
Unusual solvent polarity dependent excitation relaxation dynamics of a bis[p-ethynyldithiobenzoato]Pd-linked bis[(porphinato)zinc] complex†
Jaehong
Park
 *abc,
Tae-Hong
Park
bd,
Louise E.
Sinks
b,
Pravas
Deria
be,
Jiyong
Park
f,
Mu-Hyun
Baik
f and
Michael J.
Therien
*c
*abc,
Tae-Hong
Park
bd,
Louise E.
Sinks
b,
Pravas
Deria
be,
Jiyong
Park
f,
Mu-Hyun
Baik
f and
Michael J.
Therien
*c
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: j.park@moleng.kyoto-u.ac.jp
bDepartment of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA
cDepartment of Chemistry, Duke University, French Family Science Center, 124 Science Drive, Durham, North Carolina 27708-0346, USA. E-mail: michael.therien@duke.edu
dNuclear Chemistry Research Division, Korea Atomic Energy Research Institute, Daejeon, 34057, South Korea
eDepartment of Chemistry, Southern Illinois University, 1245 Lincoln Drive, Carbondale, Illinois 62901, USA
fDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST) & Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, South Korea
First published on 29th January 2018
Abstract
We report the synthesis and excited-state dynamics of a bis[p-ethynyldithiobenzoato]Pd(II)-bridged bis[(porphinato) zinc(II)] complex (PZn–Pd(edtb)2–PZn) that exhibits unusual solvent dielectric (ε)-dependent excited-state relaxation behavior. In nonpolar toluene solvent, PZn–Pd(edtb)2–PZn manifests an ultrafast S1 → T1 intersystem crossing time constant (τISC ≈ 2 ps), a broad, high-oscillator strength T1 → Tn transient absorption manifold (λmax(T1 → Tn) = 940 nm), and a near unity triplet-state formation quantum yield (ΦT ≈ 1; τT = 2.2 μs). In contrast, in moderately polar solvents (e.g., dichloromethane (DCM) or THF), the S1 → T1 intersystem crossing quantum yield is significantly suppressed (ΦT ≈ 0.2; τF ≈ 60 ps in DCM). Comparative femtosecond transient absorption studies in DCM and mixed DCM/toluene solvent systems reveal a new low-energy stimulated emission signal, the λmaxem of which is highly sensitive to solvent polarity. The lack of spectral signatures for radical species, and the emergence of intense stimulated emission indicate an additional low energy electronically excited-state (S*), populated via S1-state relaxation, that also possesses substantial singlet character. As solvent polarity is progressively increased, the energy of S* progressively decreases, eventually becoming lower than the S1 state and providing an excited-state relaxation channel that bypasses T1 state formation. These data show that the nature of the PZn–Pd(edtb)2–PZn excited-state dynamics is strongly influenced by the solvent dielectric, and that this Pd(II)-based linker motif offers new opportunities to engineer excited-state spin distributions and lifetimes in strongly conjugated chromophore assemblies.
Design, System, ApplicationTransition metals define molecular design tools to modulate chromophore optical, electronic, electrochemical, and magnetic properties in conjugated organic frameworks. Palladium-containing ligand frameworks that can form square or rectangular planar structures define a strategy to augment the electronic coupling between transition metals and π-conjugation organic system; in general, however, Pd-containing bridges do not guarantee significant electronic coupling between the metal ions and the organic π-conjugative systems. A model compound (PZn–Pd(edtb)2–PZn), composed of a bis[p-ethynyldithiobenzoato]Pd(II) linker (Pd(edtb)2) and zinc porphyrin (PZn) monomers was synthesized; spectroscopic studies of this complex revealed that the PZn–Pd(edtb)2–PZn design thus successfully couples a Pd(edtb)2 core and meso-ethynylated PZn chromophores to realize a π-conjugated supermolecule in which Pd(II) d orbitals participate in electronic coupling over a large porphyrin center-to-center distance (∼29.4 Å). This work demonstrates that the Pd(edtb)2 bridge is an effective transition metal-containing motif: i) to modulate the photophysics of porphyrin arrays, ii) to enhance the intersystem crossing rate constant, and iii) to introduce functionality that tunes the triplet-state formation quantum yield as a function of solvent polarity without modifying porphyrin structure. |
Introduction
Given their unique optical, electronic, electrochemical, excited-state properties, a great deal of research effort has been devoted to the molecular engineering of porphyrin arrays. The most compelling of these arrays feature molecular bridges that give rise to substantial electronic coupling between the porphyrin units, driving optical, electronic, electrochemical, and magnetic properties having utility in applications that span photovoltaics,1,2 nonlinear optics,3–7 molecular electronics,8–11 biological imaging,12,13 and photodynamic therapy.14,15 In the molecular design of such porphyrin arrays, earlier work highlights the importance of the bridging units in the modulation of key properties of these supermolecular systems.16,17Multiple organic conjugative motifs have been employed in porphyrin array design, and the photophysics of these systems have been established.16–26 On the other hand, porphyrins that take advantage of transition metal-containing bridging moieties represent an area of growing interest, as these motifs can play profound roles in modulating excited-dynamics, spintronic functionality, and charge transport. For example, porphyrin arrays linked by metal-containing bridges can facilitate enhanced triplet-state formation quantum yields, and augmented charge transfer interactions in these supermolecules.27–39 Such intimate connections between heavy metal centers and organic frameworks inspire new designs for engineering materials for optical, electronic and spintronic applications.4,40–50
In particular, palladium- or platinum-containing ligand frameworks that can form square or rectangular planar structures define an interesting strategy to electronically couple porphyrin planes via building blocks that enable electronic communication that involves d orbitals.27,30,31,34–36,38,39 In general, porphyrin arrays consisting of Pd-/Pt-containing bridges do not guarantee significant electronic coupling between the metal ions and the porphyrin π-ligand framework. Fundamental structure–property relationships for such supermolecular chromophores that manifest strong electronic coupling between the porphyrin units and metal-containing bridges are under-developed, and thus motivate the studies described below.
