
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic label-free selection of mesenchymal stem cell subpopulation during culture expansion extends the chondrogenic potential in vitro†
Lu
Yin‡
 a,
Yingnan
Wu‡
b,
Zheng
Yang
bc,
Ching Ann
Tee
bc,
Vinitha
Denslin
bc,
Zhangxing
Lai
d,
Chwee Teck
Lim
aef,
Eng Hin
Lee
*bc and
Jongyoon
Han
*ag
a,
Yingnan
Wu‡
b,
Zheng
Yang
bc,
Ching Ann
Tee
bc,
Vinitha
Denslin
bc,
Zhangxing
Lai
d,
Chwee Teck
Lim
aef,
Eng Hin
Lee
*bc and
Jongyoon
Han
*ag
aBioSystems and Micromechanics Interdisciplinary Research Group, Singapore-MIT Alliance in Research and Technology, 1 Create Way, #04-13/14, 138602 Singapore. E-mail: jyhan@mit.edu
bNUS Tissue Engineering Program, Life Sciences Institute, National University of Singapore, 27 Medical Drive1, DSO (Kent Bridge) Building, Level 4, 117510 Singapore. E-mail: dosleeeh@nus.edu.sg
cDepartment of Orthopaedic Surgery, National University of Singapore, 1E Kent Ridge Road, NUHS Tower block 11, 119288 Singapore
dDepartment of Mechanical Engineering, National University of Singapore, 9 Engineering Drive 1, Block EA, #07-08, 117575 Singapore
eMechanobology Institute Singapore, National University of Singapore, 5A Engineering Drive 1, T-Lab, #10-01, 117411 Singapore
fDepartment of Biomedical Engineering, National University of Singapore, 4 Engineering Drive 3, Engineering Block 4, #04-08, 117583 Singapore
gDepartment of Electrical Engineering and Computer Science, and, Department of Biological Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
First published on 14th February 2018
Abstract
Mesenchymal stem cells (MSCs) have been shown as potential candidates for cell-based therapies for a diverse range of tissue regenerative applications. Therapeutic use of MSCs usually requires culture expansion, which increases the heterogeneity of MSCs in vitro, thus affecting the potency of the MSCs for more specific indications. The capacity for identifying and isolating special subsets of MSCs for treatment of specific diseases therefore holds great clinical significance. An important therapeutic application of MSC is for the regeneration of cartilage tissue. We and others have previously developed label-free microfluidic means to isolate subpopulations of culture expanded MSCs based on distinct biophysical characteristics. Here we utilize a spiral micro-channel device to separate culture expanded MSCs into five subgroups according to cell size, and study their proliferation and chondrogenesis at early, middle and late passages. Results show that in all passages, the medium-size subpopulation (cell size of 17–21 μm), compared to other subpopulations, displays significantly higher proliferation rate and chondrogenic capacity in terms of cartilage extracellular matrix formation. Also, the small cell subpopulation (average cell size of 11–12 μm) shows lower viability, and large cell subpopulation (average cell size 23–25 μm) expresses higher level of senescence-associated β-galactosidase. Finally, we show that repeated microfluidic exclusion of MSCs larger than 21 μm and smaller than 17 μm at every passage during continuous culture expansion result in selected MSCs with faster proliferation and better chondrogenic potential as compared to MSC derived from conventional expansion approach. This study demonstrates the significant merit and utility of size-based cell selection for the application of MSCs in cartilage regeneration.
Introduction
Human mesenchymal stem cells (MSCs) have long been considered as promising candidates in tissue regenerative therapies.1–3 MSCs not only function as self-renewable multipotent stem cells to replace damaged tissue,4,5 but also regulate immune responses to limit immune mediated disorder6,7 and produce bioactive molecules to promote tissue regeneration.8,9 Numerous studies have shown the potential of MSCs in the treatment of a diverse range of diseases.10–24Clinical use of MSCs usually requires extensive culture expansion to acquire therapeutically relevant numbers, regardless of whether the cells function through direct engraftment or via its trophic factors,25 because the yield from primary tissue is insufficient.26 Studies on MSC expansion have mainly focused on the viability27–29 and scalability30–32 of the cells, however, culture expanded MSCs have been shown to be morphologically and functionally heterogeneous.33 The in vitro heterogeneity may be implicitly related to the inherent phenotype and potency variability of MSCs derived from different tissue microenvironments,34 which could have further perpetuated under different culture conditions.35 Studies have suggested that subgroups of MSCs with specific potency or production of specific secretome could be more therapeutically effective in the regeneration of specific tissues, when compared to the total heterogeneous population.36,37 Thus, the potential of generating large quantities of MSCs manipulated towards specific states for treatment of specific diseases holds great clinical significance.
The need for large quantities of MSCs enhanced with strong chondrogenic potential for therapeutic use is compelling. MSC-based cartilage regeneration is one of the most well-established therapeutic applications, for the treatment of trauma-induced cartilage lesion, as well as degenerative osteoarthritic cartilage.38,39 Cell numbers required for implantation in cartilage repair are large, mostly in the range of millions to a hundred million,40 which entail extensive expansion. However, as MSCs grow larger over time during expansion,41 there is a tendency of decreasing chondrogenic potential.42 As a result, the therapeutic efficacy of MSCs for regenerating cartilage would greatly benefit from manipulating them towards “chondrogenic competent” state during extensive culture expansion.
A seemingly straightforward way to achieve this is to induce MSC commitment to the desired state, however it is practically difficult, not only because of the tight restrictions on chemical treatment to MSCs for therapeutic use, but also because the favourable states of MSCs for different therapeutic uses are difficult to standardise and maintain in culture. An alternative approach is to isolate the “chondrogenic competent” subpopulation of MSCs from the heterogenous population after culture expansion. We and others have previously characterized MSC subpopulations using biophysical parameters,42,43 and developed high-throughput label-free microfluidic technique for the isolation of MSC subpopulations based on cell size differences.36,44 However, the remaining problem is the uncertainty in the ratio of targeted subpopulation cell numbers to the total population cell numbers at the end of culture expansion. It is very likely that the “chondrogenic competent” subpopulation would drastically reduce due to the increasing heterogeneity along with extensive expansion41,42 which overwhelms the culture conditions. A possibly more efficacious approach is to enrich the “chondrogenic competent” subpopulation over the extended expansion period by repeated subpopulation selection, to maintain the homogeneity of culture throughout expansion process.
The first step of this approach was to identify the subpopulation of MSCs that's “chondrogenic competent”. We first separated culture expanded human bone marrow-derived MSCs into five size-dependent subgroups through serial sorting using a spiral micro-channel device,36,45 based on Dean flow fractionation principle.46–48 Briefly, cells flowing in micro-channel experience shear-induced and wall-induced lift forces in the cross-sectional plane perpendicular to the flow direction. The spiral geometry of the micro-channel adds a Dean drag force to the cells due to centrifugal acceleration. As a consequence of the lift and drag forces, while the cells flow through the spiral micro-channel, they also migrate on the cross-sectional plane of the channel, and reach an equilibrium position where the net force is zero. This equilibrium position is determined by cell size when other conditions (such as channel dimensions, flow rate, density and viscosity of the medium, etc.) are fixed. Also, the equilibrium position of a cell alters when it flows through the spiral micro-channel at different flow rates. This principle enabled us to use a spiral micro-channel device with two outlets to sort five size-dependent subgroups of MSCs one at a time at different flow rates. We then profiled the proliferation and chondrogenesis capacity of each subgroup at early, middle and late passages during culture expansion, and identified the “chondrogenic competent” subpopulation by considering the chondrogenic capacity, proliferation rate, as well as the ratio of cell number in the subpopulation to total population. This subpopulation was selectively expanded using the spiral micro-channel device to remove other MSCs of bigger or smaller size range from the culture repeatedly at every passage/subculture over 2 month culture expansion period. The efficacy of this selective expansion to enrich proliferative “chondrogenic competent” MSC was compared to normal expansion approach. We found that the most competent MSCs for cartilage repair were medium-size cells in culture between 17 to 21 μm, and selective expansion maintained the proliferation rate and chondrogenic potential of the expanded cells for significantly prolonged culture period, and produced higher cell yield as compared to the normal MSC culture expansion approach.
