
Janus DNA orthogonal adsorption of graphene oxide and metal oxide nanoparticles enabling stable sensing in serum†
Biwu
Liu
 a,
Lingzi
Ma
a,
Zhicheng
Huang
a,
Hao
Hu
b,
Peng
Wu
b and
Juewen
Liu
*a
a,
Lingzi
Ma
a,
Zhicheng
Huang
a,
Hao
Hu
b,
Peng
Wu
b and
Juewen
Liu
*a
aDepartment of Chemistry, Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario, N2L 3G1, Canada. E-mail: liujw@uwaterloo.ca
bAnalytical & Testing Centre, Sichuan University, Chengdu, 610064, China
First published on 27th October 2017
Abstract
While DNA/graphene oxide (GO) conjugates have been widely used for DNA detection, they suffer from non-specific DNA displacement by proteins, making their application in biological samples difficult. To find new materials tightly adsorbing DNA but not proteins, we screened seven metal oxide nanoparticles, all interacting with the phosphate backbone of DNA, while DNA uses its nucleobases to interact with GO. In this regard, DNA is a Janus polymer orthogonally adsorbing GO and metal oxides. The DNA adsorption affinity ranks CoO > NiO > Cr2O3 > Fe2O3 > Fe3O4 > TiO2 > CeO2 based on a phosphate displacement assay. Among them, CoO is nearly fully resistant to protein displacement, while NiO has the best limit of detection of 0.24 nM DNA. This study provides fundamental insights into the biointerface chemistry of DNA, and reveals new materials useful for bioanalytical chemistry, DNA separation, and DNA-directed assembly.
Conceptual insightsFor the first time, DNA is described as a Janus polymer in this work for orthogonally interacting with graphene oxide (GO) using its nucleobases and with metal oxides with its phosphate backbone. At the same time, non-phosphorylated proteins do not have a favourable mechanism for interacting with metal oxides, but proteins can be strongly adsorbed on GO. This understanding has led to the screening of various metal oxides for DNA binding, and we discovered excellent selectivity of NiO and CoO for adsorbing DNA over proteins. These oxides enable highly sensitive DNA detection with detection limits similar to that with GO, but the oxide conjugates are much more resistant to non-specific probe DNA displacement by proteins, allowing detection in serum. |
DNA as a programmable polymer has been extensively used for building nanostructures,1,2 directed assembly of nanoparticles,3,4 and biosensing.5–8 The precise control of DNA-related biointerfaces is critical for ensuring its function in hybrid materials.6,9–17 Single-stranded (ss)DNA has two main functional building blocks: the phosphate backbone and the nucleobases. DNA uses its nucleobases to π–π stack and hydrogen bond on graphene oxide (GO).10,18 This adsorption complex works as a highly popular DNA sensor.19–24 In a typical reaction, a fluorescently labeled ssDNA probe is adsorbed on GO with quenched fluorescence. Addition of its complementary DNA (cDNA) as the target leads to enhanced fluorescence due to the formation of a double-stranded (ds)DNA that has lower affinity on GO.19 With aptamer probes, analytes beyond DNA were also detected.25–30
Being simple and elegant, this method however encounters difficulties in a complex sample matrix such as serum. Since DNA probes are only physisorbed, they might be displaced by proteins, leading to increased background as well as false positive signals.31,32 Proteins compete with DNA since they rely on the same mechanism of π–π stacking and hydrogen bonding with GO. While attempts were made by co-adsorbing inactive internal reference probes,33 or by covalently attaching DNA,34–36 the advantage of simplicity is lost.
We reason that this problem might be solved by exploring the other structural element of DNA: its phosphate backbone. Since phosphate is abundant in DNA but not in proteins, phosphate binders might better distinguish DNA from most protein competitors. Phosphate oxygen is a hard Lewis base that binds strongly to hard Lewis acids (such as Mg2+, Ca2+ and lanthanides from the Hard–Soft–Acid–Base theory).37–43 This is in sharp contrast to GO adsorbing nucleobases.10Fig. 1a depicts the orthogonal adsorption of GO and metal oxide (MO) by DNA. Herein, we explore this fundamental difference and harness the phosphate interaction for suppressing non-specific DNA displacement by proteins to achieve highly sensitive DNA detection in serum.
