
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of selective, fluorescent cannabinoid type 2 receptor ligands based on a 1,8-naphthyridin-2-(1H)-one-3-carboxamide scaffold†
Anna G.
Cooper
a,
Caitlin R. M.
Oyagawa
 b,
Jamie J.
Manning
b,
Sameek
Singh
a,
Sarah
Hook
a,
Natasha L.
Grimsey
b,
Michelle
Glass
b,
Joel D. A.
Tyndall
a and
Andrea J.
Vernall
*a
b,
Jamie J.
Manning
b,
Sameek
Singh
a,
Sarah
Hook
a,
Natasha L.
Grimsey
b,
Michelle
Glass
b,
Joel D. A.
Tyndall
a and
Andrea J.
Vernall
*a
aSchool of Pharmacy, University of Otago, 18 Frederick Street, Dunedin 9054, New Zealand. E-mail: andrea.vernall@otago.ac.nz; Tel: +64 3 479 4518
bDepartment of Pharmacology and Clinical Pharmacology, and Centre for Brain Research, School of Medical Sciences, University of Auckland, Auckland, New Zealand
First published on 23rd October 2018
Abstract
Cannabinoid type 2 (CB2) receptor has been implicated in several diseases and conditions, however no CB2 receptor selective drugs have made it to market. The aim of this study was to develop fluorescent ligands as CB2 receptor tools, to enable an increased understanding of CB2 receptor expression and signalling and thereby accelerate drug discovery. Fluorescent ligands have been successfully developed for other receptors, however none with adequate subtype selectivity or imaging properties have been reported for CB2 receptor. A series of 1,8-naphthyridin-2-(1H)-one-3-carboxamides with linkers and fluorophores appended in the N1 and C3-positions were developed. Molecular modelling indicated the C3 cis-cyclohexanol-linked compounds directed the linker out of the CB2 receptor between transmembrane helices 1 and 7. Herein we report fluorescent ligand 32 (hCB2 pKi = 6.33 ± 0.02) as one of the highest affinity, selective CB2 receptor fluorescent ligands reported. Despite 32 displaying poor specific labelling of CB2 receptor, the naphthyridine scaffold with this linker remains highly promising for future development of CB2 receptor tools.
Introduction
Cannabinoid type 2 (CB2) receptor is a class A G protein-coupled receptor (GPCR) and is highly expressed in immune cells and lymphoid tissues,1 and in lower levels in the central nervous system.2 Along with cannabinoid type 1 (CB1) receptor, the endogenous agonists anandamide and 2-arachidonoylglycerol and various regulatory enzymes, CB2 receptor is part of the highly regulated endocannabinoid system.3 CB2 receptor modulates cytokine release and immune cell migration, thereby regulating immune responses and inflammatory pathways.4,5 CB2 receptor has been shown to play a role in neurodegenerative disorders,6 pain,7 atherosclerosis8 and cancer.9 As such, ligands of CB2 receptor hold promise as therapeutic interventions. However, to date no CB2 receptor selective drugs have made it to market, though a few have undergone clinical trials.10 Drug development efforts would be greatly aided by an increased understanding of CB2 receptor expression and signalling. The aim of this study was therefore to develop high affinity, selective CB2 receptor fluorescent ligands and evaluate these ligands as imaging tools. Fluorescent ligands have been successfully developed for other Class A GPCRs11 and used, for example, to visualise receptor,12 track receptor internalisation13,14 and to study single-cell ligand binding kinetics.15In order to be useful imaging tools, fluorescent GPCR ligands require high affinity and selectivity, and should exhibit low levels of non-specific membrane interactions. A typical strategy for developing fluorescent ligands is to select a known high affinity and selective ligand as a scaffold to which a linker and fluorophore can be appended. It is crucial to identify a suitable position on the scaffold for linker attachment in order to retain affinity for the target receptor and minimise disruption of the binding orientation of the pharmacophore.11 It is particularly challenging to develop fluorescent ligands with minimal plasma membrane interactions for cannabinoid (CB) receptors due to the typically lipophilic nature of CB ligands.
There have been several reports of fluorescent ligands for CB2 receptor, however, these lack either receptor subtype selectivity or display high levels of non-specific binding/interactions rendering these ligands unsuitable for use in techniques such as confocal microscopy.16–21 A scaffold based on SR144528 (Fig. 1) termed ‘mbc94’ has most commonly been utilised for development of CB2 receptor fluorescent ligands, for example NIR-mbc94 (Fig. 1, mCB2 receptor Ki = 260 nM).18 Fluorescent ligands based on an aminoalkylindole scaffold have also been attempted, however despite the ligand-linker conjugates retaining CB receptor affinity the fluorescent conjugates showed little CB receptor binding.22,23 The bifunctional, photoreactive scaffold LEI121 (Fig. 1) has recently been reported as an alternative strategy to a ‘pre-assembled’ CB2 receptor fluorescent ligand. Upon photoactivation, non-fluorescent LEI121 covalently bound to CB2 receptor, which was then labelled via reaction of an azide-fluorophore to the alkyne of LEI121.24 This is a promising strategy for interrogating CB2 receptor, however a ‘pre-assembled’ non-covalent fluorescent ligand is still very desirable for many competition-based and kinetic experiments.
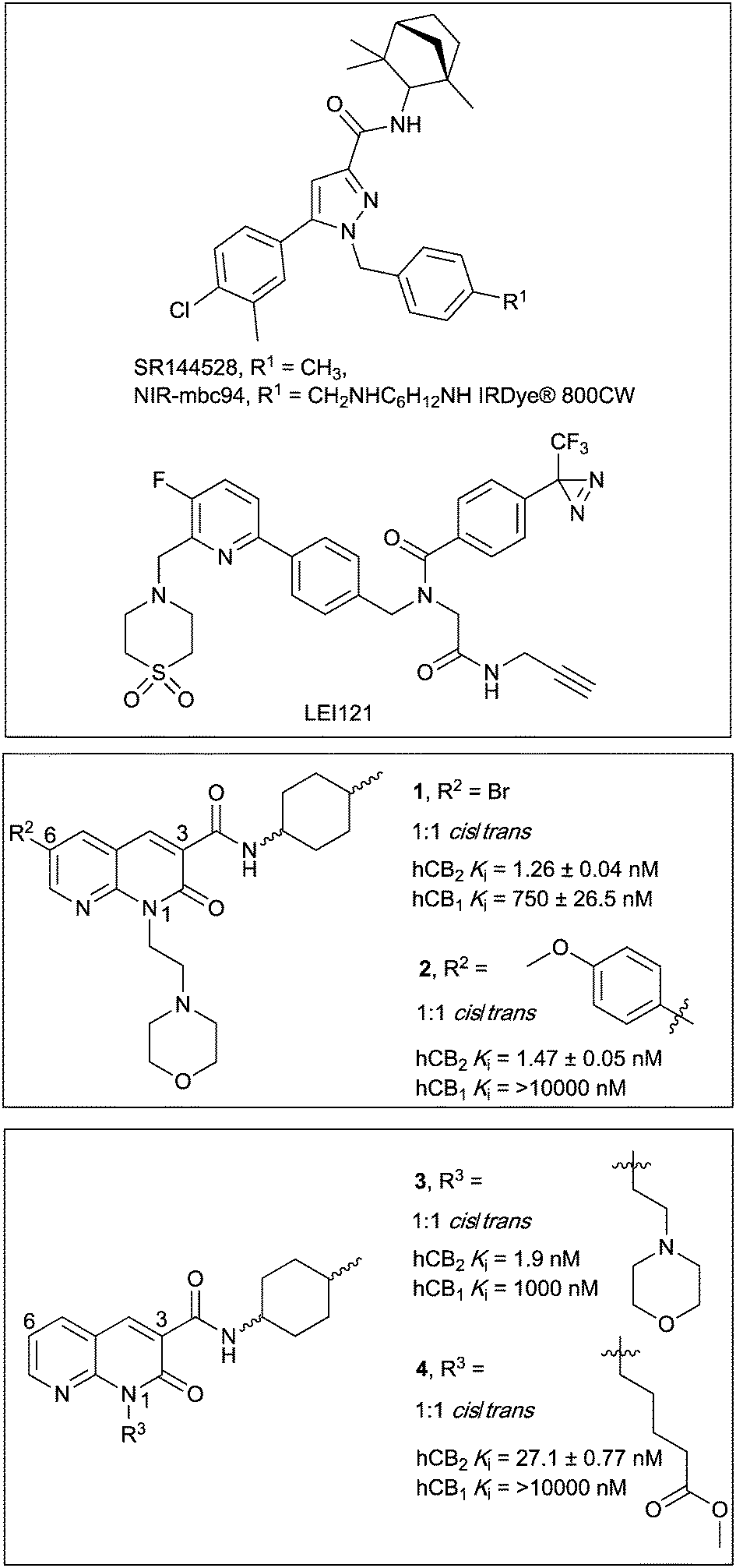 | ||
| Fig. 1 Diarylpyrazole SR144528, and 1,8-naphthyridin-2-(1H)-one-3-carboxamides 1–4, with previously reported binding affinities.25 | ||
In this paper we report the development of CB2 receptor fluorescent ligands based on the 1,8-naphthyridin-2-(1H)-one-3-carboxamide scaffold. Many derivatives of this scaffold are reported to have very high affinity and subtype selectivity for CB2 receptor, (e.g.1 and 4,25Fig. 1) and there are structure–activity-relationships (SAR) reporting the effect of substitution at N1, C3 and C6 (ref. 26–29) (e.g.1–4,25Fig. 1). Additionally, the 1,8-naphthyridin-2-(1H)-one-3-carboxamide scaffold was selected because it is less lipophilic than many other cannabinoids (e.g. clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of 2 is 2.93 compared to SR144528 clogP of 7.13), which is beneficial in terms of reducing the overall lipophilicity of a fluorescent ligand.
