oxalate | 710.8 | 3.7 | |
| Formate | 644.0 | 1.7 | |
| Phenol | 295.8 | 1.6 | |
| 7 | Blank | 350.6 | 1.5 |
| Oxalate | 695.5 | 2.2 | |
| Formate | 503.9 | 3.3 | |
| Phenol | 606.8 | 3.9 | |
Concluding that a photochemical redox cycle is possible, and considering that eumelanin has been reported to possess photoconductivity,23,24 it follows that it may also be possible to have a photoelectrochemical catalytic behavior. In this case, photoexcited electrons participate in oxygen reduction, or hydrogen evolution, while carriers originating from the underlying electrode neutralize the resultant positive holes. To this end, we deposited eumelanin films on fluorine-doped tin oxide (FTO), on which we found eumelanin had good adhesion. Photoelectrolysis was carried out with a three-electrode potentiostat using FTO/eumelanin as the working electrode, and the same light source as for the aforementioned photocatalysis experiments. The cathode chamber, containing an O2 inlet and also an Ag/AgCl reference electrode, was separated from the anodic chamber (Pt wire anode) with a Nafion membrane (Fig. S2, ESI†). The Nafion membrane is critical to prevent the diffusion of H2O2 to the anode chamber, as it would otherwise be oxidized on the counter electrode. In chronoamperometric conditions at a potential of 0 V, the eumelanin electrode was found to give a steady photocathodic current of about 8 μA cm−2, which was found to decline by about 50% over 6 hours of continuous illumination. Measurement of aliquots from the cathode chamber confirmed that the photocathodic product was hydrogen peroxide, which was produced with a constant Faraday yield of ≈90% (Fig. 3a). Fig. 3a shows a long photoelectrolysis with a few periods where the light was turned off to check the level of the dark current. Regular frequency on/off cycles, with 45 s illumination, are plotted in Fig. 3b, evidencing a rapid photoelectrochemical response from the eumelanin film. A turnover number of 5.13 was calculated for the eumelanin film used as the photocathode. This TON is higher than any of the photocatalytic cases, indicating that holes must be quenched in the eumelanin layer by the FTO electrode more efficiently than they oxidize the various donor molecules discussed previously. Since the films are 150 nm thick, conductivity/photoconductivity of the eumelanin plays a key role.