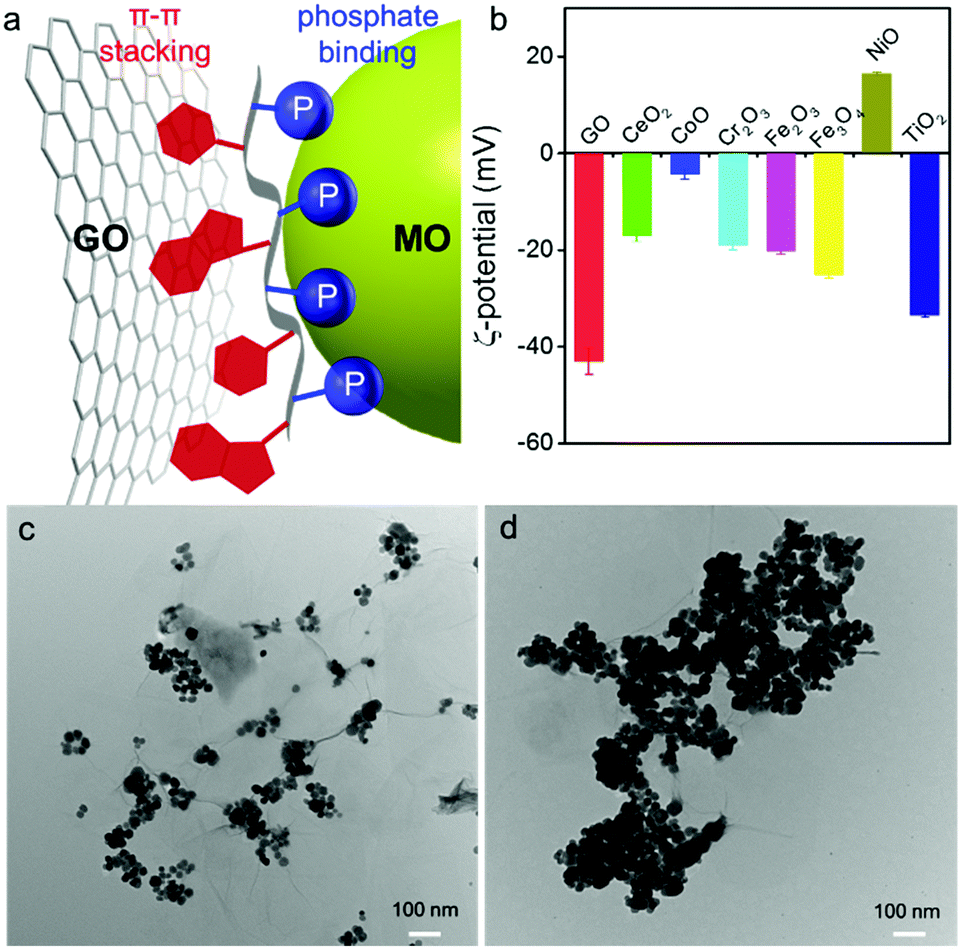 | ||
| Fig. 1 (a) A schematic showing ssDNA as a Janus polymer interacting orthogonally with GO (via nucleobases) and with MO (via backbone phosphate). (b) ζ-Potential of the nanomaterials in 10 mM HEPES buffer (pH 7.6). TEM micrographs of GO/Fe3O4 NP hybrids prepared (c) without DNA, and (d) with DNA. | ||
Many types of MOs are available, and each may have a different surface charge and affinity for DNA binding. A few MOs such as TiO2,41 Fe2O3,44 CoOOH,45,46 and MnO247 have been interfaced with DNA reported in the literature. We herein screened seven common MO nanoparticles (MONPs) between 5 and 30 nm, except for Cr2O3 (>100 nm) (Fig. S1, ESI†). Our single-layered GO was ∼1 nm thick (Fig. S2, ESI†), with a lateral size of several micrometers (Fig. S1, ESI†). Since DNA is a polyanion, we measured the ζ-potential of these materials to understand electrostatic interactions. Most of them were negatively charged at physiological pH, except for NiO which was cationic and CoO which was nearly charge neutral (Fig. 1b).
To confirm orthogonal adsorption of DNA by GO and MONPs, we mixed GO and Fe3O4 NPs. In the absence of DNA, Fe3O4 NPs mainly sat on the oxygen-rich edges of GO forming sparse clusters (Fig. 1c). With DNA, in contrast, the Fe3O4 NPs densely packed on the entire GO sheet (Fig. 1d), suggesting that DNA can bind to both GO and Fe3O4 NPs non-competitively and supporting the Janus model in Fig. 1a. To further confirm their orthogonal adsorption with chemical details, we then used DNA-based sensors.