P of 2 is 2.93 compared to SR144528 clogP of 7.13), which is beneficial in terms of reducing the overall lipophilicity of a fluorescent ligand.
The N1, C3 and C6 naphthyridine positions were considered for linker and fluorophore attachment because there is SAR reported for these positions and all are synthetically accessible. Since SAR indicated groups of varying length and bulk were tolerated in the N1-position,25,29 including a ‘linker-like’ methyl valerate (4, Fig. 1), one series of conjugates were developed linked at this position. A range of factors were considered when analysing if the C3 cyclohexyl carboxamide position might be amenable to linker and fluorophore attachment. The stereochemistry of the 4-methylcyclohexyl moiety at the C3 carboxamide has been shown to be influential on CB2 receptor binding, with cis derivatives showing improved receptor affinity compared to trans derivatives.25,27 This sensitivity of the methylcyclohexyl group could translate to a position not tolerant of much chemical change/variation, however ligand docking of 1 into a CB2 receptor homology model (as is discussed for 28 in Modelling section) positioned the cyclohexyl group close to exiting CB2 receptor between transmembrane helix (TMH) 1 and TMH7. This therefore made the C3 cyclohexyl carboxamide an appealing linker attachment position, especially in light of previously reported molecular dynamics simulations indicating that cannabinoids may enter into CB receptors via the lipid membrane between TMH6 and TMH7 or between TMH1 and TMH7.30–32 Cyclohexanol and cyclohexylamine derivatives were designed to allow linker extension and both cis and trans isomers (of the cyclohexanol) were prepared since the previously established methylcyclohexyl cis/trans SAR could not be assumed to be the same.
The C6 position was not selected for linker attachment based on SAR that showed that functional activity can be controlled by the C6 substituent. For example, compound 3 (Fig. 1) behaved as a CB2 receptor agonist in β-arrestin 2 and cAMP assays while 1 and 2 (Fig. 1) behaved as antagonists/inverse agonists in a β-arrestin 2 assay.25 It has been postulated with docking studies that this C6 substituent orientates deep into a receptor binding pocket and modulates a CWFP flexible hinge motif on TMH6.25 Linker substitution at C6 was therefore deemed most likely non-tolerable and likely would be detrimental to ligand affinity for CB2 receptor. This was also the reason that the two fluorescent ligand series (N1-linked and C3-linked) were therefore developed with a small C6 substituent present since the goal was to develop high affinity, CB2 receptor selective fluorescent ligands that do not activate CB2 receptor.
Results and discussion
Synthetic chemistry
The N1-linked series was assembled as a 1:1 cis/trans mix at the C3 cyclohexyl carboxamide as the first goal was to determine if the N1-position was tolerant of linker and fluorophore attachment. Commercially available 2-amino-3-pyridinecarboxaldehyde was converted to 5 in 3 steps following previously reported syntheses.25 Alkylation of 5 with 4-(2-chloroethyl)morpholine hydrochloride afforded the previously reported 1 (ref. 25) (Scheme 1), which was used as a pharmacological control. Alkylation of 5 with methyl 5-bromovalerate or methyl 4-bromomethylbenzoate gave 6 or 7 in low yield, due to incomplete conversion of 5 and challenging separation of products from unreacted 5. Both 6 and 7 are amenable to linker extension following methyl ester deprotection, however it was decided to proceed with valerate linked-6 and only synthesise further benzoyl derivatives if 7 showed high affinity for CB2 receptor. Bromo-6 was subjected to Suzuki coupling with 4-methoxyphenylboronic acid to afford 8, to enable comparison of the C6 bromo to methoxyphenyl substituent.
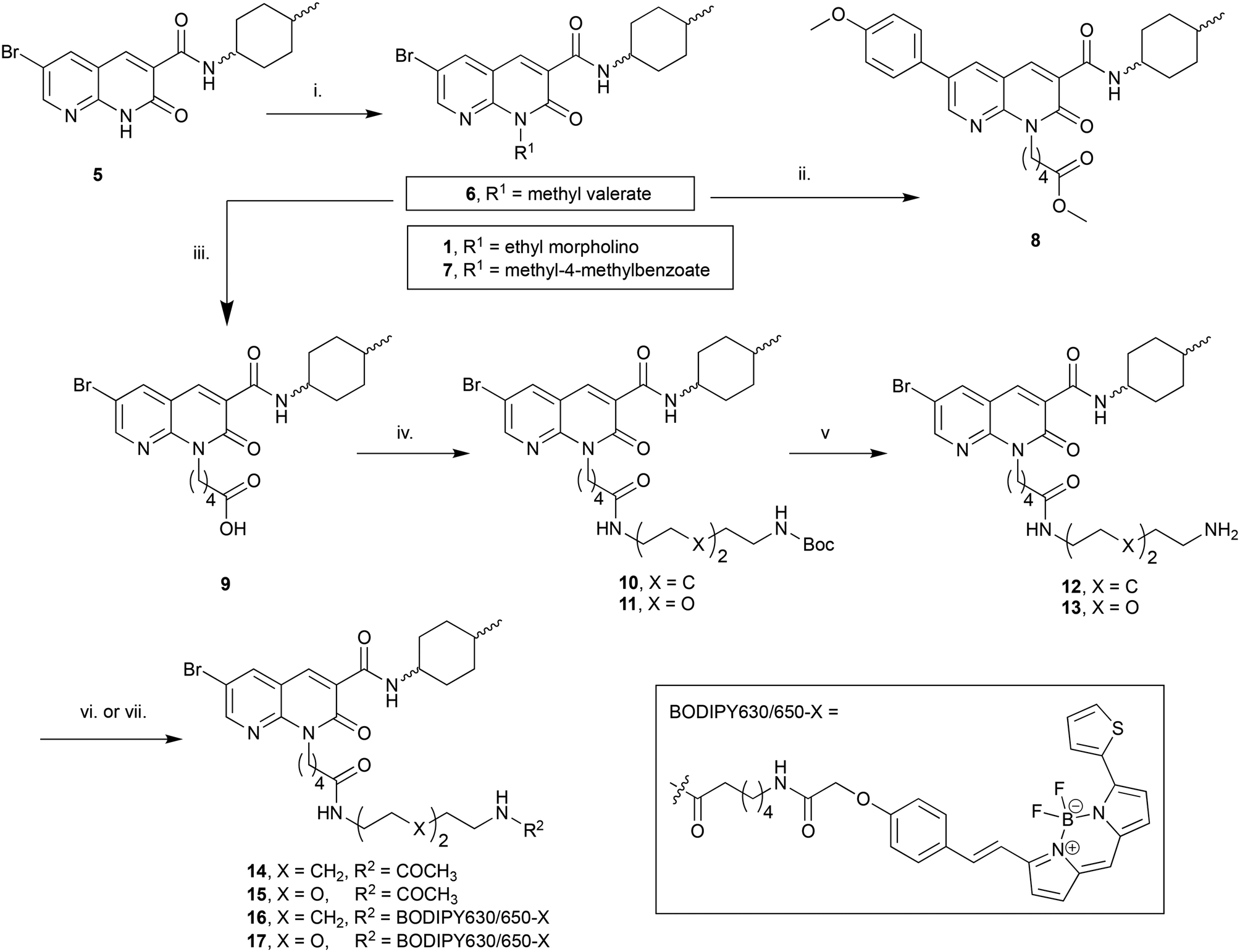 | ||
| Scheme 1 Reagents and conditions: (i) 4-(2-Chloroethyl)morpholine hydrochloride, methyl 5-bromovalerate, or methyl 4-bromomethylbenzoate, Cs2CO3, DMF, 50 °C, 12 h, 22–37%; (ii) 4-methoxyphenylboronic acid, Pd(OAc)2, Na2CO3, H2O:DMF 1:4 v:v, 110 °C, 3 h, 55%; (iii) 10% aq. NaOH, EtOH, 110 °C, 5 h, 43%; (iv) N-Boc-1,8-octanediamine or N-Boc-2,2′-(ethylenedioxy)diethylamine, DIPEA, HATU, DMF, rt, 14 h, 71–77%; (v) TFA, DCM, rt, 1 h, 86–91%; (vi) Ac2O, DIPEA, DCM, rt, 1 h, 95–96%; (vii) BODIPY 630/650-X-OSu, DIPEA, DMF, rt, 15 h, 88–92%. Compounds 1:1 cis/trans. | ||
To further extend the distance of the naphthyridine core from the fluorophore the methyl ester of 6 underwent saponification to reveal carboxylic acid 9, which was coupled to either N-Boc-1,8-octanediamine or N-Boc-2,2′-(ethylenedioxy)diethylamine to yield 10 and 11 respectively. These alkyl and short PEG-like linkers of the same atom length were chosen as a way to compare different linker lipophilicity, since it was hypothesised that the lipophilic alkyl linker may be preferable for CB2 receptor affinity, whereas the PEG-like linker may impart more hydrophilicty (than the equivalent alkyl linker) to the overall fluorescent ligand and lead to better imaging properties (e.g. lower non-receptor specific membrane interactions). Boc deprotection of 10 and 11 yielded unprotected amines 12 and 13, which were then each acetylated using acetic anhydride to give 14 and 15. In a separate procedure these were reacted with 6-(((4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-sindacene-3-yl)styryloxy) acetyl) aminohexanoic acid, succinimidyl ester (BODIPY630/650-X-OSu) to yield fluorescent ligands 16 and 17. The BODIPY630/650-X fluorophore has been used successfully to develop fluorescent ligands for other Class A GPCRs,14,33,34 and has favourable properties (such as a good quantum yield, intense absorption and high chemical and photo stability) and as a red emitting fluorophore there is minimal detection interference from cellular autofluorescence.35 A red-shifted fluorescent ligand also allows co-localisation experiments with, for example, green-fluorescent-protein-tagged receptors or proteins, to be carried out.
