
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A synthetic diphosphoinositol phosphate analogue of inositol trisphosphate†
Andrew M.
Riley
 a,
Judith E.
Unterlass
b,
Vera
Konieczny
c,
Colin W.
Taylor
c,
Thomas
Helleday
b and
Barry V. L.
Potter
*a
a,
Judith E.
Unterlass
b,
Vera
Konieczny
c,
Colin W.
Taylor
c,
Thomas
Helleday
b and
Barry V. L.
Potter
*a
aMedicinal Chemistry and Drug Discovery, Department of Pharmacology, University of Oxford, Mansfield Road, Oxford OX1 3QT, UK. E-mail: barry.potter@pharm.ox.ac.uk; Fax: +44 (0)1865 271853; Tel: +44 (0)1865 271945
bScience for Life Laboratory, Department of Oncology-Pathology, Karolinska Institutet, SE-171 21 Solna, Sweden
cDepartment of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge CB2 1PD, UK
First published on 4th June 2018
Abstract
Diphosphoinositol phosphates (PP-InsPs) are inositol phosphates (InsPs) that contain PP (diphosphate) groups. Converting a phosphate group in an InsP into a diphosphate has been reported to enhance affinity for some binding proteins. We synthesised 1-PP-Ins(4,5)P2, the first diphosphate analogue of the intracellular signalling molecule InsP3, and examined its effects on InsP3 receptors, which are intracellular Ca2+ channels. 1-PP-Ins(4,5)P2 was indistinguishable from InsP3 in its ability to bind to and activate type 1 InsP3 receptors, indicating that the diphosphate modification of InsP3 affected neither affinity nor efficacy. Nevertheless, 1-PP-Ins(4,5)P2 is the most potent 1-phosphate modified analogue of InsP3 yet identified. PP-InsPs are generally hydrolysed by diphosphoinositol polyphosphate phosphohydrolases (DIPPs), but 1-PP-Ins(4,5)P2 was not readily metabolised by human DIPPs. Differential scanning fluorimetry showed that 1-PP-Ins(4,5)P2 stabilises DIPP proteins, but to a lesser extent than naturally occurring substrates 1-PP-InsP5 and 5-PP-InsP5. The non-hydrolysable InsP7 analogues 1-PCP-InsP5 and 5-PCP-InsP5 showed comparable stabilising abilities to their natural counterparts and may therefore be promising substrate analogues for co-crystallisation with DIPPs.
Introduction
The myo-inositol phosphates (InsPs) are a family of intracellular signalling molecules containing monophosphate (P) and diphosphate (PP) groups arranged around the hexahydroxycyclohexane ring of myo-inositol (Ins).1 InsPs regulate many cellular processes, the best known being the release of Ca2+ from intracellular stores by D-myo-inositol 1,4,5-trisphosphate (InsP3), which binds to receptors on the endoplasmic reticulum.2 InsP3 is converted via a series of enzymatic phosphorylations3 into InsP6 (Fig. 1), which can then be further phosphorylated to give highly charged PP-InsPs containing diphosphate (pyrophosphate) groups.4,5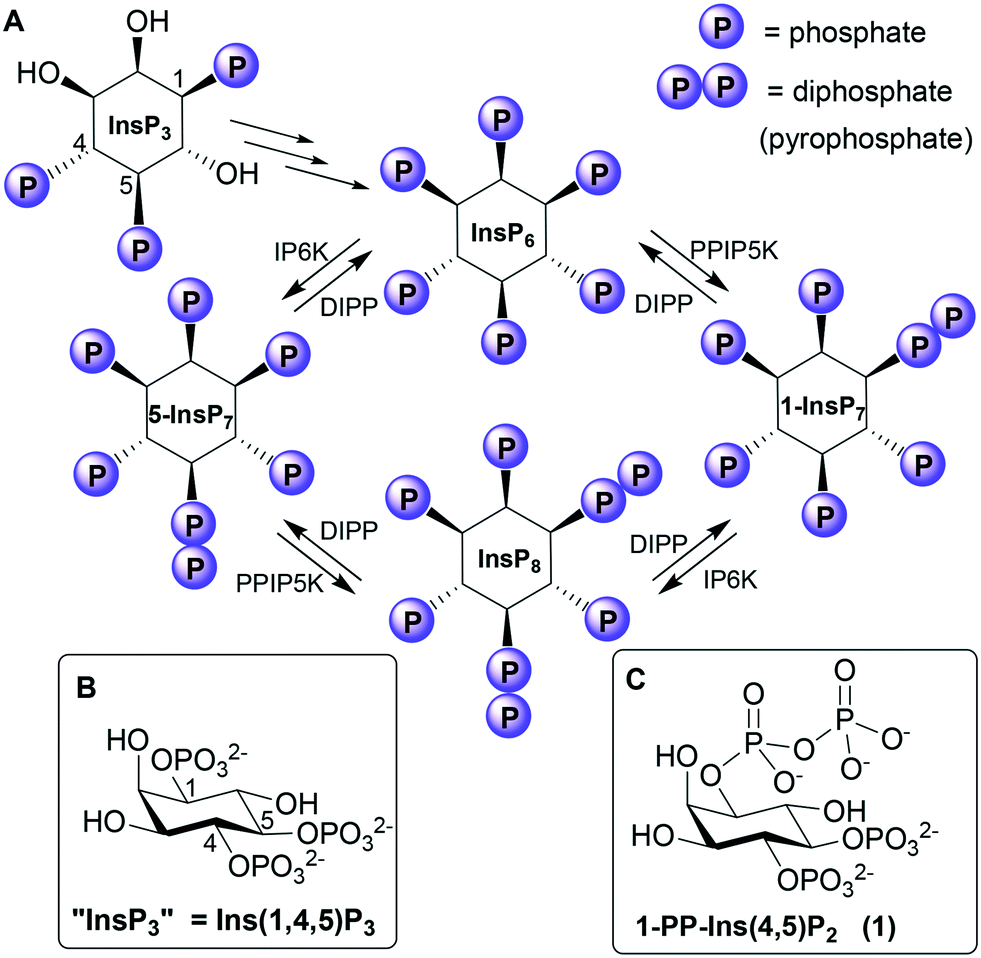 | ||
| Fig. 1 A. Biosynthetic pathway connecting Ins(1,4,5)P3 to the PP-InsPs. IP6K, inositol hexakisphosphate 5-kinase; PPIP5K, diphosphoinositol pentakisphosphate kinase; DIPP, diphosphoinositol polyphosphate phosphohydrolase. “InsP3”, “5-InsP7”, “1-InsP7” and “InsP8” are alternative names for Ins(1,4,5)P3, 5-PP-InsP5, 1-PP-InsP5 and 1,5-[PP]2-InsP4, respectively. B. Structure of Ins(1,4,5)P3. C. Structure of the synthetic analogue 1-PP-Ins(4,5)P2 (1). | ||
InsP3 receptors (IP3Rs) are tetrameric intracellular Ca2+ channels, expressed in most animal cells.2 When InsP3 binds to the N-terminal InsP3-binding core (IBC) of all four IP3R subunits,6 conformational changes propagate to the central pore. The pore then opens, allowing Ca2+ to flow into the cytosol, where it regulates many intracellular processes. The vicinal 4,5-bisphosphate structure of InsP3 is crucial (if not absolutely essential7) for activating IP3Rs because it cross-links the two domains of the clam-like IBC, pulling them together and initiating the conformational changes. The 1-phosphate group has a less direct, but enhancing, effect on activity.8
Although PP-InsP signalling is thought to be more evolutionarily ancient than InsP3-mediated mobilisation of Ca2+,9 much less is known about the functions and protein targets of PP-InsPs. Nevertheless, evidence is accumulating that PP-InsPs play important roles at the interface of cell signalling and metabolism in the regulation of bioenergetic and phosphate homeostasis.4,5 Possible receptors for PP-InsPs include the PH (pleckstrin homology) domains10,11 and SPX (SYG1/Pho81/XPR1) domains12,13 of proteins. PP-InsPs may also exert some of their effects by direct non-enzymatic diphosphorylation of target proteins.14
Phosphorylating a phosphate monoester in an InsPn to give a PP-InsPn−1 not only increases the overall negative charge of the molecule, but also changes its shape, solvation and metal complexation properties. Unsurprisingly, therefore, a diphosphate group may alter ligand affinity for protein binding sites.4 For example, some PH domains that bind InsP6 bind 5-InsP7 with higher affinity,10,11 while 1-InsP7 and InsP8 are weaker.11 In contrast, both 1-InsP7 and 5-InsP7 stimulate synthesis of inorganic polyphosphate (polyP) by the vacuolar transporter chaperone (VTC) by binding to its SPX domain, while InsP6 is inactive and InsP8 is 20-fold more potent.13 PP-InsPs can be dephosphorylated back to InsPs by diphosphoinositol polyphosphate phosphohydrolases (DIPPs, Fig. 1), which specifically hydrolyse the diphosphate group, leaving a phosphate monoester and liberating inorganic phosphate.3,15
Given that introducing a diphosphate into an InsP may modify its interaction with proteins, we were interested in the possible effects of converting one of the phosphate groups in InsP3 into a diphosphate. The 1-phosphate group of InsP3 has been a popular target for synthetic elaboration of InsP3 since early structure–activity studies showed that it is much more tolerant of modification than the 4- or 5-phosphate groups.8 Interest in the role of the 1-phosphate group was further stimulated by the discovery in 1993 of the adenophostins, fungal metabolites that are highly potent InsP3 receptor ligands.16 The adenophostins contain a glucopyranoside 3,4-bisphosphate structure that mimics the myo-inositol 4,5-bisphosphate of InsP3 but intriguingly, their third phosphate group is located on a separate (ribofuranoside) ring, suggesting that repositioning this phosphate group may enhance affinity.17
The X-ray structure18 of the IBC of type 1 InsP3 receptor bound to InsP3 confirmed the area of the binding pocket around the 1-phosphate of bound InsP3 to be relatively open. Our molecular docking experiments using this structure suggested that a 1-diphosphate should bind well to this region. We therefore set out to synthesise the 1-diphosphate analogue of InsP3, i.e. 1D-diphospho-myo-inositol 4,5-bisphosphate [1-PP-Ins(4,5)P2 (1), Fig. 1] and examine its interaction with InsP3 receptors.