Here, we report the synthesis and excited-state dynamics of a bis[p-ethynyldithiobenzoato]Pd(II)-bridged bis[(porphinato)zinc] complex (PZn–Pd(edtb)2–PZn; Scheme 1). PZn–Pd(edtb)2–PZn consists of two (porphinato)zinc(II) (PZn) units and a bis[p-ethynyldithiobenzoato]palladium(II) (Pd(edtb)2) bridge that covalently connects the PZn moieties via the macrocycle meso-positions. This PZn–Pd(edtb)2–PZn design takes advantage of a Pd(dtb)2 core that provides substantial conjugation between the phenyl and CS2 entities.51,52 Previous optical and computational studies highlight the enhanced conjugative interactions between the terminal phenyl units made possible by the square planar palladium(II) coordination environment.53 Porphyrin arrays with bridging motifs involving ethynes, and related conjugative spacers such as 1,3-butadiyne, 4,7-diethynylbenzo[c][1,2,5]thiadiazole and 4,8-diethynylbenzo[1,2-c:4,5-c′]bis([1,2,5]thiadiazole) that take advantage of linkage connectivity involving the porphyrin meso-positions, display augmented ground- and excited-state electronic interactions.23 This enhanced electronic communication between the antipodal porphyrin units derives in part from a modest degree of quinoidal character made possible by the ethynyl connectivity (Scheme 1).16–18,20,23 The PZn–Pd(edtb)2–PZn design thus couples a Pd(dtb)2 core and meso-ethynylated PZn chromophores to realize a π-conjugated supermolecule in which Pd(II) d orbitals participate in electronic coupling over a large porphyrin center-to-center distance (∼29.4 Å). The PZn–Pd(edtb)2–PZn electronic absorption spectrum (Fig. 1) demonstrates transition manifolds that derive from extensive mixing of the porphyrin B- (Soret) (S0 → S2) and Q-band (S0 → S1) transitions.16,18,23,54–58 Pump–probe transient absorption (TA) spectroscopy of PZn–Pd(edtb)2–PZn in nonpolar toluene (Tol) solvent shows: i) a near-unity triplet-state formation quantum yield (ΦT) and a 2.0 ps of S1 → T1 intersystem crossing time constant (τISC), and ii) a T1 → Tn induced absorption (IA) band (λmax(T1 → Tn) = 940 nm) in the NIR spectral domain that is absent for the (porphinato)Zn(II) and Ph–Pd(edtb)2–Ph building block chromophores (Scheme 1). While 3[PZn–Pd(edtb)2–PZn]* features a T1-state lifetime (τT) of 2.2 μs in toluene solvent, excited-state dynamical data acquired in moderately polar solvents such as dichloromethane (DCM) and tetrahydrofuran (THF) demonstrate dramatically reduced excited-state lifetimes, with an extensive fraction of the excited state population (∼80–85%) exhibiting picosecond timescale relaxation dynamics, giving rise to ΦT values ≈ 0.2. The lack of spectral signatures indicative of radical species, and the emergence of intense stimulated emission in these transient spectral data, indicate a low energy electronically excited-state (S*) that possesses substantial singlet character. The extraordinary sensitivity of the energy of this new electronically excited-state that possesses substantial singlet character (S*) to the nature of the solvent dielectric relative to that exhibited by the T1 state suggests that bis[p-ethynyldithiobenzoato]Pd(II) and related conjugated bridges offer new opportunities to engineer excited-state spin distributions and lifetimes.
 | ||
| Scheme 1 Conjugated porphyrin arrays and quinoidal resonance contribution made possible by the macrocycle meso-ethynyl connectivity. | ||
 | ||
| Fig. 1 Comparative electronic absorption spectra of PZn–Pd(edtb)2–PZn in toluene (Tol; black) and CH2Cl2 (DCM; red) solvents, relative to PZnE (green), and Ph–Pd(edtb)2–Ph (blue) benchmarks. Inset shows normalized corrected room-temperature emission spectra of PZn–Pd(edtb)2–PZn [black: toluene; brown: DCM/Tol (χmol = 0.40); purple: DCM/Tol (χmol = 0.62); red: DCM]. χmol corresponds to the DCM mole fraction = (mole of DCM)/(total moles of solvent). Experimental conditions: λex = 652 nm for toluene; λex = 640 nm for DCM/Tol (χmol = 0.40), DCM/Tol (χmol = 0.62), and DCM solvents. | ||
Results and discussion
Scheme 2 outlines the synthesis of bis[4-[(10′,20′-bis(2′,6′-bis(3,3-dimethyl-1-butyloxy)phenyl)porphinato)zinc(II)ethyn-5′-yl]dithiobenzoato]palladium(II) (PZn–Pd(edtb)2–PZn); detailed synthetic procedures and characterization data are described in the ESI.† The deprotection of precursor (3)59 and subsequent metalation with PdCl2 induced precipitation of a dark solid; filtration followed by acetone/MeOH washing afforded analytically pure PZn–Pd(edtb)2–PZn. | ||
| Scheme 2 Synthesis of PZn–Pd(edtb)2–PZn and molecular structures of PZnE, and Ph–Pd(edtb)2–Ph. | ||
Comparative solvent-dependent electronic absorption spectra of PZn–Pd(edtb)2–PZn are displayed in Fig. 1 and S2,† along with those for the 5-ethynyl(porphinato)zinc(II) (PZnE) and bis[4-[(3′,5′-di-t-butylphenyl)ethynyl]dithiobenzoato]palladium(II) (Ph–Pd(edtb)2–Ph) reference compounds; the corresponding electronic absorption spectroscopic data are tabulated in Table 1 and S1.† The absorption spectral features of PZn–Pd(edtb)2–PZn differ markedly from those of the reference compounds (PZnE (ref. 60) and Ph–Pd(edtb)2–Ph (ref. 61)). Electronic absorption spectra of PZn–Pd(edtb)2–PZn display a new, low energy electronic transition manifold that features a substantial extinction coefficient (εabs = ∼105 M−1 cm−1 near 650 nm), contrasting those of PZnE or Ph–Pd(edtb)2–Ph.53,61 This absorption band centered at 650 nm resembles those of highly conjugated bis[(porphinato)metal] complexes, and derives from symmetry breaking of the porphyrin structural units, and oscillator strength redistributions that stem from conjugation expansion.16,18,23,54–58 DFT computational studies that explore dihedral angle-dependent conformational energies between hypothetical planar 5-ethynylporphyrin and bis(dithiobenzoato)Pd(II) units, or between related 5-phenylethynylporphyrin and the bis(dithioate)Pd(II) moieties (Fig. S3†) for a DFT-optimized PZn–Pd(edtb)2–PZn structure, suggest a substantial population of conformers having modest porphyrin-bridge-porphyrin interplanar torsional angles at ambient temperature.
| Solvent | PZn–Pd(edtb)2–PZn | ||
|---|---|---|---|
| Toluene | CH2Cl2 | THF | |
| a Numbers in parentheses correspond to spectral breadths (FWHM) of the respective transitions in units of cm−1. b The Stokes shift was taken as the energy difference between the λmaxem and λmaxem values. c Fluorescence lifetimes were determined from time-resolved emission experiments. d Excited singlet-state lifetimes derive from femtosecond transient absorption experiments. e The quantum yield of triplet formation (ΦT) was calculated from femtosecond transient experimental data (see ESI for details). f The time-resolved fluorescence signals of PZn–Pd(edtb)2–PZn in toluene decays within our instrumental temporal resolution of ∼15 ps. Therefore, the τF should be less than 15 ps. | |||
| λ abs (S0 → S1) [nm] | 652 | 645 | 687 |
| λ em [nm] | 692 (1656) | 809 (3205) | 869 (2581) |
| Stokes shiftb [cm−1] | 887 | 3143 | 3049 |
| τ F [ps] | <15f | 60 | 141 |
| (ΦF, %) | (<0.1) | (0.5) | (1.3) |
| τ es [ps] | 2.0 | 54 | 144 |
| τ T [μs] | 2.2 | 2.9 | — |
| (ΦT, %)e | (∼100) | (∼20) | (∼15) |
Room-temperature steady-state emission spectra of PZn–Pd(edtb)2–PZn in various solvent systems are shown in Fig. 1 (inset); corresponding spectroscopic data are compiled in Table 1. PZn–Pd(edtb)2–PZn in toluene shows an emission band with a λmaxem at 692 nm. Note that PZnE displays two emission peaks at 607 and 662 nm that mirror its Q-band absorption spectrum; notably these fluorescence bands feature no solvent polarity dependence.60Ph–Pd(edtb)2–Ph does not emit at room temperature. These emission data for PZnE and Ph–Pd(edtb)2–Ph indicate that PZn units and the bis[p-ethynyldithiobenzoato]Pd(II) bridge are strongly coupled in PZn–Pd(edtb)2–PZn. The Fig. 1 (inset) solvent-dependent emission spectra for PZn–Pd(edtb)2–PZn are characterized by broad emission bands (full-width-at-half-maximum (FWHM) = 1656 to 3205 cm−1), the λmaxem values of which vary from ∼690 to ∼810 nm [λmaxem = 692 nm, toluene (ε = 2.38); λmaxem = 809 nm, DCM (ε = 9.1)],62 stand in sharp contrast to corresponding electronic absorption spectra which vary little as a function of solvent. These PZn–Pd(edtb)2–PZn emission spectra highlight: i) a broader FWHM of the DCM emission band (3205 cm−1) relative to toluene (1656 cm−1), ii) a DCM Stokes shift (3143 cm−1) that is more than three-fold larger than that in toluene (887 cm−1), and iii) a fluorescence quantum yield (ΦF) that is substantially larger in DCM (ΦF ∼0.5%) relative to toluene solvent (ΦF < 0.1%). These marked disparate absorption and emission spectral characteristics suggest that the ground state is largely non-polar, while the emissive excited-state is dipolar in character. Time-resolved fluorescence spectroscopic measurements determine respective PZn–Pd(edtb)2–PZn fluorescence lifetimes (τF values) of 60 ps and <15 ps (unresolved) in DCM and toluene solvents (Table 1, Fig. S4†). Note that the magnitude of the τF value determined for PZn–Pd(edtb)2–PZn in toluene corresponds to a timescale similar to the instrumental temporal resolution limit (∼15 ps), and therefore the τF cannot be accurately determined and its upper limit is 15 ps. The τF less than 15 ps for PZn–Pd(edtb)2–PZn in toluene is consistent with the low ΦF value (<0.1%) in this medium.
The excited-state dynamics of PZn–Pd(edtb)2–PZn was further interrogated via femtosecond (fs) and nanosecond transient absorption (TA) spectroscopic experiments. Fig. 2a displays representative fs TA spectra of PZn–Pd(edtb)2–PZn in toluene at selected time delays following photoexcitation (λex = 650 nm). Conventional (porphinato)Zn(II) complexes are characterized by nanosecond (ns) timescale lowest singlet excited state (S1-state) lifetimes, prominent induced absorption (IA) bands in the 400–650 nm spectral domain, and a broad low oscillator strength IA band in the 650–900 nm spectral region that is ascribed to an S1 → Sn transition manifold.63–65 On the other hand, the fs TA spectra acquired for PZn–Pd(edtb)2–PZn in toluene (Fig. 2a) manifest the following features: i) ground state bleaching (GSB) bands at ∼550 nm and ∼650 nm, ii) an IA band, peaking at ∼500 nm that is evident at tdelay ∼1 ps and overlaps with the GSB bands at ∼550 nm and ∼650 nm, iii) an IA band peaking at ∼510 nm that is evident at tdelay > ∼50 ps, and iv) an intense IA band at the near-infrared (NIR) spectral domain (720–1200 nm, λmaxabs ∼940 nm) that grows in intensity over time delays spanning several tens of picoseconds, and persists beyond our instrumental delay limit (up to 3 ns). Note that the IA bands at ∼510 nm overlap with IA bands evident at tdelay = 1 μs in the corresponding nanosecond-to-microsecond time domain TA spectroscopic experiments (Fig. S5a†). As the decay dynamics evident on this timescale are sensitive to molecular oxygen exposure, we attribute these IA bands to T1 → Tn transitions of 3[PZn–Pd(edtb)2–PZn] (λmax(T1 → Tn) = 940 nm; vide infra). Over time delays that extend up through 3 ns, no GSB recovery was observed, indicating a near-unity quantum yield of S1 → T1 intersystem crossing (ΦT). Nanosecond (ns)-to-microsecond (μs) time domain TA spectroscopic data acquired in degassed toluene (Fig. S5a and b†) determine a T1-state lifetime (τT) of 2.2 μs.
 | ||
| Fig. 2 Representative femtosecond transient absorption spectra acquired for PZn–Pd(edtb)2–PZn in: (a) toluene, (b) DCM, (c) DCM/Tol (χmol = 0.14), and (d) DCM/Tol (χmol = 0.62), at time delays noted. Experimental conditions: λex = 650, 671, 675, 690 nm for (a)–(d), respectively; pump energy = ∼0.3 μJ per pulse; temperature = 20 °C. Inverted steady-state absorption (black dashed line) and emission spectra (red dotted line) are displayed for comparison. | ||
PZn–Pd(edtb)2–PZn pump-probe transient absorptive dynamical data acquired in toluene solvent were analyzed using multiwavelength global fitting (Fig. S7†). The NIR IA signals characteristic of electronically excited 3[PZn–Pd(edtb)2–PZn] exhibit two rise time components of 2.0 ps and 16 ps, which are attributed to S1 → T1 intersystem crossing and chromophore–chromophore torsional relaxation, respectively. The 2.0 ps time constant is associated with the decay of the S1 → Sn IA signal at 743 nm in Fig. 2a and the stimulated emission (SE) signal evident at 677 nm, which probes the depletion dynamics of the S1-state. Thus, we can assign the S1 → T1 intersystem crossing time constant (τISC) to be 2.0 ps. Note that the 16 ps time constant is associated with a substantial increase in the intensity of the T1 → Tn NIR IA band; previous photophysical studies that interrogate ethyne-linked (porphinato)Zn(II)-based multichromophore systems indicate that this process is linked to structural equilibration governed by chromophore–chromophore torsional dynamics.22,65–68 This spectral evolution probed in toluene underscores that the initially prepared PZn–Pd(edtb)2–PZn excited state undergoes fast S1 → T1 intersystem crossing (τISC ∼2.0 ps), congruent with the low ΦF value (<0.1%) and the unresolved fluorescence lifetime within our temporal resolution (τF < 15 ps) in this solvent; Fig. 4C data highlight these dynamics.
In earlier work, Duncan et al. reported palladium(II)-containing porphyrin arrays featuring a meso-to-meso ethyne-bridged linkage topology, showing that this design accelerates S1 → T1 intersystem crossing (τISC = 8.6 and 52.0 ps for Pd-porphyrin dimers and trimers relative to analogous arrays that featured a porphyrin central zinc ion.55 These data acquired for PZn–Pd(edtb)2–PZn suggest that the Pd(edtb)2 bridge provides an even more effective strategy to accelerate the S1 → T1 intersystem crossing rate constant in these related supermolecules.
PZn–Pd(edtb)2–PZn excited-state dynamics were further explored in moderately polar DCM solvent (Fig. 2b). The TA spectral data acquired in DCM at ∼0.3 ps < tdelay < ∼3 ns are clearly distinguished from those in toluene (Fig. 2a); note in this regard that the spectral evolution in DCM, occurring within the initial 3 ns following optical excitation is highly complicated, showing marked evolution of IA and SE signals.
To help clarify this complex TA spectral evolution, Fig. 3 highlights the excited-state dynamics that characterize four distinct time domains: a) 0.1 ps < tdelay < 0.4 ps, b) 0.4 ps < tdelay < 4 ps, c) 10 ps < tdelay < 100 ps, d) 100 ps < tdelay < 3 ns.