Our sensors were prepared by adsorbing a 6-carboxyfluorescein (FAM)-labeled probe DNA on GO or MONPs (Scheme S1, ESI†). We respectively mixed DNA1, 2 or 3 (see Table S1, ESI† for DNA sequences) with each material followed by centrifugation to remove free DNA. The concentration of each material was individually optimized to achieve full fluorescence quenching (Fig. 2a, top panel, Fig. S3, ESI†), and this was also confirmed by measuring the supernatant fluorescence (Fig. S4, ESI†). The quenching appeared to be dynamic quenching likely due to energy or electron transfer based on the fluorescence lifetime measurement (Fig. S5, ESI†).
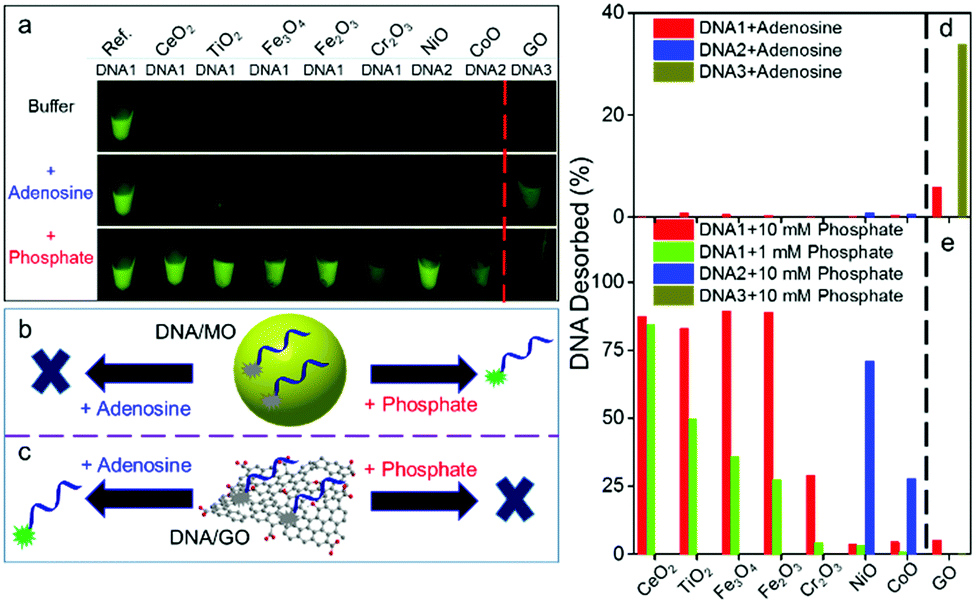 | ||
| Fig. 2 Orthogonal DNA adsorption by GO and by the MONPs. (a) Fluorescent photographs showing FAM-labeled DNA adsorption by nanomaterials in buffer A (10 mM HEPES, pH 7.6 with 300 mM NaCl) and its displacement after adding adenosine (5 mM) or inorganic phosphate (10 mM). Schemes showing the displacement of DNA from (b) MONPs and (c) GO by adenosine or phosphate. Percentage of DNA displaced by (d) adenosine or (e) inorganic phosphate. For each material, a few DNAs were used to rank the adsorption affinities: DNA1 (24-mer); DNA2 (12-mer); and DNA3 (5-mer). | ||
To test orthogonal adsorption, we added 5 mM adenosine as a competitor to study DNA base adsorption, and only the GO sample showed fluorescence recovery (Fig. 2a, d and Fig. S6, ESI†), suggesting that DNA bases are responsible for adsorption on GO. Next, we added inorganic phosphate, and all the MOs showed fluorescence recovery, but not GO (Fig. 2a, bottom panel). Fig. 2b and c illustrate this displacement reaction supportive of the orthogonal adsorption concept. Within this context, DNA is a Janus polymer with two faces: the phosphate face interacting with MO and the nucleobase face interacting with GO (Fig. 1a).
Note that different DNA sequences were used in Fig. 2a for GO and the MOs. This is because different MOs have quite different DNA adsorption affinities, and no single DNA sequence can differentiate all the MOs. Among the seven MONPs, four (CeO2, Fe2O3, Fe3O4, and TiO2) showed a significant fluorescence enhancement with DNA1 (24-mer) by adding 10 mM phosphate (Fig. S4, ESI† and Fig. 2e, red bars). By adding a lower phosphate concentration of 1 mM, we can rank their DNA adsorption affinity (Fig. 2e, green bars, and Fig. S7, ESI†). Since NiO and CoO can still adsorb DNA1 even with 10 mM phosphate (Fig. S4, ESI†), we also used DNA2, a 12-mer DNA (Fig. S8 and S9, ESI†). Combining the above data, DNA adsorption affinity is ranked as CoO > NiO > Cr2O3 > Fe2O3 > Fe3O4 > TiO2 > CeO2. CoO is nearly charge neutral and NiO is positively charged (Fig. 1b), which may contribute to their strong DNA adsorption. Cr3+ has a very slow ligand exchange rate,48 which may explain the strong adsorption on Cr2O3. Note that we cannot fully rule out minor interactions between DNA bases and some MONPs, especially those containing borderline metals such as Ni2+, Co2+ and Cr3+. The fact that phosphate can displace most of the DNA suggests at least that phosphate binding is a main factor.