We were also interested to examine the interaction of 1-PP-Ins(4,5)P2 with DIPPs. Although DIPPs can hydrolyse the PP groups of highly phosphorylated PP-InsPs (Fig. 1), inorganic polyphosphate, 5-phosphoribosyl 1-pyrophosphate and nucleotide dimers,3,15 their catalytic mechanisms are poorly understood. 1-PP-Ins(4,5)P2 contains the target 1-PP structure found in the known DIPP substrate 1-PP-InsP5, but presented in the context of a molecule with only two phosphate monoester groups. There are no reports in the literature on whether “lower” PP-InsPs such as 1-PP-Ins(4,5)P2 could be recognised by the active sites of DIPPs.
Results and discussion
Chemistry
The synthesis of 1-PP-Ins(4,5)P2 (1) begins from the known alcohol 2 (ref. 19 and 20) (Scheme 1). To construct the diphosphate unit at O-1, we employed a modification of a recently described strategy,21,22 in which a temporarily protected phosphate group is introduced and then selectively deprotected to reveal a phosphate monoester. This phosphate is then phosphitylated to give a mixed P(III)–P(V) anhydride, which is oxidised to a partially protected pyrophosphate unit. Removal of all protecting groups by catalytic hydrogenolysis then yields the target PP-InsP. We reasoned that it might be possible to employ methylsulfonylethyl (MSE)23,24 as a temporary phosphate protecting group in this sequence. The MSE group can be removed by β-elimination, similar to the better-known β-cyanoethyl (β-CE)22,25 group. However, the MSE group is unaffected by catalytic hydrogenation, affording greater synthetic versatility, and the required phosphitylating reagent, phosphoramidite 5, is a stable crystalline solid.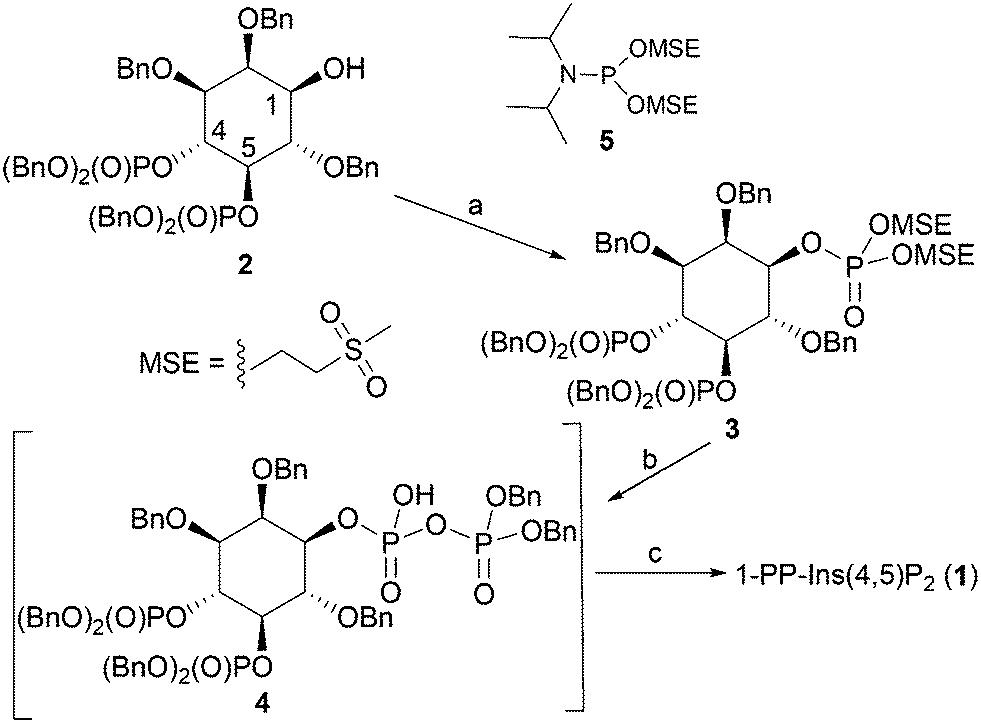 | ||
| Scheme 1 Synthesis of 1-PP-Ins(4,5)P2 (1). Reagents and conditions: a. i. 5-phenyl-1H-tetrazole, 5, CH2Cl2; ii. mCPBA, CH2Cl2, 89%; b. i. DBU, BSTFA, CDCl3; ii. MeOH, then TFA; iii. 5-phenyl-1H-tetrazole, (BnO)2PNPri2, CH2Cl2; iv. mCPBA, CH2Cl2, 90%; c. i. H2, Pd(OH)2/C, MeOH, H2O, NaHCO3; ii. Ion-exchange chromatography on Q-Sepharose Fast Flow resin, 57%. Bn, benzyl. | ||
Thus, the 1-OH group in 2 was reacted with 5 in the presence of 5-phenyl-1H-tetrazole to give an intermediate MSE-protected phosphite triester. Oxidation using mCPBA then gave 3, containing the MSE-protected phosphate triester at O-1. The diphosphate unit at O-1 was then constructed using a sequence of transformations carried out as described previously,21,22,25 with slight modifications. The progress of each step was carefully monitored by 31P NMR spectroscopy (see Experimental section and ESI†). The protected diphosphate 4 was found to be rather unstable and was immediately deprotected by catalytic hydrogenolysis at atmospheric pressure. A final purification step by gradient elution anion exchange chromatography on Q-Sepharose Fast Flow resin gave 1-PP-Ins(4,5)P2 (1) as the triethylammonium salt, which was accurately quantified by total phosphate assay.