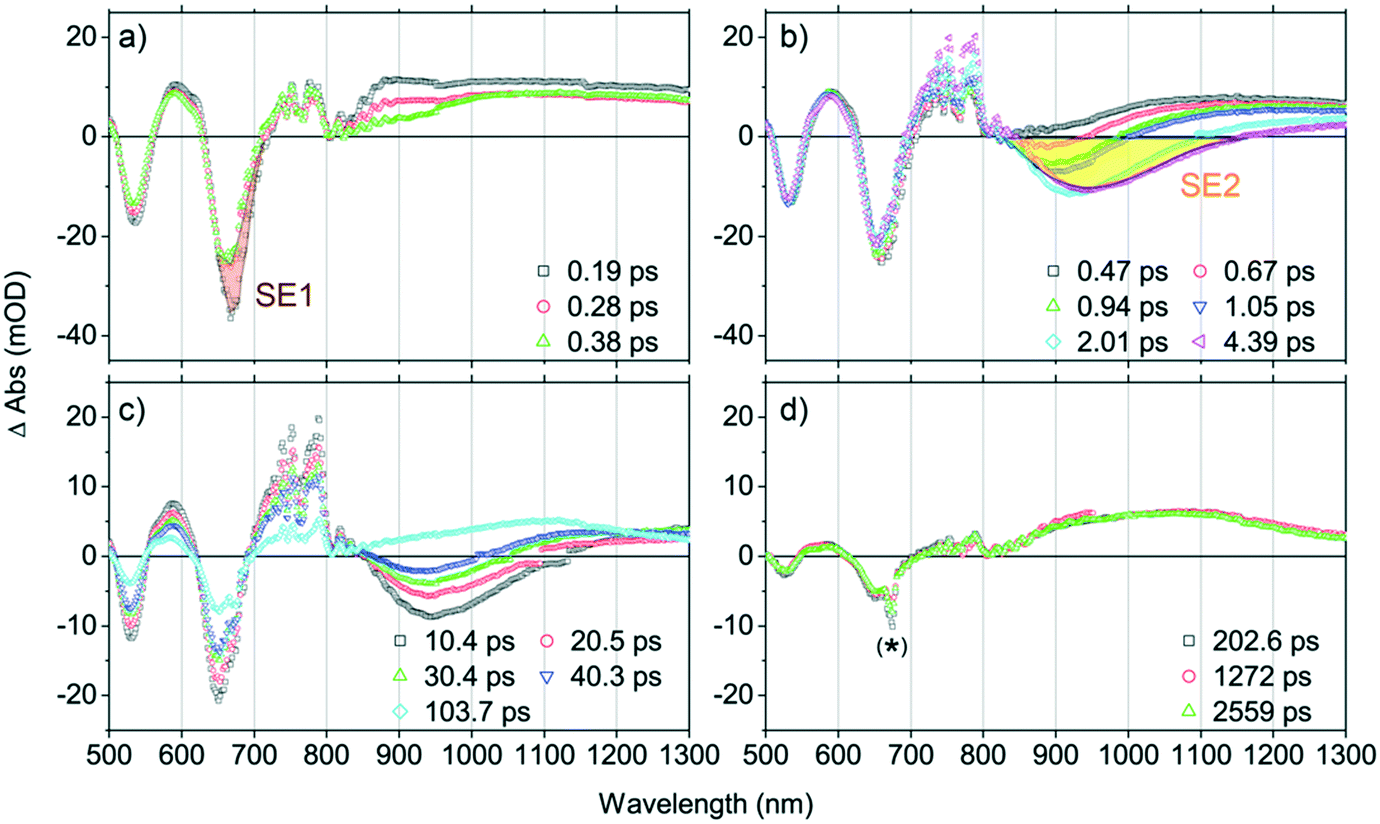 | ||
| Fig. 3 Representative femtosecond transient absorption spectral evolution of PZn–Pd(edtb)2–PZn in DCM solvent, recorded at the time delays noted: (a) tdelay = 0.1–0.4 ps, (b) 0.4–4 ps, (c) 10–100 ps, and (d) 200–2500 ps. Experimental conditions: λex = 671 nm; pump energy = 300 nJ per pulse; magic angle polarization; ambient temperature. The artifact at ∼671 nm (denoted with *) derives from scattering of the excitation beam. The spectral signatures corresponding to SE1 and SE2 are highlighted in brown and yellow, respectively, in panels a) and b). | ||
1. At tdelay < 0.4 ps (Fig. 3a): the TA spectra in this time domain are characterized by a broad IA band spanning the 720–1300 nm spectral regime; note that this IA spectral feature is identical to that manifest in toluene solvent over similar delay times (e.g., tdelay < 1 ps, Fig. 2a) and is assigned to the S1 → Sn transition. Also evident is a pronounced blue shift of the Q-state GSB signal (from 664 nm to 660 nm), along with an apparent intensity decrease at the earliest time delays: based on the corresponding position of the Q-state absorption maximum in the steady-state spectrum (645 nm), these spectral data indicate a significant contribution of S1 state stimulated emission (SE1) over these time delays, congruent with the Fig. 2a steady state and transient dynamical data acquired in toluene at early time delays (e.g., ∼665 nm at tdelay < 1 ps).
2. At 0.4 ps < tdelay < 4 ps (Fig. 3b): these TA spectra exhibit: i) a broad negative spectral feature (800–1100 nm), that displays a dynamic red-shift through time delays up to ∼4 ps; ii) an abrupt decrease of the SE1 contribution at ∼660 nm, suggesting the depletion of the initially prepared S1-state population; and iii) a conspicuous lack of GSB recovery over this time domain, indicating migration of the S1-state population into another excited-state. Given the steady-state emission spectrum of PZn–Pd(edtb)2–PZn in DCM (Fig. 1) and the corresponding Q-state absorption band position (645 nm), we attribute this broad negative spectral feature (800–1100 nm) to an additional SE signal (SE2) characteristic of an excited-state (S*) that is significantly lower in energy than the initially prepared S1-state. Note that the ground-state electronic absorption spectrum acquired in DCM solvent exhibits a low energy absorption manifold centered at 645 nm and that SE1 (∼660 nm) shows a mirror-image relationship to this manifold. As no ground-state electronic absorption is observed over the 800–1100 nm spectral domain, the S* state must be accessible only from S1, and not from a direct S0 → S* transition. In this regard, PZn–Pd(edtb)2–PZn displays excited-state dynamics that resemble those of several push–pull polyenes and carotenoids such as peridinin: in polar solvents, these chromophores access an emissive excited nuclear conformation that does not resemble the ground state, and thereby manifest an additional SE band evident at a longer wavelength than that for S1 → S0 emission. These chromophores, akin to PZn–Pd(edtb)2–PZn, also display no direct S0 → S* electronic transition.69–72 Note that this NIR SE2 is also observed in THF (Fig. S8†) but is absent in toluene (Fig. 2a), suggesting that production of S* depends on solvent polarity (vide infra).
3. At 10 ps < tdelay < 100 ps (Fig. 3c): over this time domain, TA spectral evolution highlights the time-dependent diminution of the SE2 signal at ∼950 nm along with the corresponding recovery of GSB bands at ∼550 nm and ∼650 nm. These data contrast those acquired in toluene, where no measurable excited-state relaxation is evident over the initial 3 ns following optical excitation (Fig. 2a). As S* → S0 stimulated emission (SE2) diminishes at tdelay > 100 ps in DCM, an IA band emerges over the NIR spectral domain.
4. At 100 ps < tdelay (Fig. 3d): no TA spectral evolution is observed from 100 ps through the tdelay limit of the instrument (∼3 ns); note that these TA spectra resemble those acquired at tdelay > 10 ps in toluene solvent (Fig. 2a). This NIR spectral signature decays with a time constant (τT) of 2.9 μs, determined from ns-TA spectroscopy (Fig. S5c and d†) and is therefore assigned as a T1 → Tn transition manifold. The ΦT in DCM was determined to be ∼0.2 from the methods described in the ESI.†
A multiwavelength global fit of the TA spectroscopic data acquired for PZn–Pd(edtb)2–PZn in DCM solvent determines time constants for SE1 decay (τS1 = 0.44 ps), SE2 decay (τS* = 54 ps), and GSB recovery (τGSB = 61 ps). Note that the 54 ps SE2 decay time constant is consistent with τGSB, as well as the PZn–Pd(edtb)2–PZn emission lifetime (τF = 60 ps; Fig. S4†). Therefore, the S* → S0 relaxation rate corresponds to (54 ps)−1.
