
Narrow-spectrum antibacterial agents
Roberta J.
Melander
 a,
Daniel V.
Zurawski
b and
Christian
Melander
*a
a,
Daniel V.
Zurawski
b and
Christian
Melander
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC, USA. E-mail: ccmeland@ncsu.edu
bWound Infections Department, Bacterial Diseases Branch, Walter Reed Army Institute of Research, Silver Spring, MD, USA
First published on 6th November 2017
Abstract
While broad spectrum antibiotics play an invaluable role in the treatment of bacterial infections, there are some drawbacks to their use, namely selection for and spread of resistance across multiple bacterial species, and the detrimental effect they can have upon the host microbiome. If the causative agent of the infection is known, the use of narrow-spectrum antibacterial agents has the potential to mitigate some of these issues. This review outlines the advantages and challenges of narrow-spectrum antibacterial agents, discusses the progress that has been made toward developing diagnostics to enable their use, and describes some of the narrow-spectrum antibacterial agents currently being investigated against some of the most clinically important bacteria including Clostridium difficile, Mycobacterium tuberculosis and several ESKAPE pathogens.
Introduction
Antimicrobials are arguably one of the greatest success stories in the history of medicine, and the advent of the antibiotic era with the availability of effective, non-toxic broad-spectrum antibiotics brought immeasurable benefits. The ability to treat infections early, without identification of the causative agent, resulted in the saving of countless lives and enabled many avenues of modern medicine such as surgery, premature infant care, organ transplantation, and cancer chemotherapy.1 However, rising antibiotic resistance is rapidly eroding these benefits, and in 2014 the World Health Organization (WHO) warned that a post-antibiotic era is a very real possibility.2 If antibiotic resistance continues to rise at the current rate, it is estimated that by 2050, antibiotic resistant infections will account for over 10 million deaths annually and cost the global economy up to 100 trillion USD.3,4 Tackling the antibiotic resistance crisis will require a multi-faceted approach that includes improvements in antibiotic stewardship,4 the development of novel antibiotics, the development of alterative therapeutics such as host-directed therapies and anti-virulence drugs,5 and the development of adjuvants that suppress bacterial resistance mechanisms.6 Another part of this antibacterial strategy may include the development of narrow-spectrum antibacterial agents, i.e. agents that are genus or species specific. In this review we discuss the advantages and challenges to this strategy, summarize the progress that has been made toward developing diagnostics to enable this concept, and describe some of the research avenues that have been investigated toward the identification of narrow-spectrum and pathogen-specific antibacterial agents. This review is not meant to be comprehensive, but rather to highlight some of the recent advances in this field, including: small molecule approaches, the use of bacteriocins and antimicrobial peptides, and touch on the development of bacteriophage and monoclonal antibody therapeutics.Advantages of narrow-spectrum anti-bacterial agents
The overwhelming majority of compounds used to treat bacterial infections have activity against multiple species, and alongside the vast benefits this brings, there lie a number of drawbacks to the use of broad-spectrum antibiotics. Perhaps the most obvious drawback to the use of broad-spectrum antibiotics is selection for resistance, which can occur in both the causative agent of the infection being treated, as well as in other bacteria, both pathogenic and non-pathogenic, that are exposed to the antibiotic. Selection for resistance in non-pathogenic commensal bacteria can still have detrimental consequences, as these bacteria can act as a reservoir for resistance genes that can persist for years and subsequently be transferred to pathogenic bacteria.7Another disadvantage of the use of indiscriminate broad-spectrum antibiotics is the deleterious effect they can have upon the host microbiome. These effects are not limited to the duration of antibiotic treatment, as even short-term (seven-day) antibiotic exposure has been shown to result in an altered makeup of the gut microbiota up to two years post-treatment.8 In cases of repeated antibiotic exposure, it is possible that the microbiota may never return to its initial composition.9 Disruption of the microbiome can affect the vital role it plays in numerous functions including: nutrient supply, vitamin production, and protection from pathogens.10 Some of the disparate array of health problems associated with disruption of these functions include: increased susceptibility to infectious disease11 (particularly colonization by Clostridium difficile),12 metabolic disorders such as obesity and diabetes,13,14 asthma,14 irritable bowel syndrome,15 and immune disorders such as allergies.16 Exposure to broad-spectrum antibiotics during infancy and early childhood is particularly detrimental as the early microbiota lacks diversity and stability, making it uniquely sensitive to disruption, and the developing immune system is in part shaped by the gut microbiota.17 In addition to the gut microbiome, the complex oral microbiome also plays a key role in maintaining both oral and systemic health, and its disruption has been linked to an array of health issues including respiratory, cardiovascular, and cerebrovascular diseases.18
The development of narrow-spectrum antibiotics that do not select for cross-resistance in non-targeted pathogens, and elicit abrogated or reduced collateral damage upon the host microbiome, is therefore an attractive approach in the fight to overcome multidrug-resistant (MDR) bacterial infections. While such agents may not take the place of prophylactic therapy, or initial broad-spectrum therapy in the case of a patient presenting with a life-threatening condition such as sepsis or pneumonia, a switch to narrow-spectrum antibiotics would be beneficial following identification of the causative pathogen. Narrow-spectrum antibiotics could also be utilized for infections such as: urinary tract infections, abscesses or skin and soft-tissue infections, and other non-life-threatening cases, particularly where recurrence is common.
Challenges faced in developing narrow-spectrum antibacterial agents
There are of course many challenges to the utility of a narrow-spectrum approach. Perhaps the most significant challenge is the requirement for rapid, accurate, and sensitive diagnostic assays for the identification of bacterial pathogens. Additionally, the clinical adoption of narrow-spectrum antibiotics will depend upon education of physicians to enable a culture shift away from empirical to more tailored therapy.19 The identification and development of novel narrow-spectrum antibiotics also faces economic challenges. Market size for a drug is of course proportional to the prevalence of the disease,1 and by definition, narrow-spectrum antibacterial agents have a more limited application compared to their broad-spectrum counterparts. However, the reduced revenue resulting from the smaller number of infections that can be treated by narrow-spectrum antibacterial agents may be countered by the potentially longer shelf life conferred by the reduced rates of resistance. The case could potentially also be made for higher prices for narrow-spectrum antibacterial agents, if these agents lead to reductions in mortality and morbidity along with reductions in the length of hospitalization. Indeed, in the case of Acinetobacter baumannii, where mortality rates have been reported to be as high as 60% in intensive care units (ICU), the case has already been argued for higher prices for narrow-spectrum drugs developed against this species.20A shift to the search for narrow-spectrum or pathogen-specific antibacterial agents will also require a paradigm shift within the pharmaceutical industry. Antibiotic discovery in the pharmaceutical industry for many years centered exclusively on the search for broad-spectrum agents with activity against multiple Gram-positive and Gram-negative bacterial species, and compounds that did not meet this metric were triaged from antibiotic discovery programs.21 Some of these previously rejected compounds could potentially now serve as leads for new narrow-spectrum or pathogen-specific discovery initiatives. Essential to this shift are regulatory changes,22 and the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) have recently included antibacterial agents that are active against a narrow-spectrum of MDR-pathogens in a list of treatments that qualify for a shorter route to registration.23 With the addition of the GAIN Act and Fast Track incentives, pursuing these compounds could now be more feasible and profitable than in the past.