Interactions of 1-PP-Ins(4,5)P2 with type 1 InsP3 receptors
Both InsP3 and 1-PP-Ins(4,5)P2 (1) stimulated a concentration-dependent release of Ca2+ from the intracellular stores of permeabilised DT40 cells expressing type 1 InsP3 receptors (Fig. 2A). The maximal Ca2+ release evoked by each ligand and the half-maximally effective concentration (EC50) were similar for 1 and InsP3 (Fig. 2A). Membranes from Sf9 cells expressing rat type 1 InsP3 receptors were used for equilibrium competition binding studies with 3H-InsP3, because these membranes express full-length type 1 InsP3 receptors at ∼20-fold higher levels than cerebellar membranes, the richest endogenous source. The experiments were carried out in cytosol-like medium (CLM, pH 7.3) containing 1.5 mM Mg-ATP to match the conditions used for Ca2+-release assays.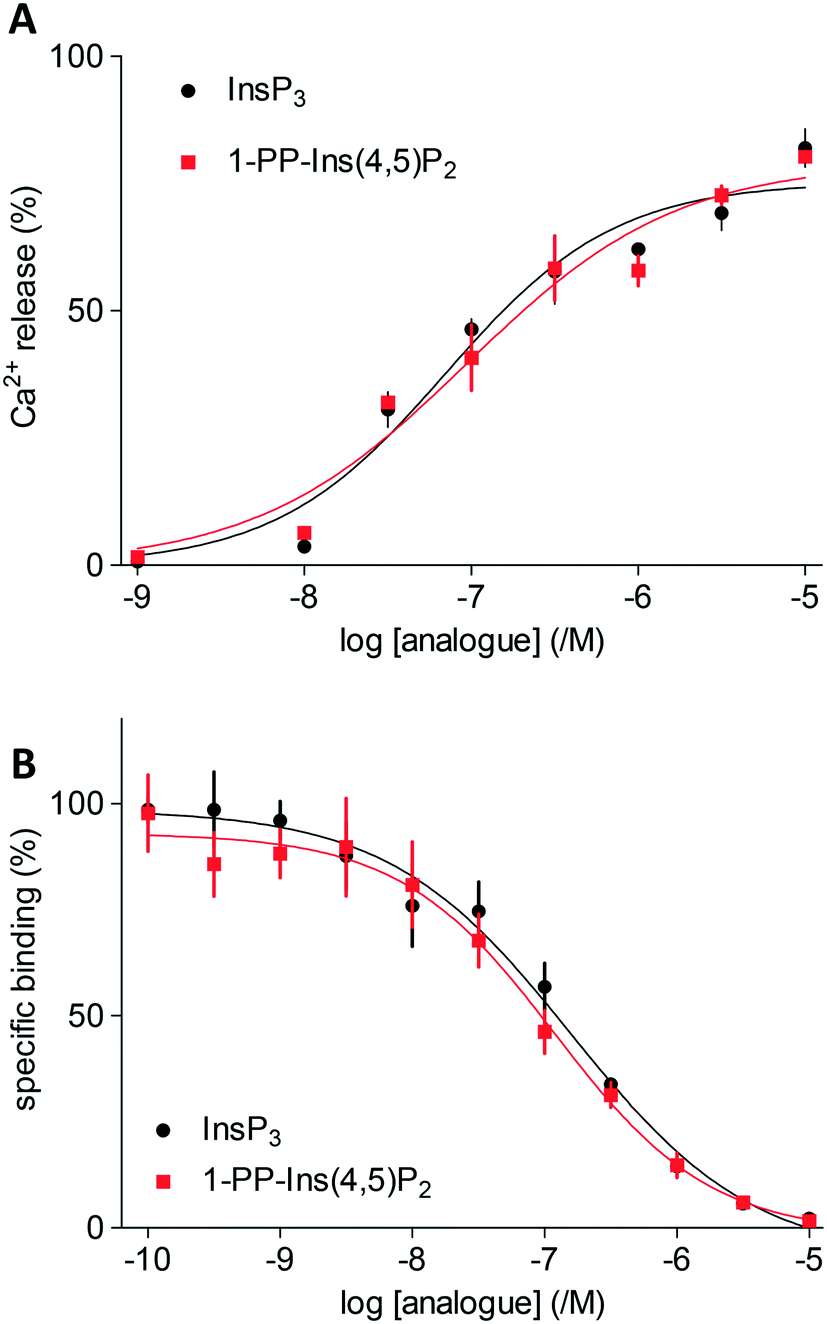 | ||
| Fig. 2 A. Ca2+ release from intracellular stores of DT40 cells expressing type 1 InsP3 receptors stimulated by InsP3 and 1-PP-Ins(4,5)P2 (1). Results are shown as % of Ca2+ content of intracellular stores. B. Equilibrium competition binding with 3H-InsP3 and InsP3 or 1-PP-Ins(4,5)P2 (1) using membranes from Sf9-IP3R1 cells in CLM containing 1.5 mM MgATP. Results are means ± s.e.m., n = 3. | ||
In agreement with the Ca2+-release assays, 1-PP-Ins(4,5)P2 (1) bound with the same affinity as InsP3 to InsP3 receptors (Fig. 2B). Thus, the two compounds were essentially indistinguishable in both functional and binding assays (Table 1). Rapid chemical hydrolysis of 1 could in principle explain the similar behaviour of InsP3 and 1, but we saw no evidence that 1 is unstable. The 31P NMR spectrum of 1 in D2O (see ESI†) was unchanged after the sample solution had been kept for several days at room temperature, followed by one year at 4 °C.
| Ca2+ release | Bindinga | ||||
|---|---|---|---|---|---|
| pEC50/M | EC50/nM | % release | pKd/M | K d/nM | |
| a Binding was done using Sf9 cell membranes overexpressing rat type 1 InsP3 receptors in CLM (pH 7.3) containing 1.5 mM Mg-ATP to match the conditions used in the Ca2+ release assay. | |||||
| InsP3 | 7.21 ± 0.08 | 62 | 82 ± 4 | 6.89 ± 0.07 | 128 |
| 1-PP-Ins(4,5)P2 (1) | 7.17 ± 0.11 | 68 | 80 ± 1 | 6.96 ± 0.05 | 110 |
Molecular docking experiments (see Experimental section and ESI† for details) using the X-ray crystal structure of the IBC of type 1 InsP3 receptor18 suggested that the diphosphate group in 1 should be well-tolerated by the InsP3-binding pocket and may be capable of forming additional hydrogen bonds with residues in the binding site (Fig. 3). Nevertheless, it is well known that attempts to optimise drug candidates by adding polar groups may fail because the expected enthalpic gains from new polar interactions are opposed by ligand desolvation penalties and unfavourable entropic effects, resulting in no gain in binding affinity.26 Such compensatory effects may underlie the similar affinities of 1 and InsP3 for type 1 InsP3 receptors.