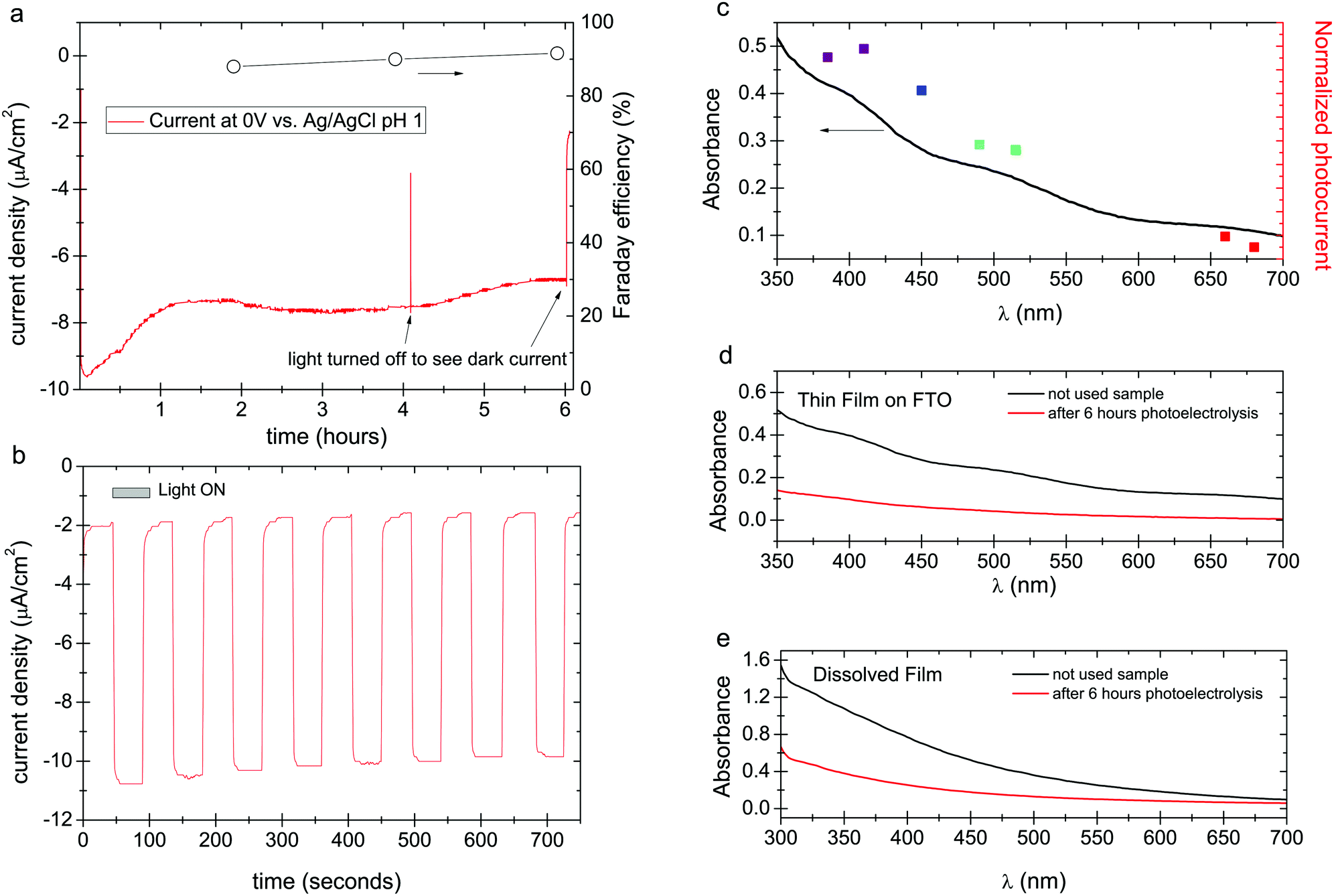 | ||
| Fig. 3 Photocathodic experiments in oxygenated conditions. (a) Chronoamperometric photocurrent density for an FTO/eumelanin working electrode under solar simulator illumination, pH = 1, 0 V vs. Ag/AgCl. Faraday efficiency of O2 reduction to H2O2 is plotted on the right axis. (b) Chronoamperometry of a eumelanin photocathode with 45 s light on/off cycles. (c) Photocurrent action spectrum measurement. Eumelanin film on FTO absorption spectrum with accompanying photocurrents for seven different LEDs. (d) UV-VIS comparison between eumelanin thin film on FTO used as blank vs. eumelanin thin film on FTO used in photo(electro)catalytic oxygen reduction reaction for H2O2 evolution. (e) UV-VIS comparison between dissolved eumelanin thin film used as blank vs. dissolved eumelanin thin film used in photo(electro)catalytic oxygen reduction reaction for H2O2 evolution. | ||
The distinctive absorption of eumelanins has been seen as evidence that it is an amorphous semiconductor analogous to amorphous silicon, for instance; or it can be interpreted as originating from a mix of different discrete chromophores overlaying to create the absorption envelope of eumelanin.13 One can question which of these chromophoric species is responsible for the photocatalytic processes. We measured the photocathodic current using a series of different narrow-band LEDs (details and chronoamperometric scans as shown Fig. S3, ESI†), calibrated to the same irradiance, in order to construct a photocurrent action spectrum (Fig. 3c). These results show that at all wavelengths where the eumelanin optically absorbs, a photocurrent is generated, and its magnitude follows the optical absorption of the eumelanin film. This behavior could be explained by a more complex picture in which there is a cooperative contribution of excited states of different potentials contributing to a single higher potential excited state as resultant after disproportionation/comproportionation reactions of different pigment constituents.21,35,36 In any case this finding indicates that all wavelengths where eumelanin absorbs result in photochemistry.