After confirming the adsorption mechanism, we then studied DNA sensing. NiO NPs were used to illustrate our experiment (Fig. 3a). After adsorbing a probe DNA, we monitored fluorescence recovery after adding various concentrations of its cDNA as the target (Fig. 3b), showing a dynamic range up to 60 nM. We then tested the selectivity by adding the non-labeled probe DNA (called same DNA or sDNA) and also a few homo-DNAs (A30, T30, C30). None of them showed much signal (Fig. 3c), indicating high specificity.
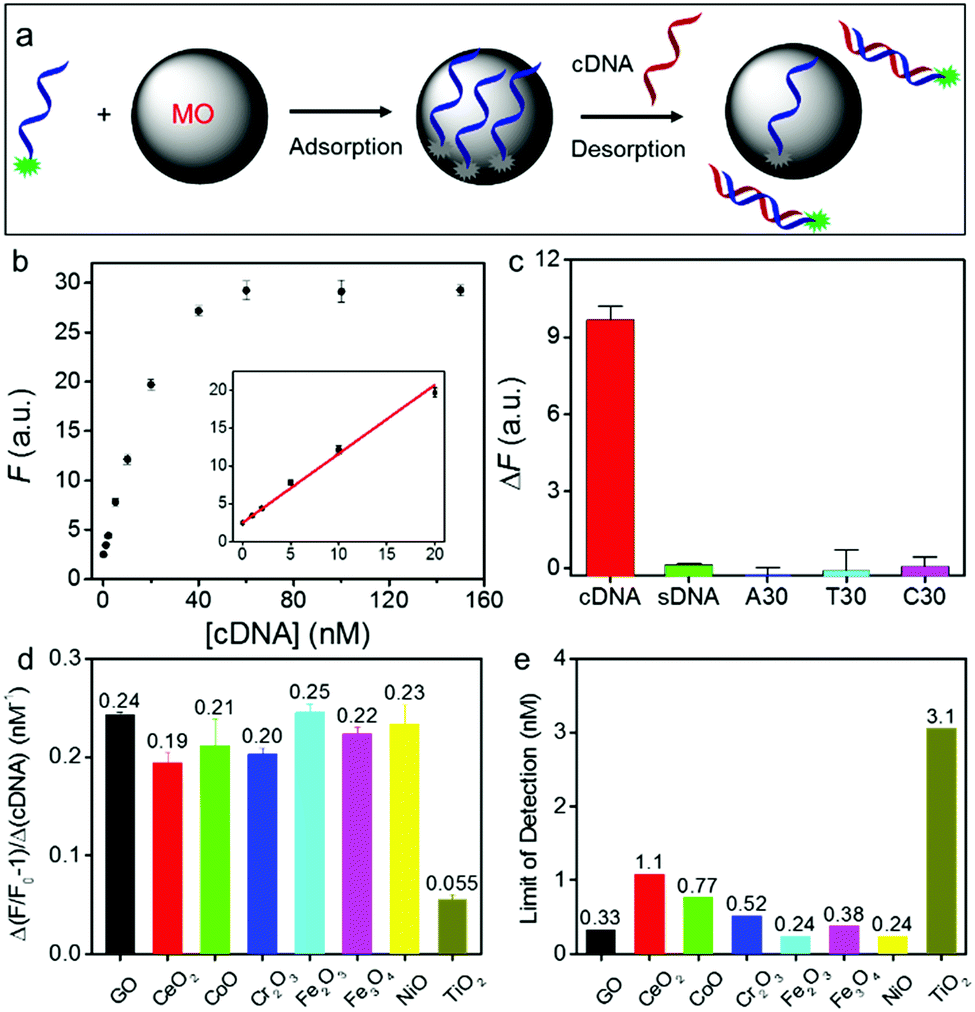 | ||
| Fig. 3 (a) A scheme showing the sensing mechanism using DNA adsorbed on an MONP such as NiO. (b) A calibration curve of the NiO/DNA sensor for cDNA detection (probe DNA = 20 nM). (The inset is the initial linear range.) (c) Comparison of fluorescence recovery in the presence of different DNA sequences (10 nM each). Comparison of (d) the sensitivity (the slope of the calibration curves) and (e) LOD of GO/DNA and MO/DNA sensors. | ||
Using this method, we then measured the sensitivity of all the DNA/MONP sensors, and made comparisons with GO (Fig. S10, ESI†). All the samples exhibited a cDNA-dependent fluorescence recovery. The slope of these fitted lines represents the sensitivity of the sensors (Fig. 3d). Except for TiO2, all the materials, including GO, had a similar sensitivity. The limit of detection (LOD) was also calculated (LOD = 3σ/slope, where σ is standard deviation of background variation in the absence of the target DNA) (Fig. 3e). Most MONPs also showed similar LODs to GO (∼0.33 nM). Therefore, despite their very different DNA adsorption mechanisms, GO and MO had a similar analytical performance. Without signal amplification, an LOD of ∼0.3 nM is already approaching the physical limit for DNA detection under ambient conditions.49,50
The basis of DNA detection on GO is the formation of duplex DNA to hide DNA bases. However, the DNA phosphate backbone is still exposed even in the duplex form and it can still interact with MONPs. The weakened adsorption of duplex DNA by MONPs might be due to its rigidity, thus reducing the number of contacting points on MONPs. In addition, the exposed DNA bases on MONPs might facilitate its hybridization with cDNA. It is also interesting to note that most of the MONPs had a similar