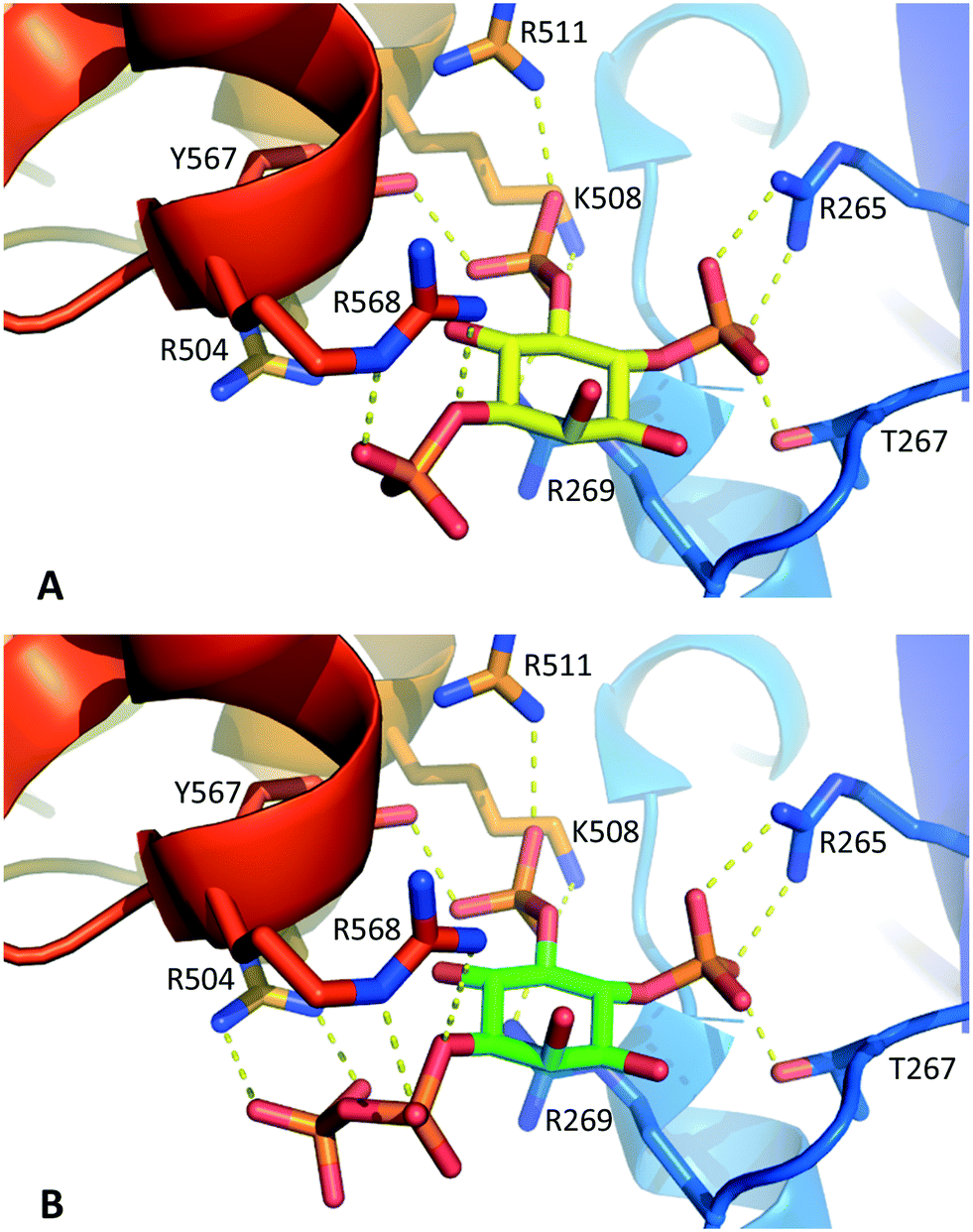 | ||
| Fig. 3 A. Interactions of InsP3 with the IBC of type 1 InsP3 receptors, based on the X-ray crystal structure of IP3R1 in complex with InsP3 (ref. 18) (1N4K). B. Model of 1-PP-Ins(4,5)P2 (1) in the IBC produced by molecular docking (see Experimental section and ESI† for details). For clarity, water molecules are not shown. | ||
Interaction of 1-PP-Ins(4,5)P2 with DIPPs
The dephosphorylation of PP-InsPs is catalysed by diphosphoinositol polyphosphate phosphohydrolases (DIPPs), which selectively cleave the diphosphate (PP) to give a phosphate monoester and inorganic phosphate (Pi).3 Humans express four DIPP types: DIPP-1 is the product of the NUDT3 gene; DIPP-2 (of which there are two isoforms, DIPP-2α and DIPP-2β, produced by alternative splicing) is the product of NUDT4; DIPP-3α is the product of NUDT10 and DIPP-3β is the product of NUDT11.3 We examined the interaction of 1-PP-Ins(4,5)P2 (1) with all four DIPPs in comparison with two naturally-occurring substrates 1-PP-InsP5 and 5-PP-InsP5 (“1-InsP7” and “5-InsP7”, respectively) and also with the alternative substrates diadenosine polyphosphates Ap3A and Ap5A. Non-hydrolysable InsP7 analogues 1-PCP-InsP5 (ref. 27) and 5-PCP-InsP5 (ref. 28) were independently synthesised and included as controls.With Mg2+ present in the buffer, 1-PP-InsP5 and 5-PP-InsP5 were rapidly metabolised by all four DIPPs (Fig. 4A). The rate of hydrolysis of 1-PP-InsP5 was significantly higher than that for 5-PP-InsP5 in each case. This finding is in agreement with a previous study.15 As expected, the PCP analogues were not metabolised, confirming that DIPPs can hydrolyse only the diphosphate unit and not the phosphate monoesters. Ap3A and Ap5A were unaffected by all four enzymes in Mg2+-containing buffer, an observation that had been reported for NUDT10 and NUDT11, but not for NUDT3 and NUDT4.29 Perhaps surprisingly, 1-PP-Ins(4,5)P2 (1) was also not metabolised under these conditions. The presence of a divalent cation is required for the activity of NUDT10 and NUDT11 and also for NUDT3.3 When Mg2+ in the buffer was replaced by Mn2+, 1 was now hydrolysed by the DIPPs, while 1-PP-InsP5 and 5-PP-InsP5 resisted hydrolysis. In addition, Ap5A now also behaved as a substrate for all four DIPPs (Fig. 4B). In the absence of enzyme none of the compounds, including 1, showed any sign of hydrolysis during the time course of the experiment in the presence of either Mg2+ or Mn2+-containing buffers. This further supports our conclusion above that 1 was not hydrolysed to InsP3 during the InsP3 receptor assays.
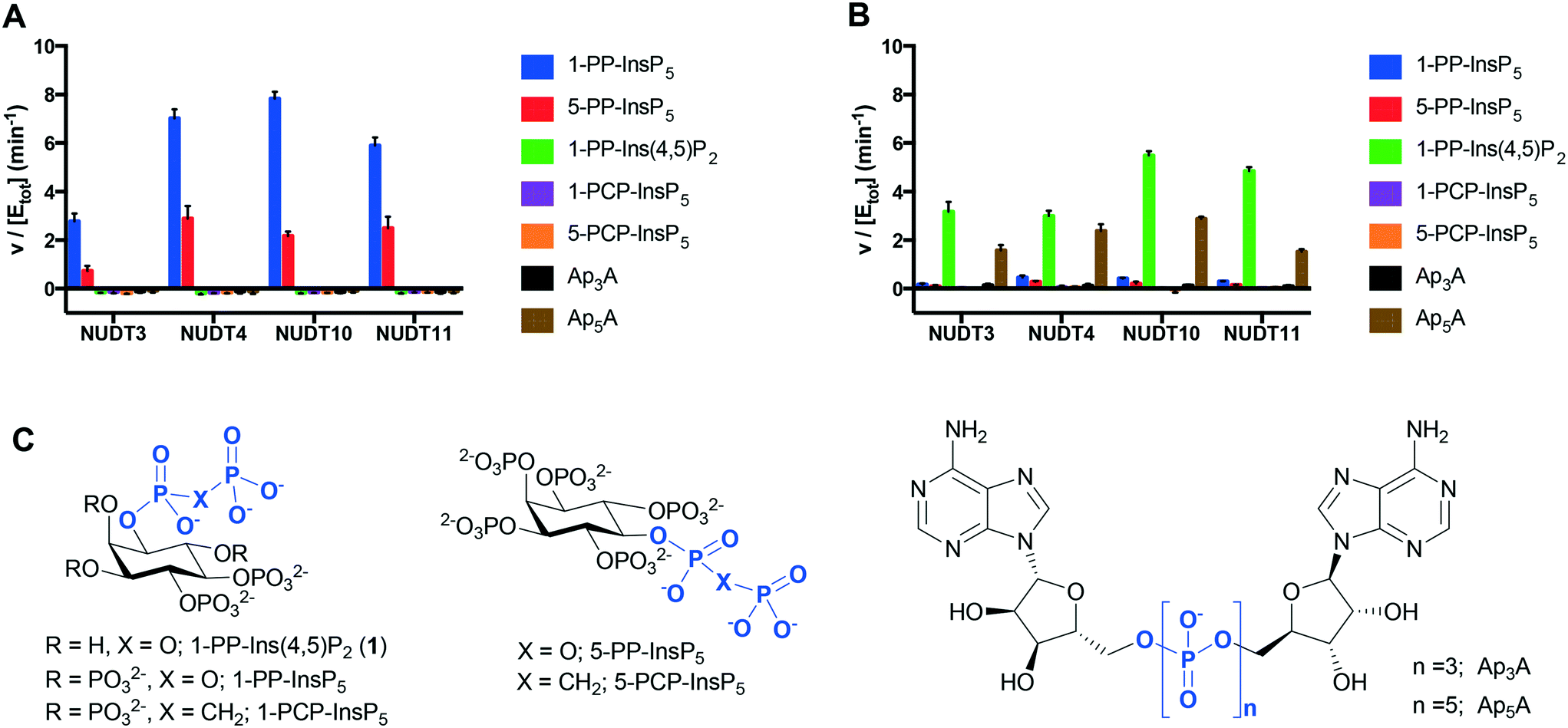 | ||
| Fig. 4 Specific activities of DIPPs with 1-PP-Ins(4,5)P2 (1), known substrates 1-PP-InsP5 and 5-PP-InsP5 and controls. Experiments were conducted in buffer containing Mg2+ (A) or Mn2+ (B). Data shown represent the formed concentration of Pi (micromolar) per enzyme concentration (micromolar) per minute. A630 was converted to Pi concentration (micromolar) using the equation A630 = 0.01897[Pi] − 0.5877 (Mg2+ containing buffer) or A630 = 0.01923[Pi] + 0.1053 (Mn2+ containing buffer). C. Structures of compounds examined, including methylenebisphosphonate (PCP) analogues of InsP7, and diadenosine polyphosphates Ap3A and Ap5A. | ||
Next, we used differential scanning fluorimetry (DSF) to measure the ability of the compounds to stabilise NUDT3 (DIPP1). While the effects of Ap3A and Ap5A were not significantly different from control (Fig. 5A), 1-PP-Ins(4,5)P2 (1) raised the melting temperature (Tm) of NUDT3 by approx. 5 °C at a concentration of 0.1 mM. As expected, the more highly phosphorylated 1-PP-InsP5 had much stronger effects, resulting in a Tm-shift of 20–25 °C. Similar DSF experiments were then carried out for NUDT4, NUDT10 and NUDT11. Ap3A did not stabilise any of the DIPPs, which supports our results for the activity assay. The results are summarised in Fig. 5B.
