
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Screening of the Pathogen Box reveals new starting points for anti-trypanosomal drug discovery†
Clinton G. L.
Veale
 a and
Heinrich C.
Hoppe
b
a and
Heinrich C.
Hoppe
b
aSchool of Chemistry and Physics, Pietermaritzburg Campus, University of KwaZulu-Natal, Private Bag X01, Scottsville, 3209, South Africa. E-mail: VealeC@ukzn.ac.za; Tel: +27 33 260 6365
bDepartment of Biochemistry and Microbiology, Rhodes University, Grahamstown, 6140, South Africa. E-mail: H.Hoppe@ru.ac.za; Tel: +27 46 603 8262
First published on 26th October 2018
Abstract
This study aimed to uncover new starting points for anti-trypansomal drug discovery through the screening of the Pathogen Box against Trypanosoma brucei brucei. Our study identified compounds 35, 39, 46, 53 and 56 whose activity and selectivity highlighted them as promising candidates with potential for further study and optimisation.
Introduction
Human African trypanosomiasis (HAT), commonly referred to as sleeping sickness whose causative agent is Trypanosoma brucei (T.b.), impacts roughly 70 million people living in sub-Saharan Africa, affecting these populations from both a health and economic perspective.1,2 With no current vaccine and limited access to appropriate facilities for early diagnosis, coupled to treatment regimens which are limited in their scope and efficacy,3,4 the importance of stimulating new drug discovery programs through cost effective means is of grave importance.5 HAT is caused by the T.b. sub-species T.b. rhodesiense and T.b. gambiense respectively, while African animal trypanosomiasis (AAT) is typically caused by the T.b. brucei subspecies.6,7 While T.b. sub-species infections are clinically distinct, they share large similarities in both their molecular and cellular biology,8 which has allowed for T.b. brucei to be used as a competent proxy for the identification of hits and leads for HAT.9–12 The Pathogen Box like the Malaria Box represents a 400 strong library of compounds with demonstrated biological activity against a range of neglected tropical diseases,13 which has formed the basis of numerous screening campaigns to identify chemical starting points for hit and lead optimisation campaigns.14–16 Van Voorhis et al.13 reported a meta-analysis of multiple screens of the Malaria Box, which included anti-trypanosomal hit compounds. Similarly, in our efforts to identify new starting points for anti-trypanosomal drug discovery, we screened the Pathogen Box against T.b. brucei and identified a handful of compounds with encouraging and selective in vitro activity. During the process of our screening campaign, we were alerted to a study by Duffy et al. in which they performed a comprehensive screen of the Pathogen Box against multiple protozoan parasites including T.b. brucei.17 This current study and that of Duffy et al. used different criteria, from which to select compounds for dose-dependent analysis, therefore, in some instances an IC50 value may not have been determined for a compound from either study. However, a lack of an IC50 value does not necessarily mean that a compound was inactive, but rather that it was likely a moderate inhibitor. In addition, false positives and negatives are a burden of screening campaigns,18,19 therefore, for the purposes of robust analysis, we took this opportunity to compare our data with that obtained by Duffy et