Materials and methods
MSC culture
Bone marrow derived human MSCs were purchased from Lonza Pte. Ltd. (Lonza PT 2501) and RoosterBio Inc. MSCs from four donors (Lonza Lot no. 000047198, 0000482966 and 0000483199, RoosterBio Lot no. 00022) were culture expanded in DMEM with 1% Glutamax, 10% FBS, 1% Pen Strep, and 1 ng ml−1 FGF-2 (Thermo Fisher Scientific Pte. Ltd., Singapore) from the original vial for 6–7 doublings before cryopreservation. The cryopreserved cells were then thawed and culture expanded in 175 cm2 cell culture flask (Thermo Fisher Scientific Pte. Ltd., Singapore) at seeding density of 1.14 × 103 cell per cm2. Since continuous culture expansion of MSCs started from 6–7 doublings in this study, we define MSCs underwent 8–10 total doublings in culture as “early passage”, 13–15 doublings as “middle passage”, 18–20 doublings as “late passage” (all doubling numbers were counted by denoting the cells from the original commercial source as 0 doubling). The population doubling number (PD) was estimated from the formula n = 3.32 × (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) EC − logIC) + X, where n is the PD at end of a given subculture, EC is the cell count at end of subculture, IC is the cell count at beginning of subculture, and X is the PD at beginning of subculture.
EC − logIC) + X, where n is the PD at end of a given subculture, EC is the cell count at end of subculture, IC is the cell count at beginning of subculture, and X is the PD at beginning of subculture.
MSCs were harvested at 90% confluency for assays and subcultured by 2 minute incubation with 0.05% trypsin-EDTA (Thermo Fisher Scientific Pte. Ltd., Singapore) at 37 °C. The cell count and viability were measured using disposable haemocytometer (INCYTO Co. Ltd. Korea) after 1:1 mixing with Trypan Blue (Thermo Fisher Scientific Pte. Ltd., Singapore). The seeding density used for subculture was 1.14 × 103 cell per cm2.
MSC colonies were formed by seeding unsorted (S0) and sorted subgroups (S1–5) of MSCs in 6-well plate at seeding density of 500 cell per well. After 5 days culture, the colonies formed was Giemsa stained and counted under light microscope. The colony forming efficiency (CFE), defined as the percentage of cells inoculated at low density that give rise to colonies, was then calculated as CFE = Nc/Ni × 100%, where Nc is the number of colonies formed, Ni is the number of cells inoculated.
Formation of cartilage pellet was achieved by seeding MSCs in U-bottom 96-well plate (NEST Biotechnology Co. Ltd. China) with 200 μl of chondrogenic medium (high glucose DMEM supplemented with 10−7 M dexamethasone 1% ITS+ Premix supplement 50 mg ml−1 ascorbic acid 1 mM sodium pyruvate and 4 mM proline and 10 ng ml−1 TGFβ-3)49 at density of 105 cell per well and centrifuged at 300g for 5 minutes. Medium was changed every alternative day for 3 weeks before the pellets were imaged and weighed.
Spiral micro-channel device design and fabrication
The spiral micro-channel device was designed and fabricated in the same way as previously described.45 Briefly, it was casted from polydimethylsiloxane (10:1 mixture of base and curing agent. Sylgard 184, Dow Corning Inc., USA) using a micro-milled aluminium mold (Whits Technologies Inc., Singapore). The device had one inlet and two outlets and 8-loops with radius decreasing from 12 mm to 4 mm (from inlet to outlet). The cross-section of the channel was trapezoidal with 580 μm width, 85 μm/133 μm inner/outer height respectively (Fig. 1A). The ratio of the inner outlet width to outer outlet width was about 5:7. The inner to outer fluid ratio was about 1:2 at input rate of 1.5 mL min−1.
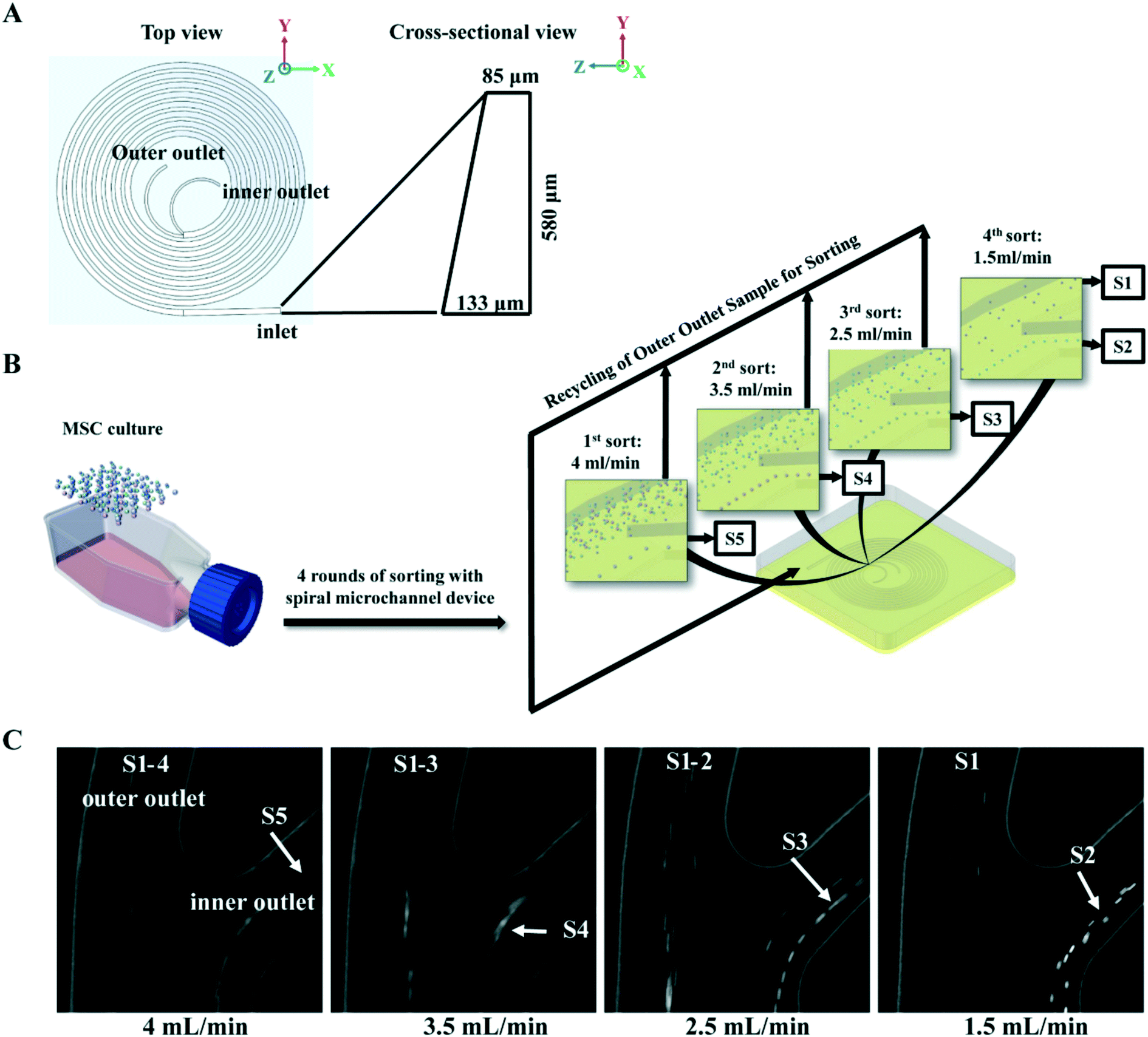 | ||
| Fig. 1 Isolation of size-dependent MSC subgroups with spiral micro-channel device. (A) Schematic of an 8-loop spiral micro-channel device with trapezoidal cross-section. (B) Flow chart of MSC inertial sorting strategy. Culture expanded MSCs are sequentially separated into five subgroups, S5–S1 with decreasing size, by four rounds of sorting at different flow rates. Colored spheres represent MSCs with various sizes. (C) Phase contrast images captured by high speed camera on separation of MSC subgroups at the branching point before the outlets of the spiral micro-channel device. Arrows indicate the S1–S5 subgroups being isolated at different flow rates. | ||
MSC sorting with spiral micro-channel device
Buffers and MSCs suspensions in 4 mL medium (0.2–1 million MSCs per mL) were loaded in syringes (Thermo Fisher Scientific Pte. Ltd., Japan), and connected to the inlet of spiral micro-channel device with precision tips (Nordson Corp. USA) and Tygon tubing (Spectra Teknik Pte, Ltd, Singapore). Tygon tubings were also connected to the two outer outlets for collection of sorted subgroups. A syringe pump (PHD 2000, Harvard Apparatus Inc., USA) was used for accurate pumping. Before sorting, 1% poloxamer 188 (Sigma-Aldrich Co. LLC. Singapore) in Milli-Q water was pumped into the spiral micro-channel device for 2 min at 1 mL min−1 to prevent cell sticking followed by 1× PBS to wash off the water.In the MSC subgroup characterization experiment, MSCs at each passage were first pumped into the device at 4 mL min−1, and subgroup S5 was collected from the inner outlet. Then subgroups S4, S3 and S2 were sequentially collected from the inner outlet by pumping the MSCs collected from outer outlet back into the device at 3.5 mL min−1, 2.5 mL min−1, and 1.5 mL min−1 respectively. In the last round of sorting at 1.5 mL min−1, subgroup S1 was collected from the outer outlet while S2 was collect from inner (Fig. 1B).