 | ||
| Fig. 5 A. Effect of 1-PP-Ins(4,5)P2 (1) and other compounds shown in Fig. 4C on the melting temperature (Tm) of NUDT3, measured using differential scanning fluorimetry (DSF). B. Comparison of melting temperature shifts (delta Tm) induced by all compounds for all four DIPPs examined. | ||
We obtained further DSF data over a range of ligand concentrations for 1-PP-InsP5 and 1-PP-Ins(4,5)P2 (1), constructing dose–response curves for the two compounds (Fig. 6). It is interesting to note that the effect of 1 on NUDT10 was significantly lower compared to the other DIPPs and especially compared to NUDT11 (Fig. 6B). NUDT10 and NUDT11 have identical protein sequences apart from residue 89, which is either proline (NUDT10) or arginine (NUDT11).
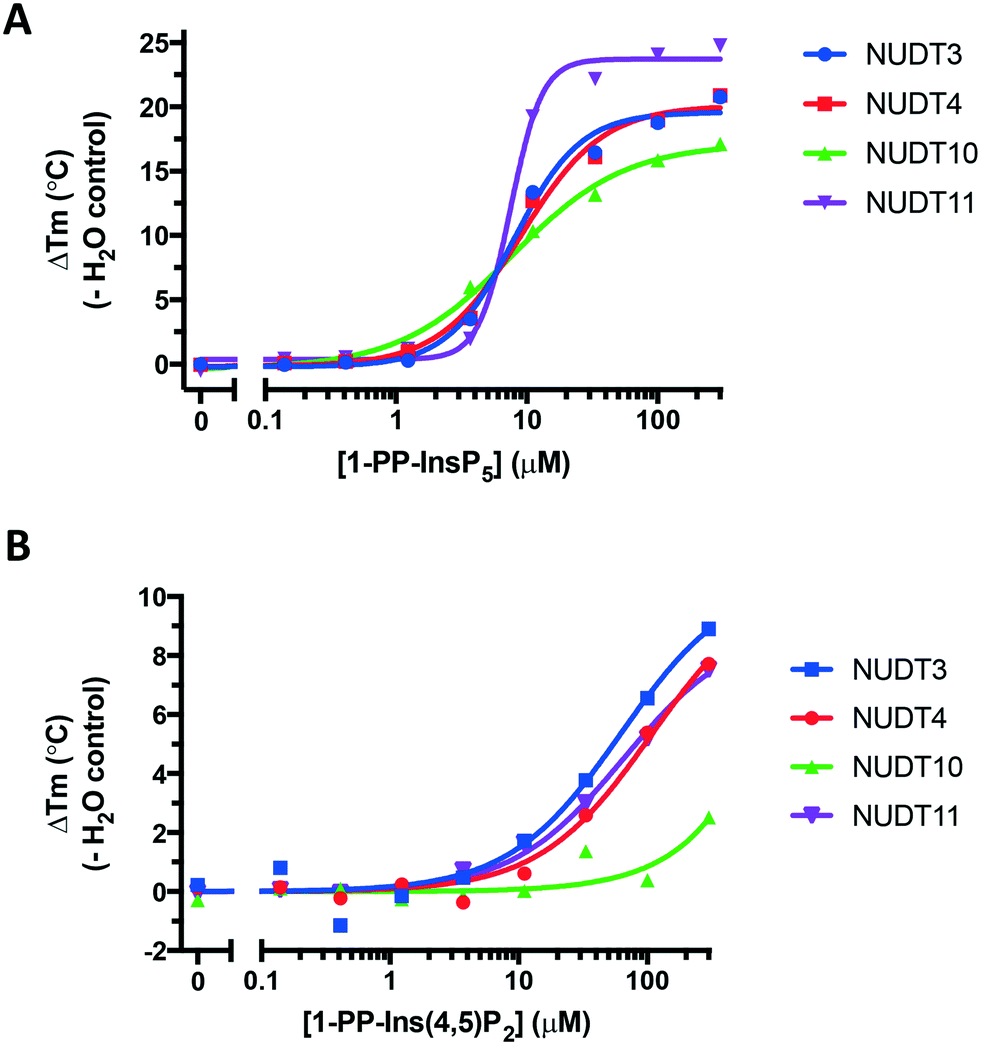 | ||
| Fig. 6 Dose–response curves showing stabilisation of all four DIPPs by A. 1-PP-InsP5 and B. 1-PP-Ins(4,5)P2 (1). Note the different y-axis scales in A and B. | ||
Noting the strong stabilisation of all the proteins by the PCP analogues, we obtained further DSF data over a range of ligand concentrations for 1-PCP-InsP5 and 5-PCP-InsP5 (ESI† Fig. S4 and S5) and calculated KD values from these curves (ESI† Tables S1 and S2). We found that, in some cases, the PCP analogues had binding affinities comparable to those of their natural PP-containing ligands.
Conclusions
Replacing a phosphate group in an inositol phosphate ligand with a diphosphate (PP) group can modify the interaction of the ligand with target proteins.10–13 Structure–activity studies have previously shown that the 1-phosphate group of InsP3 is amenable to synthetic modification, and molecular docking experiments suggested that a 1-diphosphate group should be well-tolerated by the binding site of the InsP3 receptor. We therefore synthesised 1-PP-Ins(4,5)P2 (1), the first PP-containing analogue of InsP3. Using assays of Ca2+-release through type 1 InsP3 receptors, we found that 1 was equipotent to InsP3 and in binding assays its affinity was indistinguishable from that of InsP3. Thus, the 1-diphosphate modification of InsP3 does not affect its affinity for or activity at type 1 InsP3 receptors. Nevertheless, 1 is the first Ca2+-releasing PP-InsP and also the most potent P-1 modified ligand of InsP3 receptors yet identified.‡The novel diphosphate compound 1 was not metabolised by DIPPs in the presence of Mg2+-containing buffer, while the naturally-occurring InsP7 isomers, 5-PP-InsP5 and 1-PP-InsP5 were rapidly hydrolysed. Conversely, in the presence of Mn2+, 1 was hydrolysed while the two InsP7 isomers were unaffected. Synthetic PCP-containing analogues of the InsP7s were not hydrolysed under any conditions examined, but when evaluated for their ability to stabilise DIPP proteins using differential scanning fluorimetry (DSF), they gave temperature shifts comparable to their natural PP-containing equivalents. This strongly suggests that 1-PCP-InsP5 and 5-PCP-InsP5 could be promising ligands for co-crystallisation studies with DIPPs.
Could 1-PP-Ins(4,5)P2 be an endogenous molecule? The mammalian enzymes known to synthesise PP-InsPs are 5-diphosphoinositol pentakisphosphate kinases (PPIP5Ks) and inositol hexakisphosphate kinases (IP6Ks). Inositol phosphate multikinase (IPMK) has also been reported to synthesise PP-InsP4 from InsP5in vitro,30 but the products of InsP3 phosphorylation by IPMK are Ins(1,3,4,5)P4 and/or Ins(1,4,5,6)P4.31 Phosphorylation of lower InsPs by PPIP5Ks seems unlikely, considering the constraints of the catalytic site32 and the recently discovered capture site;22 even Ins(1,3,4,5,6)P5 is not phosphorylated.32 Recombinant Kcs1p, a yeast homologue of IP6K1, was reported to phosphorylate InsP3 slowly, although the identities of the products could not be determined.33 Later work confirmed that InsP3 was phosphorylated by Kcs1 and the product was identified as Ins(1,3,4,5)P4 (i.e. in this case, Kcs1 functioned as a 3-kinase).34 More recently, a study found that EhIP6KA, an IP6K homologue from Entamoeba histolytica, was capable of slowly phosphorylating InsP3, although the products were identified as Ins(1,4,5,6)P4 and Ins(1,2,4,5)P4.35 On this basis, naturally occurring 1-PP-Ins(4,5)P2 seems unlikely. However, in both studies where the identities of the enzyme products were assigned,34,35 resistance to hydrolysis by DIPP1 was used to exclude the possibility that the products contained diphosphate groups. The present work shows that this criterion may not always be valid; in our hands, 1-PP-Ins(4,5)P2 was not metabolised in the presence of Mg2+ by any of the DIPPs, yet it does contain a diphosphate group.
Notwithstanding the evidence for PP-InsPs playing physiological roles,4,5 the present work indicates that a physiological function for 1-PP-Ins(4,5)P2, at least in relation to the regulation of InsP3 receptor-mediated Ca2+ release, may be unlikely. Converting the 1-phosphate of InsP3 into a diphosphate neither attenuates nor enhances the ability of the ligand to activate InsP3R. As the first example of a diphosphate analogue of a second messenger, however, the results add a new component to structure–activity relationships. Co-crystallisation studies with DIPPs using some of the non-hydrolysable substrate analogues discussed here are currently in progress.
Experimental
General chemistry methods
General methods were as previously reported.36 Alcohol 2 = 1D-2,3,6-tri-O-benzyl-myo-inositol 4,5-bis-O-(dibenzylphosphate) was synthesised according to the literature19 and crystallised from diethyl ether/light petroleum; m.p. 90–91 °C; Lit.19 90–91 °C; [α]20D = −18.2, (c = 2, CHCl3), Lit.19 [α]25D = −15.6, (c = 1, CHCl3); Lit.20 [α]20D = −17.8, (c = 1.7, CHCl3). N,N-Diisopropylamino-bis-[2-(methylsulfonyl)ethyloxy]-phosphine (5) was synthesised according to the literature24 and recrystallized from dichloromethane/ether; m.p. 75.5–77.0 °C; 1H NMR (CDCl3, 400 MHz) δ 1.20 (12 H, d, 3JHP 6.8 Hz, 4 × CHCH3), 3.01 (6 H, s, 2 × SCH3), 3.22–3.34 (4 H, m, 2 × OCH2CH2S), 3.59 (2 H, dh, 3JHP 10.4 Hz, 3JHH 6.8 Hz, 2 × CHCH3), 4.01–4.15 (4 H, m, 2 × OCH2CH2S); 13C NMR (CDCl3, 101 MHz) δ 24.62 (3JCP 7.3 Hz, 4 × CHCH3), 42.85 (2 × SCH3), 43.31 (2JCP 12.4 Hz, 2 × CHCH3), 56.17 (3JCP 8.3 Hz, 2 × OCH2CH2S), 57.58 (2JCP 20.0 Hz, 2 × OCH2CH2S); 31P NMR (CDCl3, 162 MHz, 1H-decoupled) δ 148.98; HRMS (m/z) [M + H]+ calcd. for C12H28O6NPS2; 378.11684; found 378.11687. 5-PP-InsP5, 1-PP-InsP5 and their PCP analogues were synthesised using similar methods to those previously described.21,22,27,28,36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 1:1) showed total conversion of 2 (Rf 0.56) into a more polar product (Rf 0.24). The mixture was then cooled to −78 °C, before mCPBA (70%, 100 mg, 0.406 mmol) was added. The mixture was allowed to warm to room temperature and then diluted with EtOAc (30 mL). The clear, colourless solution was washed with 10% aq. Na2SO3 solution (2 × 30 mL) and 1.0 mold per m3 HCl (30 mL), then dried over MgSO4 and concentrated. The residue was purified by flash chromatography on silica, eluting with methanol in ethyl acetate (0 to 15%) to give 3 as a colourless oil (225 mg, 0.178 mmole, 89%); TLC (ethyl acetate:methanol 10:1): Rf = 0.50; [α]20D = −10.3, (c = 1.4, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 2.76 (3 H, s, SCH3), 2.82 (3 H, s, SCH3), 2.79–2.87 (1 H, m, OCH2CHCHS), 2.92–3.00 (1 H, m, OCH2CHCHS), 3.03–3.13 (2 H, m, 2 × OCH2CHCHS), 3.62 (1 H, dd, J 9.8, 1.9 Hz, H-3), 4.10 (1 H, dd, J 9.5, 9.5 Hz, H-6), 4.07–4.40 (6 H, m, H-1, H-2 and 2 × OCH2CH2CHS), 4.58–4.73 (6 H, m, H-5 and 2.5 AB systems of OCH2Ph), 4.82–5.03 (9 H, m, H-4 and 4 × OCH2Ph), 5.09, 5.11 (1 H, 2JAB 11.9 Hz, 3JHP 7.0 Hz, 0.5 ABX system of POCH2Ph), 6.95–6.97 (2 H, m, Ph), 7.09–7.40 (33 H, m, Ph); 13C NMR (CDCl3, 100 MHz) δ 42.34 (2 × CH3), 54.03 (3JCP 7.7 Hz, POCH2CH2S), 54.20 (3JCP 7.7 Hz, POCH2CH2S), 61.16–61.24 (overlapping signals with 2JCP couplings, POCH2CH2S), 69.14–69.55 (overlapping signals with 2JCP couplings, POCH2Ph), 72.64 (OCH2Ph), 74.75 (OCH2Ph), 75.08 (OCH2Ph), 75.19 (C-2), 77.78–78.12 (overlapping signals with JCP couplings, C-1, C-3, C-4 and C-6), 78.81 (C-5), 127.32–128.36 (CH of Ph), 135.51 (3JCP 7.4 Hz, ipso-C of POCH2Ph), 135.98–136.06 (overlapping signals with 3JCP couplings, 3 × ipso-C of POCH2Ph), 137.53 (ipso-C of OCH2Ph), 138.13 (ipso-C of OCH2Ph), 138.19 (ipso-C of OCH2Ph); 31P NMR (CDCl3, 162 MHz) δ −3.37 (1 P), −1.94 (1 P), −1.59 (1 P); HRMS (m/z) [M + Na]+ calcd. for C61H69O19P3S2; 1285.2980; found 1285.3011.
:methanol 10:1): Rf = 0.30; 31P NMR (CDCl3, 162 MHz, 1H-decoupled) δ −1.75 (1 P, s), −2.35 (1 P, s), −11.08 (1 P, broad s, P-1β), −12.32 (1 P, broad s, P-1α); HRMS (m/z) [M + Na]+ calcd. for C69H70O18P4; 1333.3405; found 1333.3377. In earlier trials, this material had been found to be unstable after flash chromatography; a portion of it was therefore deprotected immediately as follows.
:ethyl acetate 1:1) showed total conversion of 2 (Rf 0.56) into a more polar product (Rf 0.24). The mixture was then cooled to −78 °C, before mCPBA (70%, 100 mg, 0.406 mmol) was added. The mixture was allowed to warm to room temperature and then diluted with EtOAc (30 mL). The clear, colourless solution was washed with 10% aq. Na2SO3 solution (2 × 30 mL) and 1.0 mold per m3 HCl (30 mL), then dried over MgSO4 and concentrated. The residue was purified by flash chromatography on silica, eluting with methanol in ethyl acetate (0 to 15%) to give 3 as a colourless oil (225 mg, 0.178 mmole, 89%); TLC (ethyl acetate:methanol 10:1): Rf = 0.50; [α]20D = −10.3, (c = 1.4, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 2.76 (3 H, s, SCH3), 2.82 (3 H, s, SCH3), 2.79–2.87 (1 H, m, OCH2CHCHS), 2.92–3.00 (1 H, m, OCH2CHCHS), 3.03–3.13 (2 H, m, 2 × OCH2CHCHS), 3.62 (1 H, dd, J 9.8, 1.9 Hz, H-3), 4.10 (1 H, dd, J 9.5, 9.5 Hz, H-6), 4.07–4.40 (6 H, m, H-1, H-2 and 2 × OCH2CH2CHS), 4.58–4.73 (6 H, m, H-5 and 2.5 AB systems of OCH2Ph), 4.82–5.03 (9 H, m, H-4 and 4 × OCH2Ph), 5.09, 5.11 (1 H, 2JAB 11.9 Hz, 3JHP 7.0 Hz, 0.5 ABX system of POCH2Ph), 6.95–6.97 (2 H, m, Ph), 7.09–7.40 (33 H, m, Ph); 13C NMR (CDCl3, 100 MHz) δ 42.34 (2 × CH3), 54.03 (3JCP 7.7 Hz, POCH2CH2S), 54.20 (3JCP 7.7 Hz, POCH2CH2S), 61.16–61.24 (overlapping signals with 2JCP couplings, POCH2CH2S), 69.14–69.55 (overlapping signals with 2JCP couplings, POCH2Ph), 72.64 (OCH2Ph), 74.75 (OCH2Ph), 75.08 (OCH2Ph), 75.19 (C-2), 77.78–78.12 (overlapping signals with JCP couplings, C-1, C-3, C-4 and C-6), 78.81 (C-5), 127.32–128.36 (CH of Ph), 135.51 (3JCP 7.4 Hz, ipso-C of POCH2Ph), 135.98–136.06 (overlapping signals with 3JCP couplings, 3 × ipso-C of POCH2Ph), 137.53 (ipso-C of OCH2Ph), 138.13 (ipso-C of OCH2Ph), 138.19 (ipso-C of OCH2Ph); 31P NMR (CDCl3, 162 MHz) δ −3.37 (1 P), −1.94 (1 P), −1.59 (1 P); HRMS (m/z) [M + Na]+ calcd. for C61H69O19P3S2; 1285.2980; found 1285.3011.
:methanol 10:1): Rf = 0.30; 31P NMR (CDCl3, 162 MHz, 1H-decoupled) δ −1.75 (1 P, s), −2.35 (1 P, s), −11.08 (1 P, broad s, P-1β), −12.32 (1 P, broad s, P-1α); HRMS (m/z) [M + Na]+ calcd. for C69H70O18P4; 1333.3405; found 1333.3377. In earlier trials, this material had been found to be unstable after flash chromatography; a portion of it was therefore deprotected immediately as follows.
Compound 4 (37 mg, 28 μmol) was dissolved in methanol (4 mL) and deionised water (1 mL). Powdered NaHCO3 (14 mg, 168 μmol) was added followed by Pd(OH)2/C (30 mg). The suspension was stirred vigorously under H2 (balloon) for 24 h, after which time more water (4 mL) was added. A fresh balloon of H2 was attached and stirring was continued for a further 72 h. The catalyst was then removed by filtration through a PTFE filter, giving a colourless solution, which was concentrated under reduced pressure to give a solid white residue. Analysis of this residue by 31P and 1H NMR in D2O showed that deprotection was complete. The residue was purified by anion-exchange chromatography on Q-Sepharose Fast Flow resin, eluting with a gradient of 0 to 1.5 M triethylammonium bicarbonate (TEAB). The target compound 1 eluted at 70 to 77% 1.5 M TEAB. Fractions containing the target were identified using the Briggs phosphate assay, combined and evaporated under reduced pressure. De-ionised water was repeatedly added and evaporated until the triethylammonium salt of 1 remained as a colourless glassy solid (14 mg, 16 μmol, 57%). This material was accurately quantified using total phosphate assay37 before biological evaluation. For 31P and 1H NMR analysis of 1, a small amount of EDTA (sodium salt, approx. 0.1 mg) was added to a sample of 1 (2.0 μmole in 0.4 mL D2O) to give sharper signals. This NMR sample containing EDTA was kept as the solution in D2O for >1 year at 4 °C with no sign of deterioration. 1H NMR (D2O, 500 MHz, EDTA added) δ 3.77 (1 H, dd, J 9.8, 2.9 Hz, H-3), 3.95 (1 H, t, J 9.6 Hz, H-6), 4.08 (1 H, apparent q, J 9.1 Hz, H-5), 4.15 (1 H, ddd, J 9.9, 8.3, 2.8 Hz, H-1), 4.32 (1 H, apparent q, J 9.4 Hz, H-4), 4.35 (1 H, apparent t, J 2.8 Hz, H-2); 13C NMR (D2O, 101 MHz) δ 70.15 and 70.31 (C-2 and C-3), 70.88 (C-6), 76.28 (2JCP 5.6 Hz, C-1), 76.82 (with JCP couplings, C-4) and 78.11 (with JCP couplings, C-5); 31P NMR (D2O, 202 MHz, EDTA added, 1H-decoupled) δ 1.11 (1 P), 0.45 (1 P), −10.48 (1 P, d, J 20.9 Hz, P-1β), −11.96 (d, J 20.9 Hz, P-1α); 31P NMR (D2O, 162 MHz, EDTA added, 1H-coupled) δ 1.13 (1 P, d, 3JHP 8.8 Hz), 0.47 (1 P, d, 3JHP 8.9 Hz), −10.46 (1 P, d, 2JPP 20.5 Hz, P-1β), −11.94 (1 P, dd, 2JPP 20.5, 3JHP 8.3 Hz, P-1α); HRMS (m/z) M− calcd. for C6H16O18P4; 498.9209; found 498.9214.
:10 in the assay mixture. The heat denaturation curves with a temperature increase of 1 °C min−1 from 25 °C to 95 °C were recorded on a CFX96 real-time PCR machine (Bio-Rad) by measuring the fluorescence of Sypro Orange with excitation and emission wavelengths of 470 and 570 nm, respectively. The Boltzmann equation was used to analyse the denaturation curves in GraphPad Prism. The determined melting temperature (Tm) is the inflection point of the sigmoidal denaturation curve.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
BVLP (grant 101010) and CWT (grant 101844) are Wellcome Trust Senior Investigators. We also acknowledge funding by the Swedish Pain Relief Foundation and the Swedish Cancer Society (TH). We thank Dr Stephen B. Shears for helpful discussions on the enzymes that are known to synthesise PP-InsPs.Notes and references
- M. P. Thomas, S. J. Mills and B. V. L. Potter, Angew. Chem., Int. Ed., 2016, 55, 1614–1650 CrossRef PubMed.
- C. W. Taylor and S. C. Tovey, Cold Spring Harbor Perspect. Biol., 2010, 2, a004010 Search PubMed.
- M. P. Thomas and B. V. L. Potter, FEBS J., 2014, 281, 14–33 CrossRef PubMed.
- S. B. Shears, J. Cell. Physiol., 2018, 233, 1897–1912 CrossRef PubMed.
- S. G. Thota and R. Bhandari, J. Biosci., 2015, 40, 593–605 CrossRef PubMed; M. S. C. Wilson, T. M. Livermore and A. Saiardi, Biochem. J., 2013, 452, 369–379 CrossRef PubMed.
- K. J. Alzayady, L. W. Wang, R. Chandrasekhar, L. E. Wagner, F. Van Petegem and D. I. Yule, Sci. Signaling, 2016, 9, ra35 CrossRef PubMed.
- K. M. Sureshan, A. M. Riley, M. P. Thomas, S. C. Tovey, C. W. Taylor and B. V. L. Potter, J. Med. Chem., 2012, 55, 1706–1720 CrossRef PubMed.
- B. V. L. Potter and D. Lampe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1933–1972 CrossRef.
- T. M. Livermore, C. Azevedo, B. Kolozsvari, M. S. C. Wilson and A. Saiardi, Biochem. Soc. Trans., 2016, 44, 253–259 CrossRef PubMed.
- H. B. R. Luo, Y. E. Huang, J. M. C. Chen, A. Saiardi, M. Iijima, K. Q. Ye, Y. F. Huang, E. Nagata, P. Devreotes and S. H. Snyder, Cell, 2003, 114, 559–572 CrossRef PubMed; A. Chakraborty, M. A. Koldobskiy, N. T. Bello, M. Maxwell, J. J. Potter, K. R. Juluri, D. Maag, S. Kim, A. S. Huang, M. J. Dailey, M. Saleh, A. M. Snowman, T. H. Moran, E. Mezey and S. H. Snyder, Cell, 2010, 143, 897–910 CrossRef PubMed.
- N. A. Gokhale, A. Zaremba, A. K. Janoshazi, J. D. Weaver and S. B. Shears, Biochem. J., 2013, 453, 413–426 CrossRef PubMed.
- R. Wild, R. Gerasimaite, J. Y. Jung, V. Truffault, I. Pavlovic, A. Schmidt, A. Saiardi, H. J. Jessen, Y. Poirier, M. Hothorn and A. Mayer, Science, 2016, 352, 986–990 CrossRef PubMed.
- R. Gerasimaite, I. Pavlovic, S. Capolicchio, A. Hofer, A. Schmidt, H. J. Jessen and A. Mayer, ACS Chem. Biol., 2017, 12, 648–653 CrossRef PubMed.
- A. Saiardi, R. Bhandari, A. C. Resnick, A. M. Snowman and S. H. Snyder, Science, 2004, 306, 2101–2105 CrossRef PubMed; R. Bhandari, A. Saiardi, Y. Ahmadibeni, A. M. Snowman, A. C. Resnick, T. Z. Kristiansen, H. Molina, A. Pandey, J. K. Werner Jr., K. R. Juluri, Y. Xu, G. D. Prestwich, K. Parang and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15305–15310 CrossRef PubMed.
- R. S. Kilari, J. D. Weaver, S. B. Shears and S. T. Safrany, FEBS Lett., 2013, 587, 3464–3470 CrossRef PubMed.
- M. Takahashi, K. Tanzawa and S. Takahashi, J. Biol. Chem., 1994, 269, 369–372 Search PubMed.
- H. Hotoda, K. Murayama, S. Miyamoto, Y. Iwata, M. Takahashi, Y. Kawase, K. Tanzawa and M. Kaneko, Biochemistry, 1999, 38, 9234–9241 CrossRef PubMed.
- I. Bosanac, J. R. Alattia, T. K. Mal, J. Chan, S. Talarico, F. K. Tong, K. I. Tong, F. Yoshikawa, T. Furuichi, M. Iwai, T. Michikawa, K. Mikoshiba and M. Ikura, Nature, 2002, 420, 696–700 CrossRef PubMed.
- T. Desai, J. Gigg, R. Gigg and E. Martin-Zamora, Carbohydr. Res., 1994, 262, 59–77 CrossRef PubMed.
- Z. Y. Lim, J. W. Thuring, A. B. Holmes, M. Manifava and N. T. Ktistakis, J. Chem. Soc., Perkin Trans. 1, 2002, 1067–1075 RSC.
- S. Capolicchio, D. T. Thakor, A. Linden and H. J. Jessen, Angew. Chem., Int. Ed., 2013, 52, 6912–6916 CrossRef PubMed; S. Capolicchio, H. C. Wang, D. T. Thakor, S. B. Shears and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 9508–9511 CrossRef PubMed.
- H. C. Wang, H. Y. Godage, A. M. Riley, J. D. Weaver, S. B. Shears and B. V. L. Potter, Chem. Biol., 2014, 21, 689–699 CrossRef PubMed.
- E. R. Wijsman, O. van den Berg, E. Kuyl-Yeheskiely, G. A. van der Marel and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1994, 113, 337–338 CrossRef; S. B. Engelsma, N. J. Meeuwenoord, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, Angew. Chem., Int. Ed., 2017, 56, 2955–2959 CrossRef PubMed.
- B. Bradshaw, A. Dinsmore, W. Ajana, D. Collison, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 2001, 3239–3244 Search PubMed.
- I. Pavlovic, D. T. Thakor, L. Bigler, M. S. C. Wilson, D. Laha, G. Schaaf, A. Saiardi and H. J. Jessen, Angew. Chem., Int. Ed., 2015, 54, 9622–9626 CrossRef PubMed.
- V. Lafont, A. A. Armstrong, H. Ohtaka, Y. Kiso, L. M. Amzel and E. Freire, Chem. Biol. Drug Des., 2007, 69, 413–422 Search PubMed; E. Freire, Drug Discovery Today, 2008, 13, 869–874 CrossRef PubMed.
- M. Wu, L. S. Chong, S. Capolicchio, H. J. Jessen, A. C. Resnick and D. Fiedler, Angew. Chem., Int. Ed., 2014, 53, 7192–7197 CrossRef PubMed.
- M. Wu, B. E. Dul, A. J. Trevisan and D. Fiedler, Chem. Sci., 2013, 4, 405–410 RSC.
- N. R. Leslie, A. G. McLennan and S. T. Safrany, BMC Biochem., 2002, 3, 20 CrossRef PubMed.
- T. Zhang, J. J. Caffrey and S. B. Shears, FEBS Lett., 2001, 494, 208–212 CrossRef PubMed; A. Saiardi, E. Nagata, H. B. R. Luo, A. Sawa, X. J. Luo, A. M. Snowman and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2306–2311 CrossRef PubMed.
- J. Monserrate and J. York, Curr. Opin. Cell Biol., 2010, 22, 365–373 CrossRef PubMed.
- H. Wang, J. R. Falck, T. M. Hall and S. B. Shears, Nat. Chem. Biol., 2012, 8, 111–116 CrossRef PubMed.
- E. Dubois, B. Scherens, F. Vierendeels, M. M. W. Ho, F. Messenguy and S. B. Shears, J. Biol. Chem., 2002, 277, 23755–23763 CrossRef PubMed.
- A. M. Seeds, R. J. Bastidas and J. D. York, J. Biol. Chem., 2005, 280, 27654–27661 CrossRef PubMed.
- H. C. Wang, E. F. DeRose, R. E. London and S. B. Shears, Nat. Commun., 2014, 5, 4178 CrossRef PubMed.
- A. M. Riley, H. C. Wang, S. B. Shears and B. V. L. Potter, Chem. Commun., 2015, 51, 12605–12608 RSC.
- B. N. Ames and D. T. Dubin, J. Biol. Chem., 1960, 235, 769–775 Search PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef PubMed.
- M. L. Verdonk, G. Chessari, J. C. Cole, M. J. Hartshorn, C. W. Murray, J. W. M. Nissink, R. D. Taylor and R. Taylor, J. Med. Chem., 2005, 48, 6504–6515 CrossRef PubMed.
- A. M. Vibhute, V. Konieczny, C. W. Taylor and K. M. Sureshan, Org. Biomol. Chem., 2015, 13, 6698–6710 Search PubMed.
- A. A. Baykov, O. A. Evtushenko and S. M. Avaeva, Anal. Biochem., 1988, 171, 266–270 CrossRef PubMed.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212–2221 CrossRef PubMed.
- W. Nakanishi, K. Kikuchi, T. Inoue, K. Hirose, M. Iino and T. Nagano, Bioorg. Med. Chem. Lett., 2002, 12, 911–913 CrossRef PubMed.
- H. Saleem, S. C. Tovey, T. Rahman, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2013, 8, e54877 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, additional DSF data and details of molecular docking experiments. See DOI: 10.1039/c8md00149a |
| ‡ A synthetic InsP3 derivative featuring 4-carboxy-malachite green conjugated to the 1-phosphate group was reported to have ∼170-fold higher affinity than InsP3 for an N-terminal fragment of type 1 InsP3 receptors.43 In our hands, this compound was ∼5-fold less potent than InsP3 at each InsP3 receptor subtype and had an affinity ∼7-fold less than InsP3 for type 1 InsP3 receptors.44 |
| This journal is © The Royal Society of Chemistry 2018 |