Despite the higher TON in eumelanin under oxygenated photocathodic conditions, it still degrades. The spectra obtained for pristine samples versus used samples, both as thin films (Fig. 3d), and after dissolution (Fig. 3e), confirm this. Neither the film nor the solution shows any additional peaks or otherwise changes in the shape of the spectra following irradiation. Only a monotonic decrease of each spectrum is seen, indicating that the eumelanin is uniformly degrading and not obviously producing another distinct chromophoric byproduct. Degradation proceeds as in the photocatalytic case from the action of unquenched positive moieties, holes, leading to autooxidation. It is possible that the action of accumulating H2O2 may also play an additional role in accelerating degradation as the photoelectrolysis proceeds and the concentration of peroxide increases.
In oxygenated electrolytes, the reaction of oxygen to hydrogen peroxide dominates. However, thorough deoxygenation of the electrolyte with argon for several hours followed by measurement under argon shows that a smaller photocurrent persists (Fig. 4a), on the order of 4–5 μA cm−2 at a cathodic polarization of −0.25 V. The photocurrent action spectrum follows the broad absorption of the eumelanin, exactly as in the oxygenated photocathode case (Fig. 4b and Fig. S3, ESI†). Sustained photocurrent in fully deoxygenated conditions can only be attributed to the hydrogen evolution reaction, where the photoexcited electrons reduce protons instead of oxygen. We find a relatively stable photocurrent over 32 hours. Over this time, the eumelanin film was found to degrade by the same amount as the oxygenated conditions gave after 6 hours. This indicates that despite the photocurrent density being about 70% of the oxygenated photocurrent, the eumelanin is about five times more stable (Fig. 4c and d). These photocurrent densities are too small to provide sufficient quantities of H2 for reliable quantification, however the only possible alternative hypothesis for the observed photocurrent is a degradation reaction of the eumelanin itself, which is not consistent with the observation of substantially improved stability. From this we infer that the hydrogen evolution reaction is indeed occurring, albeit with very limited efficiency.
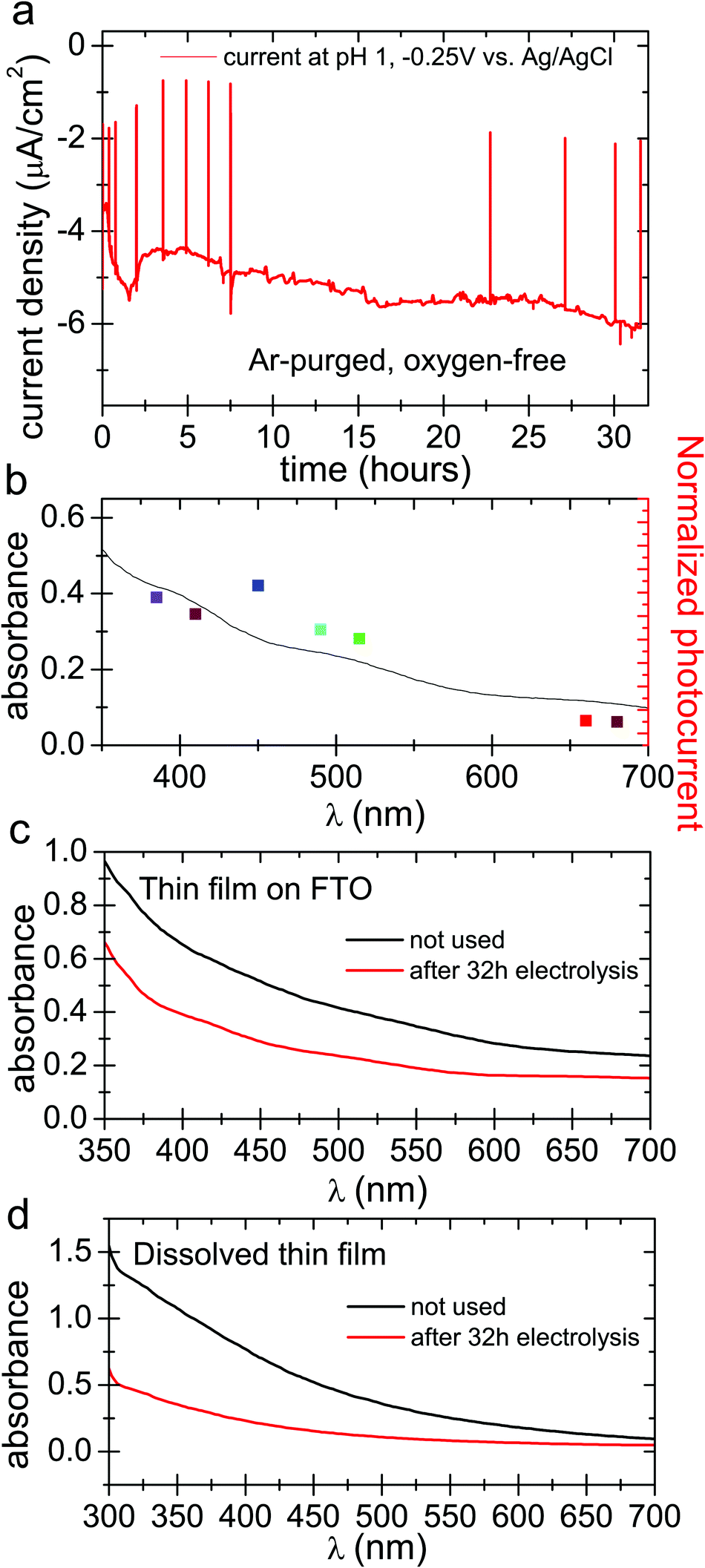 | ||
| Fig. 4 Photocathodic experiments in oxygen-free electrolyte. (a) Chronoamperometry with solar simulator illumination over 32 hours at −0.25 V vs. Ag/AgCl and deoxygenated pH 1 electrolyte. The positive spikes in the current trace are turning off of the light source in order to see the level of dark current in the sample. (b) Photocurrent action spectrum overlaid with the absorption spectrum of eumelanin on FTO for deoxygenated photocurrent. (c) Direct UV-VIS comparison between DHI eumelanin thin film used as a blank (150 nm) vs. DHI eumelanin thin film used in the 32 hour photo(electro)catalytic experiment (150 nm) and (d) UV-VIS comparison between dissolved eumelanin thin film used as a blank vs. dissolved eumelanin thin film used in the same experiment. | ||