al. as well as that originally reported in the Pathogen Box. The majority of data we obtained in our study correlated well with that reported previously, particularly with respect to compounds from the kinetoplastid category of the Pathogen Box, which provided confidence in our method as well as for the previously reported data. However, inhibitory data for a small cohort of compounds from other categories of the pathogen differed to that reported by Duffy et al. Accordingly, we report our findings as an independent analysis of the Pathogen Box, which highlights potential new starting points for anti-trypanosomal drug discovery.Results and discussion
Prior to subjecting compounds to dose-dependent assessments, the Pathogen Box was screened for T.b. brucei inhibitory activity at a single concentration (10 μM). Compounds which inhibited cell viability to below 20% (i.e. reduced cell viability by 80% or more) at this concentration were put forward for dose dose-dependent assessment.We opted for fairly stringent exclusion criteria, since the principal aim of this study was to identify or confirm compounds with activity in the sub-micromolar range. This data is supplied in the supplementary information. Of the 70 compounds categorised in the Pathogen Box as active against kinetoplastids, 34 were classified as inhibitors of T.b. brucei. Of these, 29 compounds (1–22, 25, 28–31, 33, 34) met our exclusion threshold and were subjected for IC50 evaluation (Table 1). In order to assess the robustness of our screen, we plotted pIC50 values (M) from the Pathogen Box vs. our experimentally obtained values (Fig. 1). Inhibitory data which was not provided as a precise value, but rather as an approximation below a threshold (i.e. <0.13 μM) was left out of our initial plot. Gratifyingly, this data showed that in all instances where IC50 data was obtained, the difference in pIC50 was lower than 1 (an order of magnitude). However, five compounds (15, 17, 19, 20, 21) differed by greater than 0.5 log units. Five compounds (23, 24, 26, 27, 32) appeared on the base on the x-axis, and represent compounds which did not sufficiently reduce cell viability in our single concentration screen, and were not subjected to IC50 analysis. However, of these compounds, only compound 24 (MMV688776) had activity in the sub-micromolar range reported in the Pathogen Box. The same plot was obtained by comparing the data of Duffy et al. and ours (Fig. 1). In this instance a change in pIC50 of greater than 0.5 log units was observed for six compounds (1, 4, 7, 15, 21, 29) with only compound 1 (MMV688180) displaying a change in pIC50 greater than 1. However, this was a significant outlier. Compound 1 has previously been reported as a moderate inhibitor of T. brucei culture through the selective inhibition of T. brucei N-myristoyl transferase.20,21
| Cpd no. | ID | T. brucei brucei IC50 (μM) | HeLa IC50 (μM) | ||
|---|---|---|---|---|---|
| Duffy et al.a | Pathogen Boxb | This study | |||
| IC50 values are the mean of experiments conducted in technical triplicate. a Data reported in ref. 17. b Data available at https://www.pathogenbox.org/. c ND = not determined. | |||||
| 1 | MMV688180 | 0.01 | <0.13 | 11 | 2.8 |
| 2 | MMV688372 | 0.02 | <0.13 | 0.049 | 0.8 |
| 3 | MMV652003 | 0.11 | 0.15 | 0.14 | >25 |
| 4 | MMV688550 | 0.13 | <0.13 | 0.030 | >25 |
| 5 | MMV688797 | 0.13 | 0.13 | 0.050 | >25 |