Diagnostic tests for the identification of bacterial pathogens
The availability of broad-spectrum antibiotics meant that for many years physicians were able to effectively and safely treat many bacterial infections without the need for a microbiological diagnosis. The resultant culture of empiricism minimized the importance of diagnostic clinical microbiology, and the development of diagnostic tests with the necessary speed, accuracy, and sensitivity to enable effective narrow-spectrum therapies (even those previously available such as antibody therapies) was not widely pursued.1,24 Furthermore, the increasing prevalence of pathogens that harbor acquired resistance determinants, such as methicillin-resistant Staphylococcus aureus (MRSA), and carbapenem-resistant Enterobacteriaceae (CRE), has led to even broader empiric treatment regimens in recent years.25 Such regimens often involve “last resort” agents such as glycopeptide, carbapenem, and polymyxin antibiotics, further exacerbating the emergence and spread of MDR organisms.With the ever-shrinking number of effective antimicrobials resulting from the rise in antibiotic resistance in recent years however, it has become apparent that the development of diagnostic tests that can rapidly identify a specific pathogen is vital.26 Culture-based methods take long periods of time, initial cultures take 24–48 h (during which time broad-spectrum antibiotics are typically initiated), then following identification of the microbe, antimicrobial susceptibility determination requires another 24–48 h.27 In a 2015 survey of infectious disease practitioners, 31% reported that they believed patients are treated with incorrect antibiotics during the wait for blood culture results.28 In addition to long-turnaround times, culture-based methods also have the potential to return false-negative results when samples are obtained after antimicrobial therapy has begun. Other techniques that have been traditionally used alongside culture methods to aid in microbial identification include: Gram-staining, microscopy, the use of fluorescently labeled monoclonal antibodies, ELISA, and radiometric detection of labeled metabolites as in the example of 14C-labeled carbon dioxide produced in the metabolism of palmitic acid by Mycobacterium tuberculosis. While these methods are less time consuming than culture-based methods, they are not any more sensitive, and do not allow for antimicrobial susceptibility determination.26
In more recent years the improvement of nucleic acid-based amplification technologies (NAATs) such as polymerase chain reaction (PCR) and next generation sequencing (NGS), and the introduction of additional rapid, higher resolution technologies such as MALDI-TOF mass spectrometry fingerprinting into the clinical diagnostic setting, have transformed approaches toward pathogen identification.25
NAAT based approaches for pathogen detection are now ubiquitous in clinical microbiology laboratories, and the more recent introduction of commercial multiplex PCR assays has enabled rapid and specific pathogen detection from a single specimen. These approaches hold particular promise for bloodstream infections, as in addition to accelerating pathogen detection compared to culture-based approaches (species identification can be achieved within 3–6 h),25 PCR based approaches allow pathogen detection in cases in which blood cultures remain negative, cases in which it is difficult to obtain an adequate volume of blood for culture based identification, and cases in which empirical antibiotic treatment has already been employed,29 though direct analysis of blood using PCR-based approaches can be hampered by the presence of PCR-inhibitors such as iron, heparin, and immunoglobulins.25 Additionally, multiplex PCR-based assays that can detect certain bacterial pathogens implicated in lower respiratory tract infections are also commercially available.25 The major drawback of PCR-based techniques is that DNA from an organism can be detected even after the organism has been killed or cleared, so there is a potential for false positives. Also, since the primers are directed against certain genes, there is the potential that there could be a mutation or loss of the gene during infection, especially in antibiotic resistant determinants that are under evolutionary pressure, which in turn could provide a false negative.
Traditional sequencing techniques such as capillary electrophoresis sequencing or pyrosequencing have long been paired with NAAT based approaches for pathogen identification, and can provide identification to the genus level for upwards of 90%, to and species-level identification for 65–85% of isolates tested (all microbes). NGS exhibits increased accuracy, and has the potential to detect virulence markers antimicrobial resistance determinants, but is subject to the need for extensive bioinformatics to enable data interpretation.26 NGS could also suffer some of the same limitations as PCR-based tests as DNA could be present in a sample when the living organism itself is no longer present.
MALDI-TOF MS fingerprinting for bacterial pathogen identification has been widely embraced by clinical microbiology laboratories around the world.30 It is rapid (turnaround times are typically reduced by at least one working day, up to several days for slower growing species), accurate, and cost effective.25,30 Identification can be carried out using direct colony testing, in which a bacterial colony is directly placed on the MALDI plate and the generated spectrum compared to a database of reference spectra. Direct testing of clinical samples is not feasible in most cases due to an inadequate limit of detection; however certain clinical samples, such as urine (though this must be processed prior to testing), and cerebrospinal fluid, contain high enough numbers of bacteria to allow direct testing.30 MALDI-TOF MS can also be used to identify microorganisms from blood cultures; this is accomplished by either subculture from positive blood culture bottle onto solid media and following a short (2–4 h) incubation period, analysis by MALDI-TOF MS, or by direct testing of blood culture bottles following removal of exogenous macromolecules. Direct identification of pathogens from positive blood cultures results in a reduction in turnaround time of at least one day compared to conventional processing, and enables species level identification of the causative pathogen within the critical phase of septic illness.25
Limitations of MALDI-TOF fingerprinting for bacterial pathogen identification include the inability to reliably identify polymicrobial infections, or to reliably distinguish between organisms with similar spectral profiles, for example E. coli and Shigella species cannot be reliably distinguished using currently available algorithms.30 This is also the case with some species of Acinetobacter,31 however databases are constantly being updated so this issue may eventually be overcome. Another recent approach exploits MALDI-TOF to detect bacterial lipids and polysaccharides, and can better distinguish between different species in mixed culture and may provide better accuracy and sensitivity as this technology further develops.32
Other approaches that have been utilized for bacterial identification include fluorescent in situ hybridization (FISH), electrochemical biosensor assays, and rapid antigen testing. FISH based assays have been used in bloodstream infection diagnosis for many years and probes specific for bacterial ribosomal RNA have been designed for greater than 95% of pathogens typically associated with such infections. In one study of 115 positive blood cultures (both bacterial and fungal), identification of 111 samples to the family, genus, or species level was achieved within 2.5 h compared to 1 to 3 days by conventional culture-based methods.33
Electrochemical biosensor arrays have been developed to enable identification of urinary tract infection (UTI) pathogens. The working electrode of the sensor contains a biotin-modified capture probe that is specific for a clinically relevant bacterial urinary pathogen, to which the bacterial 16S rRNA target hybridizes. A fluorescein-modified detector probe then hybridizes to the rRNA target and detection of the target-probe hybrids is achieved through binding of a horseradish peroxidase (HRP)-conjugated anti-fluorescein antibody. It was reported that species-specific detection of pathogens from clinical urine samples could be achieved in less than 6 h, identification was demonstrated for 98% of Gram-negative bacteria for which species-specific probes were available, and causative agents in polymicrobial infections could also be identified.34,35
Rapid antigen testing utilizes a visible readout upon antibody–antigen binding and has been clinically successful for the identification of group A beta-hemolytic streptococcus (GABHS), the most common causative agent of acute pharyngitis. This test provides results in approximately 15 min at the point-of-care, as compared to up to two days for culture based identification, and has a specificity of greater than 95% and a sensitivity of greater than or equal to 90%.36,37 Rapid antigen testing is limited in scope and is currently only available for a small number of select pathogens, but possibilities for extension to additional pathogens exist, particularly for indications in which clinical signs point to a specific type of infection.37
Finally, there are companies that have recognized that the use of narrow spectrum agents for certain clinical indications will require species-specific identification, and they have invested in diagnostic technologies. For example, in 2016 MedImmune, Inc. partnered with Cepheid, who developed Xpert® tests, PCR-based technology, to detect Pseudomonas aeruginosa and S. aureus early in patients to enable the use of monoclonal antibodies for treatment (see below). Another example is the purchase of GeneWEAVE BioSciences, Inc. by Roche Ltd. GeneWEAVE is developing pathogen-specific diagnostics using non-replicating bacteriophage.38 With the strain specificity of phage, this technology holds considerable promise, as it may be more sensitive in detecting live bacteria than current techniques. Recently, this technology was employed to detect M. tuberculosis in a phage/ELISA-based assay.39
Identification of narrow-spectrum antibacterial agents
Approaches that have been taken toward developing antibacterial agents with specificity for a particular species or genus include: the targeting of proteins and pathways that are specific to the bacteria of interest, the use of bacteriocins and other antimicrobial peptides that are specific for a particular bacteria, and whole cell phenotypic screening. Progress toward the development of narrow-spectrum antibacterial agents to combat some of the most threatening human pathogens including ESKAPE pathogens, C. difficile and M. tuberculosis is described below.Narrow spectrum antibacterial agents for M. tuberculosis
M. tuberculosis is the causative agent of tuberculosis (TB) and infects approximately one-third of the global population. TB was the leading infectious disease killer in 2015, causing an estimated 1.8 million deaths – more than HIV and malaria combined.40,41 Three of the four first-line anti-TB drugs are narrow-spectrum, isoniazid 1, pyrazinamide 2, and ethambutol 3 (Fig. 1), having little or no activity outside the mycobacterial genus.42,43 However, even drug-susceptible TB requires treatment durations of six months with a combination of these mycobacterial specific antibiotics, along with the broad-spectrum antibiotic rifampin.43 In the case of MDR strains, which are becoming increasingly prevalent, multiple broad-spectrum antibiotics including fluoroquinolones and aminoglycosides are employed, and treatment regimens for multi-drug resistant strains that require these second-line broad-spectrum drugs typically last for two years.40 These lengthy treatment durations represent some of the most extensive exposures of humans to antibiotics,43 and will undoubtedly have considerable effects upon the commensal microbiota. Given the lengthy treatment regimens required for the treatment of TB, the development of additional narrow-spectrum anti-TB agents that could potentially reduce the dependence on broad-spectrum antibiotics would be particularly valuable.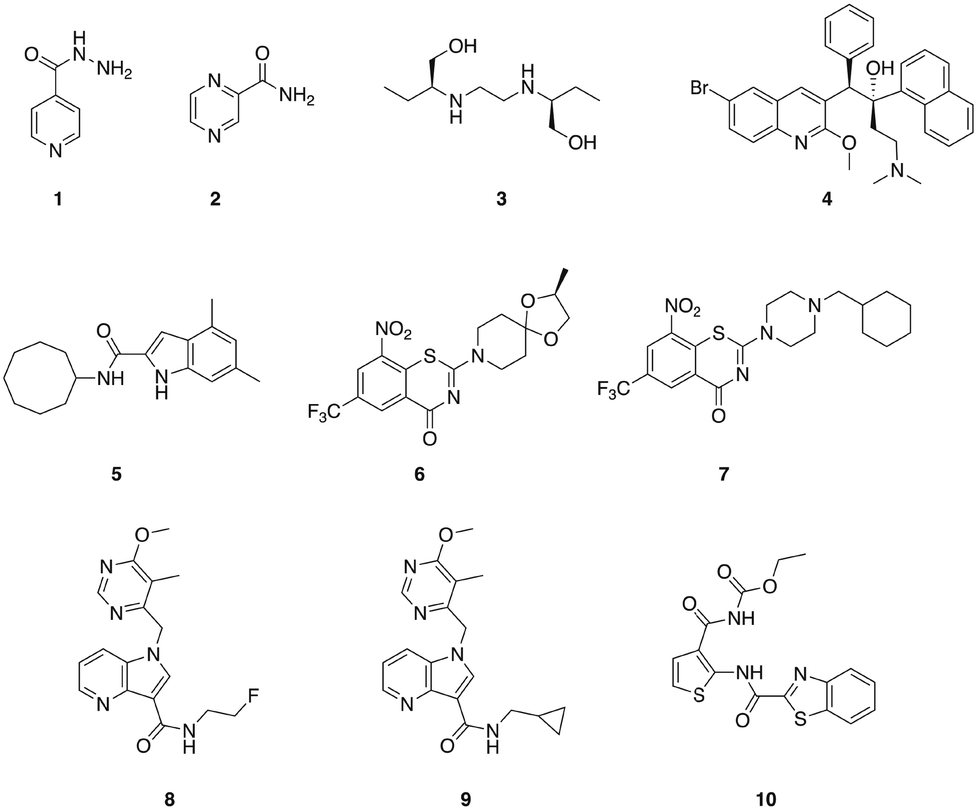 | ||
| Fig. 1 Compounds that display narrow-spectrum antibacterial activity against M. tuberculosis. | ||
The diarylquinolone bedaquiline 4 (Fig. 1), which was approved by the FDA in 2012 for the treatment of MDR TB,44 exhibits narrow-spectrum anti-mycobacterial activity as a result of selectively targeting the mycobacterial ATP synthase.45 Bedaquiline demonstrates activity against both non-replicating and replicating mycobacteria, and against both drug-sensitive and drug resistant isolates. However, bedaquiline is subject to CYP3A4 metabolism,46 and exhibits potent hERG channel inhibition,47 and as such is subject to a limited indication of use in patients for which there is considerable unmet need and a positive benefit-risk balance.44
The mycobacterial membrane protein large 3 (MmpL3) is a transporter that is required for the translocation of the mycolic acid of trehalose-monomycolate (TMM), a mycobacterial outer membrane component precursor, to the outer membrane. This transporter is essential in mycobacteria, and is specific to Actinobacteria,40,48 making it a promising target for the development of narrow spectrum antibacterial agents for mycobacterial pathogens. A series of indole-containing compounds have been developed as inhibitors of this transporter, and have demonstrated potent in vitro antibacterial activity against M. tuberculosis49 along with several species of non-tuberculosis mycobacteria (NTM), which are a growing healthcare problem. The lead compound from this series, 5 (Fig. 1), exhibited potent antibacterial activity against M. tuberculosis and also against a panel of NTM pathogens that included M. avium complex (MAC) pathogens, which are the most common human NTM pathogens. Compound 5 displayed selectivity for mycobacteria, with no bacterial growth inhibition observed against either P. aeruginosa or S. aureus at concentrations up to 160 μg mL−1.40 This compound demonstrated dose-dependent activity when administered orally in a mouse model of M. tuberculosis infection, suggesting that this class of compounds has potential for development as anti-mycobacterial agents.50
The essential flavo-enzyme decaprenylphosphoryl-β-D-ribose 2′-epimerase 1 (DprE1) plays a role in the synthesis of the precursor to the mycobacterial cell wall component arabinogalactan, and has been marked as a promising target for the identification of novel selective anti-TB drugs.46 Several inhibitors of DprE1 have been described, the first of which were a series of 1,3-benzothiazin-4-ones (benzothiazones), including BTZ043 6, and PBTZ169 7 (Fig. 1).51,52 BTZ043 exhibits high antimycobacterial selectivity, with no activity observed against representative Gram-positive (S. aureus and Micrococcus luteus) and Gram-negative (P. aeruginosa and A. baumannii) strains.53 Another class of DprE1 inhibitors is the 1,4-azaindole class, which unlike the benzothiazones, inhibit DprE1 via a non-covalent mechanism. The lead compounds from this series, 8 and 9 exhibit in vivo efficacy in both acute and chronic mouse TB models.54 TCA1 10 (Fig. 1) is another inhibitor of DprE1 that was identified from a whole cell phenotypic screen for inhibitors of biofilm formation, and also inhibits MoeW, an enzyme involved in the biosynthesis of the molybdenum cofactor (MoCo). TCA1 is selective for mycobacteria, displaying no activity against E. coli, S. aureus, or P. aeruginosa. This compound has activity against both replicating and non-replicating M. tuberculosis, including drug-resistant strains, and demonstrated in vivo efficacy in acute and chronic rodent mouse models of TB making a promising potential lead for the development of a new class of narrow-spectrum anti-TB agents.55
Narrow spectrum antibacterial agents for C. difficile
As mentioned earlier, the use of broad-spectrum antibiotics that eradicate commensal gut bacteria can result in colonization by the opportunistic pathogen C. difficile. C. difficile is responsible for an estimated 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 infections that either require hospitalization or affect already hospitalized patients per year in the United States, 14000 of which are fatal. Infections caused by this bacterium also generate at least $1 billion in excess medical costs per year.56C. difficile is a spore forming bacterium, which makes it difficult to remove from surfaces and enables it to spread rapidly.57 Despite receiving high-dose extended duration treatment with multiple antibiotics, typically metronidazole and vancomycin, up to 65% of patients that become colonized by C. difficile suffer a relapse, an outcome that is correlated with the presence of a low-diversity microbiome.10 Additionally, treatment with both oral vancomycin and metronidazole has been associated with colonization of the gut by vancomycin resistant Enterococcus faecium (VRE).58 Narrow-spectrum antibacterial agents would be hugely beneficial in the treatment of C. difficile infections, as they would enable rapid restoration of the commensal gut microbiota, and several different scaffolds have been investigated.
000 infections that either require hospitalization or affect already hospitalized patients per year in the United States, 14000 of which are fatal. Infections caused by this bacterium also generate at least $1 billion in excess medical costs per year.56C. difficile is a spore forming bacterium, which makes it difficult to remove from surfaces and enables it to spread rapidly.57 Despite receiving high-dose extended duration treatment with multiple antibiotics, typically metronidazole and vancomycin, up to 65% of patients that become colonized by C. difficile suffer a relapse, an outcome that is correlated with the presence of a low-diversity microbiome.10 Additionally, treatment with both oral vancomycin and metronidazole has been associated with colonization of the gut by vancomycin resistant Enterococcus faecium (VRE).58 Narrow-spectrum antibacterial agents would be hugely beneficial in the treatment of C. difficile infections, as they would enable rapid restoration of the commensal gut microbiota, and several different scaffolds have been investigated.
The narrow-spectrum macrolide fidaxomicin 11 (Fig. 2) was approved by the FDA for treatment of C. difficile infections in 2011. Fidaxomicin is selective for Gram-positive anaerobes, exhibiting only limited bactericidal activity against Gram-positive aerobes including staphylococci and enterococci, and a lack of activity against Gram-negative bacteria.59 High concentrations of fidaxomicin can be achieved in the colon with very little concomitant systemic exposure. In addition to its antibiotic activity, fidaxomicin also inhibits spore formation and toxin production by C. difficile. Treatment with fidaxomicin leads to greater preservation of the gut microbiota, reduced acquisition of VRE, and has also been reported to decrease the recurrence of C. difficile infections compared to treatment with vancomycin.60
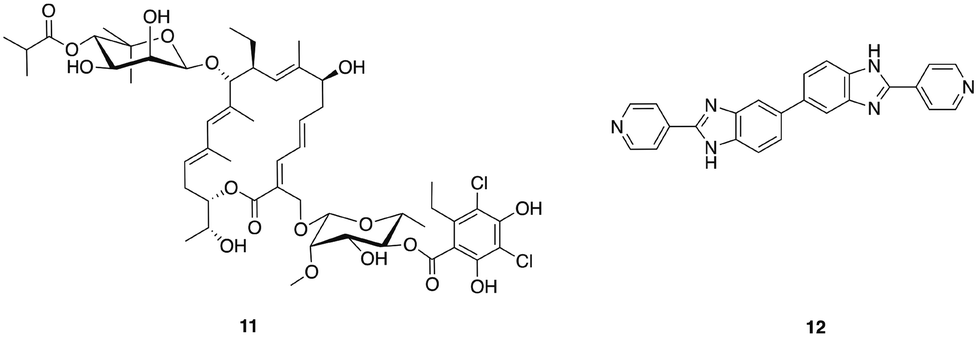 | ||
| Fig. 2 Narrow-spectrum antibacterial agents for C. difficile. | ||
Other narrow-spectrum antibiotics that have been investigated for combatting C. difficile include ridinilazole (formerly SMT19969) 12, a novel antibiotic that acts by an as yet not fully understood mechanism of action that may involve impairment of cell division.61 Ridinilazole exhibits comparable or greater in vitro antibacterial activity against C. difficile to that of fidaxomicin, and more potent activity than either vancomycin or metronidazole, it also exhibits a prolonged post-antibiotic effect and has low systemic absorption.61 The spectrum of activity of ridinilazole is particularly narrow and it exhibits reduced activity against Gram-negative anaerobes than vancomycin and metronidazole, is inactive against Gram-positive aerobes including S. aureus, E. faecium, Enterococcus faecalis, and several Streptococcus species, and is more selective than fidaxomicin against other Gram-positive anaerobes, with activity only observed against Clostridium innocuum and some Lactobacillus species.62 In a hamster model of clindamycin-induced C. difficile infection, ridinilazole exhibited increased efficacy in comparison to vancomycin, and comparable to that of fidaxomicin, with 90–100% survival at day 28 compared to only 10% survival in vancomycin treated animals.61 A recent phase 2 clinical trial (completed August 2015) affirmed that ridinilazole was well tolerated, established non-inferiority compared to vancomycin and demonstrated statistical superiority at the 10% level.63
Another approach in the search for narrow-spectrum antimicrobials for the treatment of C. difficile infection is the use of bacteriocins – small ribosomally synthesized peptides produced by bacteria that inhibit the growth of other bacteria, often closely related species. One bacteriocin that has shown promise is thuricin CD, which is produced by a strain of Bacillus thuringiensis. Thuricin CD consists of two distinct peptides, Trn-α and Trn-β,64 and exhibits comparable in vitro antimicrobial activity to vancomycin and metronidazole against clinically significant strains, and did not significantly impact the composition of the commensal gut microbiota in a human distal colon model.65,66
Narrow spectrum antibacterial agents for ESKAPE pathogens
E. faecium, S. aureus, Klebsiella pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter species have been collectively termed the ESKAPE pathogens,67 and account for almost one-third of all nosocomial infections in the United States.68 Clinical isolates of ESKAPE species that are resistant to every available antibiotic have now been isolated,69 and new treatment options are desperately needed.Enterococci are important clinical pathogens that have the potential for resistance to virtually all clinically used antibiotics through both intrinsic and acquired resistance mechanisms.70 This genus is responsible for an estimated 66000 healthcare-associated infections in the United States each year, 20000 of which are caused by strains resistant to vancomycin, resulting in approximately 1300 deaths.56 Vancomycin resistant E. faecium (VRE) accounts for 25% of enterococci infections in intensive-care units, and resistance to both antibiotics approved by the FDA for the treatment VRE (quinupristin-dalfopristin and linezolid) has been observed, meaning new treatments for this bacterium are much needed.