In the selective MSC culture expansion experiment, MSCs were expanded normally to passage 3, and then the same number of cells were cultured in parallel using either selective culture expansion or conventional culture expansion up to passage 10 in 2 months. For selective MSC culture, cells at each passage were first pumped into the device at 3.5 mL min−1 to remove large cell population (>21 μm) from the inner outlet. MSCs collected from the outer outlet were pumped through the device again at 1.5 mL min−1 to further remove small cell population (<17 μm) from the outer outlet (Fig. 6A).
Real-time cell separation within the spiral micro-channel device was recorded by an inverted microscope (IX71, Olympus Co., Japan) equipped with a high-speed CCD camera (Phantom v9, Vision Research Inc., USA). The recorded sorting videos were processed for snapshot images using ImageJ software (NIH, USA).
Estimation of theoretical total MSC yield after continuous culture expansion
In the selective MSC culture expansion experiment, only a fraction (0.5–1 million) of harvested MSCs were passaged for subculture, and the theoretical total cell yield (TTC), assuming all cells were passaged, was estimated from the formula c = EC × Y/IC × α, where c is the TTC at the end of a passage, EC is the cell count at the end of a give subculture, Y is the TTC at the end of previous passage, IC is the cell count seeded at the beginning of the subculture, α is the proportion of 17–21 μm MSC over the total number of MSC at this passage. The value of α equals to 1 when MSCs are cultured without subpopulation selection, and equals to the recovery rate of 17–21 μm MSCs at each passage (Table S2†) during selective culture expansion.MSC size analysis
MSCs were measured by Moxi Z Mini automated cell counter with type M cassette (Orflo Technologies Inc., USA). MATLAB (The MathWorks Inc., USA) was then used to calculate the statistics from the original CSV files generated by the cell counter. MSCs with measured diameter less than 8 μm were considered as artifacts and excluded from study. An imaging-based MATLAB algorithm using circle Hough Transforms to identify cells (developed by Prof. Krystyn Van Vliet's group from MIT) was also used to measure MSC sizes, and validate the readings from Moxi Z Mini (Table S1†).RT-PCR
RT-PCRs for collagen I (Col I), collagen II (Col II), collagen X (Col X), aggrecan (Aggr), Ki-67, and GAPDH were conducted using the SYBR green system according to previous published protocol.50 Expression of Col I, Col II, Col X, Aggr and Ki-67 were normalized to GAPDH. Col I, Col II, Col X and Aggr were presented as fold changes with reference to the undifferentiated MSCs.Histology staining
Formalin fixed cartilage pellet samples were dehydrated with xylene, and embedded in paraffin blocks. Sections of 5 μm thickness were cut with a microtome, and mounted on polylysine-coated slides. The paraffin sections were de-waxed and rehydrated in ethanol using standard protocol before staining. For detection of chondrogenesis, the sections were stained with Alcian Blue (Sigma-Aldrich Pte. Ltd., Singapore) for 30 minutes, washed with 1× PBS and counter-stained with Nuclear Fast Red (Sigma-Aldrich Pte. Ltd., Singapore) for 5 minutes.Senescence assay
Unsorted MSCs (S0) and sorted subgroups (S1–5) were cultured in 6-well plate for 3–4 days at seeding density of 3 × 104 cell per well. Senescence β-galactosidase staining kit (Cell Signalling Technology, USA) was used to stain the cells according to the manufacturer's guide. The cells were then stained for nucleus with Hoechst 33253 solution (Thermo Fisher Scientific Pte. Ltd., USA, Cat. no. 86145) at 1 μg mL−1 in PBS for 5 min in dark. An inverted microscope (U-TB 190, Olympus Co., Japan) equipped with a CCD camera (Retiga 2000R, Qimaging Inc., Canada.) was used to capture bright field images with senescence staining (the green colour precipitate resulted from cleavage of X-gal substrate) and fluorescent images with nucleus staining at the same spot. Five to ten random spots were imaged for each subgroup, and the images were processed with MATLAB (The MathWorks Inc., USA) for computing the senesces index (SI). SI was defined as the average level of senescence signal per cell in all images taken from the same subgroup. The senescence staining appeared to be green/blue colour in the bright field images, and the level of greenness/blueness indicated the level of senescence. The positive staining areas in the images were reflected as areas lack of red color component. Thus, the positive staining areas were selected based on the criteria that the red color intensity in the area was lower than 65 (maximum 255 in unit8 RGB image format), and lower than both green and blue content; meanwhile, green and blue color intensities are higher than 50. Extremely small areas fulfilling the criteria were ignored as artefacts. Next, the total level of senescence in an image was calculated as the sum of the complement of red intensity (255 – red intensity) in all the selected areas. The total number of cells in the image was computed based on the number of Hoechst positive areas in the corresponding fluorescent image. Then SI of the image was computed as the ratio of total senescence level to the total cell number in the image. The final SI of a subgroup of MSCs was normalized by subtracting the SI of the total MSCs at the beginning of the culture expansion.