sensitivity and detection limit for target DNA, despite the fact that they had quite large differences in DNA adsorption affinity. This is rationalized by the fact that for high affinity MONPs, they had a higher probe DNA density (e.g. a lower concentration of nanoparticles was sufficient to fully adsorb the probe DNA). Although it is more difficult to desorb its probe DNA, this is compensated by the high probe concentration on the surface.
All the above studies were performed in clean buffers. A main goal of this study is to achieve DNA detection in biological samples. We first tested the stability of DNA/GO in the presence of a model protein, bovine serum albumin (BSA). On GO, 100 μg mL−1 BSA released more than 30% of the probe, although the sample with the cDNA released slightly more (Fig. 4a). The signal-to-background ratio was only 1.6. In fact, even 10 μg mL−1 BSA induced significant probe desorption (Fig. S11, ESI†).51–54 For NiO, interestingly, BSA barely induced any desorption, and the signal-to-background ratio reached 19.2, a 12-fold improvement (Fig. 4b). Therefore, the NiO system was resistant to protein, but still sensitive to DNA.
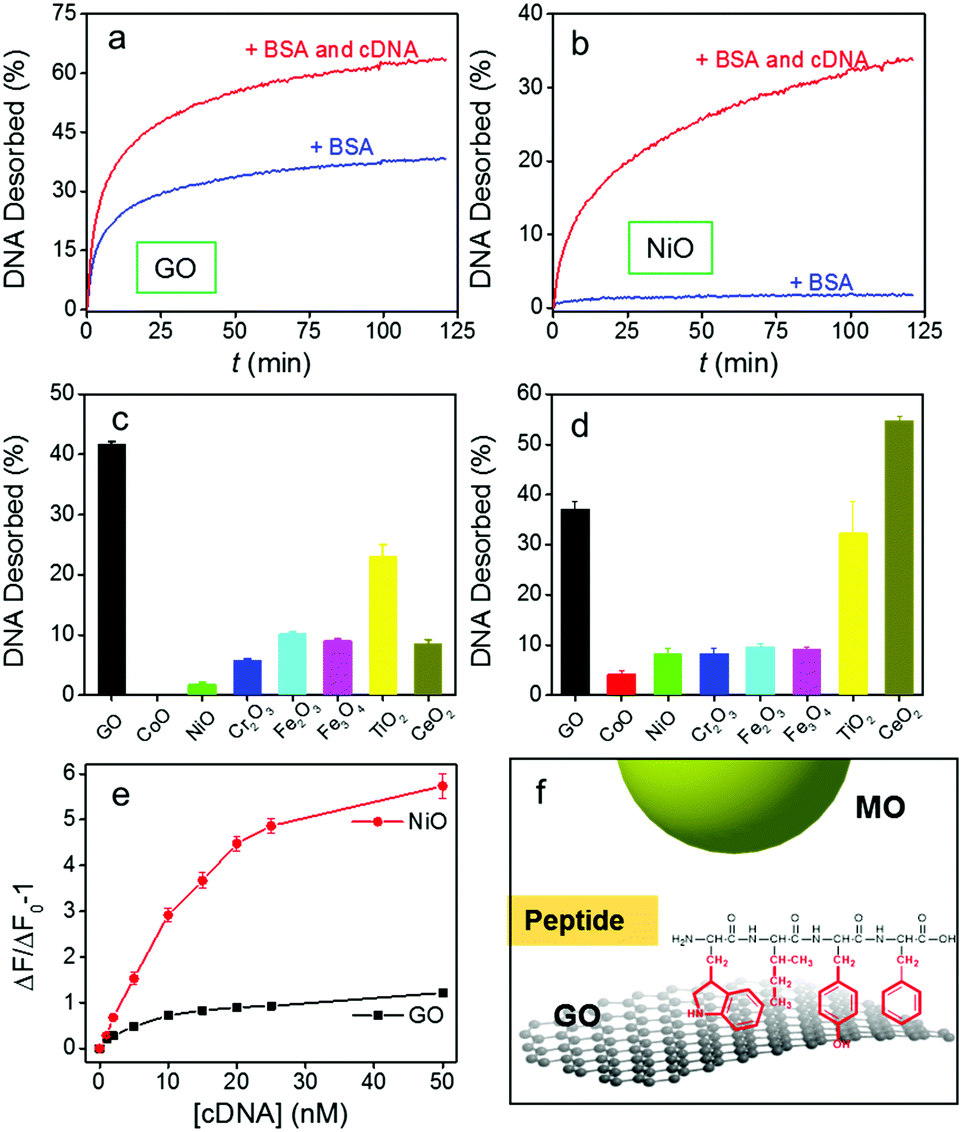 | ||
| Fig. 4 Kinetics of DNA1 signaling induced by BSA (100 μg mL−1) or a BSA/cDNA (100 nM) mixture from (a) GO and (b) NiO. Percentage of DNA1 desorbed from GO and the MOs after 1 h by (c) 100 μg mL−1 of BSA and (d) 0.5% FBS. (e) Detection of cDNA in 0.5% FBS using DNA1/GO and DNA1/NiO. (f) A scheme showing peptide adsorption on GO but not on MO. | ||
In general, much less DNA was desorbed by BSA from MONPs than from GO (Fig. 4c, and Fig. S12, ESI†). CoO resisted BSA displacement the most, followed by NiO, Cr2O3, CeO2, Fe3O4, Fe2O3, and TiO2. This order roughly correlates with their DNA adsorption affinity in Fig. 2e. Therefore, BSA directly competes with DNA bases on GO, but it does not compete effectively with the phosphate backbone adsorbed on MONPs. This can be rationalized by the aromatic amino acid residues in BSA interacting with GO (Fig. 2f),55–57 similar to those of DNA bases. On the other hand, it is more difficult to adsorb proteins on MONPs. In fact, only phosphorylated proteins can be adsorbed by TiO2 NPs, which has been used for their enrichment.58–60