The C3-linked series was assembled in a different order to allow for more efficient variation of the C3 substituent. The previously reported 18 (ref. 25) was N1-alkylated, substituted at C6 with methoxyphenyl and the methyl ester saponified to afford 19 (Scheme 2). Carboxylic acid 19 was coupled in separate reactions to cis- or trans-4-aminocyclohexanol to give alcohols 20 and 21 respectively, or to 1-N-Boc-cis-1,4-cyclohexyldiamine to give cis-Boc-protected amine 22.
 | ||
| Scheme 2 Reagents and conditions: (i) 4-(2-Chloroethyl)morpholine hydrochloride, Cs2CO3, DMF, 50 °C, 12 h; (ii) 4-methoxyphenylboronic acid, Na2CO3, Pd(OAc)2, H2O, DMF, 110 °C, 3 h; (iii) 0.2 M aq. LiOH·H2O, THF, 0 °C, 1 h, 20% over three steps; (iv) cis-4-aminocyclohexanol or trans-4-aminocyclohexanol or 1-N-Boc-cis-1,4-cyclohexyldiamine, HATU, DIPEA, DMF, rt, 14 h, 79–82%; (v) N-Boc-glycine or N-Boc-7-aminoheptanoic acid, TFFH, Et3N, DMAP, DCM, rt, 14 h, 19–80%; (vi) 1.Trifluoroacetic acid (TFA), DCM, rt, 1 h, quantitative; 2. N-Boc-glycine, HATU, DIPEA, DMF, rt, 14 h, 31%; (vii) TFA, DCM, rt, 1 h, 45–57%; (viii) Ac2O, DIPEA, DCM, rt, 1 h, 96–98%; (ix) BODIPY 630/650-X-OSu, DIPEA, DMF, rt, 14 h, 72–97%. | ||
The intention was to introduce an ether linkage via alkylation of alcohols 20 and 21. However, despite several attempts using alkyl bromides such as tert-butyl bromoacetate or 2-(Boc)-amino-ethylbromide with either NaH or CsCO3 as the base, varying equivalents of each base and addition order, and varying temperature the intended ether product was either detected only in trace amounts or not at all. An alternative synthetic strategy was envisaged, whereby the ether bond to the cyclohexyl could instead be preassembled and then coupled to 19. Attempts to alkylate carboxybenzyl and dibenzyl protected 4-aminocyclohexanol with various alkyl bromides using a range of bases (NaH, CsCO3, K2CO3, tBuOK and lithium bis(trimethylsilyl)amide), temperatures, solvents and reaction times were all unsuccessful. A Mitsunobu reaction to form the ether bond was not attempted due to the calculated pKa (∼15) of the cyclohexanol alcohol.
Since 20 and 21 were already prepared it was decided to proceed with formation of an ester bond to enable pharmacological evaluation of the C3-cyclohexyl-linker position, which could then be revisited for a more stable bond if the C3-position proved suitable. Activation of Boc-glycine or N-Boc-7-aminoheptanoic acid with tetramethylfluoroformamidinium hexafluorophosphate (TFFH) followed by addition of 20 or 21 yielded esters 23–26. TFFH was used rather than a carbodiimide reagent in an attempt to counteract the poor nucleophilicity of alcohols 20 and 21. Instead of using linkers analogous to the alkyl and PEG-like linkers of the N-substituted series, it was decided to use a commercially available single ‘glycine’ and a longer 7-aminoheptanoic acid linker to allow exploration of how varying linker length might effect receptor affinity. Boc-deprotection of 22 followed by HATU-mediated coupling of Boc-glycine afforded 27, the analogous compound to 23 but with an amide replacing the ester bond. Boc-deprotection of 23–27 followed by either acylation or reaction with BODIPY630/650-X-OSu gave 28–31 or 32–36 respectively.
Pharmacology
A radioligand competition binding screen was carried out to determine the ability of 10 μM of 1, 6–8, 14–17, 20, 21, 28–36 to displace [3H]CP-55940 (2.5 nM or 1 nM) from CB2 or CB1 receptor (data not shown). Compounds that displaced [3H]CP55940 by more than 50% were then evaluated at varying concentrations to determine pKi values (Tables 1 and 2). Compounds with pKi > 5 at CB2 receptor were also analysed for function at CB2 and CB1 receptors in a cAMP assay (Tables 1 and 2).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| R 1 | R 2 | CB2 receptor | CB1 receptor | CB2 selectivitye | |||||
| pKia (±SEM) | pIC50b,c (±SEM) | E max , (% ± SEM) | pKi (±SEM)a | pIC50 (±SEM)b,c | E max , (% ± SEM) | ||||
|
a Radioligand binding performed with [3H]CP55940 (2.5 nM) and HEK293-hCB2 or -hCB1 membranes. Data is the mean ± SEM of at least three individual experiments performed in triplicate. Compounds which at 10 μM significantly displaced [3H]CP55940 but with <50% displacement are annotated as having pKi >5 M.
b cAMP levels measured in a BRET assay using a CAMYEL biosensor, in either HEK-293-hCB2 or hCB1 cells. Data is the mean ± SEM of at least three independent experiments conducted in duplicate.
c pIC50 calculated by concentration response.
d
E
max response (at 10 μM for compounds without pIC50 determined), normalised to basal (0%) and forskolin only (100%) levels of cAMP. Emax >100% is consistent with inverse agonism.
e CB2 receptor selectivity is calculated by: 10^(pKi CB2–pKi CB1) Naphthyridine compounds all 1:1 cis:trans mixture.
f Except (6) which is two independent experiments performed in duplicate. ‘—’ Indicates that pIC50 or Emax was not determined.
g Indicates no significant difference from forskolin only (100%), indicating no measurable response.
|
|||||||||
| 1 | Br | Ethyl morpholino | 7.26 ± 0.04 | 7.41 ± 0.17 | 166 ± 5.6 | <5 | No responseg | 89 ± 5.6g | >182 |
| 6 | Br | (CH2)4CO2Me | 5.96 ± 0.04 | 6.24 ± 0.05f | 161 ± 5.0f | <5 | 5.73 ± 0.14 | 134 ± 4.4 | >9 |
| 7 | Br | Methyl-4-methylbenzoate | <5 | — | — | <5 | — | — | — |
| 8 | Me-OPh | (CH2)4CO2Me | 6.59 ± 0.05 | 7.19 ± 0.27 | 139 ± 4.2 | 5.06 ± 0.03 | 6.02 ± 0.25 | 126 ± 3.2 | 33 |
| 14 | Br | (CH2)4CO | <5 | — | — | <5 | — | — | — |
| NH(CH2)8NHAc | |||||||||
| 15 | Br | (CH2)4CO | <5 | — | — | <5 | — | — | — |
| NH(C2H4O)2NHAc | |||||||||
| 16 | Br | (CH2)4CO | <5 | — | — | <5 | — | — | — |
| NH(CH2)8NH-BODIPY-630/650-X | |||||||||
| 17 | Br | (CH2)4CO | <5 | — | — | <5 | — | — | — |
| NH(C2H4O)2NH-BODIPY-630/650-X | |||||||||
| SR144528 | — | 7.29 ± 0.03 | 6.90 ± 0.08 | 153 ± 3.8 | 5.40 ± 0.2 | No responseg | 108 ± 3.6g | 78 | |
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| R | CB2 receptor | CB1 receptor | CB2 selectivitye | ||||||
| pKia (±SEM) | pIC50b,c (±SEM) | E max , (% ± SEM) | pKia (± SEM) | pIC50b,c (±SEM) | E max , (% ± SEM) | ||||
|
a Radioligand binding performed with [3H]CP55940 (1 nM) and HEK293-hCB2 or -hCB1 membranes. Data is the mean ± SEM of at least three experiments performed in triplicate. Compounds which at 10 μM significantly displaced [3H]CP55940 but with <50% displacement are annotated as having pKi >5 M.