FACS-based cell cycle analysis
Unsorted MSCs (S0) and sorted subgroups (S1–5) were suspended in 500 μl 1× PBS at 0.1 million cells per mL. Five μl of 50× SYBR green stain (Thermo Fisher Scientific Pte. Ltd., USA, Cat. no. S7563) were added to the suspension and incubated on ice for 15 min in dark. The stained cells were then washed with 1 mL 1× PBS for three times and filtered with 40 μm Nylon mesh cell strainer (Thermo Fisher Scientific Pte. Ltd., China) before loading to Accuri C6 flow cytometer (BD biosciences, Singapore). Ten thousand events were recorded for each measurement, and green fluorescence signal was detected as a measurement of DNA content. Flow data were processed by CFlow Plus software (BD biosciences, Singapore) for analysing cells under mitosis.Results and discussion
Microfluidic sorting of size-dependent MSC subgroups
To identify the most competent subpopulation of culture expanded MSCs for chondrogenesis, total MSCs population at early, middle and late passages (refer to Materials and methods for the definition of early, middle and late passage) were sorted respectively into five subgroups based on cell size. Original population of MSCs (Pre-sorted, S0) were pumped into a spiral micro-channel device (Fig. 1A), and the subgroups (S1–S5, S1: smallest size) were sequentially collected from the outlets of the device after four rounds of sorting (Fig. 1B). Separation of the size-dependent MSC subgroups at the branching point of the spiral micro-channel was recorded with high speed camera. The cell size difference among the subgroups can be observed in the images (Fig. 1C). Analysis showed that the average size of MSCs increased from early to late passages, and the total MSCs were successfully separated into subgroups with distinct size differences (p < 0.05) at all passages (Fig. 2A). The biggest size difference between adjacent subgroups was 5–7 μm, occurred between S1 and S2; the smallest difference was 1–3 μm, between S4 and S5. All the cell sizes were measured using an automated Coulter counter in this study. A comparison between the cell size measured by the Coulter counter and an automated imaging-based algorithm is shown in Table S1.† Majority of the MSCs belonged to subgroups S2 and S3, corresponding to average diameter of 17–21 μm, and comprising 60–70% of the total population in all passages. The composition of S2 and S3 decreased from early to late passage, while S4 and S5 increased, which was consistent with the increasing average cell size. | ||
| Fig. 2 Cell size and composition of size-dependent MSC subgroups. (A) Average sizes of total MSC (S0) and MSC subgroups (S1–5) at early, middle and late passages during culture expansion. The three horizontal lines in each box represent first quartile, median, and third quartile. The error bars represent standard deviations. The dots represent the means. Statistical significance is determined by Student's t-test, * indicates p < 0.05, ** indicates p < 0.001. (B) Cell count composition of each subgroups in total population at early, middle and late passages during culture expansion. | ||
Profiling proliferation of size-dependent MSC subgroups during culture expansion
The efficiency of producing MSCs in large quantity is determined by the proliferation rate of the cells. We assayed the proliferation rate of the five sorted MSC subgroups in early, middle and late passages by PCR analysis of proliferation marker Ki-67 (Fig. 3A). Results showed that the proliferation significantly reduced from middle to late passage, and was mainly contributed by the decrease in subgroup S4 and S5. Subgroups S1–3 had faster overall proliferation rate as compared to S4 and S5 in all passages. Notably, proliferation of S2 and S3 subgroups was maintained from early to late passages.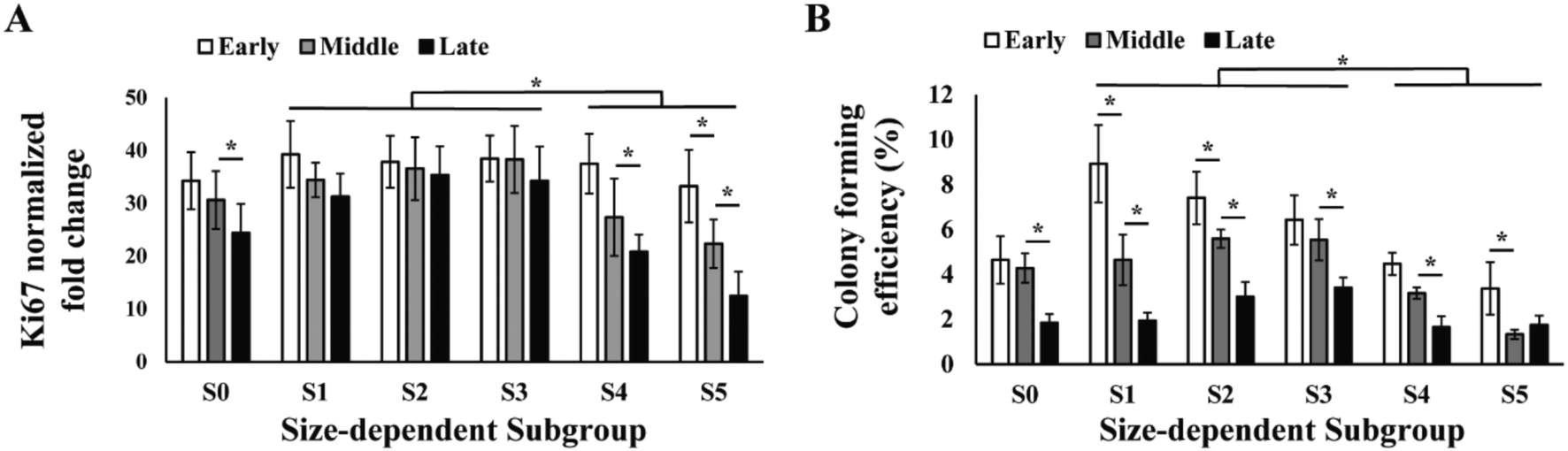 | ||
| Fig. 3 Proliferation of size-dependent MSC subgroups. (A) RT-PCR analysis on Ki-67 and (B) colony forming efficiency of total MCS (S0) and MSC subgroups (S1–5) at early, middle and late passages during culture expansion. All error bars represent standard deviations. Statistical significance is determined by Student's t-test, * indicates p < 0.05. | ||
Similar to the proliferation rate, colony forming efficiency (CFE) of MSCs also decreased from 5% in early passage to 2% in late passage, and subgroups S1–3 had significantly higher overall CFE comparing to S4 and S5 in all passages (Fig. 3B). It was also observed that the subgroups with highest CFE gradually shifted from S1 in early passage to S3 in late passage (Fig. 3B).
Profiling chondrogenesis of size-dependent MSC subgroups during culture expansion
MSCs with stronger chondrogenic capacity are preferred in the therapeutic use for cartilage repair. We investigated the chondrogenic capacity of the sorted MSC subgroups in early, middle and late passages by inducing chondrogenesis in pellet culture. We observed that S2 and S3 were able to form spherical cartilage pellets in culture after 3 weeks. However, S1, S4 and S5 either failed to form spherical pellets or formed much smaller pellets (Fig. 4A). The sizes and weights of the cartilage pellets were measured. Results showed that the pellets formed by S2 and S3 were significantly larger and heavier as compared to those of other subgroups over all passages (Fig. 4B and C).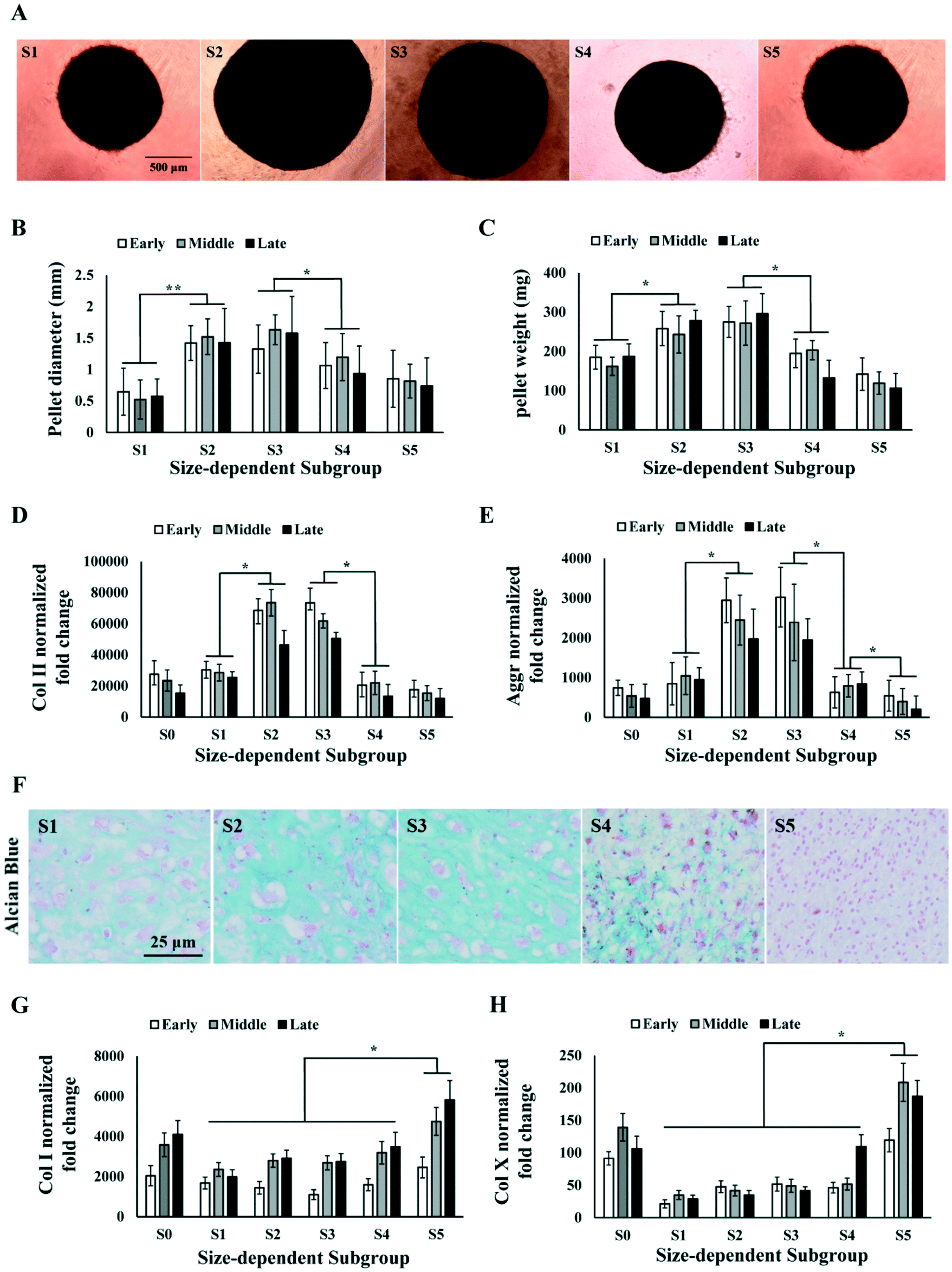 | ||
| Fig. 4 Chondrogenesis of size-dependent MSC subgroups. (A) Images of cartilage pellets formed by MSC subgroups after 3 weeks of differentiation. Scale bar: 500 μm. (B and C) Measurement of cartilage pellet size (B) and weight (C) formed by total MSC (S0) and MSC subgroups (S1–5) at early, middle and late passages. (D and E) RT-PCR analysis on Col II (D) and Aggrecan (E) level in cartilage pellet formed by MSC (S0) and subgroups (S1–5) at early, middle and late passages. (F) Alcian blue staining of cartilage pellets formed by MSC subgroups. Scale bar: 25 μm. (G and H) RT-PCR analysis on Col I (G) and Col X (H) level in cartilage pellet formed by MSC (S0) and subgroups (S1–5) at early, middle and late passages. All error bars represent standard deviations. Statistical significance is determined by Student's t-test, * indicates p < 0.05, ** indicates p < 0.001. | ||