Encouraged by this result, we further challenged the sensors in serum (Fig. 4d and Fig. S13, ESI†). In this case, CeO2 had the highest desorption of the probe, even higher than GO, while the other MONPs still showed much lower desorption than GO. The high signal of CeO2 might be related to its strong affinity to some small molecules in serum such as ascorbate.39,61–63
Based on our screen of all these materials, NiO has a very low LOD and it is also quite resistant to displacement. We then used it to detect DNA in serum (Fig. 4e). The NiO/DNA system still had a higher sensitivity with an LOD of 0.25 nM. More importantly, the nonspecific desorption of probe DNA from GO increased the background, yielding only ca. 1-fold fluorescence enhancement, while the NiO/DNA still had 6-fold enhancement, making the latter a better candidate for detection in serum.
Conclusions
In summary, this work has linked the structural features of DNA and proteins to the surface properties of GO and MONPs with useful analytical applications. (1) For the first time, DNA is described here as a Janus polymer with a phosphate face and a nucleobase face for respectively adsorbing GO and MONPs. Without a phosphate backbone, proteins can only be adsorbed strongly by GO but not by MONPs. Within this context, we explored DNA sensing in the presence of competing proteins and serum using NiO. (2) We systematically compared seven types of MONPs with GO, ranking DNA adsorption affinity by the MONPs from low (CeO2) to very high (NiO and CoO). Using NiO and CoO has retained the simplicity and sensitivity of GO, while the specificity and robustness is significantly improved. Such improvement is rooted in fundamental insights of DNA for orthogonal adsorption of GO and MONPs, and the chemical difference between DNA and proteins. This understanding will further the design of DNA-based biosensors in complex biological samples and new smart nanomaterials based on DNA nanotechnology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work is from the Natural Sciences and Engineering Research Council of Canada (NSERC), and the National Natural Science Foundation of China (21522505).Notes and references
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- H. Wang, R. Yang, L. Yang and W. Tan, ACS Nano, 2009, 3, 2451–2460 CrossRef CAS PubMed.
