
A fully-integrated and automated testing device for PCR-free viral nucleic acid detection in whole blood†
 b
Shana O.
Kelley
*abce
b
Shana O.
Kelley
*abce
Abstract
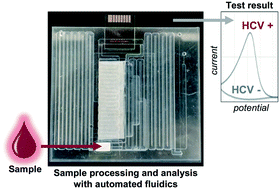
Integrated devices for automated nucleic acid testing (NAT) are critical for infectious disease diagnosis to be performed outside of centralized laboratories. The gold standard methods for NAT are enzymatic amplification methods like the polymerase chain reaction that typically require expensive equipment and highly-trained personnel, limiting use in low-resource settings. A low-cost, integrated, rapid, portable and user-friendly point-of-care (POC) nucleic acid diagnostic device will improve the accessibility of NAT. Here, we present a fully integrated and simple-to-use POC device operated by a passive fluidic method that is able to perform a sequential multi-step assay to detect viral nucleic acids in blood. This simple device enabled the rapid detection of hepatitis C virus in blood in approximately 30 minutes with minimal sample handling by the user.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