The cartilage pellets formed by different MSC subgroups were processed for PCR and histology analysis on chondrogenic markers. PCR results showed that the average expression level of collagen II (Col II) and aggrecan (Aggr) over all passages were significantly higher in S2 and S3 comparing to S1, S4 and S5 (Fig. 4D and E), which was consistent with the observed difference in cartilage pellet sizes. Alcian Blue staining on the cartilage pellet sections also indicated the occurrence of chondrogenesis in all subpopulations of MSCs, but significantly higher level of chondrogenesis in S2 and S3 (Fig. 4F). Collagen I (Col I) expressions in different subgroups did not follow the same pattern as Col II or Aggr. Col I expression was found to be similar in S1 to S4, but significantly higher in S5, at all passage points (Fig. 4G). The ratio of Col II to Col I in different subgroups followed similar trend as Col II expression, with S2 and S3 being the highest subgroups (Fig. S1A†). We further analysed the expression of hypertrophic marker collagen X (Col X) in the cartilage pellets. Significantly higher level of Col X was found in S5 in all passages, and in S4 in the late passage (Fig. 4H). Chondrocyte hypertrophy is associated with enlarged cell size,50 which is consistent with the larger size of MSCs in S5. The high Col II, low Col I and Col X expression in S2 and S3 indicated formation of more hyaline and less fibrous cartilage tissues in these subgroups. The different trends in Col I, Col II and Col X expression from S1 to S5 ruled out the possibility that observed higher Col II in S2 and S3 was merely caused by higher metabolic activity.
Analysis of viability and senescence of size-dependent MSC subgroups
Characterization of the size-dependent MSC subgroups indicated that the medium-size MSCs (S2 and S3), with average size ranging from 17 to 21 μm, had faster proliferation rate and chondrogenic capacity in all passages as compared to the smaller (S1) and bigger (S4 and S5) MSCs. Both the proliferation and differentiation of MSCs could be affected by the cell viability and senescence,51 and studies have reported the correlation of cell viability,52,53 as well as MSC senescence,45,58 with cell size in culture.41,54 Thus, we investigated the cell viability and senescence level in the size-dependent subgroups in early, middle and late passages. Results showed that average viabilities of each subgroup over all the passages were greater than 90%, and no significant change in viability was found from early to late passages. However, S1 has an average viability of 93% over all passages, which was significantly lower than 98% in other subgroups (Fig. 5A). Senescence-related β-galactosidase staining of MSCs subgroups at different passages was performed. It was observed that the percentage of senescent cells increased from small (S1) to big (S5) size, and from early to late passages (Fig. 5B). The senescence level of MSCs was quantified as senescence index (SI), which was computed as the amount of senescence-related β-galactosidase staining per cell in culture using an automated image processing algorithm (refer to Materials and methods). SI increased from early to middle, and from middle to late passages (p < 0.05). In all passages, senescent cells were not observed in S1; very few appeared in S2 and increased from S3 to S5. Significantly higher level of senescence was observed in S5 than the rest of the subgroups in middle and late passages, indicating increase in the cell size of aging cells (Fig. 5C). However, even in S5 of late passage MSC, not all the cells in S5 were senescent. FACS analysis on the cellular amount of DNA showed that 25% of cells in S5 were at G2/M phase of the cell cycle over all passages (Fig. 5D and Fig. S2†), despite the overall reduction of dividing cells from 19% in early passage to 10% in late passage (Fig. S2†).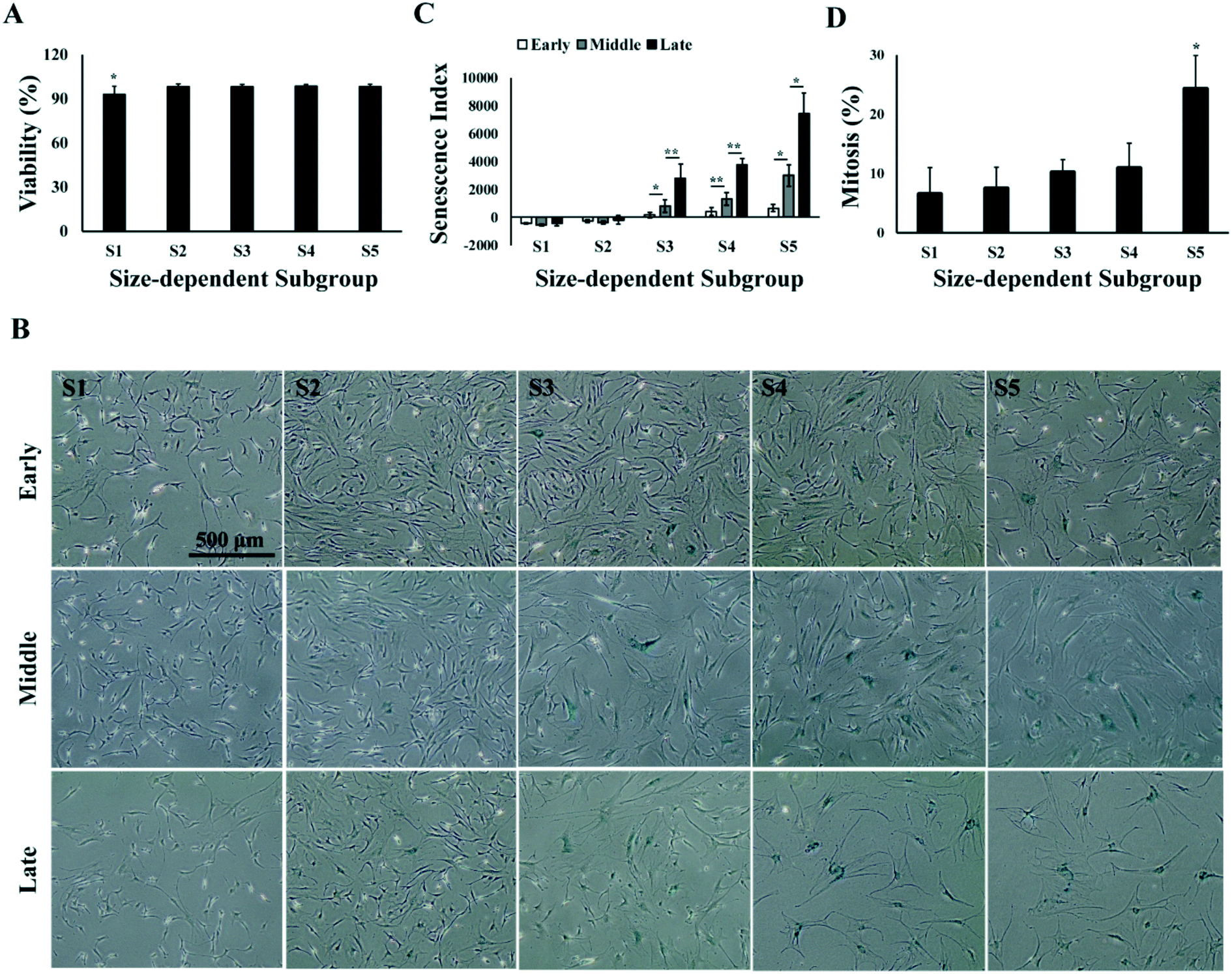 | ||
| Fig. 5 Viability and senescence of size-dependent MSC subgroups. (A) Average viability of size-dependent MSC subgroups over all passages. (B) Senescence index of size-dependent MSC subgroups. (C) Phase contrast images of size-dependent MSC subgroups with senescence staining at different passages. Scale bar: 500 μm. (D) Average percentage of dividing cells in each size-dependent MSC subgroups over all passages. All error bars represent standard deviations. Statistical significance is determined by Student's t-test, * indicates p < 0.05, ** indicates p < 0.001. | ||
Selective MSC culture expansion with repeated microfluidic subpopulation selection
To identify the most competent subpopulation of MSCs for therapeutic use in articular cartilage repair, we considered several properties of the cells. Firstly, this subpopulation should possess strong chondrogenic capacity in order to differentiate into chondrocytes and secrete corresponding ECM to form hyaline cartilage tissue. Secondly, they should proliferate to a large enough cell number needed therapeutically. Lastly, for technical applicability, this subpopulation should account for enough high percentage of cell numbers in the total MSC population so as to be efficiently isolated and expanded to adequate numbers for therapeutic use. Our investigation concluded that the combination of S2 and S3 MSC subgroups matched all three aspects (Fig. 2B–4), indicating that the most competent MSCs for cartilage repair are medium-size cells in culture between 17 to 21 μm (Fig. 2A). Colter et al. has identified an extremely small-sized (7 μm) population of MSCs with stronger multipotency than the normal-size MSCs (15–50 μm),42 and Poon et al. has further suggested that smaller MSCs (15 μm) are at multipotent state, whereas larger MSCs (20 μm) commit towards osteoprogenitor state with reduced multipotency.36 The “chondrogenic competent” subpopulation we identified fell in-between these sizes.We next designed a simplified sorting strategy with the same spiral micro-channel device to enrich MSCs of 17–21 μm from the total population by excluding the larger and smaller cells respectively with two rounds of sorting (see Materials and methods). This sorting strategy was applied to each subculture up to 10 passages as a “selective expansion” approach to enrich only MSCs of high chondrogenic competence (Fig. 6A). MSCs from the selective expansion approach were compared to normal expansion approach in terms of population doubling time (PDT) and total cell yield within same period of culture time.