- H. Sun, J. Ren and X. Qu, Acc. Chem. Res., 2016, 49, 461–470 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Zhu, Q. Huang and C. Fan, Acc. Chem. Res., 2014, 47, 550–559 CrossRef CAS PubMed.
- J. Li, L. Mo, C.-H. Lu, T. Fu, H.-H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- J. Liu, Phys. Chem. Chem. Phys., 2012, 14, 10485–10496 RSC.
- B. Liu, S. Salgado, V. Maheshwari and J. Liu, Curr. Opin. Colloid Interface Sci., 2016, 26, 41–49 CrossRef CAS.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed.
- L. H. Tan, Y. Yue, N. S. R. Satyavolu, A. S. Ali, Z. Wang, Y. Wu and Y. Lu, J. Am. Chem. Soc., 2015, 137, 14456–14464 CrossRef CAS PubMed.
- N. S. R. Satyavolu, L. H. Tan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 16542–16548 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- J. Wu, L. H. Tan, K. Hwang, H. Xing, P. Wu, W. Li and Y. Lu, J. Am. Chem. Soc., 2014, 136, 15195–15202 CrossRef CAS PubMed.
- L. H. Tan, H. Xing and Y. Lu, Acc. Chem. Res., 2014, 47, 1881–1890 CrossRef CAS PubMed.
- D. Zhu, P. Song, J. Shen, S. Su, J. Chao, A. Aldalbahi, Z. Zhou, S. Song, C. Fan, X. Zuo, Y. Tian, L. Wang and H. Pei, Anal. Chem., 2016, 88, 4949–4954 CrossRef CAS PubMed.
- J. S. Park, H.-K. Na, D.-H. Min and D.-E. Kim, Analyst, 2013, 138, 1745–1749 RSC.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS PubMed.
- S. He, B. Song, D. Li, C. Zhu, W. Qi, Y. Wen, L. Wang, S. Song, H. Fang and C. Fan, Adv. Funct. Mater., 2010, 20, 453–459 CrossRef CAS.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- M. Liu, H. Zhao, S. Chen, H. Yu and X. Quan, ACS Nano, 2012, 6, 3142–3151 CrossRef CAS PubMed.
- C.-H. Lu, J. Li, J.-J. Liu, H.-H. Yang, X. Chen and G.-N. Chen, Chem. – Eur. J., 2010, 16, 4889–4894 CrossRef CAS PubMed.
- W. Qiang, W. Li, X. Li, X. Chen and D. Xu, Chem. Sci., 2014, 5, 3018–3024 RSC.
- Z. Liu, B. Liu, J. Ding and J. Liu, Anal. Bioanal. Chem., 2014, 406, 6885–6902 CrossRef CAS PubMed.
- X.-H. Zhao, R.-M. Kong, X.-B. Zhang, H.-M. Meng, W.-N. Liu, W. Tan, G.-L. Shen and R.-Q. Yu, Anal. Chem., 2011, 83, 5062–5066 CrossRef CAS PubMed.
- H. Pei, J. Li, M. Lv, J. Wang, J. Gao, J. Lu, Y. Li, Q. Huang, J. Hu and C. Fan, J. Am. Chem. Soc., 2012, 134, 13843–13849 CrossRef CAS PubMed.
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- M. Liu, J. Song, S. Shuang, C. Dong, J. D. Brennan and Y. Li, ACS Nano, 2014, 8, 5564–5573 CrossRef CAS PubMed.
- H. Zhang, F. Li, B. Dever, X.-F. Li and X. C. Le, Chem. Rev., 2013, 113, 2812–2841 CrossRef CAS PubMed.
- M. Wu, R. Kempaiah, P.-J. J. Huang, V. Maheshwari and J. Liu, Langmuir, 2011, 27, 2731–2738 CrossRef CAS PubMed.
- J. S. Park, N.-I. Goo and D.-E. Kim, Langmuir, 2014, 30, 12587–12595 CrossRef CAS PubMed.
- X. Tan, T. Chen, X. Xiong, Y. Mao, G. Zhu, E. Yasun, C. Li, Z. Zhu and W. Tan, Anal. Chem., 2012, 84, 8622–8627 CrossRef CAS PubMed.