With respect to the mechanism of S1 state depletion and T1 state formation, two possibilities can be considered: i) sequential population transfer from S* to T1 (S1 → S* → T1), or ii) parallel processes that allow for both S1 → T1 intersystem crossing and S1 → S*. The combination of the T1-state formation quantum yield (ΦT ∼0.2) in DCM, the lifetimes of the S1 and S* states (τS1 = 0.44 ps; τS* = 54 ps), and the absence of a slow rise of the T1 → Tn transition beyond tdelays > 200 ps (Fig. 3d) strongly indicate that the T1-state population derives from S1 → T1 intersystem crossing that competes with S1 → S* relaxation.‡ The S1 → T1 intersystem crossing time constant (τISC) is therefore determined to be 2.2 ps (eqn (1) and (2)):
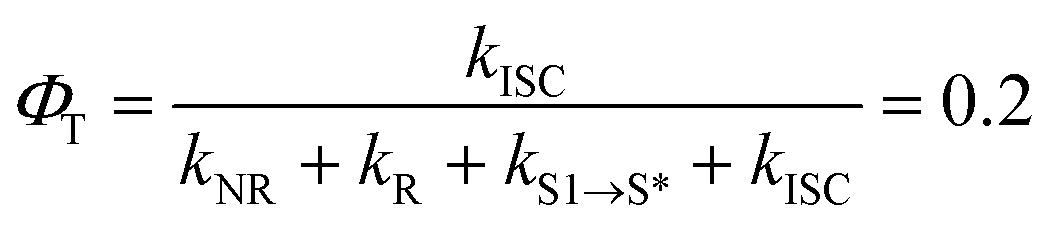 | (1) |
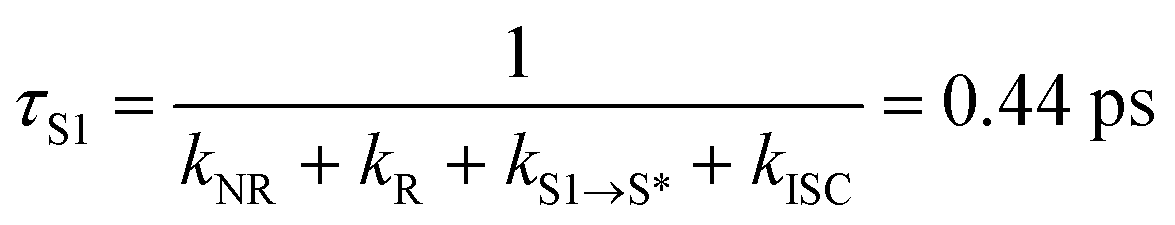 | (2) |
What clearly differs as a function of solvent polarity is the S*-state accessibility from S1. Because GSB recovery is negligible in DCM at tdelays < ∼10 ps, τS1→S* can be determined to be ∼0.6 ps, as the magnitudes of kNR and kR are significantly smaller than those for both kS1→S* and kISC. This 0.6 ps time constant for S1 → S* relaxation agrees with the emergence of the SE2 signal on the sub-picosecond timescale, shown in Fig. 3b. PZn–Pd(edtb)2–PZn excited-state relaxation in DCM thus occurs predominantly through dynamical channels defined by S1 → S* and S* → S0, with only a fraction (∼0.2) of the initially prepared excited state returning to ground via the S1 → T1 and T1 → S0 pathway (Fig. 4A). The fast S1 → T1 intersystem crossing time constant (∼2 ps), and the even faster time constant for S1 → S* dynamics (∼0.6 ps) determined for PZn–Pd(edtb)2–PZn in DCM solvent suggests little or no contribution of structural dynamics of this large supermolecular system playing a role in these dynamics, as previous studies of porphyrin dimers featuring ethyne and butadiyne bridges have shown that the singlet-state structural relaxation time constants range between 10–30 ps.22,65,73 In this regard, the solvent nature thus likely plays a crucial role in modulating excited state dynamics.
 | ||
| Fig. 4 Summary of the solvent polarity dependent excited state dynamics of PZn–Pd(edtb)2–PZn in: (A) DCM, (B) a DCM/Tol mixture (χmol = 0.62), and (C) toluene. | ||
PZn–Pd(edtb)2–PZn excited-state dynamics determined in DCM/toluene solvent mixtures provide insights into how relative S1 and S* state energies vary as a function of solvent polarity. Fig. 1b, 2c and d, and S9† show steady-state emission and fs-TA spectra of PZn–Pd(edtb)2–PZn recorded in solvents composed of various DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Tol molar ratios; in these solvents, DCM/Tol (χmol) represents the DCM mole fraction, the moles of DCM/(total moles of DCM and toluene in a mixture). In Fig. 2c, the PZn–Pd(edtb)2–PZn TA spectra recorded in DCM/Tol (χmol = 0.14) resemble those acquired in neat toluene, and lack the SE2 signature. In contrast, the PZn–Pd(edtb)2–PZn TA spectral evolution in DCM/Tol (χmol = 0.62) resembles that determined in neat DCM (Fig. 2d). Note, however, that in the DCM/Tol (χmol = 0.62) mixture, the SE2 signal emerges at ∼900 nm, blue-shifted with respect to that observed in neat DCM (∼950 nm, in Fig. 2b). Additional experiments further confirm that the SE2 peak position is clearly solvent-dependent: for example, in DCM/Tol (χmol = 0.40; Fig. S9†) the SE2 signal appears at ∼850 nm.
:Tol molar ratios; in these solvents, DCM/Tol (χmol) represents the DCM mole fraction, the moles of DCM/(total moles of DCM and toluene in a mixture). In Fig. 2c, the PZn–Pd(edtb)2–PZn TA spectra recorded in DCM/Tol (χmol = 0.14) resemble those acquired in neat toluene, and lack the SE2 signature. In contrast, the PZn–Pd(edtb)2–PZn TA spectral evolution in DCM/Tol (χmol = 0.62) resembles that determined in neat DCM (Fig. 2d). Note, however, that in the DCM/Tol (χmol = 0.62) mixture, the SE2 signal emerges at ∼900 nm, blue-shifted with respect to that observed in neat DCM (∼950 nm, in Fig. 2b). Additional experiments further confirm that the SE2 peak position is clearly solvent-dependent: for example, in DCM/Tol (χmol = 0.40; Fig. S9†) the SE2 signal appears at ∼850 nm.
This increasing blue shift of the SE2 signal with decreasing solvent polarity agrees with steady-state emission spectra highlighted in Fig. 1 (inset). The different positions of the steady-state emission spectra λmaxem values and the SE2 signature stem from both a dynamic Stokes-shift contribution to SE2, and the overlap of SE2 and IA signals in the TA spectra (Fig. 3). As a result, in non-polar solvents such as toluene and DCM/Tol (χmol = 0.14), S1 → S0 radiative relaxation is responsible for the steady-state emission highlighted in the Fig. 1 inset, whereas in moderately polar solvents (DCM, DCM/Tol (χmol = 0.40 and 0.62), or THF), the emission originates from the S*→S0 radiative relaxation. Approximation of the dielectric constant (ε) of the DCM/Tol mixtures§ allows the emission peak energy (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) em) to be displayed as a function of the Onsager function (f(ε) = 2(ε − 1)/(2ε + 1); Fig. S10†). These data (χmol = 0.40, 0.62, 1.0) highlight a linear increase of em with decreasing f(ε). Note, however, that em for χmol = 0.0 (neat toluene) deviates from the linear fit line (red solid line in Fig. S10†): this dependence derives from the fact that in moderately polar solvent two different PZn–Pd(edtb)2–PZn emitting states are manifest (S1 and S*), while in neat toluene, only emission from S1 is detected. From the linear fit line, the S* state in neat toluene is estimated to lie ∼15900 cm−1 above the ground state and higher in energy than S1. For χmol = 0.14, where PZn–Pd(edtb)2–PZn excited-state dynamical data (Fig. 2c) show no evidence for SE2, this analysis determines an S* state energy of ∼14700 cm−1, which is also higher than the em in toluene (em = 14400 cm−1) and congruent with the near-unit ΦT and similarity of the excited-state dynamics evinced in DCM/Tol (χmol = 0.14) and neat toluene solvent systems.
em) to be displayed as a function of the Onsager function (f(ε) = 2(ε − 1)/(2ε + 1); Fig. S10†). These data (χmol = 0.40, 0.62, 1.0) highlight a linear increase of em with decreasing f(ε). Note, however, that em for χmol = 0.0 (neat toluene) deviates from the linear fit line (red solid line in Fig. S10†): this dependence derives from the fact that in moderately polar solvent two different PZn–Pd(edtb)2–PZn emitting states are manifest (S1 and S*), while in neat toluene, only emission from S1 is detected. From the linear fit line, the S* state in neat toluene is estimated to lie ∼15900 cm−1 above the ground state and higher in energy than S1. For χmol = 0.14, where PZn–Pd(edtb)2–PZn excited-state dynamical data (Fig. 2c) show no evidence for SE2, this analysis determines an S* state energy of ∼14700 cm−1, which is also higher than the em in toluene (em = 14400 cm−1) and congruent with the near-unit ΦT and similarity of the excited-state dynamics evinced in DCM/Tol (χmol = 0.14) and neat toluene solvent systems.
A multiexponential global fit for PZn–Pd(edtb)2–PZn in DCM/Tol (χmol = 0.62) yields time constants of 0.6 ps, 3 ps, 211 ps, and a long component (>3 ns). The 0.6 ps component, related to the decay of SE1, corresponds to τS1. In both toluene and DCM, τISC is ∼2 ps; as τISC shows little solvent dependence, ΦT can be determined to be ∼0.27 in DCM/Tol (χmol = 0.62). This ΦT value determined from eqn (1) and (2) agrees with that evaluated from the TA spectral data (ΦT = ∼0.3), as described in the ESI.† From the ΦT, τS1, and τISC values, we determine τS1→S* to be ∼0.8 ps. PZn–Pd(edtb)2–PZn excited-state dynamics determined in DCM/Tol (χmol = 0.62) are summarized in Fig. 4B.
Fig. 4 highlights the fact that the S* energy level is sensitive to small changes of solvent polarity, suggesting that this state may possess charge-transfer character; certainly, as S* may be destabilized relative to both S1 and T1, its electronic structure must diverge from both the initially prepared singlet and relaxed triplet states. Previously, the excited-state dynamics of other highly conjugated organic chromophoric systems such as peridinin69–71 and push–pull type polyenes72 have shown closely related solvent-polarity-dependent excited-state dynamical behaviors. TA spectroscopic studies of peridinin, for example, revealed two singlet excited states, one of which corresponded to a highly delocalized intramolecular charge transfer (ICT) state, which gives rise to sub-optical band gap stimulated emission only in polar solvents and no spectral signatures consistent with cation or anion radical species.70 While the exact nature of the PZn–Pd(edtb)2–PZn S* state manifest in polar solvents is obscure, congruent with the transient spectral signatures that characterize peridinin69–71 and push–pull type polyenes,72 these PZn–Pd(edtb)2–PZn excited-state dynamical studies evince no spectroscopic hallmarks congruent with the light-driven formation of cation or anion radical species in polar solvents.74
Experimental
Materials and methods
All manipulations were carried out under Ar unless otherwise stated. CH2Cl2 and tetrahydrofuran (THF) were distilled over CaH2 and Na/benzophenone under N2, respectively. Triethylamine (Et3N) was distilled over KOH under N2. All NMR solvents were used as received. Pd(PPh3)4 was purchased from Strem Chemicals and used as received. Tetrabutylammonium fluoride (TBAF) (1 M THF solution, Aldrich) was further diluted to 0.1 M. For optical spectroscopy, HPLC grade toluene, CH2Cl2, and THF were purchased from Sigma Aldrich and purified by passing through alumina columns in a Puresolv solvent purification system (Innovative Technology, Inc., Amesbury, MA) before use. ACS spectrophotometric grade CHCl3 was purchased from Sigma Aldrich and used as received. Synthetic details and characterization data are described in the ESI.†Instrumentation
Electronic absorption spectra were collected using a Shimadzu UV-1700 UV-vis spectrophotometer or a Varian Cary-5000 UV-vis-NIR spectrophotometer. Steady-state emission spectra were obtained on FLS920P spectrophotometer (Edinburgh Instruments Ltd. Livingston, UK) in 1 cm quartz optical cells. A set of appropriate long-pass filters were used to remove second order scattering light from the lamp and grating for the excitation beam and to eliminate scattered excitation light on the emission collection pathway. Steady-state emission spectra were corrected using the correction factor generated by the manufacturer. Details of material characterization and time-resolved photoluminescence spectroscopy and femtosecond and nanosecond-microsecond transient absorption apparatus75,76 are described in the ESI.†Conclusion
In summary, we describe the synthesis of a bis[p-ethynyldithiobenzoato]Pd(II)-bridged bis[(porphinato)zinc(II)] complex (PZn–Pd(edtb)2–PZn). Electronic absorption spectra of PZn–Pd(edtb)2–PZn suggest extensive electronic delocalization in this supermolecule and highlight a new, low energy electronic transition manifold that features a substantial extinction coefficient (εabs = ∼105 M−1 cm−1 near 650 nm), contrasting benchmark spectral data for simple (porphinato)zinc monomers and Pd(edtb)2-bridged aryl units. Excited-state dynamical data acquired for PZn–Pd(edtb)2–PZn demonstrate that this structure exhibits unusual solvent dielectric (ε)-dependent excited-state relaxation behavior. In nonpolar toluene solvent, PZn–Pd(edtb)2–PZn manifests an ultrafast S1 → T1 intersystem crossing time constant (τISC ≈ 2 ps), a broad, high-oscillator strength T1 → Tn transient absorption manifold in the NIR (λmax(T1 → Tn) = 940 nm), and a near unit triplet-state formation quantum yield (ΦT ≈ 1; τT = 2.2 μs). The Pd(edtb)2 bridge thus demonstrates an effective motif to accelerate the S1 → T1 intersystem crossing dynamics in organic molecular systems. In contrast, in moderately polar solvents (e.g., CH2Cl2 (DCM) or THF), the S1 → T1 intersystem crossing quantum yield is significantly suppressed (ΦT ≈ 0.2; τF ≈ 60 ps in DCM) despite the similarly ultrafast τISC (≈2.2 ps). Comparative femtosecond transient absorption studies in DCM and mixed DCM:Tol solvent systems reveal a new low-energy stimulated emission signal, the λmaxem of which is highly sensitive to solvent polarity. The lack of spectral signatures for radical species and the emergence of intense stimulated emission indicate that this additional low-energy electronically excited-state (S*), populated via S1-state relaxation, also possesses substantial singlet character. As solvent polarity is progressively increased from the toluene benchmark, transient dynamical data indicate that the energy of S* progressively decreases, eventually becoming lower than the S1 state, providing an excited-state relaxation channel that bypasses T1 state formation. These data show that the nature of the PZn–Pd(edtb)2–PZn excited-state dynamics is strongly influenced by the solvent dielectric, and that this Pd(II)-based linker motif offers new opportunities to engineer excited-state spin distributions and lifetimes in strongly conjugated chromophore assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, of the U.S. Department of Energy through Grant DE-SC0001517. J. P. acknowledges The Kyoto University Research Fund for Young Scientists (Start-up) and JSPS KAKENHI Grant-in-Aid for Research Activity (Start-up Grant Number 17H06791). T.-H. P. acknowledges support from the National Research Foundation of Korea (2017M2A8A5014710). J.-Y. P. and M.-H. B. acknowledge support from the Institute for Basic Science (IBS-R10-D1) in Korea.Notes and references
- H.-P. Wu, Z.-W. Ou, T.-Y. Pan, C.-M. Lan, W.-K. Huang, H.-W. Lee, N. M. Reddy, C.-T. Chen, W.-S. Chao, C.-Y. Yeh and E. W.-G. Diau, Energy Environ. Sci., 2012, 5, 9843–9846 CAS.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- T. Ishizuka, L. E. Sinks, K. Song, S.-T. Hung, A. Nayak, K. Clays and M. J. Therien, J. Am. Chem. Soc., 2011, 133, 2884–2896 CrossRef CAS PubMed.
- G.-J. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC.
- M. de Torres, S. Semin, I. Razdolski, J. Xu, J. A. A. W. Elemans, T. Rasing, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2015, 51, 2855–2858 RSC.
- S. U. Hassan, H. M. Asif, Y. Zhou, L. Zhang, N. Qu, J. Li and Z. Shi, J. Phys. Chem. C, 2016, 120, 27587–27599 CAS.
- N. Fukui, T. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2017, 139, 9075–9088 CrossRef CAS PubMed.
- G. Sedghi, L. J. Esdaile, H. L. Anderson, S. Martin, D. Bethell, S. J. Higgins and R. J. Nichols, Adv. Mater., 2011, 24, 653–657 CrossRef PubMed.
- Z. Li, T.-H. Park, J. Rawson, M. J. Therien and E. Borguet, Nano Lett., 2012, 12, 2722–2727 CrossRef CAS PubMed.
- M. Noori, H. Sadeghi and C. J. Lambert, Nanoscale, 2017, 9, 5299–5304 RSC.
- M. D. Peeks, C. E. Tait, P. Neuhaus, G. M. Fischer, M. Hoffmann, R. Haver, A. Cnossen, J. R. Harmer, C. R. Timmel and H. L. Anderson, J. Am. Chem. Soc., 2017, 139, 10461–10471 CrossRef CAS PubMed.
- P. P. Ghoroghchian, P. R. Frail, K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922–2927 CrossRef CAS PubMed.
- J. E. Raymond and T. Goodson III, J. Phys. Chem. Lett., 2011, 2, 329–333 CrossRef CAS.
- S. Achelle, P. Couleaud, P. Baldeck, M.-P. Teulade-Fichou and P. Maillard, Eur. J. Org. Chem., 2011, 2011, 1271–1279 CrossRef.
- Z. Xiang, L. Zhu, L. Qi, L. Yan, Y. Xue, D. Wang, J.-F. Chen and L. Dai, Chem. Mater., 2016, 28, 8651–8658 CrossRef CAS.
- V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105–1111 CAS.
- T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- V. S.-Y. Lin and M. J. Therien, Chem. – Eur. J., 1995, 1, 645–651 CrossRef CAS.
- P. J. Angiolillo, V. S.-Y. Lin, J. M. Vanderkooi and M. J. Therien, J. Am. Chem. Soc., 1995, 117, 12514–12527 CrossRef CAS.
- H. L. Anderson, Chem. Commun., 1999, 2323–2330 RSC.
- N. Aratani, A. Osuka, H. S. Cho and D. Kim, J. Photochem. Photobiol., C, 2002, 3, 25–52 CrossRef CAS.
- I. V. Rubtsov, K. Susumu, G. I. Rubtsov and M. J. Therien, J. Am. Chem. Soc., 2003, 125, 2687–2696 CrossRef CAS PubMed.
- K. Susumu, T. V. Duncan and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 5186–5195 CrossRef CAS PubMed.
- T. V. Duncan, K. Susumu, L. E. Sinks and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 9000–9001 CrossRef CAS PubMed.
- N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922–1934 CrossRef CAS PubMed.
- D. Kim, Multiporphyrin Arrays: Fundamentals and Applications, Pan Stanford Publishing, 2012 Search PubMed.
- A. Ferri, G. Polzonetti, S. Licoccia, R. Paolesse, D. Favretto, P. Traldi and M. V. Russo, J. Chem. Soc., Dalton Trans., 1998, 4063–4069 RSC.
- S. Richeter, C. Jeandon, J.-P. Gisselbrecht, R. Ruppert and H. J. Callot, J. Am. Chem. Soc., 2002, 124, 6168–6179 CrossRef CAS PubMed.
- L. Yu, K. Muthukumaran, I. V. Sazanovich, C. Kirmaier, E. Hindin, J. R. Diers, P. D. Boyle, D. F. Bocian, D. Holten and J. S. Lindsey, Inorg. Chem., 2003, 42, 6629–6647 CrossRef CAS PubMed.
- R. D. Hartnell and D. P. Arnold, Organometallics, 2004, 23, 391–399 CrossRef CAS.
- Y.-J. Chen, S.-S. Chen, S.-S. Lo, T.-H. Huang, C.-C. Wu, G.-H. Lee, S.-M. Peng and C.-Y. Yeh, Chem. Commun., 2006, 1015–1017 RSC.
- D. Bellows, S. M. Aly, C. P. Gros, M. El Ojaimi, J.-M. Barbe, R. Guilard and P. D. Harvey, Inorg. Chem., 2009, 48, 7613–7629 CrossRef CAS PubMed.
- P. J. Chmielewski, B. Durlej, M. Siczek and L. Szterenberg, Angew. Chem., Int. Ed., 2009, 48, 8736–8739 CrossRef CAS PubMed.
- Y. Matano, K. Matsumoto, H. Hayashi, Y. Nakao, T. Kumpulainen, V. Chukharev, N. V. Tkachenko, H. Lemmetyinen, S. Shimizu, N. Kobayashi, D. Sakamaki, A. Ito, K. Tanaka and H. Imahori, J. Am. Chem. Soc., 2012, 134, 1825–1839 CrossRef CAS PubMed.
- M. Abdelhameed, P.-L. Karsenti, A. Langlois, J.-F. Lefebvre, S. Richeter, R. Ruppert and P. D. Harvey, Chem. – Eur. J., 2014, 20, 12988–13001 CrossRef CAS PubMed.
- M. Abdelhameed, A. Langlois, P.-L. Karsenti, S. Richeter, R. Ruppert and P. D. Harvey, Chem. Commun., 2014, 50, 14609–14612 RSC.
- J. Haumesser, J.-P. Gisselbrecht, L. Karmazin-Brelot, C. Bailly, J. Weiss and R. Ruppert, Organometallics, 2014, 33, 4923–4930 CrossRef CAS.
- H.-W. Jiang, T. Tanaka and A. Osuka, Chem. Sci., 2015, 6, 6102–6105 RSC.
- N. Fukui, H.-W. Jiang and A. Osuka, Org. Chem. Front., 2017, 4, 767–772 RSC.
- U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC.
- J. L. Segura, Acta Polym., 1998, 49, 319–344 CrossRef CAS.
- G. R. Whittell and I. Manners, Adv. Mater., 2007, 19, 3439–3468 CrossRef CAS.
- E. Holder, B. M. W. Langeveld and U. S. Schubert, Adv. Mater., 2005, 17, 1109–1121 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- W.-Y. Wong and C.-L. Ho, Acc. Chem. Res., 2010, 43, 1246–1256 CrossRef CAS PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530–2541 CrossRef CAS.
- J. Rawson, P. J. Angiolillo, P. R. Frail, I. Goodenough and M. J. Therien, J. Phys. Chem. B, 2015, 119, 7681–7689 CrossRef CAS PubMed.
- R. Wang, A. M. Brugh, J. Rawson, M. J. Therien and M. D. E. Forbes, J. Am. Chem. Soc., 2017, 139, 9759–9762 CrossRef CAS PubMed.
- M.-C. Dul, E. Pardo, R. Lescouëzec, Y. Journaux, J. Ferrando-Soria, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, D. Cangussu, C. L. M. Pereira, H. O. Stumpf, J. Pasán and C. Ruiz-Pérez, Coord. Chem. Rev., 2010, 254, 2281–2296 CrossRef CAS.
- M. Bonamico and G. Dessy, Chem. Commun., 1968, 483–484 RSC.
- M. Bonamico, G. Dessy, V. Fares and L. Scaramuzza, J. Chem. Soc., Dalton Trans., 1975, 2250–2256 RSC.
- C. Furlani and M. L. Luciani, Inorg. Chem., 1968, 7, 1586–1592 CrossRef CAS.
- R. Shediac, M. H. B. Gray, H. T. Uyeda, R. C. Johnson, J. T. Hupp, P. J. Angiolillo and M. J. Therien, J. Am. Chem. Soc., 2000, 122, 7017–7033 CrossRef CAS.
- T. V. Duncan, P. R. Frail, I. R. Miloradovic and M. J. Therien, J. Phys. Chem. B, 2010, 114, 14696–14702 CrossRef CAS PubMed.
- S. M. LeCours, S. G. DiMagno and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 11854–11864 CrossRef CAS.
- S. M. LeCours, H.-W. Guan, S. G. DiMagno, C. H. Wang and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 1497–1503 CrossRef.
- S. M. LeCours, C. M. Philips, J. C. de Paula and M. J. Therien, J. Am. Chem. Soc., 1997, 119, 12578–12589 CrossRef CAS PubMed.
- T.-H. Park and M. J. Therien, Org. Lett., 2007, 9, 2779–2782 CrossRef CAS PubMed.
- K. Susumu and M. J. Therien, J. Am. Chem. Soc., 2002, 124, 8550–8552 CrossRef CAS PubMed.
- T.-H. Park, PhD Dissertation, University of Pennsylvania, 2008 Search PubMed.
- in CRC, Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press/Taylor and Francis, Boca Raton, FL, 93rd edn., 2012-2013, pp. 8–127 Search PubMed.
- J. Rodriguez, C. Kirmaier and D. Holten, J. Am. Chem. Soc., 1989, 111, 6500–6506 CrossRef CAS.
- A. Nakano, Y. Yasuda, T. Yamazaki, S. Akimoto, I. Yamazaki, H. Miyasaka, A. Itaya, M. Murakami and A. Osuka, J. Phys. Chem. A, 2001, 105, 4822–4833 CrossRef CAS.
- R. Kumble, S. Palese, V. S.-Y. Lin, M. J. Therien and R. M. Hochstrasser, J. Am. Chem. Soc., 1998, 120, 11489–11498 CrossRef CAS.
- T. V. Duncan, I. V. Rubtsov, H. T. Uyeda and M. J. Therien, J. Am. Chem. Soc., 2004, 126, 9474–9475 CrossRef CAS PubMed.
- T. V. Duncan, T. Ishizuka and M. J. Therien, J. Am. Chem. Soc., 2007, 129, 9691–9703 CrossRef CAS PubMed.
- T. V. Duncan, S. P. Wu and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 10423–10435 CrossRef CAS PubMed.
- H. A. Frank, J. A. Bautista, J. Josue, Z. Pendon, R. G. Hiller, F. P. Sharples, D. Gosztola and M. R. Wasielewski, J. Phys. Chem. B, 2000, 104, 4569–4577 CrossRef CAS.
- D. Zigmantas, T. Polívka, R. G. Hiller, A. Yartsev and V. Sundström, J. Phys. Chem. A, 2001, 105, 10296–10306 CrossRef CAS.
- D. Zigmantas, R. G. Hiller, V. Sundström and T. Polívka, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16760–16765 CrossRef CAS PubMed.
- P. Plaza, D. Laage, M. M. Martin, V. Alain, M. Blanchard-Desce, W. H. Thompson and J. T. Hynes, J. Phys. Chem. A, 2000, 104, 2396–2401 CrossRef CAS.
- G. E. O'Keefe, G. J. Denton, E. J. Harvey, R. T. Phillips, R. H. Friend and H. L. Anderson, J. Chem. Phys., 1996, 104, 805–811 CrossRef.
- J. Koo, J. Park, A. Tronin, R. Zhang, V. Krishnan, J. Strzalka, I. Kuzmenko, H. C. Fry, M. J. Therien and J. K. Blasie, Langmuir, 2012, 28, 3227–3238 CrossRef CAS PubMed.
- J. Park, P. Deria and M. J. Therien, J. Am. Chem. Soc., 2011, 133, 17156–17159 CrossRef CAS PubMed.
- A. Nayak, J. Park, K. De Mey, X. Hu, T. V. Duncan, D. N. Beratan, K. Clays and M. J. Therien, ACS Cent. Sci., 2016, 2, 954–966 CrossRef CAS PubMed.
- A. Jouyban and S. Soltanpour, J. Chem. Eng. Data, 2010, 55, 2951–2963 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of materials, synthetic procedures, characterization data, spectroscopic data acquisition and analysis, additional optical and transient dynamical data, electronic structure computational data. See DOI: 10.1039/c8me00001h |
‡ If the S* → T1 process is responsible for T1-state formation, its S* → T1 time constant  is determined to be ∼300 ps, from the following equations: is determined to be ∼300 ps, from the following equations:  and and  , where , where  is the quantum yield of triplet formation from S* → T1, and is the quantum yield of triplet formation from S* → T1, and 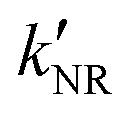 , , 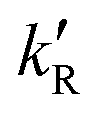 , and , and 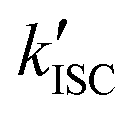 are the decay rates of S*-state through nonradiative relaxation (NR), radiative relaxation (R) and S* → T1 intersystem crossing processes, respectively. are the decay rates of S*-state through nonradiative relaxation (NR), radiative relaxation (R) and S* → T1 intersystem crossing processes, respectively. |
| § Since the ε values of DCM/Tol mixtures are not available, these values were calculated from ε(χmol) = χmolε(DCM) + (1 – χmol)ε(Tol).77 |
| This journal is © The Royal Society of Chemistry 2018 |