| 6 | MMV676604 | 0.14 | 0.26 | 0.084 | 1.2 |
| 7 | MMV676602 | 0.17 | <0.13 | 0.048 | 4.0 |
| 8 | MMV688796 | 0.23 | 0.10 | 0.11 | >25 |
| 9 | MMV688371 | 0.24 | <0.13 | 0.49 | 7.5 |
| 10 | MMV688958 | 0.24 | 0.15 | 0.18 | >25 |
| 11 | MMV689028 | 0.24 | 0.14 | 0.12 | >25 |
| 12 | MMV675998 | 0.27 | 0.25 | 0.54 | >25 |
| 13 | MMV688795 | 0.27 | 0.15 | 0.21 | >25 |
| 14 | MMV688798 | 0.50 | 0.56 | 1.2 | >25 |
| 15 | MMV690027 | 0.50 | 0.02 | 0.12 | >25 |
| 16 | MMV688271 | 0.52 | 0.60 | 0.65 | 17 |
| 17 | MMV202553 | 1.0 | 2.46 | 0.38 | >25 |
| 18 | MMV689029 | 1.1 | 0.50 | 0.44 | >25 |
| 19 | MMV688467 | 1.2 | 0.26 | 1.3 | >25 |
| 20 | MMV688793 | 1.2 | 2.07 | 0.62 | >25 |
| 21 | MMV676600 | 1.3 | 1.02 | 0.30 | >25 |
| 22 | MMV188296 | 1.5 | 0.49 | 0.71 | >25 |
| 23 | MMV687762 | 1.8 | 7.58 | ND | ND |
| 24 | MMV688776 | 1.8 | 0.50 | ND | ND |
| 25 | MMV690028 | 2.3 | 0.79 | 1.7 | >25 |
| 26 | MMV688514 | 3.5 | 4.06 | ND | ND |
| 27 | MMV595321 | 4.3 | 5.30 | ND | ND |
| 28 | MMV099637 | 4.4 | 1.94 | 4.6 | >25 |
| 29 | MMV688279 | 4.5 | 0.98 | 0.98 | 15 |
| 30 | MMV687706 | 5.1 | 0.99 | 2.3 | 11 |
| 31 | MMV688283 | 5.3 | 3.78 | 2.2 | >25 |
| 32 | MMV688179 | 5.7 | 1.03 | ND | ND |
| 33 | MMV001561 | 6.2 | 3.97 | 4.1 | 10 |
| 34 | MMV1236379 | NDc | 1.22 | 0.41 | >25 |
| Pentamidine | 0.0031 | ||||
| Emetine | 0.4 | ||||
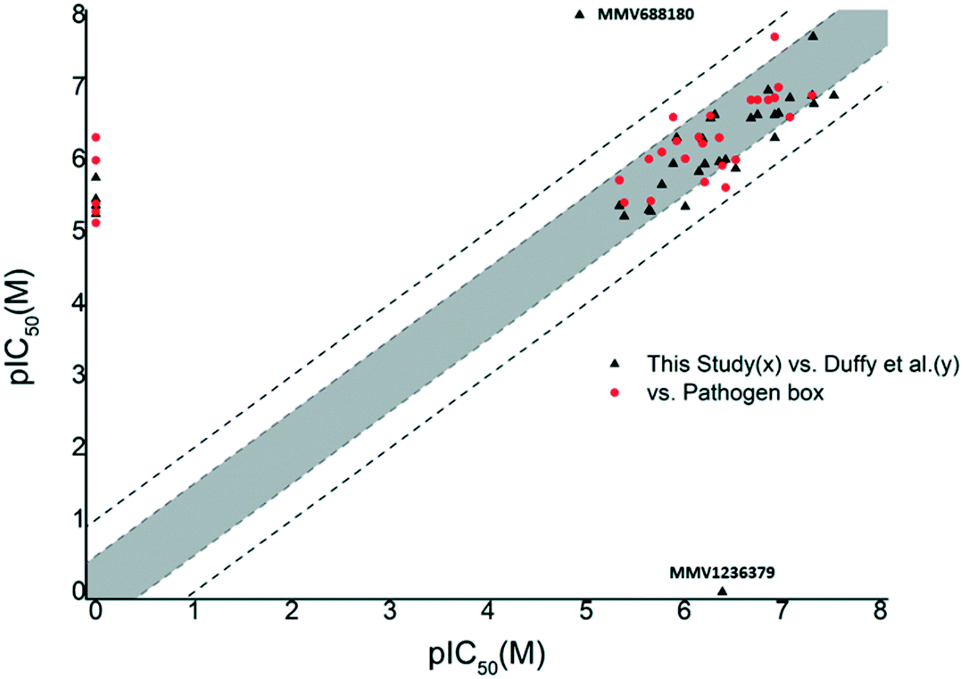 | ||
| Fig. 1 Plot of pIC50 (T.b. brucei) data obtained in this study (x) vs. the Pathogen Box or Duffy et al. (y) from compounds in the kinetoplastid category of the Pathogen Box. Compounds falling outside the grey shaded area differed in pIC50 by 0.5 log units or more. Compounds falling outside the dashed lines differed by 1 log unit or more. This figure indicated that our data was in good agreement with previous reports. | ||

While Duffy et al. did not obtain an IC50 value for compound 34 (MMV1236379), our data suggested that 34 was a mid-nanomolar inhibitor of T.b. brucei, while the Pathogen Box placed it at the low micromolar range. While there were several instances, where compound potency differed by more than 0.5 log units, the comparative data obtained in this screen indicated that for the large majority of compounds, the pIC50 values we obtained fall within a 0.5–1 log range when compared to the Pathogen Box and Duffy et al. which we defined as an acceptable degree of similarity with previously reported data. With the exception of 1, 24 and 34, the majority of compounds whose data did not match to at least within an order of magnitude were compounds which were not subjected to dose-dependent analysis due to those compounds not conforming to our criteria in the single concentration screen (cell viability ≤20%). The data of Duffy et al. as well as the Pathogen Box, show that these were all low micromolar inhibitors, of T.b. brucei possibly accounting for their moderate performance in the single concentration screen, which indicated that overall, our screening method was robust. The correlation of our screening results and that of Duffy et al. highlights the complexities associated with the confirmation of biological activity and should also be seen in the context of differences in the detailed assay methodology employed. We used 96-well as opposed to 384-well plates, and compounds were incubated with parasites for 48 hours at 37 °C, while Duffy et al. added an additional 2 hours at 37 °C and 22 hours at room temperature.