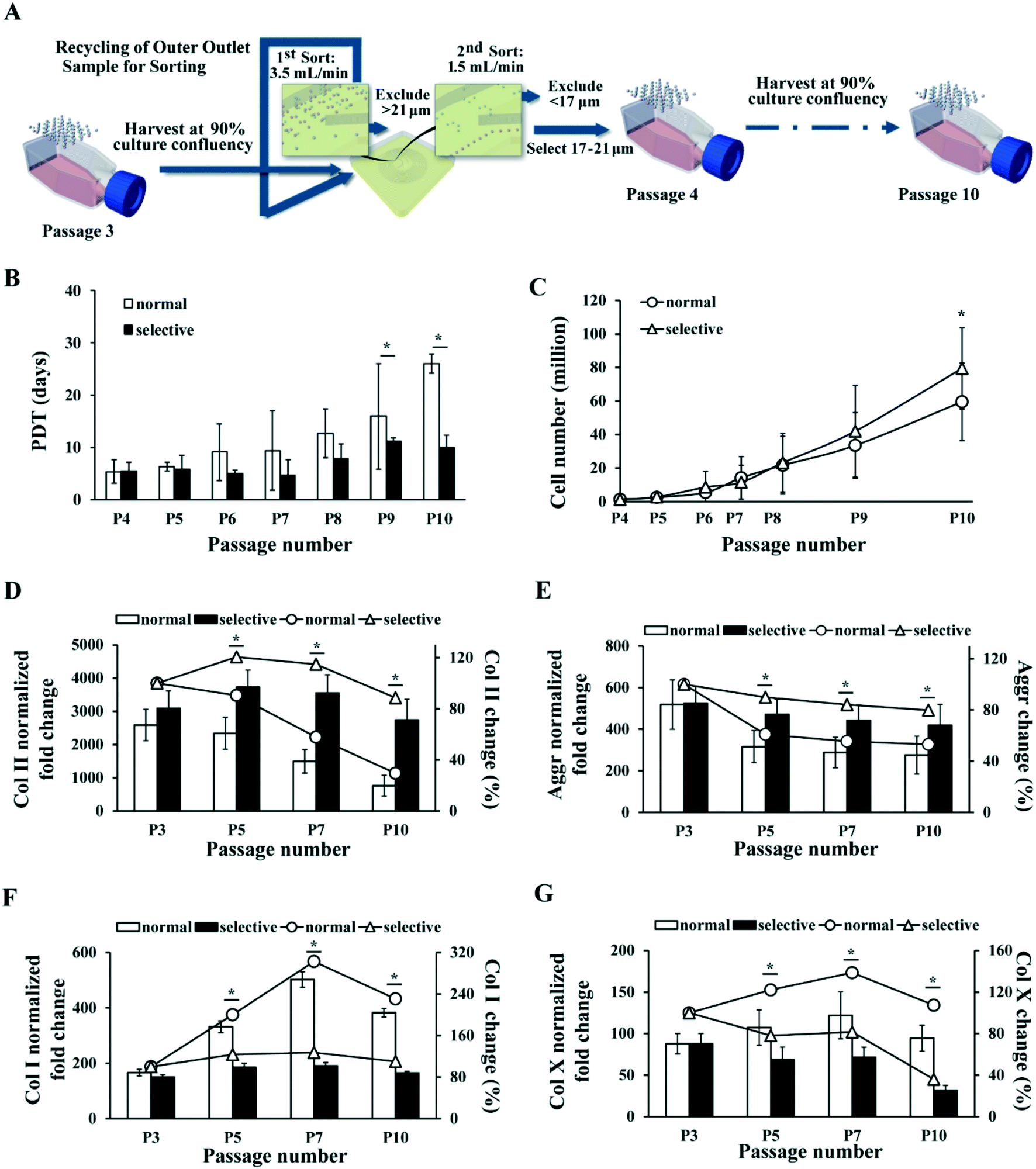 | ||
| Fig. 6 MSC selective culture expansion with repeated inertial sorting maintains proliferation rate and chondrogenic potential in late passages. (A) Schematic of selective culture expansion strategy. MSCs were expanded normally in TCP and subcultured at 90% confluency. MSCs larger than 21 μm or smaller than 17 μm were excluded at every subculture and only 17–21 μm ones were passaged. (B) MSC population doubling time in normal (black bar) and selective (white bar) culture expansion at each passage. (C) Total number of MSCs produced by normal (line with round dots) and selective (line with triangle dots) culture expansion at different passages. The inconsistent gaps among the passage numbers in the x-axis represent different days of culture among the passages. Corresponding days of culture post initial seeding at each passage is provided in Table S2.† (D–G) RT-PCR analysis on Col II (D), aggrecan (E), Col X (F) and Col I (G) level in cartilage pellet formed by MSCs at different passages after normal (white bar) and selective (black bar) culture expansion. The fold changes are normalized to the corresponding levels in undifferentiated MSCs. The percentage changes of Col II (D), aggrecan (E), Col I (F) and Col X (G) levels as compared to initial culture (P3) at different passages after normal (line with round dots) and selective (line with triangle dots) culture expansion are plotted simultaneously. All error bars represent standard deviation. Statistical significance is determined by Student's t-test, * indicates p < 0.05. | ||
Results showed that PDT of the MSCs with normal expansion gradually increased from about 5 days to 26 days by passage 10, whereas PDT of the MSCs with selective expansion remained below 11 days (Fig. 6B). In this experiment, MSCs cultured with both methods were passaged at the same time points for the convenience of comparison. Thus, the confluency of normal culture expanded MSCs at every passage decreased gradually, as the difference in PDT of MSCs between the cultures increased. The corresponding number of days post initial seeding for every passage performed is provided in Table S2.† The average recovery of 17–21 μm MSCs decreased from 67% to 46% of total cells, with an average of about 60% (Table S2†).
As selective culture yielded shorter PDT than normal culture in all passages, we then calculated the theoretical total MSC yield from the two culture expansion approaches (see Materials and methods). The calculated result showed that selective culture expansion led to approximately 33% more cell yield comparing to the normal expansion by passage 10 (Fig. 6C).