From our results it emerges that synthetic eumelanin can produce peroxide not only from autooxidation, as previously established,33 but also as part of a redox cycle where a donor molecule is oxidized, or where holes are neutralized by an electrode. This behavior is analogous to a classic semiconductor photocatalyst model, from the point of view of photochemical processes the semiconductor picture holds macroscopically. The mechanisms at play specifically in eumelanin are more complex, and still need to be resolved in detail. Sarna et al. have proven that eumelanin can perform a single-electron photoreduction of oxygen to superoxide, which then becomes further reduced and protonated to yield peroxide.32 These findings however are for conditions of UV irradiation. We would speculate that with lower energy photons, as in our case, the superoxide mechanism may be surpassed by the thermodynamically-favored two-electron reduction of oxygen directly to peroxide. Our present results on photocathodic current are consistent with either the one-electron, two-electron, or coexistence of both pathways. Our observation of eumelanin-catalyzed photooxidation of various electron-donors, as well as the stabilizing effect of having an electrode injecting electrons into eumelanin photocathodes, highlights the important role of the photogenerated positive species on eumelanin. In the traditional semiconductor model of photocathodes, photoinduced electrons reduce a species in solution, while the corresponding holes are transported away to the electrode. Previous studies have established that protons are the positive charge carriers in eumelanin, while semiquinones carry mobile negative charges.20,23 This means that for eumelanin the photocathode model must be understood differently. A picture consistent with the prevailing knowledge on eumelanin is that photoreduction occurs, leaving behind a positive charge. This can be quenched by electron-carrying semiquinone species, which can be restored via injection of electrons from an underlying electrode. In the pure photocatalytic case, the positively-charged eumelanin moiety oxidizes electron donors directly. Our results confirm that oxygen accelerates photodegradation, and that in oxygen-free conditions a redox cycle involving hydrogen evolution is possible, with apparently better eumelanin stability.
Conclusions
These results are remarkable from the point of view of catalysis – eumelanin is a photocatalytic biomaterial capable of peroxide evolution, hydrogen evolution, and photooxidation of various organic substrates. This not only invites exploration in sustainability technologies with eumelanin as a bio-origin photocatalyst, but also has implications in the biological/bioelectronic sciences. Photodynamic therapies exploiting these photoprocessess in eumelanin are intriguing. Recently, eumelanin particles have been explored for photodynamic therapy which harnesses photothermal heating of the particles,37 however our results suggest that photofaradaic reactions could also be used to achieve various physiological effects. Mechanistic understanding must still be improved. More detailed investigation of eumelanin photochemistry should be conducted to elucidate the role of one- and two-electron processes. Overall, our results open a new side of understanding of the physical chemistry of eumelanins.Experimental methods
Eumelanin sample preparation
All commercially available reagents were used as received and all the solvents were of analytical grade quality. Anhydrous solvents were purchased from commercial sources. 5,6-Dihydroxindole (DHI) was prepared according to a reported procedure.38 DHI thin films were prepared by spin coating with a Laurell WS-650MZ-23NPP/LITE coater on round PET foils of 3.14 cm2 and on square FTO glasses of 3.75 cm2 from concentrated methanol solutions (30 mg mL−1) after filtering through a 0.2 μm Whatman membrane using a speed gradient of 2500 rpm for 30 s. Appropriate volume deposition (100–150 μL) was used. The DHI films then were chemically polymerized following the AISSP procedure: eumelanin thin films were obtained by exposing the DHI films (150–200 nm thickness) for 1 h to air-equilibrated gaseous ammonia from an ammonia solution (28% in water) in a sealed chamber at 1 atm pressure at controlled temperature (25–40 °C).Photocatalysis
Eumelanin films were put in vials in 1 mL solutions at two different pH values (pH = 2, pH = 7) in the presence and in the absence of different sacrificial electron donors (phenol, formate, and oxalate) to test the possibility to increase, with suitable donors, the amount of peroxide produced during the photocatalytic oxygen reduction reaction. A white LED solar simulator with intensity of 1.05 sun was used to illuminate the substrates for a total time of 12 h. Before the exposure of these films to the light, every vial was filled with O2 at the beginning and after every single H2O2 assay made every 4 h. The quantification of the produced hydrogen peroxide was done spectrophotometrically via the method of horseradish peroxidase/3,3′,5,5′-tetramethylbenzidine (TMB) H2O2 quantification by following the oxidation of TMB34 at 653 nm using a synergy/H1 microplate reader, BioTek®. Aliquots of every single solution (10 μL) were afterwards added into a mixture of 30 μg mL−1 TMB (Sigma-Aldrich) and 0.750 ng mL−1 HRP (Sigma-Aldrich) in 0.1 M citrate–phosphate buffer pH 6.0, giving a volume of 1 mL. Quantification of the produced H2O2 was performed using extinction coefficient values for the TMB dimer and cross-checked with a calibration curve made by measuring solutions of known H2O2 (Merck). The TON was calculated with respect to the amount of DHI monomer in the sample. Eumelanin was quantified by dissolving thin films in 1 M NaOH solution, and comparing the absorption to a standard curve. The standard curve was produced by polymerizing DHI by dropcasting varying known starting quantities of DHI onto glass, and subsequently converting these polymerized films into solutions by adding NaOH and measuring the absorbance of these solution in quartz cuvettes. This gives a linear relationship between absorbance and DHI content.Photoelectrocatalysis
Photoelectrochemical measurements were conducted using an H-cell configuration as shown in Fig. S2 (ESI†), using a Redox.me MM PEC H-CELL. Cyclic voltammetry and chronoamperometry experiments were carried out using an Ivium technologies Vertex One potentiostat, with the eumelanin-modified FTO as the working electrode, a Pt wire as the counter electrode, and an Ag/AgCl (3 M KCl) reference electrode. The electrodes were illuminated with a white LED light source with the power of 255 mW cm−2. In our case we compared both oxygenated and deoxygenated (O2-bubbled, Ar-purged) conditions.Conflicts of interest
No competing financial interests have been declared.Acknowledgements
The authors gratefully acknowledge financial support from the Knut and Alice Wallenberg Foundation within the framework of the Wallenberg Centre for Molecular Medicine at Linköping University and the Italian Project RELIGHT (PON02_00556_3306937). L. M. and M. G. conducted all photo(electrochemical) experiments. V. D. optimized the sample preparation and measurement setups. A. P. and E. D. G. conceived and supervised the project. The manuscript was written with contribution from all coauthors.References
- D. D. Ordinario, L. Phan, W. G. Walkup IV, J.-M. Jocson, E. Karshalev, N. Hüsken and A. A. Gorodetsky, Nat. Chem., 2014, 6, 596–602 CrossRef PubMed.
- D. D. Ordinario, L. Phan, Y. Van Dyke, T. Nguyen, A. G. Smith, M. Nguyen, N. M. Mofid, M. K. Dao and A. A. Gorodetsky, Chem. Mater., 2016, 28, 3703–3710 CrossRef.
- N. Amdursky, X. Wang, P. Meredith, D. D. C. Bradley and M. M. Stevens, Adv. Mater., 2016, 28, 2692–2698 CrossRef PubMed.
- D. Porath, A. Bezryadin, S. de Vries and C. Dekker, Nature, 2000, 403, 635–638 CrossRef PubMed.
- C. Zhong, Y. Deng, A. F. Roudsari, A. Kapetanovic, M. P. Anantram and M. Rolandi, Nat. Commun., 2011, 2, 476 CrossRef PubMed.
- E. E. Josberger, Y. Deng, W. Sun, R. Kautz and M. Rolandi, Adv. Mater., 2014, 26, 4986–4990 CrossRef PubMed.
- C. W. Tang, J. Chem. Phys., 1975, 62, 2139–2149 CrossRef.
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef PubMed.
- E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef PubMed.
- P. Meredith, C. J. Bettinger, M. Irimia-Vladu, A. B. Mostert and P. E. Schwenn, Rep. Prog. Phys., 2013, 76, 034501 CrossRef PubMed.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef PubMed.
- M. Ischia, K. Wakamatsu, S. Briganti, D. Kovacs, P. Meredith, A. Pezzella, T. Sarna, J. D. Simon and S. Ito, Pigm. Cell Melanoma Res., 2013, 26, 616–633 CrossRef PubMed.
- M. D’Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed.
- A. Huijser, A. Pezzella and V. Sundström, Phys. Chem. Chem. Phys., 2011, 13, 9119–9127 RSC.
- P. Meredith and T. Sarna, Pigm. Cell Res., 2006, 19, 572–594 CrossRef PubMed.
- J. McGinness, P. Corry and P. Proctor, Science, 1974, 183, 853–855 CrossRef PubMed.