From screening the remainder of the Pathogen Box listed for indications other than kinetoplastids, we identified 28 compounds (35–62), which satisfied our single concentration screening criteria, while several compounds (63–69) for which Duffy et al. obtained IC50 data, did not fit our criteria. (Table 2). The remainder of the Pathogen Box was inactive in both screens. Of compounds 63–69, only the anti-mycobacterial 66 (MMV687807) whose MOA has been speculated to occur via disruption of mitochondrial proton gradient22 was reported by Duffy et al. as a sub-micromolar inhibitor of T.b. brucei. Similarly, we obtained IC50 data for 13 compounds (38, 39, 43–47, 49, 53, 55, 59, 60, 62), which were not reported by Duffy et al. likely due to their own cut off criteria of 5 μM in the primary screen. Of these compounds, 39 (MMV676477), 46 (MMV687812) and 53 (MMV010576) were identified as sub-micromolar inhibitors of T.b. brucei. The anti-mycobacterial compound 39 (ref. 23) has yet to be reported as an anti-trypanosomal. However, the closely related analogue TCMDC 142497 (70) was identified from a screen of 1.8 million compounds as a potent kinetoplastid inhibitor.24
| Cpd no. | ID | T. brucei brucei IC50 (μM) | Selectivity indexc | ||
|---|---|---|---|---|---|
| Duffy et al.a | This study | HeLa IC50 (μM) | |||
| IC50 values are the mean of experiments conducted in technical triplicate. a Data reported in ref. 17. b ND = not determined. c Selectivity index calculated as IC50(HeLa)/IC50(T.bb). | |||||
| 35 | MMV021013 | 3.51 | 0.83 | >25 | 30 |
| 36 | MMV024311 | 5.15 | 2.2 | 8.9 | 4.0 |
| 37 | MMV153413 | 2.99 | 11 | >25 | 2.2 |
| 38 | MMV461553 | NDb | 11 | >25 | 2.2 |
| 39 | MMV676477 | ND | 0.59 | 2.6 | 4.4 |
| 40 | MMV676411 | 3.22 | 0.76 | 8.4 | 11 |
| 41 | MMV676512 | 2.87 | 1.1 | 5.1 | 4.6 |
| 42 | MMV687273 | 2.28 | 2.3 | 15 | 6.5 |
| 43 | MMV676409 | ND | 2.9 | >25 | 8.6 |
| 44 | MMV687703 | ND | 4.2 | 22 | 5.2 |
| 45 | MMV687765 | ND | 5.1 | >25 | 4.9 |
| 46 | MMV687812 | ND | 0.87 | 4.8 | 5.5 |
| 47 | MMV688844 | ND | 29 | >25 | 0.8 |
| 48 | MMV675968 | 2.07 | 1.2 | >25 | 21 |
| 49 | MMV687776 | ND | 1.2 | 10 | 8.3 |
| 50 | MMV688417 | 6.10 | 4.3 | >25 | 5.8 |
| 51 | MMV024035 | 2.72 | 1.0 | 7.5 | 7.5 |
| 52 | MMV006901 | 3.36 | 4.7 | >25 | 5.3 |
| 53 | MMV010576 | ND | 0.33 | >25 | 76 |
| 54 | MMV019189 | 4.68 | 1.0 | 6.6 | 6.6 |
| 55 | MMV020391 | ND | 6.5 | 12 | 1.8 |
| 56 | MMV022029 | 2.38 | 0.22 | 13 | 59 |
| 57 | MMV023233 | 3.29 | 1.2 | 5.4 | 4.5 |
| 58 | MMV028694 | 2.39 | 1.5 | >25 | 17 |
| 59 | MMV1030799 | ND | 7.1 | >25 | 3.5 |
| 60 | MMV002817 | ND | 2.0 | >25 | 13 |
| 61 | MMV688761 | 3.49 | 11 | >25 | 2.3 |
| 62 | MMV637229 | ND | 3.5 | >25 | 7.1 |
| 63 | MMV272144 | 4.12 | ND | ND | |
| 64 | MMV495543 | 5.45 | ND | ND | |
| 65 | MMV687248 | 1.05 | ND | ND | |
| 66 | MMV687807 | 0.66 | ND | ND | |
| 67 | MMV022478 | 1.45 | ND | ND | |
| 68 | MMV026490 | 6.02 | ND | ND | |
| 69 | MMV688768 | 1.50 | ND | ND | |
| Pentamidine | 0.0031 | ||||
| Emetine | 0.4 | ||||

Comparison of remaining pIC50 values again showed that the majority of compounds did not differ by more than 0.5 log units from the values reported by Duffy et al. In our screen, the pIC50 of compound 37 (MMV153413) was found to be more than 0.5 log units lower than that reported by Duffy et al. (Fig. 2). However, our moderate inhibitory data for 37 matched that of its close analogue 38 (MMV461553), for which Duffy et al. did not obtain IC50 data. Both of these compounds specifically inhibit mycobacterial cell wall synthesis39 likely through the inhibition of mycolic acid biosynthesis.40 The remaining four compounds whose pIC50 values differed by more than 0.5 log units (35, 40, 54, 56) were all found to have pIC50 values greater than six, corresponding to sub-micromolar inhibition. Compounds 35 (MMV021013)23 and 56 (MMV022029)41 had been identified as promising anti-mycobacterial and anti-plasmodial compounds respectively from large scale screening, without compelling evidence for a biological target.
 | ||
| Fig. 2 Plot of pIC50 (T.b. brucei) data obtained in this study (x) vs. Duffy et al. (y) from the reminder of the Pathogen Box. Compounds falling outside the grey shaded area differed in pIC50 by 0.5 log units or more. Compounds falling outside the dashed lines differed by 1 log unit or more. The pink shaded area highlights compounds which inhibited T.b. brucei in the sub-micromolar range in our screen. Compounds 35, 39, 40, 46, 53, 54 and 56 were all found in this area and either differed 0.5 log units or more, or IC50 data was not determined for them by Duffy et al. | ||

Compound 54 (MMV019189) had also previously been reported as an anti-plasmodial agent.41 While to date there have been no reports of a potential mechanism of action for 54, the structurally related compounds 74 (Lipofermata) and 75 have been identified as inhibitors of human fatty acid transport protein (FATP2)45 and metallo-β-lactamase.46 However, it is unclear whether analogous targets are present in T. brucei.
Having determined cytotoxicity against a HeLa cell line, we were able to derive the selectivity index (SI) for all active compounds (Table 2). A plot of pIC50vs. SI allowed us to rapidly identify which compounds combined good activity (pIC50 > 6) with acceptable selectivity (SI > 4) (Fig. 3). Compounds 35, 39, 40, 46, 53, 54, and 56 which have been discussed previously, all satisfied these criteria. Of these compounds, 39 and 46 are respectively reported to have CC50 values of 1.3 μM and 3.9 μM against HepG2 cells by the Pathogen Box, which agrees with our respective values of 2.6 μM and 4.8 μM against HeLa cells. While these compounds are very close to our selectivity index cut-off of 4, their encouraging activity may warrant further investigation and SAR analysis. None of the other compounds were reported as cytotoxic against HepG2 cells in the Pathogen Box dataset. Furthermore, antimalarial compound 5141 (MMV024035) was identified as a candidate that selectively inhibited T.b. brucei, albeit moderately. This compound also showed moderate inhibition in the screen of Duffy et al.
 | ||
| Fig. 3 Plot of pIC50 (T.b. brucei) data obtained in this study (x) vs. selectivity index (y) from the reminder of the Pathogen Box. Compounds appearing in the pink shaded area combined sub-micromolar activity with acceptable selectivity over a HeLa cell line. Compounds 35, 39, 40, 46, 51, 53, 54, and 56 were all deemed to be promising compounds for further study based on this assessment. | ||
Conclusions
In conclusion, this study sought to identify new starting points for anti-trypanosomal drug discovery, through the repurposing of the Pathogen Box. Our results have identified eight compounds, which may hold promise as starting points for in-depth SAR studies. Compounds 35, 53 and 56 in particular combined activity in the nanomolar range coupled to good selectively, while compounds 39, 46 and 53 again hold potential for target based programs due to their reported activity against targets for which analogous biological processes are present in T. brucei.Experimental procedures
Trypanosoma brucei assay
T.b. brucei (strain 427) bloodstream form parasites were cultured at 37 °C in a 5% CO2 incubator in IMDM medium supplemented with 25 mM HEPES, 10% fetal bovine serum, 1 mM hypoxanthine, antibiotics (penicillin/streptomycin) and HMI-9 supplement.47 For screening purposes, parasites were distributed with\ test compounds in 96-well plates at a final concentration of 10 μM compound and 2.4 × 104 parasites per well in a total volume of 200 μl per well. After a 24 hour incubation at 37 °C in a 5% CO2 incubator, 20 μl resazurin reagent (0.5 mM resazurin in phosphate-buffered saline) was added to each well and incubation continued for a further 24 hours. Resazurin conversion to resorufin was determined by measuring fluorescence (Exc560/Em590) in a plate reader. Fluorescence values obtained in drug-treated wells were converted to % parasite viability relative to readings obtained in control wells (non-treated parasites). Dose–response assays were performed by incubating parasites with 3-fold serial dilutions of test compounds, plotting % parasite viability vs. log[compound] and determining IC50 concentrations by non-linear regression analysis using GraphPad Prism. All assays were performed in technical triplicate.HeLa cell assay
HeLa cells were cultured in DMEM medium supplemented with 10% fetal bovine serum and antibiotics (penicillin/streptomycin/amphotericin B) at 37 °C in a 5% CO2 incubator. On the day prior to compound addition, cells were plated in 96-well plates at 2 × 104 cells per well. Compounds were added to the cells in 3-fold serial dilutions and incubation at 37 °C in a 5% CO2 incubator continued for 48 hours. Twenty μl resazurin reagent (0.5 mM resazurin in phosphate-buffered saline) was added to each well and, after a 2-hour incubation, fluorescence (Exc560/Em590) was measured in a plate reader. Percentage cell viability in drug-treated wells was calculated from the fluorescent readings obtained relative to those in wells containing control, untreated cells. Plots of % cell viability vs. log[compound] were used to determine IC50 values by non-linear regression analysis using GraphPad Prism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by funds from the South African Medical Research Council. A copy of the Pathogen Box was provided by the Medicines for Malaria Venture.References
- P. Büscher, G. Cecchi, V. Jamonneau and G. Priotto, Lancet, 2017, 390, 2397–2409 CrossRef.