Besides the actively excluded large and small MSCs, some cells were lost during the sorting process. Part of the loss was attributed to the dead-end volume of cell suspension in the syringe, tubing, micro-channel, and collection tubes, especially during repeated transferring of cell suspension from collection tubes to syringes for sorting (estimated to be approximately 5% of total cells). It was also possible that weaker and unhealthy cells ruptured during the sorting process due to hydraulic shear. Nevertheless, the selective culture expansion was able to produce more MSCs than the normal approach within the same period of time despite of the cell loss (Fig. 6C). Higher cell yield could be achieved with further refinement in sorting procedure. Higher percentage of dividing cells was detected in S5 than other subgroups (Fig. 5D). It was likely that some medium-size MSCs undergoing division might have been removed together with the large cells due to their transient expansion in cell size.
The chondrogenic potential of the MSCs expanded with selective and normal expansion approach was also compared at multiple passage points. PCR analysis showed that the level of Col II gradually decreased to 29.5% of the original level by passage 10 with the normal culture expansion, but remained at 88.5% of the original level with selective culture approach (Fig. 6D). Similarly, the level of Aggr dropped to 53.1% of the original level with normal culture expansion, but remained at 79.8% with selective expansion (Fig. 6E). In addition, comparing to the drastically increased Col I and relatively higher Col X level by end of passage 10 under normal culture expansion, the level of Col I only increased by 10% (Fig. 6F), and Col X decreased to 36% under selective culture expansion. Similar to Col II level, the Col II/Col I ratio also reflected significant difference between the two culture methods (Fig. S1B†). These results clearly demonstrated the advantage of the selective expansion approach over normal approach in maintaining the chondrogenic potential of MSC during culture expansion.
Given that small and large MCSs that were recursively removed from the culture comprised up to 30–40% of total cells at each passage (Fig. 2B and Table S2†), the observed enhancement in proliferation (PDT), total cell yield, and chondrogenic potential of the new selective culture expansion protocol is of high clinical significance and utility. Although the biological mechanism of how the small and large MSCs influence the medium-size MSCs in culture was not investigated in detail, we noticed the difference in viability and senescence among the populations. Dead cells were mainly observed in the small (S1) MSC population (Fig. 5A), suggesting their disadvantage in growth. However, the slightly lower viability (93%) in small MSC population was unlikely the major contribution to the slower growth and significantly weaker chondrogenic capacity, compared to the selected medium-size population. The large MSC population (S5) comprised of more senescent cells than the small and medium-size population (Fig. 5B and C), which could directly contribute to their slow proliferation rate and weak differentiation capacity.55,56 It is highly likely that the secretome from the larger and smaller MSC could have affected the state of the medium-size cells. Reducing the potentially negative paracrine signalling from unwanted subgroups might further help to maintain the homogeneity of the culture.
Advantages of the microfluidic selection approach over other purification methods
Although other biophysical or biochemical approaches could possibly be used to purify 17–21 μm MSCs as well, our microfluidic approach possesses clear advantages in many aspects. Our approach is label-free and high-throughput as compared to FACS. Being label-free eliminates the potential influences of surface-labelling to the MSCs in therapeutic use, thus simplifying the complexity and improving the compliance with the regulations. In this study, we used about 1 million per mL MSC concentration for the sorting, with minimum sorting flow rate at 1.5 mL min−1, achieving high throughput sorting of 1.5 million cells in a minute. Ultra-high throughput can be achieved by multiplexing the spiral micro-channel device to specially cater the need for purifying extremely large quantities of cells.57 Also, different from most active microfluidic cell sorting techniques where external fields (electrical, magnetic, acoustic, optical, etc.) are required to sort the cells,58 our device requires only hydraulic pressure to operate, making it easy to operate and scale-up. The large channel dimension (comparing to cell diameter) and fast sorting flow rate of our spiral micro-channel device also minimizes the risk of cell clogging, making it much more robust than conventional size-based separation using cell filters.42Conclusion
In summary, we identified a subpopulation of medium-size (17–21 μm) “chondrogenic competent” MSCs from highly heterogeneous total population of culture expanded MSCs. We demonstrated that enriching this subpopulation of MSCs during continuous culture expansion by repeated high-throughput microfluidic exclusion of other subgroups resulted in higher yield of MSCs with better chondrogenic potential as compared to conventional expansion approach. This approach enabled manipulation of culture expanded MSCs towards “chondrogenic competent” state with minimum biophysical and no additional biochemical stimulus, which can greatly facilitate translation to clinical use. Although this study primarily illustrated the use of microfluidic subpopulation selection to produce large quantities of MSCs with enhanced chondrogenic capacity, other therapeutic needs of MSCs can potentially be fulfilled using similar approach.Conflict of interests
The authors declare no conflict of interests in this work.Acknowledgements
This research was supported by the National Research Foundation of Singapore through the Singapore MIT Alliance for Research and Technology's BioSystems and Micromechanics Interdisciplinary Research Group, and National Medical Research Council of Singapore (NMRC/CIRG/1403/2014). We also thank Frances Liu, Dr. Guneta Vipra and Prof. Krystyn Van Vliet for providing human MSCs (Lonza Pte. Ltd.) from three donors and imaging-based analysis software.References
- J. Tolar, K. Le Blanc, A. Keating and B. R. Blazar, Stem cells, 2010, 28, 1446–1455 CrossRef PubMed.
- A. Uccelli, L. Moretta and V. Pistoia, Nat. Rev. Immunol., 2008, 8, 726–736 CrossRef CAS PubMed.
- X. Wei, X. Yang, Z. P. Han, F. F. Qu, L. Shao and Y. F. Shi, Acta Pharmacol. Sin., 2013, 34, 747–754 CrossRef CAS PubMed.
- J. Shao, W. Zhang and T. Yang, Biol. Res., 2015, 48, 62 CrossRef PubMed.
- X. Wang, Y. Wang, W. Gou, Q. Lu, J. Peng and S. Lu, Int. Orthop., 2013, 37, 2491–2498 CrossRef PubMed.
- S. Zhao, R. Wehner, M. Bornhauser, R. Wassmuth, M. Bachmann and M. Schmitz, Stem Cells Dev., 2010, 19, 607–614 CrossRef CAS PubMed.
- L. C. Marti, A. A. Ribeiro and N. Hamerschlak, Einstein, 2011, 9, 224–228 CrossRef PubMed.
- P. R. Baraniak and T. C. McDevitt, Regener. Med., 2010, 5, 121–143 CrossRef PubMed.
- X. Liang, Y. Ding, Y. Zhang, H. F. Tse and Q. Lian, Cell Transplant., 2014, 23, 1045–1059 Search PubMed.
- N. Tzaribachev, M. Vaegler, J. Schaefer, P. Reize, M. Rudert, R. Handgretinger and I. Muller, Isr. Med. Assoc. J., 2008, 10, 232–234 Search PubMed.
- S. M. Richardson and J. A. Hoyland, Curr. Pain Headache Rep., 2008, 12, 83–88 CrossRef PubMed.
- E. M. Horwitz, D. J. Prockop, L. A. Fitzpatrick, W. W. Koo, P. L. Gordon, M. Neel, M. Sussman, P. Orchard, J. C. Marx, R. E. Pyeritz and M. K. Brenner, Nat. Med., 1999, 5, 309–313 CrossRef CAS PubMed.
- U. Noth, A. F. Steinert and R. S. Tuan, Nat Rev Rheumatol, 2008, 4, 371–380 Search PubMed.
- L. C. Amado, A. P. Saliaris, K. H. Schuleri, M. St John, J. S. Xie, S. Cattaneo, D. J. Durand, T. Fitton, J. Q. Kuang, G. Stewart, S. Lehrke, W. W. Baumgartner, B. J. Martin, A. W. Heldman and J. M. Hare, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11474–11479 CrossRef CAS PubMed.
- R. H. Lee, A. A. Pulin, M. J. Seo, D. J. Kota, J. Ylostalo, B. L. Larson, L. Semprun-Prieto, P. Delafontaine and D. J. Prockop, Cell Stem Cell, 2009, 5, 54–63 CrossRef CAS PubMed.
- S. H. Ranganath, O. Levy, M. S. Inamdar and J. M. Karp, Cell Stem Cell, 2012, 10, 244–258 CrossRef CAS PubMed.
- J. M. Kim, S. T. Lee, K. Chu, K. H. Jung, E. C. Song, S. J. Kim, D. I. Sinn, J. H. Kim, D. K. Park, K. M. Kang, N. Hyung Hong, H. K. Park, C. H. Won, K. H. Kim, M. Kim, S. Kun Lee and J. K. Roh, Brain Res., 2007, 1183, 43–50 CrossRef CAS PubMed.
- E. Zappia, S. Casazza, E. Pedemonte, F. Benvenuto, I. Bonanni, E. Gerdoni, D. Giunti, A. Ceravolo, F. Cazzanti, F. Frassoni, G. Mancardi and A. Uccelli, Blood, 2005, 106, 1755–1761 CrossRef CAS PubMed.
- V. V. Erokhin, I. A. Vasil'eva, A. G. Konopliannikov, V. I. Chukanov, A. F. Tsyb, T. R. Bagdasarian, A. A. Danilenko, L. A. Lepekhina, S. Kal'sina, I. V. Semenkova and E. V. Agaeva, Probl. Tuberk. Bolezn. Legk., 2008, 3–6 CAS.
- J. W. Lee, X. Fang, N. Gupta, V. Serikov and M. A. Matthay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16357–16362 CrossRef CAS PubMed.
- B. Parekkadan, D. van Poll, K. Suganuma, E. A. Carter, F. Berthiaume, A. W. Tilles and M. L. Yarmush, PLoS One, 2007, 2, e941 Search PubMed.
- L. Peng, D. Y. Xie, B. L. Lin, J. Liu, H. P. Zhu, C. Xie, Y. B. Zheng and Z. L. Gao, Hepatology, 2011, 54, 820–828 CrossRef PubMed.
- P. Kharaziha, P. M. Hellstrom, B. Noorinayer, F. Farzaneh, K. Aghajani, F. Jafari, M. Telkabadi, A. Atashi, M. Honardoost, M. R. Zali and M. Soleimani, Eur. J. Gastroenterol. Hepatol., 2009, 21, 1199–1205 CrossRef CAS PubMed.
- F. Togel, Z. Hu, K. Weiss, J. Isaac, C. Lange and C. Westenfelder, Am. J. Physiol., 2005, 289, F31–F42 CrossRef PubMed.
- S. Jung, K. M. Panchalingam, R. D. Wuerth, L. Rosenberg and L. A. Behie, Biotechnol. Appl. Biochem., 2012, 59, 106–120 CrossRef CAS PubMed.
- J. Chang and G. Gupta, Tissue engineering for the hand: research advances and clinical applications, World Scientific, New Jersey, London, 2010 Search PubMed.
- I. Dimarakis and N. Levicar, Stem cells, 2006, 24, 1407–1408 CrossRef PubMed.
- F. Mannello and G. A. Tonti, Stem cells, 2007, 25, 1603–1609 CrossRef CAS PubMed.
- N. Mizuno, H. Shiba, Y. Ozeki, Y. Mouri, M. Niitani, T. Inui, H. Hayashi, K. Suzuki, S. Tanaka, H. Kawaguchi and H. Kurihara, Cell Biol. Int., 2006, 30, 521–524 CrossRef CAS PubMed.
- J. A. King and W. M. Miller, Curr. Opin. Chem. Biol., 2007, 11, 394–398 CrossRef CAS PubMed.
- C. A. Rodrigues, T. G. Fernandes, M. M. Diogo, C. L. da Silva and J. M. Cabral, Biotechnol. Adv., 2011, 29, 815–829 CrossRef CAS PubMed.
- L. Y. Sun, S. Z. Lin, Y. S. Li, H. J. Harn and T. W. Chiou, Cell Transplant., 2011, 20, 49–62 CAS.
- D. G. Phinney, J. Cell. Biochem., 2012, 113, 2806–2812 CrossRef CAS PubMed.
- M. Al-Nbaheen, R. Vishnubalaji, D. Ali, A. Bouslimi, F. Al-Jassir, M. Megges, A. Prigione, J. Adjaye, M. Kassem and A. Aldahmash, Stem Cell Rev. Rep., 2013, 9, 32–43 CrossRef CAS PubMed.
- A. D. Ho, W. Wagner and W. Franke, Cytotherapy, 2008, 10, 320–330 CrossRef CAS PubMed.
- Z. Poon, W. C. Lee, G. Guan, L. M. Nyan, C. T. Lim, J. Han and K. J. Van Vliet, Stem Cells Transl. Med., 2015, 4, 56–65 CrossRef CAS PubMed.
- D. G. Phinney, Cell Cycle, 2007, 6, 2884–2889 CrossRef CAS PubMed.
- V. Savkovic, H. Li, J. K. Seon, M. Hacker, S. Franz and J. C. Simon, Curr. Stem Cell Res. Ther., 2014, 9, 469–488 CrossRef CAS PubMed.
- P. K. Gupta, A. K. Das, A. Chullikana and A. S. Majumdar, Stem Cell Res. Ther., 2012, 3, 25 CrossRef CAS PubMed.
- A. Goldberg, K. Mitchell, J. Soans, L. Kim and R. Zaidi, J. Orthop. Surg. Res., 2017, 12, 39 CrossRef PubMed.
- M. J. Whitfield, W. C. Lee and K. J. Van Vliet, Stem Cell Res., 2013, 11, 1365–1377 CrossRef CAS PubMed.
- D. C. Colter, I. Sekiya and D. J. Prockop, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7841–7845 CrossRef CAS PubMed.
- I. Majore, P. Moretti, R. Hass and C. Kasper, Cell Commun. Signaling, 2009, 7, 6 CrossRef PubMed.
- W. C. Lee, H. Shi, Z. Poon, L. M. Nyan, T. Kaushik, G. V. Shivashankar, J. K. Chan, C. T. Lim, J. Han and K. J. Van Vliet, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4409–4418 CrossRef CAS PubMed.
- M. E. Warkiani, G. Guan, K. B. Luan, W. C. Lee, A. A. Bhagat, P. K. Chaudhuri, D. S. Tan, W. T. Lim, S. C. Lee, P. C. Chen, C. T. Lim and J. Han, Lab Chip, 2014, 14, 128–137 RSC.
- A. A. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Lab Chip, 2008, 8, 1906–1914 RSC.
- D. Di Carlo, Lab Chip, 2009, 9, 3038–3046 RSC.
- D. Di Carlo, D. Irimia, R. G. Tompkins and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18892–18897 CrossRef CAS PubMed.
- Y. N. Wu, J. B. Law, A. Y. He, H. Y. Low, J. H. Hui, C. T. Lim, Z. Yang and E. H. Lee, Nanomedicine, 2014, 10, 1507–1516 CrossRef CAS PubMed.
- J. F. Schmitt, K. H. See, Z. Yang, J. H. Hui and E. H. Lee, J. Orthop. Res., 2012, 30, 1753–1759 CrossRef CAS PubMed.
- N. Baker, L. B. Boyette and R. S. Tuan, Bone, 2015, 70, 37–47 CrossRef CAS PubMed.
- J. A. Searles, P. Todd and D. S. Kompala, Biotechnol. Prog., 1994, 10, 198–206 CrossRef CAS PubMed.
- P. Tibayrenc, L. Preziosi-Belloy, J. M. Roger and C. Ghommidh, J. Biotechnol., 2010, 149, 74–80 CrossRef CAS PubMed.
- J. C. Estrada, Y. Torres, A. Benguria, A. Dopazo, E. Roche, L. Carrera-Quintanar, R. A. Perez, J. A. Enriquez, R. Torres, J. C. Ramirez, E. Samper and A. Bernad, Cell Death Dis., 2013, 4, e691 CrossRef CAS PubMed.
- V. Turinetto, E. Vitale and C. Giachino, Int. J. Mol. Sci., 2016, 17, 1164–1182 CrossRef PubMed.
- M. M. Bonab, K. Alimoghaddam, F. Talebian, S. H. Ghaffari, A. Ghavamzadeh and B. Nikbin, BMC Cell Biol., 2006, 7, 14 CrossRef PubMed.
- M. E. Warkiani, B. L. Khoo, D. S. Tan, A. A. Bhagat, W. T. Lim, Y. S. Yap, S. C. Lee, R. A. Soo, J. Han and C. T. Lim, Analyst, 2014, 139, 3245–3255 RSC.
- C. W. T. Shields, C. D. Reyes and G. P. Lopez, Lab Chip, 2015, 15, 1230–1249 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc01005b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |