
Rapid detection of multiple respiratory viruses based on microfluidic isothermal amplification and a real-time colorimetric method†‡
 §abc
§abc
Abstract
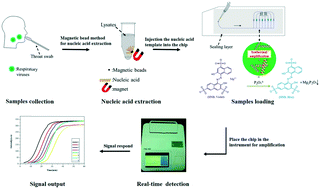
Respiratory viruses are major threats causing development of acute respiratory tract infections, which are common causes of illness and death throughout the world. Here, an integrated microsystem based on real-time colorimetry was developed for diagnosing multiple respiratory viruses. The microsystem employed magnetic beads for nucleic acid extraction and an eight-channel microfluidic array chip integrated with a loop-mediated isothermal amplification system for point-of-care screening of respiratory viruses. The overall detection process (including sample collection, nucleic acid extraction, sample loading, real-time detection, and signal output) could be completed within 1 h. Our results show that the developed method could specifically recognize influenza A virus subtypes (H1N1, H3N2, H5N1, and H7N9), influenza B virus, and human adenoviruses. The results obtained with 109 clinical samples indicate that the developed method has high specificity (100%, confidence interval 94.9–100.0) and sensitivity (96%, confidence interval 78.1–99.9). The integration of magnetic bead-based pre-treatment techniques and microfluidic isothermal amplification provides an effective solution for rapidly detecting etiological agents of respiratory diseases. The strategy of using a closed chip system and real-time colorimetry reduced aerosol contamination and ensured the accuracy of the results. The developed method provides an effective alternative for rapid point-of-care screening for viruses that cause respiratory disease syndromes and further aids in accurate and timely detection to control and prevent the spread of respiratory diseases caused by such pathogens.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