- S. Aksoy, P. Buscher, M. Lehane, P. Solano and J. Van Den Abbeele, PLoS Neglected Trop. Dis., 2017, 11, e0005454 CrossRef PubMed , 1–6.
- S. Patterson, M. S. Alphey, D. C. Jones, E. J. Shanks, I. P. Street, J. A. Frearson, P. G. Wyatt, I. H. Gilbert and A. H. Fairlamb, J. Med. Chem., 2011, 54, 6514–6530 CrossRef CAS PubMed.
- L. Ferrins, M. Gazdik, R. Rahmani, S. Varghese, M. L. Sykes, A. J. Jones, V. M. Avery, K. L. White, E. Ryan, S. A. Charman, M. Kaiser, C. A. S. Bergström and J. B. Baell, J. Med. Chem., 2014, 57, 6393–6402 CrossRef CAS PubMed.
- G. Patel, N. E. Roncal, P. J. Lee, S. E. Lee, J. Erath, A. Rodriguez, R. J. Sciotti and M. P. Pollastri, MedChemComm, 2014, 5, 655–658 RSC.
- M. Witschel, M. Rottmann, M. Kaiser and R. Brun, PLoS Neglected Trop. Dis., 2012, 6, e1805 CrossRef PubMed , 1–10.
- M. Berninger, I. Schmidt, A. Ponte-Sucre and U. Holzgrabe, MedChemComm, 2017, 8, 1872–1890 RSC.
- K. Stuart, R. Brun, S. Croft, A. Fairlamb, R. E. Gürtler, J. Mckerrow, S. Reed and R. Tarleton, J. Clin. Invest., 2008, 118, 1301–1310 CrossRef CAS PubMed.
- M. Keita, B. Bouteille, B. Enanga, J.-M. Vallat and M. Dumas, Exp. Parasitol., 1997, 85, 183–192 CrossRef CAS PubMed.
- H. B. Tatipaka, J. R. Gillespie, A. K. Chatterjee, N. R. Norcross, M. A. Hulverson, R. M. Ranade, P. Nagendar, S. A. Creason, J. McQueen, N. A. Duster, A. Nagle, F. Supek, V. Molteni, T. Wenzler, R. Brun, R. Glynne, F. S. Buckner and M. H. Gelb, J. Med. Chem., 2014, 57, 828–835 CrossRef CAS PubMed.
- M. L. Sykes, J. B. Baell, M. Kaiser, E. Chatelain, S. R. Moawad, D. Ganame, J. R. Ioset and V. M. Avery, PLoS Neglected Trop. Dis., 2012, 6, e1896 CrossRef PubMed , 1–14.
- L. A. T. Cleghorn, S. Albrecht, L. Stojanovski, F. R. J. Simeons, S. Norval, R. Kime, I. T. Collie, M. De Rycker, L. Campbell, I. Hallyburton, J. A. Frearson, P. G. Wyatt, K. D. Read and I. H. Gilbert, J. Med. Chem., 2015, 58, 7695–7706 CrossRef CAS PubMed.
- W. C. Van Voorhis, J. H. Adams, R. Adelfio, V. Ahyong, M. H. Akabas, P. Alano, A. Alday, Y. Alemán Resto, A. Alsibaee, A. Alzualde, K. T. Andrews, S. V. Avery, V. M. Avery, L. Ayong, M. Baker, S. Baker, C. Ben Mamoun, S. Bhatia and Q. Bickle, et al. , PLoS Pathog., 2016, 12, e1005763 CrossRef PubMed , 1–23.
- J. Jeong, G. Kim, C. Moon, H. J. Kim, T. H. Kim and J. Jang, PLoS One, 2018, 13, e0195595 CrossRef PubMed , 1–10.
- T. Vila and J. L. Lopez-Ribot, Antimicrob. Agents Chemother., 2017, 61, e02006-16 CrossRef PubMed , 1–9.
- S. Preston, Y. Jiao, A. Jabbar, S. L. McGee, B. Laleu, P. Willis, T. N. C. Wells and R. B. Gasser, Int. J. Parasitol.: Drugs Drug Resist., 2016, 6, 329–334 Search PubMed.
- S. Duffy, M. L. Sykes, A. J. Jones, T. B. Shelper, M. Simpson, R. Lang, S.-A. Poulsen, B. E. Sleebs and V. M. Avery, Antimicrob. Agents Chemother., 2017, 61, e00379-17 CrossRef PubMed , 1–22.
- N. Thorne, D. S. Auld and J. Inglese, Curr. Opin. Chem. Biol., 2011, 14, 315–324 CrossRef PubMed.
- R. Sink, S. Gobec and S. Pecar, Curr. Med. Chem., 2010, 17, 4231–4255 CrossRef CAS PubMed.