b cAMP levels measured in a BRET assay using a CAMYEL biosensor, in either HEK-293-hCB2 or hCB1 cells. Data is the mean ± SEM of at least three independent experiments conducted in duplicate.
c pIC50 calculated by concentration response.
d
E
max response (at 10 μM for compounds without pIC50 determined.
e CB2 receptor selectivity is calculated by: 10^(pKi CB2–pKi CB1).
f Except which are two independent experiments performed in duplicate. ‘—’ Indicates that pIC50 or Emax was not determined.
g Indicates no significant difference from forskolin only (100%), indicating no measurable response.
h Except which is at 1 μM due to high non-specific effects at 10 μ, see ESI), normalised to basal (0%) and forskolin only (100%) levels of cAMP. Emax >100% is consistent with inverse agonism.
|
|||||||||
| 20 | cis | OH | 5.66 ± 0.07 | 6.74 ± 0.16 | 156 ± 5.5 | <5 | — | 117 ± 2.4f | >4 |
| 21 | trans | OH | 5.47 ± 0.10 | — | — | No binding | — | — | — |
| 28 | cis | OC(O)CH2NHAc | 5.99 ± 0.03 | 6.99 ± 0.11 | 154 ± 5.4 | <5 | No responseg | 115 ± 6.5f,g | >9 |
| 29 | trans | OC(O)CH2NHAc | <5 | — | — | No binding | — | — | — |
| 30 | cis | OC(O)C6H12NHAc | 5.51 ± 0.02 | 5.81 ± 0.15 | 179 ± 12.1 | <5 | — | 130 ± 2.6f | >3 |
| 31 | trans | OC(O)C6H12NHAc | 5.43 ± 0.06 | — | — | <5 | — | — | >2 |
| 32 | cis | OC(O)CH2NH-BODIPY630/650-X | 6.33 ± 0.02 | 6.72 ± 0.18 | 210 ± 15.7 | <5 | — | 117 ± 3.5h | >21 |
| 33 | trans | OC(O)CH2NH-BODIPY630/650-X | 5.23 ± 0.06 | — | — | <5 | — | —g | >1.6 |
| 34 | cis | OC(O)C6H12NH-BODIPY 630/650-X | 5.11 ± 0.04 | — | — | <5 | — | — | >1.2 |
| 35 | trans | OC(O)C6H12NH-BODIPY 630/650-X | No binding | — | — | <5 | — | — | — |
| 36 | cis | NHC(O)CH2NH-BODIPY630/650-X | <5 | — | — | <5 | — | — | |

The function of N1-linked naphthyridines 1, 6 and 8 was analysed using a bioluminescence resonance energy transfer (BRET) biosensor to measure modulation of forskolin-stimulated cyclic adenosine monophosphate (cAMP) at CB2 and CB1 receptors (Table 1). All three (1, 6, 8) were found to increase the level of cAMP, consistent with inverse agonism for CB2 receptor, in agreement with the literature data for 1 and the reported trend that substitution at the C6 position results in inverse agonism.25 The most potent for CB2 receptor was 1, followed by 8, while 6 was the least potent. A much lower potency (6 and 8) or no response (1) was measured for CB1 receptor. It was concluded from these results (Table 1) that linker and fluorophore substitution at the naphthyridine N1-position is unlikely to lead to fluorescent ligands with useful affinity at CB2 receptor.
:1 cis:trans methylcyclohexyl 2 (ref. 25) (Table 1) despite the relatively small chemical difference of an alcohol or methyl moiety. The acetylated aminoheptanoate-linked 30 and 31 also showed comparable poor affinity for CB2 receptor. Cis-glycine linked-28 showed a 0.5 log unit improvement in CB2 receptor affinity over the analogous cis-aminoheptanoate 30. All cyclohexyl-linked compounds showed little or no affinity for CB1 receptor.
Amongst all the cyclohexyl-linked naphthyridines, the highest CB2 receptor affinity (and the only fluorescent ligand with any appreciable affinity) was measured for fluorescent ligand 32 (pKi = 6.33 ± 0.02 at hCB2 receptor and >21-fold selectivity over CB1 receptor). It is interesting that the larger, BODIPY 630/650-contianing 32 showed approximately 0.3 log unit better affinity for CB2 receptor than the corresponding truncated 28 without the fluorophore (Fig. 2A), implying the BODIPY 630/650 moiety contributed favourably to binding. Similar observations that Class A GPCR fluorescent ligands have higher affinity for the receptor than just the core ligand and/or ligand-linker have also been reported in the literature.33,34 Despite fluorescent ligand 36 differing from 32 only by replacement of the ester in 32 with an amide, 36 showed minimal binding (pKi < 5). This could be due to the lack of flexibility of the amide in comparison to the ester, restricting the movement of 36 and preventing favourable positioning of the linker and fluorophore relative to the naphthyridine core ligand. However, evaluation of shorter precursors to 36 would be required to draw conclusions.
 | ||
| Fig. 2 (A) Competition radioligand binding assay of 20, 28 and 32 at hCB2 receptor, using [3H]CP55940. Data shown is a single experiment conducted in triplicate with error bars showing ±SD; representative of 3 independent experiments. (B) Concentration response curve of 28 and 32 measuring forskolin (5 μM) stimulated cAMP. Area under the curve values from kinetic data were normalised so that forskolin-only response = 100% and basal (vehicle) response = 0%. Representative data from a single experiment conducted in duplicate, with error bars showing ± SD. | ||
The ability of cyclohexyl-linked naphthyridines with micromolar or nanomolar affinity for CB2 receptor to modulate cAMP was determined in a BRET cAMP assay (Table 2). All tested compounds reduced basal signalling (i.e. increased cAMP levels) consistent with inverse agonism at CB2 receptor. The inverse agonist function observed for these cyclohexyl-linked compounds, all which contain a C6-p-methoxy benzyl substituent, aligns with literature reports of inverse agonism/antagonism for C6-substituted 1,8-naphthyridin-2(1H)-one-3-carboxamides (as also described for compounds in Table 1). The potency of 20, 28, 30 and 32 (Fig. 2B shows 28, 32) was determined using concentration response assays at CB2 receptor (and at CB1 receptor for 32 only). It is interesting that despite 20, 28 and 32 all having between a 1–1.5 log unit weaker affinity for CB2 receptor compared to SR144528 (Table 1), 20, 28, 32 and SR144528 all showed similar pIC50 values at CB2 receptor. Fluorescent ligand 32 appeared to display a greater Emax at CB2 receptor (hCB2 pIC50 = 6.72 ± 0.18, Emax = 210 ± 15.7) compared to SR144528 (hCB2 pIC50 = 6.90 ± 0.08, Emax = 153 ± 3.8) and 32 was much less potent at CB1 receptor (hCB1 pIC50 = 5.26 ± 0.15, Emax = 157 ± 2.1). Compounds analysed for potency in the cAMP BRET assay (Tables 1 and 2) were also screened at 10 μM and 1 μM in the parental HEK cells that lack CB receptors (refer to ESI†). A small effect was observed in the parental HEK cells using fluorescent ligand 32 at 10 μM but not at 1 μM (Table S1†). However, due to the low potency of 32 the 10 μM data point of the concentration response assay at CB2 receptor could not be excluded and therefore the calculated potency of 32 reported at CB2 receptor is only an estimate. In addition, the higher Emax of fluorescent ligand 32 at CB2 receptor compared to SR144528 could be influenced the small non-receptor mediated effect at 10 μM, thus illustrating the importance of wild type controls to verify receptor-mediated responses.
Ligand 32 is one of the highest affinity CB2 receptor selective fluorescent ligands reported in the literature to date. It is not possible to meaningfully compare the affinity of 32 to other fluorescent ligands reported in the literature due to different experimental conditions used to measure binding. For example, the affinity of fluorescent ligand ‘NIR760-Q’ (Kd = 75.5 ± 28.0 nM) was determined in Jurkat cells using a fluorescence saturation binding assay.20 Fluorescent ligand ‘NMP6’ had a reported affinity for hCB2 receptor of Ki = 387 nM using CHO-K1 cells, but with no SEM or Kd value provided for the competing radioligand utilised ([3H]CP55940).16
Molecular modelling
The high CB2 receptor affinity of fluorescent ligand 32 validates the cyclohexyl position as suitable for linker and fluorophore extension. To explore how the ligand-linker of the cyclohexyl-substituted naphthyridines may interact with the receptor, ligand docking studies were performed using a homology model of CB2 receptor, that was generated using the crystal structure of antagonist bound CB1 receptor (PDB:5TGZ).36 To improve docking accuracy, the linker conjugate 28 rather than fluorescent ligand 32 was used. The lowest energy and most consistent docking pose showed the glycine linker of 28 exiting the receptor between TMH1 and TMH7 (Fig. 3A), thus potentially illustrating why the cyclohexyl group is a favourable linker position – allowing an exit point for the linker and fluorophore whilst allowing the core ligand to bind deep within the orthosteric pocket. This is especially interesting in light of previously reported molecular dynamics simulations where a potential ligand entry pathway between TMH1 and TMH7 was identified for anandamide and tetrahydrocannabinol binding to CB1 receptor,32 as well as with other fluorescent ligand docking studies at CB2 receptor.17,23 The phenyl group of 28 was positioned deep in the hydrophobic region of the binding pocket, in a similar pose to other previously reported docking studies with this naphthyridine scaffold.25 The methoxy group showed a hydrogen bond with S292, whilst the glycine linker hydrogen bonded with D24 of the amino-terminus and Q32 of TMH1. Favourable placement of hydrogen bond donors and acceptors in the ligand's linker that allow interactions with the polar residues located around the proposed ligand binding pocket entry/exit between TMH1 and TMH7 may be significant for receptor affinity. | ||
| Fig. 3 (A) Lowest energy docking pose of glycine-linked cis-28 (cyan carbon atoms) in hCB2 receptor homology model (grey ribbons). Hydrogen bonding (yellow dotted line) is shown between the methoxy phenyl oxygen and S292, the ester carbonyl and the glycine amide with Q32 and between the glycine amide and D24. The glycine linker is directed out of the receptor binding pocket between TMH1 and TMH7. Side chains of selected residues within 4 Å of 28 are shown as sticks (green). (B) Overlay of the lowest energy docking poses of cis-28 (cyan) and the analogous trans-29 (gold) docked into hCB2 receptor homology model (receptor is hidden for clarity), showing that trans-29 is shifted upwards in the binding pocket, which resulted in loss of hydrogen bonding with receptor residues. | ||
The cis isomers of the cyclohexyl-linked series consistently showed higher affinities for CB2 receptor than the trans isomers therefore this was explored further through ligand docking studies. In contrast to cis-28, trans-29 showed an altered pose, in which the whole ligand was shifted upwards in the binding pocket (Fig. 3B). Cyclohexyl-linked trans-29 did not show any hydrogen bonds between the methoxy and S292 or the glycine linker and the residues around the binding pocket exit of TMH1 and TMH7, which may explain the reduced affinity of the trans derivatives.
Cellular imaging
Fluorescent ligand 32 showed promise as an imaging tool due to high affinity for CB2 receptor, selectivity over CB1 receptor, and a similar pIC50 to SR144528. Unfortunately, 32 did not show ideal properties in imaging experiments (Fig. 4). There was little co-localisation of CB2 receptor (as detected by antibody labelling) with 32, pre-incubation of cells with a high concentration of non-fluorescent high affinity ligand SR144528 did not appear to prevent or reduce 32 labelling, and application of 32 to cells not transfected with CB2 receptor still showed a high level of fluorescence. Cell-associated fluorescence arising from 32 was readily detectable within two minutes of incubation, and although fluorescence intensity increased with longer incubation (tested up to 30 minutes) the pattern of staining was equivalent (data not shown). Similarly, incubation with a greater (10 μM) or lesser (1 μM) concentration of 32 influenced the overall intensity of fluorescence but not the subcellular distribution of fluorescence (data not shown). It was concluded that the ester bond of 32 was stable for the duration of the imaging experiments, based on the SAR from 32 and truncated analogues (Table 2, in particular 20 compared to 32) and from reverse phase high performance liquid chromatography (RP-HPLC) experiments that indicated 32 remained stable with no ester bond cleavage (data not shown). Therefore, reasons for the poor CB2 receptor imaging properties of 32 are likely due to high levels of membrane association and non-CB2 receptor associated intracellular accumulation.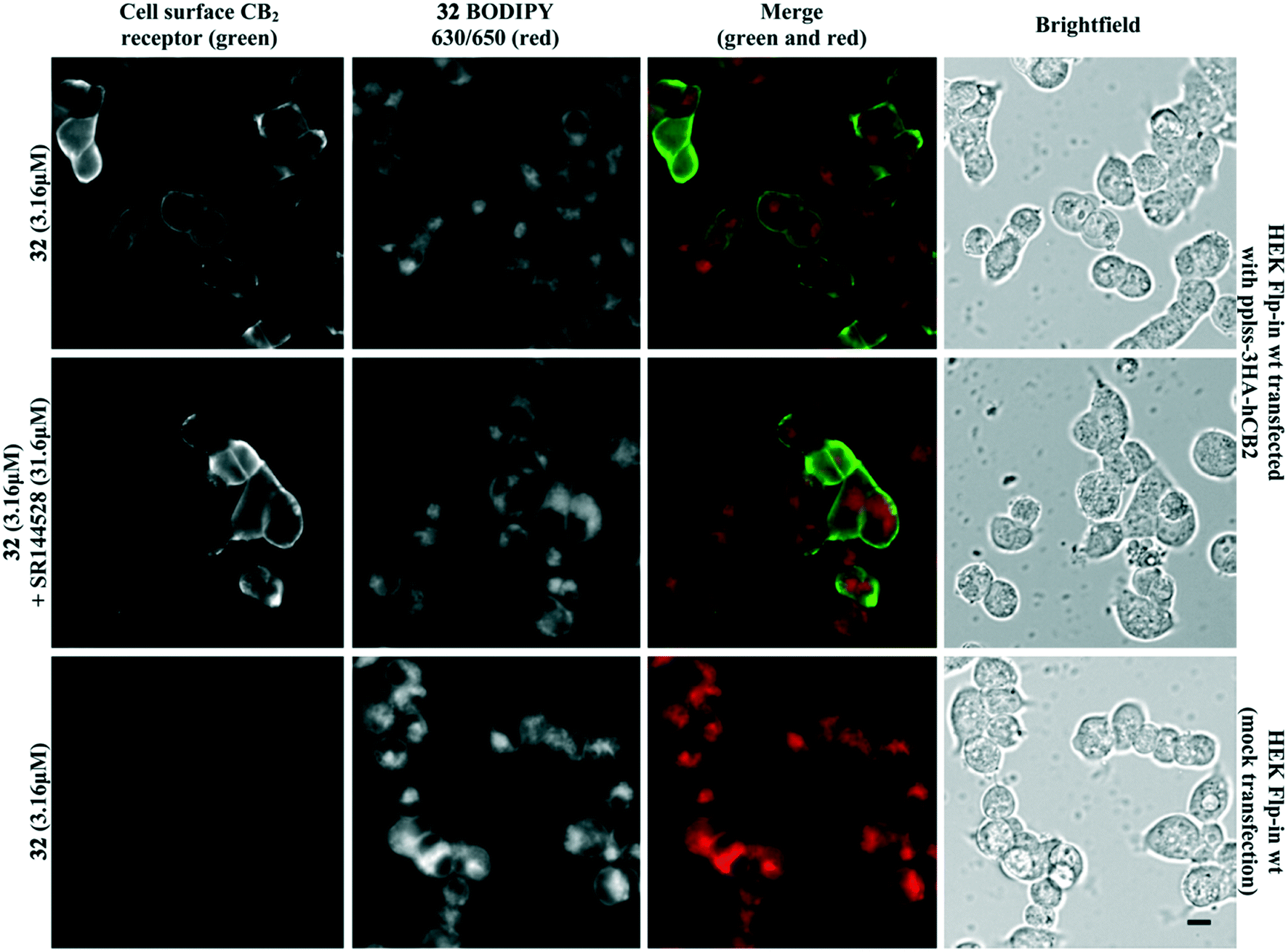 | ||
| Fig. 4 HEK Flp-in wt cells transiently transfected with pplss-3HA-hCB2 or mock transfection, preincubated with SR144528 or vehicle for 30 min, then treated with 32 + SR144528 or vehicle (2 min followed by 3 washes). Cell surface CB2 receptor visualised using mouse anti-HA and Alexa 488-conjugated goat anti-mouse. Scale bar 10 μM. Images representative of n = 3 experiments. | ||
Conclusions
A library of 1,8-naphthyridin-2(1H)-one-3-carboxamides with linker and fluorophore substitution at the N1 and C3-carboxamide cyclohexyl position were designed, synthesised and pharmacologically evaluated. The N1 position was not tolerant of linker and fluorophore attachment, however a high affinity, selective CB2 receptor fluorescent inverse agonist (32) was developed via a C3-carboxamide-cyclohexanol linkage. Despite imaging studies that showed high levels of non-CB2 receptor-specific fluorescence, 32 remains a promising lead for future fluorescent ligand development because of the affinity and selectivity of this large ligand. Molecular modelling and SAR showed the C3-carboxamide-cyclohexyl position is an excellent position for linker and fluorophore attachment.Experimental
Chemistry
Chemicals were purchased from Sigma Aldrich, Merck or AK Scientific and BODIPY 630/650-X-OSu from Life Technologies. Anhydrous grade solvents were used when a dry atmosphere was required. Unless stated, all reactions were carried out at room temperature (rt) under atmospheric pressure. Thin layer chromatography was carried out on 0.2 mm aluminium-backed silica gel plates 60 F254 and visualised using UV light (λ = 254 nM, 365 nM) and potassium permanganate. Flash column chromatography used 40–63 μm silica. RP-HPLC was carried out on an Agilent 1260 Infinity system with a YMC C8 5 μm (150 × 4.6 mm) or YMC C8 5 μm (150 × 10 mm) column, and mobile phases A: H2O (0.05% TFA) and B: 9:1 acetonitrile (ACN):H2O (0.05% TFA). Analytical RP-HPLC retention times were determined using the method −5% B/A 0–1 min, linear gradient of 5–95% B 1–27 min, 95% B 27–28 min, linear gradient of 95–5% B 28–30 min, 5% B/A 30–34 min. All compounds analysed for biological activity were >95% purity by analytical RP-HPLC UV detection at 254, 380 and 550 nm. All compounds HPLC purified as the TFA salt were neutralised using an Amberlyst A21 ion exchange resin before biological testing. High resolution electrospray ionisation mass spectra (HRMS-ESI) were obtained on a microTOFQ mass spectrometer. Proton and carbon nuclear magnetic resonance (NMR) spectra were obtained on a 400 MHz or a 500 MHz Varian MR spectrometer. Chemical shifts are listed on the δ scale in ppm, spectra are referenced to CDCl3, MeOD-d4 or DMSO-d6 residual solvent. Coupling constants (J) are recorded in Hertz (Hz) with signals assigned as: s, singlet; d, doublet; t, triplet; q, quartet; br, broad; or m, multiplet.