- C. J. Bettinger, J. P. Bruggeman, A. Misra, J. T. Borenstein and R. Langer, Biomaterials, 2009, 30, 3050–3057 CrossRef PubMed.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20912–20917 CrossRef PubMed.
- A. Pezzella, M. Barra, A. Musto, A. Navarra, M. Alfe, P. Manini, S. Parisi, A. Cassinese, V. Criscuolo and M. D’Ischia, Mater. Horiz., 2015, 2, 212–220 RSC.
- A. Bernardus Mostert, B. J. Powell, I. R. Gentle and P. Meredith, Appl. Phys. Lett., 2012, 100, 093701 CrossRef.
- S. B. Rienecker, A. B. Mostert, G. Schenk, G. R. Hanson and P. Meredith, J. Phys. Chem. B, 2015, 119, 14994–15000 CrossRef PubMed.
- J. Wünsche, Y. Deng, P. Kumar, E. Di Mauro, E. Josberger, J. Sayago, A. Pezzella, F. Soavi, F. Cicoira, M. Rolandi and C. Santato, Chem. Mater., 2015, 27, 436–442 CrossRef.
- A. B. Mostert, B. J. Powell, F. L. Pratt, G. R. Hanson, T. Sarna, I. R. Gentle and P. Meredith, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8943–8947 CrossRef PubMed.
- A. B. Mostert, S. B. Rienecker, C. Noble, G. R. Hanson and P. Meredith, Sci. Adv., 2018, 4, 1–7 Search PubMed.
- M. Sheliakina, A. B. Moster and P. Meredith, Mater. Horiz., 2018, 5, 256–263 RSC.
- J. Wunsche, F. Cicoira, C. F. O. Graeff and C. Santato, J. Mater. Chem. B, 2013, 1, 3836–3842 RSC.
- Y. J. Kim, W. Wu, S. E. Chun, J. F. Whitacre and C. J. Bettinger, Adv. Mater., 2014, 26, 6572–6579 CrossRef PubMed.
- M. K. Węcławski, M. Jakešová, M. Charyton, N. Demitri, B. Koszarna, K. Oppelt, S. Sariciftci, D. T. Gryko and E. D. Głowacki, J. Mater. Chem. A, 2017, 5, 20780–20788 RSC.
- M. Gryszel, M. Sytnyk, M. Jakesova, G. Romanazzi, R. Gabrielsson, W. Heiss and E. D. Głowacki, ACS Appl. Mater. Interfaces, 2018, 10, 13253–13257 CrossRef PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
- M. Warczak, M. Gryszel, M. Jakešová, V. Đerek and E. D. Głowacki, Chem. Commun., 2018, 54, 1960–1963 RSC.
- W. Korytowski, B. Pilas, T. Sarna and B. Kalyanaraman, Photochem. Photobiol., 1987, 45, 185–190 CrossRef PubMed.
- W. Korytowski and T. Sarna, J. Biol. Chem., 1990, 265, 12410–12416 Search PubMed.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 Search PubMed.
- A. Pezzella, O. Crescenzi, L. Panzella, A. Napolitano, E. J. Land, V. Barone and M. D’Ischia, J. Am. Chem. Soc., 2013, 135, 12142–12149 CrossRef PubMed.
- A. J. Clulow, A. B. Mostert, M. Sheliakina, A. Nelson, N. Booth, P. L. Burn, I. R. Gentle and P. Meredith, Soft Matter, 2017, 13, 3954–3965 RSC.
- M. Kim, H. S. Kim, M. A. Kim, H. Ryu, H. J. Jeong and C. M. Lee, Macromol. Biosci., 2017, 17, 1–6 Search PubMed.
- R. Edge, M. D’Ischia, E. J. Land, A. Napolitano, S. Navaratnam, L. Panzella, A. Pezzella, C. A. Ramsden and P. A. Riley, Pigm. Cell Res., 2006, 19, 443–450 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh00715b |
| This journal is © The Royal Society of Chemistry 2018 |