- S. Brand, L. A. T. Cleghorn, S. P. Mcelroy, D. A. Robinson, V. C. Smith, I. Hallyburton, J. R. Harrison, N. R. Norcross, D. Spinks, T. Bayliss, L. Stojanovski, L. S. Torrie, J. A. Frearson, R. Brenk, A. H. Fairlamb, M. A. J. Ferguson, K. D. Read, P. G. Wyatt and I. H. Gilbert, J. Med. Chem., 2012, 55, 140–152 CrossRef CAS PubMed.
- J. A. Frearson, S. Brand, S. P. McElroy, L. A. T. Cleghorn, O. Smid, L. Stojanovski, H. P. Price, M. L. S. Guther, L. S. Torrie, D. A. Robinson, I. Hallyburton, C. P. Mpamhanga, J. A. Brannigan, A. J. Wilkinson, M. Hodgkinson, R. Hui, W. Qiu, O. G. Raimi and D. M. F. van Aalten, et al. , Nature, 2010, 464, 728–734 CrossRef CAS PubMed.
- I. Y. Lee, T. D. Gruber, A. Samuels, M. Yun, B. Nam, M. Kang, K. Crowley, B. Winterroth, H. I. Boshoff and C. E. Barry, Bioorg. Med. Chem., 2013, 21, 114–126 CrossRef CAS PubMed.
- L. Ballell, R. H. Bates, R. J. Young, D. Alvarez-Gomez, E. Alvarez-Ruiz, V. Barroso, D. Blanco, B. Crespo, J. Escribano, R. González, S. Lozano, S. Huss, A. Santos-Villarejo, J. J. Martín-Plaza, A. Mendoza, M. J. Rebollo-Lopez, M. Remuiñan-Blanco, J. L. Lavandera and E. Pérez-Herran, et al. , ChemMedChem, 2013, 8, 313–321 CrossRef CAS PubMed.
- I. Peña, M. Pilar Manzano, J. Cantizani, A. Kessler, J. Alonso-Padilla, A. I. Bardera, E. Alvarez, G. Colmenarejo, I. Cotillo, I. Roquero, F. De Dios-Anton, V. Barroso, A. Rodriguez, D. W. Gray, M. Navarro, V. Kumar, A. Sherstnev, D. H. Drewry and J. R. Brown, et al. , Sci. Rep., 2015, 5, 8771 CrossRef PubMed.
- Y. Nakamura, A. Suganami, M. Fukuda, M. K. amru. Hasan, T. Yokochi, A. Takatori, S. Satoh, T. Hoshino, Y. Tamura and A. Nakagawara, Cancer Med., 2014, 3, 25–35 CrossRef CAS PubMed.
- R. Behera, S. M. Thomas and K. Mensa-Wilmot, Antimicrob. Agents Chemother., 2014, 58, 2202–2210 CrossRef PubMed.
- P. S. Shirude, P. Madhavapeddi, J. A. Tucker, K. Murugan, V. Patil, H. Basavarajappa, A. V. Raichurkar, V. Humnabadkar, S. Hussein, S. Sharma, V. K. Ramya, C. B. Narayan, T. S. Balganesh and V. K. Sambandamurthy, ACS Chem. Biol., 2013, 8, 519–523 CrossRef CAS PubMed.
- B. B. Das, T. Sengupta, A. Ganguly and H. K. Majumder, Mol. Microbiol., 2006, 62, 917–927 CrossRef CAS PubMed.
- A. Das, A. Dasgupta, T. Sengupta and H. K. Majumder, Trends Parasitol., 2004, 20, 381–387 CrossRef CAS PubMed.
- M. J. Almela, S. Lozano, J. Lelièvre, G. Colmenarejo, J. M. Coterón, J. Rodrigues, C. Gonzalez and E. Herreros, PLoS One, 2015, 10, 1–18 CrossRef PubMed.
- Y. Younis, F. Douelle, T. S. Feng, D. G. Cabrera, C. Le Manach, A. T. Nchinda, S. Duffy, K. L. White, D. M. Shackleford, J. Morizzi, J. Mannila, K. Katneni, R. Bhamidipati, K. M. Zabiulla, J. T. Joseph, S. Bashyam, D. Waterson, M. J. Witty and D. Hardick, et al. , J. Med. Chem., 2012, 55, 3479–3487 CrossRef CAS PubMed.
- J. Okombo and K. Chibale, MedChemComm, 2018, 9, 437–453 RSC.
- T. Paquet, C. Le Manach, D. G. Cabrera, Y. Younis, P. P. Henrich, T. S. Abraham, M. C. S. Lee, R. Basak, S. Ghidelli-Disse, M. J. Lafuente-Monasterio, M. Bantscheff, A. Ruecker, A. M. Blagborough, S. E. Zakutansky, A. M. Zeeman, K. L. White, D. M. Shackleford, J. Mannila and J. Morizzi, et al. , Sci. Transl. Med., 2017, 9, eaad9735 CrossRef PubMed , 1–14.