- P.-J. J. Huang and J. Liu, Anal. Chem., 2012, 84, 4192–4198 CrossRef CAS PubMed.
- Z. Liu, S. Chen, B. Liu, J. Wu, Y. Zhou, L. He, J. Ding and J. Liu, Anal. Chem., 2014, 86, 12229–12235 CrossRef CAS PubMed.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469–4476 CrossRef CAS PubMed.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- B. Liu and J. Liu, Langmuir, 2015, 31, 371–377 CrossRef CAS PubMed.
- B. Liu, Z. Sun, P.-J. J. Huang and J. Liu, J. Am. Chem. Soc., 2015, 137, 1290–1295 CrossRef CAS PubMed.
- B. Liu and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 24833–24838 CAS.
- X. Zhang, F. Wang, B. Liu, E. Y. Kelly, M. R. Servos and J. Liu, Langmuir, 2014, 30, 839–845 CrossRef CAS PubMed.
- B. Liu and J. Liu, Chem. Commun., 2014, 50, 8568–8570 RSC.
- R. Pautler, E. Y. Kelly, P.-J. J. Huang, J. Cao, B. Liu and J. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 6820–6825 CAS.
- C. Song, G.-Y. Wang and D.-M. Kong, Biosens. Bioelectron., 2015, 68, 239–244 CrossRef CAS PubMed.
- Y. Chang, Z. Zhang, H. Liu, N. Wang and J. Tang, Analyst, 2016, 141, 4719–4724 RSC.
- Y. Cen, Y. Yang, R.-Q. Yu, T.-T. Chen and X. Chu, Nanoscale, 2016, 8, 8202–8209 RSC.
- Y. Yuan, S. Wu, F. Shu and Z. Liu, Chem. Commun., 2014, 50, 1095–1097 RSC.
- W. Zhou, T. Yu, M. Vazin, J. Ding and J. Liu, Inorg. Chem., 2016, 55, 8193–8200 CrossRef CAS PubMed.
- N. Li, C. Chang, W. Pan and B. Tang, Angew. Chem., Int. Ed., 2012, 51, 7426–7430 CrossRef CAS PubMed.
- Y. Zhang, B. Zheng, C. Zhu, X. Zhang, C. Tan, H. Li, B. Chen, J. Yang, J. Chen, Y. Huang, L. Wang and H. Zhang, Adv. Mater., 2015, 27, 935–939 CrossRef CAS PubMed.
- S. F. Oliveira, G. Bisker, N. A. Bakh, S. L. Gibbs, M. P. Landry and M. S. Strano, Carbon, 2015, 95, 767–779 CrossRef CAS.
- F. De Leo, A. Magistrato and D. Bonifazi, Chem. Soc. Rev., 2015, 44, 6916–6953 RSC.
- J. G. Vilhena, P. Rubio-Pereda, P. Vellosillo, P. A. Serena and R. Pérez, Langmuir, 2016, 32, 1742–1755 CrossRef CAS PubMed.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS PubMed.
- S. N. Kim, Z. Kuang, J. M. Slocik, S. E. Jones, Y. Cui, B. L. Farmer, M. C. McAlpine and R. R. Naik, J. Am. Chem. Soc., 2011, 133, 14480–14483 CrossRef CAS PubMed.
- J. Zhou, X. Xu, W. Liu, X. Liu, Z. Nie, M. Qing, L. Nie and S. Yao, Anal. Chem., 2013, 85, 5746–5754 CrossRef CAS PubMed.
- J. Katoch, S. N. Kim, Z. Kuang, B. L. Farmer, R. R. Naik, S. A. Tatulian and M. Ishigami, Nano Lett., 2012, 12, 2342–2346 CrossRef CAS PubMed.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. D. Jørgensen, Mol. Cell. Proteomics, 2005, 4, 873–886 CAS.
- T. E. Thingholm, T. J. D. Jorgensen, O. N. Jensen and M. R. Larsen, Nat. Protoc., 2006, 1, 1929–1935 CrossRef CAS PubMed.
- M. W. H. Pinkse, P. M. Uitto, M. J. Hilhorst, B. Ooms and A. J. R. Heck, Anal. Chem., 2004, 76, 3935–3943 CrossRef CAS PubMed.
- C. Xu and X. Qu, NPG Asia Mater., 2014, 6, e90 CrossRef CAS.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- H. Cheng, S. Lin, F. Muhammad, Y.-W. Lin and H. Wei, ACS Sens., 2016, 1, 1336–1343 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mh00804j |
| This journal is © The Royal Society of Chemistry 2018 |