Synthesis of fluorescent ligand 32 is detailed below. Synthesis and characterisation of all other compounds is detailed in the ESI.†
:3 mixture (5.17 g) of the ethyl ester (ethyl 6-bromo-1-[2-(morpholin-4-yl)ethyl]-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate) and the carboxylic acid (6-bromo-1-[2-(morpholin-4-yl)ethyl]-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylic acid) as a yellow solid. This 1:3 mixture (4.07 g), 4-methoxyphenylboronic acid (2.03 g, 13.3 mmol), Na2CO3 (2.83 g, 26.7 mmol) were dissolved in H2O (25 mL) and DMF (100 mL). Pd(OAc)2 (23 mg, 0.10 mmol) was added and the reaction heated to 110 °C and stirred for 3 h. After cooling to rt, aq. HCl was added until pH 1–2, H2O (100 mL) added, and extracted with DCM (3 × 100 mL). The combined organics were washed with H2O (2 × 150 mL) and sat. aq. NaCl (150 mL), dried over MgSO4, filtered and evaporated under reduced pressure. The residue was washed with EtOH, filtered and the solid dried under reduced pressure yielding a 1:15 mixture (1.99 g) of ethyl 6-(4-methoxyphenyl)-1-[2-(morpholin-4-yl)ethyl]-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate and 6-(4-methoxyphenyl)-1-[2-(morpholin-4-yl)ethyl]-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylic acid, as a yellow solid. A stirred solution of the 1:15 mixture (1.9 g) in THF (30 mL) was cooled to 0 °C and 0.2 M LiOH·H2O (49 mL) was added dropwise. The reaction was stirred at 0 °C for 1 h and then quenched with a biphase of 0.2 M aq. HCl/EA (1:1 v:v, 200 mL). The aqueous layer was extracted with DCM (10 × 100 mL), dried over MgSO4, filtered and evaporated under reduced pressure to yield 19 (1.66 g, 4.1 mmol, 20% over three steps) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.97 (d, J = 2.4 Hz, 1H, ArH), 8.93 (s, 1H, ArH), 8.22 (d, J = 2.4 Hz, 1H, ArH), 7.58–7.51 (m, 2H, ArH MeO![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) ), 7.08–7.01 (m, 2H, ArH MeO), 4.86 (t, J = 6.8 Hz, 2H, N1–CH2), 3.87 (s, 3H, O–CH3), 3.77–3.64 (m, 4H, O–CH2 morpholino), 2.93–2.83 (m, 2H, N1–CH2C
), 7.08–7.01 (m, 2H, ArH MeO), 4.86 (t, J = 6.8 Hz, 2H, N1–CH2), 3.87 (s, 3H, O–CH3), 3.77–3.64 (m, 4H, O–CH2 morpholino), 2.93–2.83 (m, 2H, N1–CH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ), 2.82–2.56 (m, 4H, N–CH2 morpholino). 13C NMR (101 MHz, CDCl3) δ 164.63, 164.56, 160.41, 152.08, 148.49, 144.86, 135.94, 133.49, 128.33, 128.14, 119.28, 115.08, 115.07, 66.93, 56.00, 55.59, 53.96, 39.37. HRMS-ESI calculated for C22H24N3O5 [M + H]+ 410.1710, found m/z 410.1701.
), 7.10–6.99 (m, 2H, ArH MeO), 5.07–4.99 (m, 2H, NH, CH), 4.80 (t, J = 7.1 Hz, 2H, N1–C), 4.20–4.07 (m, 1H, CH), 3.93 (d, J = 5.7 Hz, 2H, CH2), 3.88 (s, 3H, O–CH3), 3.76–3.63 (m, 4H, N–CH2 morpholino), 2.76 (t, J = 7.1 Hz, 2H, N1–CH2C), 2.70–2.58 (m, 4H, O–CH2 morpholino), 1.98–1.71 (m, 8H, CH2), 1.47 (s, 9H, tBu CH3). 13C NMR (101 MHz, CDCl3) δ 169.94, 162.61, 162.24, 160.13, 150.68, 148.69, 142.25, 135.45, 132.38, 128.87, 128.27, 123.24, 114.97, 114.79, 80.11, 70.85, 67.18, 56.06, 55.59, 54.07, 46.82, 42.82, 39.17, 28.53, 28.49, 27.74 (one quaternary carbon not observed). HRMS-ESI calculated for C35H46N5O8 [M + H]+ 664.3341, found 664.3343.
), 2.82–2.56 (m, 4H, N–CH2 morpholino). 13C NMR (101 MHz, CDCl3) δ 164.63, 164.56, 160.41, 152.08, 148.49, 144.86, 135.94, 133.49, 128.33, 128.14, 119.28, 115.08, 115.07, 66.93, 56.00, 55.59, 53.96, 39.37. HRMS-ESI calculated for C22H24N3O5 [M + H]+ 410.1710, found m/z 410.1701.
), 7.10–6.99 (m, 2H, ArH MeO), 5.07–4.99 (m, 2H, NH, CH), 4.80 (t, J = 7.1 Hz, 2H, N1–C), 4.20–4.07 (m, 1H, CH), 3.93 (d, J = 5.7 Hz, 2H, CH2), 3.88 (s, 3H, O–CH3), 3.76–3.63 (m, 4H, N–CH2 morpholino), 2.76 (t, J = 7.1 Hz, 2H, N1–CH2C), 2.70–2.58 (m, 4H, O–CH2 morpholino), 1.98–1.71 (m, 8H, CH2), 1.47 (s, 9H, tBu CH3). 13C NMR (101 MHz, CDCl3) δ 169.94, 162.61, 162.24, 160.13, 150.68, 148.69, 142.25, 135.45, 132.38, 128.87, 128.27, 123.24, 114.97, 114.79, 80.11, 70.85, 67.18, 56.06, 55.59, 54.07, 46.82, 42.82, 39.17, 28.53, 28.49, 27.74 (one quaternary carbon not observed). HRMS-ESI calculated for C35H46N5O8 [M + H]+ 664.3341, found 664.3343.
Pharmacology
940 (Cayman Chemical, Michigan, USA) which was 10 mM in EtOH) were serially diluted using binding buffer containing the requisite amount of EtOH and DMSO to maintain equivalent vehicle levels throughout the dilution series and between all compounds. For vehicle control points, binding buffer containing matched concentrations of EtOH and DMSO was used in place of test ligands. [3H]CP55940 (PerkinElmer, Waltham, MA, USA) was used at a final concentration of 2.5 nM (Table 1) or 1 nM (Table 2). V-Bottom plates containing hCB2 or hCB1 membranes, [3H]CP55940 and ligand (or CP55940 or vehicle) were incubated at 30 °C for 1 h prior to harvesting and washing on filter plates (treated with PEI to minimise non-specific binding of the ligand), drying, incubation with scintillation fluid and detection. Binding experiments were performed a minimum of three independent times in technical triplicate. Data was analysed with GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA, USA) and competition binding curves fit by nonlinear regression using one site competition binding. Dissociation constants (pKi) of compounds were determined using [3H]CP55940 Kd = 2 nM (hCB1) or 3 nM (hCB2), and are expressed as mean ± standard error of the mean (SEM). In cases where less than 50% displacement of [3H]CP55940 was observed with 10 μM compound, affinity of the compound was deemed too low to be able to generate an accurate competition binding curve. Therefore, a one sample t-test (P < 0.05) was used to determine if there was significant difference between displacement in the absence (vehicle normalised to 0%) and presence of compound (with CP55940 normalised to 100%); if so, the ligand was determined to have a pKi <5, otherwise it was determined to show no significant binding.
Cells were transfected with 5 μg of pcDNA3L-His-CAMYEL plasmid (ATCC) using 30 μg of linear PEI (molecular weight 25 kDa; Polysciences, Warrington, PA, USA) in 150 mM NaCl. After 24 h, transfected cells were plated in poly-D-lysine (PDL) (0.05 mg mL−1 in PBS; Sigma-Aldrich, St Louis, MO, USA) treated 96-well solid white flat bottom polystyrene TC-treated microplates (Corning) at a density of 60–80000 cells per well in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; New Zealand-origin, Moregate Biotech, Brisbane, Australia). After 16 h, cells were serum-starved in Hank's balanced salt solution (HBSS, Thermo Fisher Scientific, Waltam, MA, USA) containing 1 mg mL−1 FAF BSA, pH 7.4 for 30 min. Cells were then treated with 7.5 μM coelenterazine-h (Nanolight Technology, Pinetop, AZ, USA) for 5 min, followed by addition of ligand or matched vehicle in HBSS plus 1 mg mL−1 FAF BSA and 5 μM forskolin (Cayman Chemical, Michigan, USA). A LUMIstar plate reader (BMG Labtech, Ortenberg, German) was used to immediately measure emission signals at 37 °C following ligand addition, which were simultaneously detected at 460/25 nM (Renilla luciferase, RLuc) and 535/25 nM (yellow fluorescent protein, YFP). Assays were carried out a minimum of three times (except where stated) in duplicate. Data analysis was performed using GraphPad Prism, with sigmoidal concentration response curves fit by nonlinear regression using values normalised to the vehicle (0%) or forskolin (100%) values for individual experiments. A t-test (P < 0.05) was used to determine if there was a significant difference in response for compounds at 10 μM in WT HEK cells to determine receptor mediated signalling.
Molecular modelling
The CB2 receptor homology model was generated using MODELLER 9.15 (ref. 41) using the structure of the antagonist-bound CB1 receptor (PDB ID: 5TGZ) as a template, based on a modified sequence alignment between hCB1 and hCB2 receptors from the T-Coffee server.42 Three dimensional models of ligands were generated using Avogadro 1.2 (ref. 43) and minimised using the universal force field. Ligand docking was performed using GOLD v5.5 (CCDC Software)44 centred on Ser285 extending for a distance of up to 15 Å and visualised in PyMOL (The PyMOL Molecular Graphics System, Version 1.8.6.0 Schrödinger, LLC.).Cellular imaging
HEK Flp-in wt cells were seeded at a density of 30000 cells per well in PDL treated Nunc™ 96-well black optical-bottom plates (Thermo Fisher Scientific). Approximately 24 h after seeding, cells were transfected with 125 ng per well of pplss-3HA-hCB2 or empty pcDNA 3.1 (for mock transfected cells) using Lipofectamine® 2000 Transfection Reagent (0.5 μL per well). All drugs and reagents for imaging assays were prepared in HBSS supplemented with 1 mg mL−1 BSA. After expressing for 18–24 h, medium was aspirated and cells incubated with mouse monoclonal anti-HA.11 (Clone 16B12, BioLegend, San Diego, CA, USA) diluted 1:500, for 30 min at room temperature. Cells were then briefly washed and co-incubated with Alexa Fluor® 488-conjugated goat anti-mouse secondary antibody (Thermo Fisher Scientific) diluted 1:300, and 31.6 μM SR144528 (kindly gifted by Roche; Basel, Switzerland) or Vehicle for 30 min at room temperature. Following a brief wash, cells were then treated with 3.16 μM 32 and SR144528 or Vehicle for 2 min, followed by 3 washes. Cells were then imaged using an ImageXpress® Micro XLS Widefield Microscope (Molecular Devices, Sunnyvale, CA, USA) (20× objective).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a University of Otago Research Grant, the School of Pharmacy, University of Otago and the Maurice Wilkins Centre for Molecular Biodiscovery. A. C. was supported by a University of Otago Doctoral Scholarship. We thank Christa Macdonald for her technical assistance with pharmacological assay preparations.References
- S. Galiegue, S. Mary, J. Marchand, D. Dussossoy, D. Carriere, P. Carayon, M. Bouaboula, D. Shire, G. L. Fur and P. Casellas, Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations, Eur. J. Biochem., 1995, 232, 54–61 CrossRef CAS.
- D.-J. Chen, M. Gao, F.-F. Gao, Q.-X. Su and J. Wu, Brain Cannabinoid Receptor 2: Expression, Function and Modulation, Nature, 2017, 38, 312–316 CAS.
- I. Katona and T. F. Freund, Multiple Functions of Endocannabinoid Signaling in the Brain, Annu. Rev. Neurosci., 2012, 35, 529–558 CrossRef CAS.
- M. E. Ferrini, S. Hong, A. Stierle, D. Stierle, N. Stella, K. Roberts and Z. Jaffar, CB2 Receptors Regulate Natural Killer Cells That Limit Allergic Airway Inflammation in a Murine Model of Asthma, Allergy, 2017, 72, 937–947 CrossRef CAS.
- C. Turcotte, M.-R. Blanchet, M. Laviolette and N. Flamand, The CB2 Receptor and Its Role as a Regulator of Inflammation, Cell. Mol. Life Sci., 2016, 73, 4449–4470 CrossRef CAS.
- T. Cassano, S. Calcagnini, L. Pace, F. De Marco, A. Romano and S. Gaetani, Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target, Front. Neurosci., 2017, 11, 30 Search PubMed.
- S. Han, L. Thoresen, J.-K. Jung, X. Zhu, J. Thatte, M. Solomon, I. Gaidarov, D. J. Unett, W. H. Yoon, J. Barden, A. Sadeque, A. Usmani, C. Chen, G. Semple, A. J. Grottick, H. Al-Shamma, R. Christopher and R. M. Jones, Discovery of APD371: Identification of a Highly Potent and Selective CB2 Agonist for the Treatment of Chronic Pain, ACS Med. Chem. Lett., 2017, 8, 1309–1313 CrossRef CAS.
- F. Carbone, F. Mach, N. Vuilleumier and F. Montecucco, Cannabinoid Receptor Type 2 Activation in Atherosclerosis and Acute Cardiovascular Diseases, Curr. Med. Chem., 2014, 21, 4046–4058 CrossRef CAS.
- A. I. Fraguas-Sánchez, C. Martín-Sabroso and A. I. Torres-Suárez, Insights Into the Effects of the Endocannabinoid System in Cancer: a Review, Br. J. Pharmacol., 2018, 175, 2566–2580 CrossRef.
- M. Aghazadeh Tabrizi, P. G. Baraldi, P. A. Borea and K. Varani, Medicinal Chemistry, Pharmacology, and Potential Therapeutic Benefits of Cannabinoid CB2 Receptor Agonists, Chem. Rev., 2016, 116, 519–560 CrossRef CAS.
- A. J. Vernall, S. J. Hill and B. Kellam, The Evolving Small-Molecule Fluorescent-Conjugate Toolbox for Class a GPCRs, Br. J. Pharmacol., 2014, 171, 1073–1084 CrossRef CAS.
- R. Lam, A. B. Gondin, M. Canals, B. Kellam, S. J. Briddon, B. Graham and P. J. Scammells, Fluorescently Labeled Morphine Derivatives for Bioimaging Studies, J. Med. Chem., 2018, 61, 1316–1329 CrossRef CAS.
- A. Tabor, D. Möller, H. Hübner, J. Kornhuber and P. Gmeiner, Visualization of Ligand-Induced Dopamine D2S and D2L Receptor Internalization by TIRF Microscopy, Sci. Rep., 2017, 1–11 CAS.
- L. A. Stoddart, A. J. Vernall, S. J. Briddon, B. Kellam and S. J. Hill, Direct Visualisation of Internalization of the Adenosine A3 Receptor and Localization with Arrestin3 Using a Fluorescent Agonist, Neuropharmacology, 2015, 98, 68–77 CrossRef CAS.
- L. A. Stoddart, L. E. Kilpatrick and S. J. Hill, NanoBRET Approaches to Study Ligand Binding to GPCRs and RTKs, Trends Pharmacol. Sci., 2017, 1–12 Search PubMed.
- R. R. Petrov, M. E. Ferrini, Z. Jaffar, C. M. Thompson, K. Roberts and P. Diaz, Design and Evaluation of a Novel Fluorescent CB2 Ligand as Probe for Receptor Visualization in Immune Cells, Bioorg. Med. Chem. Lett., 2011, 21, 5859–5862 CrossRef CAS.
- L. Martín-Couce, M. Martín-Fontecha, Ó. Palomares, L. Mestre, A. Cordomí, M. Hernangomez, S. Palma, L. Pardo, C. Guaza, M. L. López-Rodríguez and S. Ortega-Gutiérrez, Chemical Probes for the Recognition of Cannabinoid Receptors in Native Systems, Angew. Chem., Int. Ed., 2012, 51, 6896–6899 CrossRef.
- M. Sexton, G. Woodruff, E. A. Horne, Y. H. Lin, G. G. Muccioli, M. Bai, E. Stern, D. J. Bornhop and N. Stella, NIR-Mbc94, a Fluorescent Ligand That Binds to Endogenous CB2 Receptors and Is Amenable to High-Throughput Screening, Chem. Biol., 2011, 18, 563–568 CrossRef CAS.
- S. Zhang, P. Shao and M. Bai, In Vivo Type 2 Cannabinoid Receptor-Targeted Tumor Optical Imaging Using a Near Infrared Fluorescent Probe, Bioconjugate Chem., 2013, 24, 1907–1916 CrossRef CAS.
- Z. Wu, P. Shao, S. Zhang, X. Ling and M. Bai, Molecular Imaging of Human Tumor Cells That Naturally Overexpress Type 2 Cannabinoid Receptors Using a Quinolone-Based Near-Infrared Fluorescent Probe, J. Biomed. Opt., 2014, 19, 076016–076018 CrossRef.
- X. Ling, S. Zhang, P. Shao, W. Li, L. Yang, Y. Ding and C. Xu, Novel Near-Infrared Fluorescence Imaging Probe That Preferentially Binds to Cannabinoid Receptors CB2R Over CB1R, Biomaterials, 2015, 57, 169–178 CrossRef CAS.
- A. S. Yates, S. W. Doughty, D. A. Kendall and B. Kellam, Chemical Modification of the Naphthoyl 3-Position of JWH-015: in Search of a Fluorescent Probe to the Cannabinoid CB2 Receptor, Bioorg. Med. Chem. Lett., 2005, 15, 3758–3762 CrossRef CAS.
- A. G. Cooper, C. MacDonald, M. Glass, S. Hook, J. D. A. Tyndall and A. J. Vernall, Alkyl Indole-Based Cannabinoid Type 2 Receptor Tools: Exploration of Linker and Fluorophore Attachment, Eur. J. Med. Chem., 2018, 145, 770–789 CrossRef CAS.
- M. Soethoudt, S. C. Stolze, M. V. Westphal, L. van Stralen, A. Martella, E. J. van Rooden, W. Guba, Z. V. Varga, H. Deng, S. I. van Kasteren, U. Grether, A. P. Ijzerman, P. Pacher, E. M. Carreira, H. S. Overkleeft, A. Ioan-Facsinay, L. H. Heitman and M. Van der Stelt, Selective Photoaffinity Probe That Enables Assessment of Cannabinoid CB2 Receptor Expression and Ligand Engagement in Human Cells, J. Am. Chem. Soc., 2018, 140, 6067–6075 CrossRef CAS.
- V. Lucchesi, D. P. Hurst, D. M. Shore, S. Bertini, B. M. Ehrmann, M. Allarà, L. Lawrence, A. Ligresti, F. Minutolo, G. Saccomanni, H. Sharir, M. Macchia, V. Di Marzo, M. E. Abood, P. H. Reggio and C. Manera, CB2-Selective Cannabinoid Receptor Ligands: Synthesis, Pharmacological Evaluation, and Molecular Modeling Investigation of 1,8-Naphthyridin-2(1H)-One-3-Carboxamides, J. Med. Chem., 2014, 57, 8777–8791 CrossRef CAS.
- C. Manera, V. Benetti, M. P. Castelli, T. Cavallini, S. Lazzarotti, F. Pibiri, G. Saccomanni, T. Tuccinardi, A. Vannacci, A. Martinelli and P. L. Ferrarini, Design, Synthesis, and Biological Evaluation of New 1,8-Naphthyridin-4(1H)-on-3-Carboxamide and Quinolin-4(1 H)-on-3-Carboxamide Derivatives as CB2 Selective Agonists, J. Med. Chem., 2006, 49, 5947–5957 CrossRef CAS.
- C. Manera, G. Saccomanni, B. Adinolfi, V. Benetti, A. Ligresti, M. G. Cascio, T. Tuccinardi, V. Lucchesi, A. Martinelli, P. Nieri, E. Masini, V. Di Marzo and P. L. Ferrarini, Rational Design, Synthesis, and Pharmacological Properties of New 1,8-Naphthyridin-2(1H)-on-3-Carboxamide Derivatives as Highly Selective Cannabinoid-2 Receptor Agonists, J. Med. Chem., 2009, 52, 3644–3651 CrossRef CAS.
- C. Manera, G. Saccomanni, A. M. Malfitano, S. Bertini, F. Castelli, C. Laezza, A. Ligresti, V. Lucchesi, T. Tuccinardi, F. Rizzolio, M. Bifulco, V. Di Marzo, A. Giordano, M. Macchia and A. Martinelli, Rational Design, Synthesis and Anti-Proliferative Properties of New CB2 Selective Cannabinoid Receptor Ligands: An Investigation of the 1,8-Naphthyridin-2(1H)-One Scaffold, Eur. J. Med. Chem., 2012, 52, 284–294 CrossRef CAS.
- C. Manera, A. M. Malfitano, T. Parkkari, V. Lucchesi, S. Carpi, S. Fogli, S. Bertini, C. Laezza, A. Ligresti, G. Saccomanni, J. R. Savinainen, E. Ciaglia, S. Pisanti, P. Gazzerro, V. Di Marzo, P. Nieri, M. Macchia and M. Bifulco, New Quinolone- and 1,8-Naphthyridine-3-Carboxamides as Selective CB2 Receptor Agonists with Anticancer and Immuno–Modulatory Activity, Eur. J. Med. Chem., 2015, 97, 10–18 CrossRef CAS.
- T. Kimura, K. Cheng, K. C. Rice and K. Gawrisch, Location, Structure, and Dynamics of the Synthetic Cannabinoid Ligand CP-55,940 in Lipid Bilayers, Biophys. J., 2009, 96, 4916–4924 CrossRef CAS.
- D. P. Hurst, A. Grossfield, D. L. Lynch, S. Feller, T. D. Romo, K. Gawrisch, M. C. Pitman and P. H. Reggio, A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-Coupled Receptor, J. Biol. Chem., 2010, 285, 17954–17964 CrossRef CAS.
- J. Jakowiecki and S. Filipek, Hydrophobic Ligand Entry and Exit Pathways of the CB1 Cannabinoid Receptor, J. Chem. Inf. Model., 2016, 56, 2457–2466 CrossRef CAS.
- A. J. Vernall, L. A. Stoddart, S. J. Briddon, H. W. Ng, C. A. Laughton, S. W. Doughty, S. J. Hill and B. Kellam, Conversion of a Non-Selective Adenosine Receptor Antagonist Into A3-Selective High Affinity Fluorescent Probes Using Peptide-Based Linkers, Org. Biomol. Chem., 2013, 11, 5673–5682 RSC.
- A. J. Vernall, L. A. Stoddart, S. J. Briddon, S. J. Hill and B. Kellam, Highly Potent and Selective Fluorescent Antagonists of the Human Adenosine A3 Receptor Based on the 1,2,4-Triazolo[4,3-a]Quinoxalin-1-One Scaffold, J. Med. Chem., 2012, 55, 1771–1782 CrossRef CAS.
- Y. Ni and J. Wu, Far-red and near infrared BODIPY dyes: synthesis and applications for fluorescent pH probes and bio-imaging, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC.
- T. Hua, K. Vemuri, M. Pu, L. Qu, G. W. Han, Y. Wu, S. Zhao, W. Shui, S. Li, A. Korde, R. B. Laprairie, E. L. Stahl, J.-H. Ho, N. Zvonok, H. Zhou, I. Kufareva, B. Wu, Q. Zhao, M. A. Hanson, L. M. Bohn, A. Makriyannis, R. C. Stevens and Z.-J. Liu, Crystal Structure of the Human Cannabinoid Receptor CB1, Cell, 2016, 167, 750–755.e14 CrossRef CAS.
- N. L. Grimsey, C. E. Goodfellow, M. Dragunow and M. Glass, Cannabinoid Receptor 2 Undergoes Rab5-Mediated Internalization and Recycles via a Rab11-Dependent Pathway, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 1554–1560 CrossRef CAS.
- E. E. Cawston, W. J. Redmond, C. M. Breen, N. L. Grimsey, M. Connor and M. Glass, Real-Time Characterization of Cannabinoid Receptor 1 (CB 1) Allosteric Modulators Reveals Novel Mechanism of Action, Br. J. Pharmacol., 2013, 170, 893–907 CrossRef CAS.
- J. M. McPartland, C. MacDonald, M. Young, P. S. Grant, D. P. Furkert and M. Glass, Affinity and Efficacy Studies of Tetrahydrocannabinolic Acid a at Cannabinoid Receptor Types One and Two, Cannabis Cannabinoid Res., 2017, 2, 87–95 CrossRef.
- M. Soethoudt, U. Grether, J. U. R. Fingerle, T. W. Grim, F. Fezza, L. de Petrocellis, C. Ullmer, B. R. A. Usler, C. Perret, N. van Gils, D. Finlay, C. MacDonald, A. Chicca, M. D. Gens, J. Stuart, H. de Vries, N. Mastrangelo, L. Xia, G. Alachouzos, M. P. Baggelaar, A. Martella, E. D. Mock, H. Deng, L. H. Heitman, M. Connor, V. Di Marzo, J. U. R. Gertsch, A. H. Lichtman, M. Maccarrone, P. Pacher, M. Glass and M. Van der Stelt, Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity, Nat. Commun., 2016, 8, 1–14 Search PubMed.
- A. Sali and T. L. Blundell, Comparative protein modelling by satisfaction of spatial restraints, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS.
- C. Notredame, D. G. Higgins and J. Heringa, T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment, J. Mol. Biol., 2000, 302, 205–217 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform, J. Cheminf., 2012, 4, 17 CAS.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, Development and Validation of a Genetic Algorithm for Flexible Docking, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00448j |
| This journal is © The Royal Society of Chemistry 2018 |