- C. W. McNamara, M. C. S. Lee, C. S. Lim, S. H. Lim, J. Roland, A. Nagle, O. Simon, B. K. S. Yeung, A. K. Chatterjee, S. L. McCormack, M. J. Manary, A. M. Zeeman, K. J. Dechering, T. R. S. Kumar, P. P. Henrich, K. Gagaring, M. Ibanez, N. Kato and K. L. Kuhen, et al. , Nature, 2013, 504, 248–253 CrossRef CAS PubMed.
- M. J. Rodgers, J. P. Albanesi and M. A. Phillips, Eukaryotic Cell, 2007, 6, 1108–1118 CrossRef CAS PubMed.
- A. M. Gimenez, M. C. Gesumaría, A. C. Schoijet, G. D. Alonso, M. M. Flawiá, G. E. Racagni and E. E. Machado, Mol. Biochem. Parasitol., 2015, 203, 14–24 CrossRef CAS PubMed.
- C. Le Manach, A. T. Nchinda, T. Paquet, D. Gonzàlez Cabrera, Y. Younis, Z. Han, S. Bashyam, M. Zabiulla, D. Taylor, N. Lawrence, K. L. White, S. A. Charman, D. Waterson, M. J. Witty, S. Wittlin, M. E. Botha, S. H. Nondaba, J. Reader and L. M. Birkholtz, et al. , J. Med. Chem., 2016, 59, 9890–9905 CrossRef CAS PubMed.
- C. Brunschwig, N. Lawrence, D. Taylor, E. Abay, M. Njoroge, G. S. Basarab, C. Le Manach, T. Paquet, D. González Cabrera, A. T. Nchinda, C. de Kock, L. Wiesner, P. Denti, D. Waterson, B. Blasco, D. Leroy, M. J. Witty, C. Donini and J. Duffy, et al. , Antimicrob. Agents Chemother., 2018, 62, e00012 Search PubMed.
- R. Wilson, P. Kumar, V. Parashar, C. Vilchèze, R. Veyron-Churlet, J. S. Freundlich, S. W. Barnes, J. R. Walker, M. J. Szymonifka, E. Marchiano, S. Shenai, R. Colangeli, W. R. Jacobs, M. B. Neiditch, L. Kremer and D. Alland, Nat. Chem., 2013, 9, 499–506 CrossRef CAS PubMed.
- S. Thanna, S. E. Knudson, A. Grzegorzewicz, S. Kapil, S. J. Sucheck, C. M. Goins, D. R. Ronning, M. Jackson, R. A. Slayden and S. J. Sucheck, Org. Biomol. Chem., 2016, 14, 6119–6133 RSC.
- F.-J. Gamo, L. M. Sanz, J. Vidal, C. de Cozar, E. Alvarez, J.-L. Lavandera, D. E. Vanderwall, D. V. S. Green, V. Kumar, S. Hasan, J. R. Brown, C. E. Peishoff, L. R. Cardon and J. F. Garcia-Bustos, Nature, 2010, 465, 305–310 CrossRef CAS PubMed.
- M. Esposito, S. Szadocka, G. Degiacomi, B. S. Orena, G. Mori, V. Piano, F. Boldrin, J. Zemanová, S. Huszár, D. Barros, S. Ekins, J. Lelièvre, R. Manganelli, A. Mattevi, M. R. Pasca, G. Riccardi, L. Ballell, K. Mikušová and L. R. Chiarelli, ACS Infect. Dis., 2017, 3, 428–437 CrossRef CAS PubMed.
- C. Leija, F. Rijo-Ferreira, L. N. Kinch, N. V. Grishin, N. Nischan, J. J. Kohler, Z. Hu and M. A. Phillips, PLoS Pathog., 2016, 12, 1–30 Search PubMed.
- J. A. M. Ali, D. N. A. Tagoe, J. C. Munday, A. Donachie, L. J. Morrison and H. P. de Koning, PLoS One, 2013, 8, e58034 CrossRef CAS PubMed , 1–11.
- P. N. Black, C. Ahowesso, D. Montefusco, N. Saini and C. C. Dirusso, MedChemComm, 2016, 7, 612–622 RSC.
- S. B. Falconer, S. A. Reid-Yu, A. M. King, S. S. Gehrke, W. Wang, J. F. Britten, B. K. Coombes, G. D. Wright and E. D. Brown, ACS Infect. Dis., 2016, 1, 533–543 CrossRef PubMed.
- H. Hirumi and K. Hirumi, J. Parasitol., 1989, 75, 985–989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00319j |
| This journal is © The Royal Society of Chemistry 2018 |