A series of 1,2,4-triazolo[1,5-a]pyrimidines that display potent, selective activity against E. faecium was identified from an initial in silico screen of 1.2 million drug-like compounds for inhibitors of penicillin binding protein 2a (PBP 2a).71 This initial screen identified compound 13 (Fig. 3), which exhibited a minimum inhibitory concentration of 8 μg mL−1 against E. faecium, and was devoid of activity against all other ESKAPE pathogens tested. Analogue synthesis around this core scaffold delivered compound 14, which possessed comparable or increased antibacterial activity compared to compound 13 across a panel of E. faecium clinical isolates. The mechanism of action of compound 14 was determined to involve inhibition of peptidoglycan biosynthesis, though interestingly this was not through inhibition of penicillin binding proteins, despite PBP 2a being used in the initial screen that uncovered this class of antibiotics. While the specific target remains to be elucidated, this class of narrow-spectrum antibacterial agents holds promise for combatting infections caused by E. faecium.71
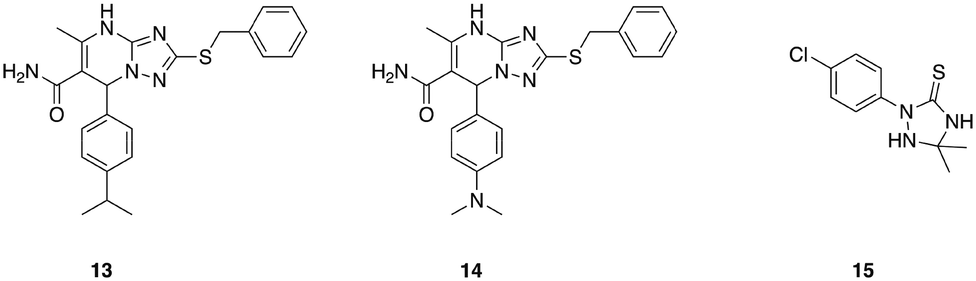 | ||
| Fig. 3 Antibacterial agents that display narrow-spectrum activity against ESKAPE pathogens. | ||
Methicillin-resistant S. aureus (MRSA) is responsible for an estimated 80461 infections and 11285 deaths anually.56 Currently, vancomycin, daptomycin or linezolid are used to treat MRSA infections, however similar to VRE, resistance to these agents has been observed.72 A novel antimicrobial peptide has been investigated as a potential narrow-spectrum antibacterial agent for the treatment of S. aureus infections. MP1106 was designed based on the fungal defensin plectasin, which had previously been shown to possess antibacterial activity against a number of Gram-positive pathogens via a mechanism that involves binding to the peptidoglycan intermediate lipid II.73 MP1106 displayed increased activity against S. aureus compared to the parent peptide, and also exhibited potent activity against the Gram-positive pathogen Streptococcus suis, as well as moderate activity against select other Gram-positive bacteria, including some Bacillus species. The peptide did not display antimicrobial activity against S. epidermidis, or any of the Gram-negative species tested,74 demonstrating its potential as a starting point for the development of narrow spectrum therapeutics for select Gram-positive pathogens including S. aureus.
When it comes to A. baumannii, the need for narrow-spectrum diagnostics and novel antibacterial approaches may be even more apparent due to the very high mortality rates (>60%) associated with this pathogen, and the fact that patients can succumb in sometimes 48–72 hours.75–77 In the United States, A. baumannii makes up only 1.8% of all hospital-acquired infections and is responsible for an estimated 45000–83000 infections per year.20 In contrast, in Asia, South America, and the Middle East, it is the most dominant nosocomial organism responsible for infections.45 The rise in the prevalence of A. baumannii infection, along with increasing drug-resistance has fostered an uptick in the development of both new A. baumannii small molecule interventions, and non-traditional approaches. Because A. baumannii has some unique biochemistry78 and unique membrane54 components when compared to other Gram-negative pathogens such as E. coli or P. aeruginosa, it may be more suited to the development of narrow-spectrum antibacterial agents.
One such class of narrow spectrum A. baumannii specific antibiotics are the 1,2,4-triazolidine-3-thiones class of small molecules that specifically target fatty acid synthesis/elongation in A. baumannii.79 These compounds display no activity against any other species tested including E. coli, P. aeruginosa, K. pneumoniae, and S. aureus, and a lead compound from this series (15, Fig. 3), was shown to exhibit activity in Galleria mellonella infected with a highly virulent A. baumannii strain (AB5075).80
It should also be noted that non-small molecule, non-traditional antibacterial approaches with regard to A. baumannii infections are also being revived. For example, a U.S. Army/Navy collaboration showed that a cocktail of bacteriophage could successfully target one strain of A. baumannii in an in vivo wound model81 and subsequently a cocktail approach was successfully used to cure an infected patient.82 Phage products, lysins, have also been successful at specifically targeting A. baumannii.83 Lastly, a monoclonal antibody, another non-traditional approach, has recently been shown to eradicate A. baumannii from bloodstream infections in mice.84 The target of the antibody is the bacteria's capsule, which is certainly species-specific, but could also be somewhat strain-specific limiting the utility unless other capsules from other strains could also be targeted for a cocktail.
P. aeruginosa is a common cause of healthcare-associated infections, an estimated 51000 of which occur in the United States each year, resulting in approximately 400 deaths.85 Increasing numbers of P. aeruginosa strains are multidrug-resistant, and novel antimicrobial strategies are continually being sought. One such strategy that has been investigated is the use of bacteriocins, whose high specificity and activity against clinically relevant pathogens makes them promising starting points for the development of narrow-spectrum antibacterial agents.85 One class of such bacteriocins are the LlpA bacteriocins, which are produced by several bacterial species including the pseudomonads: P. putida, P. protegens, and P. syringae, the plant pathogen Xanthomonas citri, and the human pathogen Burkholderia cenocepacia.86 LlpA bacteriocins such as Pyocin L1 (PyoL1) display high affinity for D-rhamnose, which is a constituent of the common polysaccharide antigen (CPA) found in the P. aeruginosa cell membrane, accounting for the specificity exhibited by this bacteriocin. PyoL1 displayed antibacterial activity against a majority of P. aeruginosa isolates tested, and displayed no activity against other Pseudomonads.86 Another pyocin, pyocin S5 was reported to exhibit antibacterial activity against several P. aeruginosa strains, and was inactive against E. coli and S. aureus even at concentrations of up to 2.16 mg mL−1.87
Finally, it would be remiss not to mention that, as discussed for A. baumannii, monoclonal antibodies can also provide a narrow-spectrum treatment for other bacterial pathogens For example, a bispecific antibody has been developed for P. aeruginosa by MedImmune LLC. MEDI3902 targets both the type III secretion system protein PcrV and the Psl polysaccharide that plays a role in immune evasion and biofilm formation. MEDI3902 protected mice from lethal P. aeruginosa infection88 and is currently being evaluated in clinical trials. This is also the case for the human immunoglobulin G1(κ) monoclonal antibody MEDI4893, which targets the S. aureus alpha-toxin. MEDI4893 was shown to be safe in humans89 and is currently in a Phase II trial for efficacy. These products developed against these two ESKAPE pathogens are following in the wake of bezlotoxumab, which was recently FDA-approved for the prevention of C. difficile infections.90 Additionally, it is possible that as more monoclonal antibodies are developed, combinations with narrow-spectrum small molecules could also be a potent approach.
Conclusions
Multiple novel approaches are required to address the antibiotic resistance crisis we are currently facing. While broad-spectrum antibiotics have bestowed numerous benefits upon society, they also suffer from drawbacks, most significantly selection for resistance, and the deleterious effects they exert upon the host microbiome. The development of narrow-spectrum antibacterial agents that exhibit selectivity for a particular genus or species has the potential to overcome some of these issues. Narrow-spectrum antibacterial agents with activity against some of the most medically relevant bacterial pathogens, including: several ESKAPE pathogens, the opportunistic gut colonizer C. difficile, and the chronic infectious agent M. tuberculosis have been presented here. There are examples of narrow-spectrum antibacterial agents that have been recently deployed into the clinic in the form of fidaxomicin for the treatment of C. difficile infections, and bedaquiline for the treatment of MDR TB. While there is still much work to be done to expand the scope of this approach clinically, the advantages narrow-spectrum agents possess and the improving diagnostic landscape should make narrow spectrum-antibacterial agents a more significant player in the antibacterial armamentarium.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense. The authors would also like to thank the National Institutes of Health for support (GM055769, AI106733, and AI10721 to CM).Notes and references
- A. Casadevall, Expert Opin. Pharmacother., 2009, 10, 1699–1703 CrossRef PubMed.
- World Health Organization, Antimicrobial Resistance - Global Report on Surveillance, 2014 Search PubMed.
- J. O'Neill, Review on AMR, Antimicrobial resistance: tackling a crisis for the health and wealth of nations, 2014 Search PubMed.
- D. A. Goff, R. Kullar, E. J. Goldstein, M. Gilchrist, D. Nathwani, A. C. Cheng, K. A. Cairns, K. Escandon-Vargas, M. V. Villegas, A. Brink, D. van den Bergh and M. Mendelson, Lancet Infect. Dis., 2017, 17, e56–e63 CrossRef PubMed.
- E. E. Gill, O. L. Franco and R. E. Hancock, Chem. Biol. Drug Des., 2015, 85, 56–78 Search PubMed.
- R. J. Melander and C. Melander, ACS Infect. Dis., 2017, 3, 559–563 CrossRef PubMed.
- G. Karam, J. Chastre, M. H. Wilcox and J. L. Vincent, Crit. Care, 2016, 20, 136 CrossRef PubMed.
- C. Jernberg, S. Lofmark, C. Edlund and J. K. Jansson, ISME J., 2007, 1, 56–66 CrossRef PubMed.
- K. Raymann, Z. Shaffer and N. A. Moran, PLoS Biol., 2017, 15, e2001861 Search PubMed.
- A. Langdon, N. Crook and G. Dantas, Genome Med., 2016, 8, 39 CrossRef PubMed.
- C. Ubeda and E. G. Pamer, Trends Immunol., 2012, 33, 459–466 CrossRef PubMed.
- V. Stevens, G. Dumyati, L. S. Fine, S. G. Fisher and E. van Wijngaarden, Clin. Infect. Dis., 2011, 53, 42–48 CrossRef PubMed.
- B. Boursi, R. Mamtani, K. Haynes and Y. X. Yang, Eur. J. Endocrinol., 2015, 172, 639–648 CrossRef PubMed.
- M. J. Blaser and S. Falkow, Nat. Rev. Microbiol., 2009, 7, 887–894 CrossRef PubMed.
- G. J. Holtmann, A. C. Ford and N. J. Talley, Lancet Gastroenterol. Hepatol., 2016, 1, 133–146 CrossRef PubMed.
- A. T. Stefka, T. Feehley, P. Tripathi, J. Qiu, K. McCoy, S. K. Mazmanian, M. Y. Tjota, G. Y. Seo, S. Cao, B. R. Theriault, D. A. Antonopoulos, L. Zhou, E. B. Chang, Y. X. Fu and C. R. Nagler, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13145–13150 CrossRef PubMed.
- A. Schulfer and M. J. Blaser, PLoS Pathog., 2015, 11, e1004903 Search PubMed.
- V. N. Stone and P. Xu, Mol. Oral Microbiol., 2017 DOI:10.1111/omi.12190.
- L. B. Rice, Clin. Infect. Dis., 2011, 52(Suppl 4), S357–S360 CrossRef PubMed.
- B. Spellberg and J. H. Rex, Nat. Rev. Drug Discovery, 2013, 12, 963 CrossRef PubMed.
- R. Tommasi, D. G. Brown, G. K. Walkup, J. I. Manchester and A. A. Miller, Nat. Rev. Drug Discovery, 2015, 14, 529–542 CrossRef PubMed.
- J. H. Rex, M. Goldberger, B. I. Eisenstein and C. Harney, Ann. N. Y. Acad. Sci., 2014, 1323, 11–21 CrossRef PubMed.
- R. Bax and S. Green, J. Antimicrob. Chemother., 2015, 70, 1281–1284 CrossRef PubMed.
- A. Casadevall, Clin. Infect. Dis., 2006, 42, 1414–1416 CrossRef PubMed.
- F. P. Maurer, M. Christner, M. Hentschke and H. Rohde, Infect. Dis. Rep., 2017, 9, 6839 Search PubMed.
- A. M. Caliendo, D. N. Gilbert, C. C. Ginocchio, K. E. Hanson, L. May, T. C. Quinn, F. C. Tenover, D. Alland, A. J. Blaschke, R. A. Bonomo, K. C. Carroll, M. J. Ferraro, L. R. Hirschhorn, W. P. Joseph, T. Karchmer, A. T. MacIntyre, L. B. Reller, A. F. Jackson and A. Infectious Diseases Society of, Clin. Infect. Dis., 2013, 57(Suppl 3), S139–S170 CrossRef PubMed.
- J. Faro, M. Mitchell, Y. J. Chen, S. Kamal, G. Riddle and S. Faro, Infect. Dis. Obstet. Gynecol., 2016, 2016, 5293034 CrossRef PubMed.
- R. C. She, S. Alrabaa, S. H. Lee, M. Norvell, A. Wilson and C. A. Petti, PLoS One, 2015, 10, e0121493 Search PubMed.
- O. Opota, K. Jaton and G. Greub, Clin. Microbiol. Infect., 2015, 21, 323–331 CrossRef PubMed.
- R. Patel, Clin. Chem., 2015, 61, 100–111 Search PubMed.
- M. Mari-Almirall, C. Cosgaya, P. G. Higgins, A. Van Assche, M. Telli, G. Huys, B. Lievens, H. Seifert, L. Dijkshoorn, I. Roca and J. Vila, Clin. Microbiol. Infect., 2017, 23, 210 Search PubMed.
- L. M. Leung, W. E. Fondrie, Y. Doi, J. K. Johnson, D. K. Strickland, R. K. Ernst and D. R. Goodlett, Sci. Rep., 2017, 7, 6403 CrossRef PubMed.
- V. A. Kempf, K. Trebesius and I. B. Autenrieth, J. Clin. Microbiol., 2000, 38, 830–838 CAS.
- J. C. Liao, M. Mastali, V. Gau, M. A. Suchard, A. K. Moller, D. A. Bruckner, J. T. Babbitt, Y. Li, J. Gornbein, E. M. Landaw, E. R. McCabe, B. M. Churchill and D. A. Haake, J. Clin. Microbiol., 2006, 44, 561–570 CrossRef CAS PubMed.
- E. Altobelli, R. Mohan, K. E. Mach, M. L. Y. Sin, V. Anikst, M. Buscarini, P. K. Wong, V. Gau, N. Banaei and J. C. Liao, Eur. Urol. Focus, 2016 DOI:10.1016/j.euf.2015.12.010.
- A. K. Leung, R. Newman, A. Kumar and H. D. Davies, Expert Rev. Mol. Diagn., 2006, 6, 761–766 CrossRef CAS PubMed.
- T. Maxson and D. A. Mitchell, Tetrahedron, 2016, 72, 3609–3624 CrossRef CAS PubMed.
- , Roche gobbles Smarticles, Nat. Biotechnol., 2015, 33, 1012 Search PubMed.
- N. Zhao, J. Spencer, M. A. Schmitt and J. D. Fisk, Anal. Biochem., 2017, 521, 59–71 CrossRef CAS PubMed.
- N. D. Franz, J. M. Belardinelli, M. A. Kaminski, L. C. Dunn, V. Calado Nogueira de Moura, M. A. Blaha, D. D. Truong, W. Li, M. Jackson and E. J. North, Bioorg. Med. Chem., 2017, 25, 3746–3755 CrossRef CAS PubMed.
- M. Pai, N. Correa, N. Mistry and P. Jha, Lancet, 2017, 389, 1174–1176 CrossRef.
- F. Bardou, A. Quemard, M. A. Dupont, C. Horn, G. Marchal and M. Daffe, Antimicrob. Agents Chemother., 1996, 40, 2459–2467 CAS.
- M. F. Wipperman, D. W. Fitzgerald, M. A. J. Juste, Y. Taur, S. Namasivayam, A. Sher, J. M. Bean, V. Bucci and M. S. Glickman, Sci. Rep., 2017, 7, 10767 CrossRef PubMed.
- E. Cox and K. Laessig, N. Engl. J. Med., 2014, 371, 689–691 CrossRef CAS PubMed.
- S. J. Tantry, V. Shinde, G. Balakrishnan, S. D. Markad, A. K. Gupta, J. Bhat, A. Narayan, A. Raichurkar, L. K. Jena, S. Sharma, N. Kumar, R. Nanduri, S. Bharath, J. Reddy, V. Panduga, K. R. Prabhakar, K. Kandaswamy, P. Kaur, N. Dinesh, S. Guptha, R. Saralaya, M. Panda, S. Rudrapatna, M. Mallya, H. Rubin, T. Yano, K. Mdluili, C. B. Cooper, V. Balasubramanian, V. K. Sambandamurthy, V. Ramachandran, R. Shandil, S. Kavanagh, S. Narayanan, P. Iyer, K. Mukherjee, V. P. Hosagrahara, S. Solapure, P. S. Hameed and S. Ravishankar, MedChemComm, 2016, 7, 1022–1032 RSC.
- D. T. Hoagland, J. Liu, R. B. Lee and R. E. Lee, Adv. Drug Delivery Rev., 2016, 102, 55–72 CrossRef CAS PubMed.
- P. J. C. Amy, S. T. Tong, A. Blaser, H. S. Sutherland, S. K. Y. Tsang, J. Guillemont, M. Motte, C. B. Cooper, K. Andries, W. Van den Broeck, S. G. Franzblau, A. M. Upton, W. A. Denny, B. D. Palmer and D. Conole, ACS Med. Chem. Lett., 2017, 8, 1019–1024 CrossRef PubMed.
- G. Degiacomi, A. Benjak, J. Madacki, F. Boldrin, R. Provvedi, G. Palu, J. Kordulakova, S. T. Cole and R. Manganelli, Sci. Rep., 2017, 7, 43495 CrossRef PubMed.
- O. K. Onajole, M. Pieroni, S. K. Tipparaju, S. Lun, J. Stec, G. Chen, H. Gunosewoyo, H. Guo, N. C. Ammerman, W. R. Bishai and A. P. Kozikowski, J. Med. Chem., 2013, 56, 4093–4103 CrossRef CAS PubMed.
- S. Lun, H. Guo, O. K. Onajole, M. Pieroni, H. Gunosewoyo, G. Chen, S. K. Tipparaju, N. C. Ammerman, A. P. Kozikowski and W. R. Bishai, Nat. Commun., 2013, 4, 2907 Search PubMed.
- V. Makarov, G. Manina, K. Mikusova, U. Mollmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni, A. P. Lucarelli, A. Milano, E. De Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801–804 CrossRef CAS PubMed.
- V. Makarov, B. Lechartier, M. Zhang, J. Neres, A. M. van der Sar, S. A. Raadsen, R. C. Hartkoorn, O. B. Ryabova, A. Vocat, L. A. Decosterd, N. Widmer, T. Buclin, W. Bitter, K. Andries, F. Pojer, P. J. Dyson and S. T. Cole, EMBO Mol. Med., 2014, 6, 372–383 CrossRef CAS PubMed.
- R. Tiwari, P. A. Miller, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2015, 6, 128–133 CrossRef CAS PubMed.
- P. S. Shirude, R. Shandil, C. Sadler, M. Naik, V. Hosagrahara, S. Hameed, V. Shinde, C. Bathula, V. Humnabadkar, N. Kumar, J. Reddy, V. Panduga, S. Sharma, A. Ambady, N. Hegde, J. Whiteaker, R. E. McLaughlin, H. Gardner, P. Madhavapeddi, V. Ramachandran, P. Kaur, A. Narayan, S. Guptha, D. Awasthy, C. Narayan, J. Mahadevaswamy, K. G. Vishwas, V. Ahuja, A. Srivastava, K. R. Prabhakar, S. Bharath, R. Kale, M. Ramaiah, N. R. Choudhury, V. K. Sambandamurthy, S. Solapure, P. S. Iyer, S. Narayanan and M. Chatterji, J. Med. Chem., 2013, 56, 9701–9708 CrossRef CAS PubMed.
- F. Wang, D. Sambandan, R. Halder, J. Wang, S. M. Batt, B. Weinrick, I. Ahmad, P. Yang, Y. Zhang, J. Kim, M. Hassani, S. Huszar, C. Trefzer, Z. Ma, T. Kaneko, K. E. Mdluli, S. Franzblau, A. K. Chatterjee, K. Johnsson, K. Mikusova, G. S. Besra, K. Futterer, S. H. Robbins, S. W. Barnes, J. R. Walker, W. R. Jacobs Jr. and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2510–E2517 CrossRef CAS PubMed.
- CDC, 2013.
- J. A. Leeds, Cold Spring Harbor Perspect. Med., 2016, 6, a025445 CrossRef PubMed.
- G. G. Zhanel, A. J. Walkty and J. A. Karlowsky, Can. J. Infect. Dis. Med. Microbiol., 2015, 26, 305–312 Search PubMed.
- D. J. Biedenbach, J. E. Ross, S. D. Putnam and R. N. Jones, Antimicrob. Agents Chemother., 2010, 54, 2273–2275 CrossRef CAS PubMed.
- D. N. Shah, F. S. Chan, N. Kachru, K. P. Garcia, H. E. Balcer, A. P. Dyer, J. E. Emanuel, M. D. Jordan, K. T. Lusardi, G. Naymick, R. S. Polisetty, L. Sieman, A. M. Tyler, M. L. Johnson and K. W. Garey, SpringerPlus, 2016, 5, 1224 CrossRef PubMed.
- R. J. Vickers, G. Tillotson, E. J. Goldstein, D. M. Citron, K. W. Garey and M. H. Wilcox, Int. J. Antimicrob. Agents, 2016, 48, 137–143 CrossRef CAS PubMed.
- E. J. Goldstein, D. M. Citron, K. L. Tyrrell and C. V. Merriam, Antimicrob. Agents Chemother., 2013, 57, 4872–4876 CrossRef CAS PubMed.
- R. J. Vickers, G. S. Tillotson, R. Nathan, S. Hazan, J. Pullman, C. Lucasti, K. Deck, B. Yacyshyn, B. Maliakkal, Y. Pesant, B. Tejura, D. Roblin, D. N. Gerding, M. H. Wilcox and D. S. G. Co, Lancet Infect. Dis., 2017, 17, 735–744 CrossRef CAS PubMed.
- M. C. Rea, C. S. Sit, E. Clayton, P. M. O'Connor, R. M. Whittal, J. Zheng, J. C. Vederas, R. P. Ross and C. Hill, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9352–9357 CrossRef CAS PubMed.
- M. C. Rea, D. Alemayehu, R. P. Ross and C. Hill, J. Med. Microbiol., 2013, 62, 1369–1378 CrossRef CAS PubMed.
- M. C. Rea, A. Dobson, O. O'Sullivan, F. Crispie, F. Fouhy, P. D. Cotter, F. Shanahan, B. Kiely, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(Suppl 1), 4639–4644 CrossRef CAS PubMed.
- L. B. Rice, J. Infect. Dis., 2008, 197, 1079–1081 CrossRef PubMed.
- M. Cei, R. Pardelli, S. Sani and N. Mumoli, Clin. Exp. Med., 2014, 14, 77–82 CrossRef CAS PubMed.
- L. Chen, R. Todd, J. Kiehlbauch, M. Walters and A. Kallen, MMWR Morb. Mortal. Wkly. Rep., 2017, 66, 33 CrossRef PubMed.
- B. L. Hollenbeck and L. B. Rice, Virulence, 2012, 3, 421–433 CrossRef PubMed.
- H. Wang, M. Lee, Z. Peng, B. Blazquez, E. Lastochkin, M. Kumarasiri, R. Bouley, M. Chang and S. Mobashery, J. Med. Chem., 2015, 58, 4194–4203 CrossRef CAS PubMed.
- S. M. Purrello, J. Garau, E. Giamarellos, T. Mazzei, F. Pea, A. Soriano and S. Stefani, J. Glob. Antimicrob. Resist., 2016, 7, 178–186 CrossRef CAS PubMed.
- T. Schneider, T. Kruse, R. Wimmer, I. Wiedemann, V. Sass, U. Pag, A. Jansen, A. K. Nielsen, P. H. Mygind, D. S. Raventos, S. Neve, B. Ravn, A. M. Bonvin, L. De Maria, A. S. Andersen, L. K. Gammelgaard, H. G. Sahl and H. H. Kristensen, Science, 2010, 328, 1168–1172 CrossRef CAS PubMed.
- X. Cao, Y. Zhang, R. Mao, D. Teng, X. Wang and J. Wang, Appl. Microbiol. Biotechnol., 2015, 99, 2649–2662 CrossRef CAS PubMed.
- H. Y. Lee, C. L. Chen, S. R. Wu, C. W. Huang and C. H. Chiu, Crit. Care Med., 2014, 42, 1081–1088 CrossRef CAS PubMed.
- M. Magret, T. Lisboa, I. Martin-Loeches, R. Manez, M. Nauwynck, H. Wrigge, S. Cardellino, E. Diaz, D. Koulenti, J. Rello and E.-V. C. S. Group, Crit. Care, 2011, 15, R62 CrossRef PubMed.
- N. Sinha, M. Niazi and D. Lvovsky, Case Rep. Infect. Dis., 2014, 2014, 705279 Search PubMed.
- U. Waack, T. L. Johnson, K. Chedid, C. Xi, L. A. Simmons, H. L. T. Mobley and M. Sandkvist, Front. Cell. Infect. Microbiol., 2017, 7, 380 CrossRef PubMed.
- B. W. Corey, M. G. Thompson, L. E. Hittle, A. C. Jacobs, E. A. Asafo-Adjei, W. M. Huggins, R. J. Melander, C. Melander, R. K. Ernst and D. V. Zurawski, ACS Infect. Dis., 2017, 3, 62–71 CrossRef CAS PubMed.
- W. M. Huggins, B. M. Minrovic, B. W. Corey, A. C. Jacobs, R. J. Melander, R. D. Sommer, D. V. Zurawski and C. Melander, ACS Med. Chem. Lett., 2017, 8, 27–31 CrossRef CAS PubMed.
- J. M. Regeimbal, A. C. Jacobs, B. W. Corey, M. S. Henry, M. G. Thompson, R. L. Pavlicek, J. Quinones, R. M. Hannah, M. Ghebremedhin, N. J. Crane, D. V. Zurawski, N. C. Teneza-Mora, B. Biswas and E. R. Hall, Antimicrob. Agents Chemother., 2016, 60, 5806–5816 CrossRef CAS PubMed.
- R. T. Schooley, B. Biswas, J. J. Gill, A. Hernandez-Morales, J. Lancaster, L. Lessor, J. J. Barr, S. L. Reed, F. Rohwer, S. Benler, A. M. Segall, R. Taplitz, D. M. Smith, K. Kerr, M. Kumaraswamy, V. Nizet, L. Lin, M. D. McCauley, S. A. Strathdee, C. A. Benson, R. K. Pope, B. M. Leroux, A. C. Picel, A. J. Mateczun, K. E. Cilwa, J. M. Regeimbal, L. A. Estrella, D. M. Wolfe, M. S. Henry, J. Quinones, S. Salka, K. A. Bishop-Lilly, R. Young and T. Hamilton, Antimicrob. Agents Chemother., 2017, 61, e00954-17 CrossRef PubMed.
- M. Thandar, R. Lood, B. Y. Winer, D. R. Deutsch, C. W. Euler and V. A. Fischetti, Antimicrob. Agents Chemother., 2016, 60, 2671–2679 CrossRef CAS PubMed.
- T. B. Nielsen, P. Pantapalangkoor, B. M. Luna, K. W. Bruhn, J. Yan, K. Dekitani, S. Hsieh, B. Yeshoua, B. Pascual, E. Vinogradov, K. M. Hujer, T. N. Domitrovic, R. A. Bonomo, T. A. Russo, M. Lesczcyniecka, T. Schneider and B. Spellberg, J. Infect. Dis., 2017, 216, 489–501 CrossRef PubMed.
- P. D. Cotter, R. P. Ross and C. Hill, Nat. Rev. Microbiol., 2013, 11, 95–105 CrossRef CAS PubMed.
- M. G. Ghequire, J. Dingemans, J. P. Pirnay, D. De Vos, P. Cornelis and R. De Mot, MicrobiologyOpen, 2014, 3, 875–884 CrossRef CAS PubMed.
- H. Ling, N. Saeidi, B. H. Rasouliha and M. W. Chang, FEBS Lett., 2010, 584, 3354–3358 CrossRef CAS PubMed.
- A. DiGiandomenico, A. E. Keller, C. Gao, G. J. Rainey, P. Warrener, M. M. Camara, J. Bonnell, R. Fleming, B. Bezabeh, N. Dimasi, B. R. Sellman, J. Hilliard, C. M. Guenther, V. Datta, W. Zhao, C. Gao, X. Q. Yu, J. A. Suzich and C. K. Stover, Sci. Transl. Med., 2014, 6, 262ra155 CrossRef PubMed.
- X. Q. Yu, G. J. Robbie, Y. Wu, M. T. Esser, K. Jensen, H. I. Schwartz, T. Bellamy, M. Hernandez-Illas and H. S. Jafri, Antimicrob. Agents Chemother., 2017, 61, e01020-16 CrossRef PubMed.
- M. H. Wilcox, D. N. Gerding, I. R. Poxton, C. Kelly, R. Nathan, T. Birch, O. A. Cornely, G. Rahav, E. Bouza, C. Lee, G. Jenkin, W. Jensen, Y. S. Kim, J. Yoshida, L. Gabryelski, A. Pedley, K. Eves, R. Tipping, D. Guris, N. Kartsonis, M. B. Dorr, I. Modify and M. I. Investigators, N. Engl. J. Med., 2017, 376, 305–317 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |