
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micro flow injection ICP-MS analysis of high matrix samples: an investigation of its capability to measure trace elements in iron meteorites†
Phil
Holdship
 *a,
Pierre
Bonnand
b,
David
Price
c and
Paul
Watson
d
*a,
Pierre
Bonnand
b,
David
Price
c and
Paul
Watson
d
aDepartment of Earth Sciences, University of Oxford, OX1 3AN, UK. E-mail: philip.holdship@earth.ox.ac.uk
bLaboratoire Magmas et Volcans, Université Clermont Auvergne, CNRS, F-63000 Clermont-Ferrand., France
cPerkinElmer, Seer Green, HP9 2FX, UK
dElemental Scientific, Warrington, WA1 4AE, UK
First published on 19th September 2018
Abstract
The coupling of a micro-flow injection sample introduction system to a highly versatile quadrupole ICP-MS (FI-ICP-MS) has been demonstrated to achieve accurate and precise trace element measurements for several iron meteorites. Measurements were made for the trace siderophile elements, Ru, Au, Rh, Pd, Ir, Pt, Ga, Cr, Cu and Re, where results present good agreements for most metals to duplicate samples also measured by a sector-field ICP-MS (ICP-SFMS). A major advantage for trace element measurements by flow-injection ICP-MS is the added flexibility to increase the total dissolved solids (TDS) concentrations of the working solutions above the 2000 μg g−1 ICP-MS limit. The FI-ICP-MS technique used in this study allowed the sample digests to be measured directly, where the TDS concentrations were 2.5 times the above limit. The data generated clearly demonstrate that the micro-flow inputs of the high matrix material never overloaded the plasma or compromised on instrument performance. Without the need for extra dilutions a better detection capability for most elements was found in comparison to those made by the ICP-SFMS, where all method detection limits were below 0.1 μg g−1, apart from Ru and Ga. Furthermore, it was evident from the reduced sample preparation time, together with measurement times of less than 7 minutes per sample, that the FI-ICP-MS analysis was a much more rapid technique. The accuracy of the FI-ICP-MS technique was tested from the measurement of two synthetic iron meteorites. A selection of elements, including Cr, Ga, Ru, Ir and Re, gained absolute error values better than ±5%, where Re, the analyte with the lowest concentration, reported the lowest absolute error, with a value of less than ±1%. Precision, determined from two times the relative standard deviation of repeat measurements of the synthetic solutions (n = 7), gained values less than 10% for all elements.
1. Introduction
For many geochemical research applications the concentration of trace elements, and more often their ratios, either to other similar trace elements or to a major element, are important proxies to improve our understanding of earth system processes. The quantity of trace elements that can be detected and their inter-variability in concentrations, which often span many orders of magnitude, offer a wide spectrum of geochemical information.1 This data highlights information about the differences in their chemical behaviours and sheds light into the occurrence of many earth system processes. Research has been conducted to interrogate the behaviours of trace elements in many geochemical systems, examples ranging from the measurement of rare earth elements in foraminifera to examine paleoceanographic and climate change,2 to the use of lithium as a proxy for chemical weathering.3Within such systems and also beyond, many of these elements are contained at levels that are extremely depleted, thus their measurement requires the use of a particularly sensitive analytical technique in order to achieve accurate detection. Inductively coupled plasma-mass spectrometry (ICP-MS); with its capability to measure elemental concentrations over a linear working range that spans over nine orders of magnitude (pg L−1 up to mg L−1), as well as being able to measure multiple elements in a single sweep, has a significant potential to be employed for many trace element applications. This includes the field of Earth Sciences, where its attributes have been harnessed as a powerful tool for high precision geochemical trace element studies.4 However, a noticeable limitation in the measurement of trace metals by ICP-MS is the concentration of total dissolved solids (TDS) that the instrument can tolerate. If the TDS of the working (or measured) sample solution exceeds 2000 μg g−1, undesirable effects such as plasma suppression and the accumulation of material onto the cones can lead to analytical drift and contamination problems.5,6 Coupled to this, space charge effects also induce further suppression and are a result of an inbalance in the net charge of the ion beam. This is caused by the excessive influx of positively charged matrix ions that produce repulsions, thus spreading and de-focussing the beam inside the mass spectrometer.7
In order to minimise these matrix effects and to make it possible to detect trace elements in samples that contain rich sample matrices, experimental processes need to be adopted. The classical approach is to dilute the solutions until the TDS concentrations are at an acceptable level. However, even with the most powerful ICP-MS instrument (e.g. magnetic-sector), if the samples are diluted significantly the trace element concentrations may fall to such low concentrations that they are rendered undetectable. Another approach is to use matrix separation to selectively separate the trace elements of interest from the major element matrix. This methodology has many advantages, as not only are the TDS concentrations reduced to negligible levels but the trace elements of interest can also be pre-concentrated for significantly improved detection. However, it should be noted that chemical separations are time consuming and that the separation method needs to be checked carefully in order that accurate concentrations are obtained. Similar studies have successfully adopted this analytical approach, such as the separation of PGEs and other siderophile elements from geological samples8 and extraterrestrial materials.9,10
Iron meteorites are an example of a sample material that contain significant TDS concentrations, particularly from the metals Fe, Ni and Co. This makes them a challenging sample material to measure their trace element compositions. However, their trace element concentrations, especially Ge and Ga, plus the more refractory element, Ir, provide significant information about the origins of iron meteorites and how they formed.11
Up until recent years trace element measurements of iron meteorites were conducted using instrumental-neutron-activation-analysis (INAA) or radiochemical-neutron-activation-analysis (RNAA).12,13 All of these studies have provided a significant amount of excellent data that has not only expanded our knowledge of extra-terrestrial materials, but have also established the iron meteorite classification.14 Although these techniques do not destroy the sample, which is particularly advantageous for precious sample types such as iron meteorites, and can offer excellent precision and accuracy, they can be time consuming, expensive and the measurements require access to a suitable activation nuclear reactor.15,16 Another consideration that needs to be addressed when using these techniques includes the safe disposal or storage of the measured materials, as the irradiated samples may remain radioactive for many years after analysis.16
In the last few years there has been an emerging trend for the development of studies to measure iron meteorites by solution ICP-MS methods, to exploit its high performance but also to exclude inaccuracies induced by the matrix elements.10,17–20 Although each of the above studies achieved high precision trace element data, several method developments had to be applied. An example is the use of matrix-matched external calibration standards, where it was reported that accurate trace element data was only found if the Fe and Ni concentrations in samples were similar to those contained in the standards.17 Also, the use of an ion exchange chemistry methodology to remove Fe from the sample solutions has been proven to improve instrument performance.10 Although a high degree of accuracy was obtained from the eluted solutions, pre-concentration of iron meteorites is ultimately unnecessary, as the concentrations of many trace metals in their solution digests (ca. diluted 100 times from the solid) can be in a measurable range for ICP-MS. Therefore, if the ICP-MS equipment can be modified to accept samples containing high matrix concentrations, the time consuming preparation and measurement methodologies described above can be avoided.
Flow injection analysis can be coupled to ICP-MS to expand its capability to measure rich matrices that have TDS concentrations greater than 2000 μg g−1. The principle of this technique was first developed by Ruzicka and Hansen, 1975,21 where the approach was applied to many analytical chemistry techniques, primarily to reduce sample consumption, but also to improve sample throughput and sample handling. PerkinElmer pioneered the development of direct flow injection for ICP-MS and launched FIAS (Flow Injection Automation System) in the 1990s, to expand the capabilities of its Elan ICP-MS, especially to improve the accuracy of trace elements in high TDS solutions and to increase through-put of such samples in the laboratory. However, there were limitations in its use, particularly with respect to dispersion of the analyte solution in the line leading to the nebuliser.22 Recent refinements to this flow injection system by Elemental Scientific have eliminated this problem in the development of the FAST-FIAS and prepFAST-FIAS systems.
In addition to the recent production of the prepFAST-FIAS system by Elemental Scientific, other similar FI-ICP-MS systems have also been employed for the direct measurement of trace elements in high matrix samples.23–25 The results presented from each study yielded excellent trace element data, which further strengthens the position of FI-ICP-MS as a powerful trace element analytical technique for challenging sample matrices.
In this study we have measured several iron meteorites, five from group IIIAB (Charcas, Henbury, Boxhole, Cumpas and Orange River), one from group IIAB (North Chile) and one from group IIIF (Clark County), for a variety of trace siderophile elements from two different techniques: (1) a flow injection ICP-MS, which incorporated the hybridization of an Elemental Scientific Flow Injection Automation System (prepFAST-FIAS) to a quadrupole ICP-MS (FI-ICP-MS), and measured the samples at TDS concentrations of 5000 μg g−1; and (2) a magnetic-sector ICP-MS (ICP-SFMS) instrument, which consisted of a conventional ICP-MS set-up where the samples were measured at TDS levels below the 2000 μg g−1 limit. The data collected from both systems will be compared, together with values found in the literature. In addition to this, performance criteria including method detection limits, accuracy, precision and robustness (a performance indicator for intra-run stability and also repeatability from one run to another) will also be investigated.
2. Methodology
2.1 Reagents and labware
The iron meteorites were digested in Savillex (Eden Prairie, U.S.A.) PFA digestion vials, which were chosen for their high heating efficiency and after acid-cleaning pre-treatment, their superior trace metal purity. AnalaR grade hydrochloric acid (HCl), purchased from Merck, was purified by sub-boiling distillation and then added to the samples prior to them being placed on the hotplate for digestion. Measurements by ICP-SFMS also required nitric acid (HNO3), which was also purchased at AnalaR grade (Thermo Fisher) and purified by sub-boiling distillation.Additionally, the acids were diluted for use as lower molarity diluent reagents for the preparation of blanks and standards. This involved mixing the above high purity acids with 18.2 MΩ cm de-ionised water, to denoted acid to water v/v ratios.
Calibration curves for both instruments were generated from stock standards that were prepared from the following standards for both measurement methods: 1000 μg mL−1 Merck Certipur single element ICP standards and 10 μg mL−1 custom-blend multi-element standards (produced by CPAchem Ltd. and distributed by VWR). The calibrants were prepared on the day of analysis by sequentially diluting from a stock standard. Details of the calibration standards that were prepared for both methods of analysis are described in the FI-ICP-MS analysis and ICP-SFMS method sections. Quality control standards (QCs), used to verify the validity of the calibrations, were also prepared for each run and were produced from separate 10 μg mL−1 custom-blend multi-element standards (produced by CPAchem Ltd. and distributed by Qmx Laboratories).
Terbium and bismuth were selected as internal standards for the FI-ICP-MS analysis. A stock standard containing these two elements was prepared from individual 1000 μg mL−1 Merck Certipur single element standards and poured into the ‘internal standard’ reservoir in the Elemental Scientific prepFAST system. Praseodymium was selected as an internal standard for the measurements by ICP-SFMS. An aliquot from a 1000 μg mL−1 Merck Certipur Pr standard was taken and diluted analogously to the FI-ICP-MS analysis and then subsequently added into the blanks, standards and samples.
2.2 Sample preparation and digestion
A diamond saw blade was used to cut up the iron meteorite samples into small pieces, where up to 3 g were prepared for measurement per sample in this study. After cleaning, the chips were accurately weighed into the PFA vials and digested in 6 M HCl on a hotplate for 96 hours. The samples were then dried down and redissolved in 6 M HCl. After digestion, aliquots corresponding to 150 mg of samples were diluted to 15 mL with de-ionised water, to a HCl concentration of 4% v/v. These solutions were decanted into trace metal clean polypropylene centrifuge tubes for measurement by FI-ICP-MS, where they were diluted two-times in-line with the Elemental Scientific prepFAST and an internal standard added. A second aliquot (for ICP-SFMS) was dried down and re-dissolved in 2% HNO3 and diluted to obtain a TDS concentration of 100 μg g−1.2.3 FI-ICP-MS analysis
The sample digests described above (in the 4% HCl matrix) were placed directly for measurement on an Elemental Scientific (Omaha, U.S.A.) SC-4 DX FAST autosampler, having only been diluted 100 times by the preparation process. Hydrochloric acid; used routinely as a matrix acid for the measurement of PGEs by ICP-MS, was selected as the matrix acid due to their high solubility in this reagent.26,27 However, a significant issue with the analysis of HCl bearing solutions by ICP-MS are the resulting chlorine-based molecular ions that form in the plasma, which can cause interferences on mass spectra. Although, in this study HCl was successfully adopted for analysis using a flow injection approach, which allowed measurement of the samples without significant impendence of the undesirable interferences. Coupled to the autosampler, an Elemental Scientific (Omaha, U.S.A.) prepFAST M5; comprising five syringes to automate rinsing, dilution, internal standard addition and sample uptake, rapidly transported the sample solutions to an Elemental Scientific P7+ FAST injection valve, which subsequently prepared each diluted working solution inside a 1 mL loop with in-line additions of the Tb and Bi internal standard (to correct for any instrument drift). These solutions were then transferred to a second, P6 FAST valve, which instead of introducing each sample as a continuous flow into the mass spectrometer, to achieve a steady-state signal, was toggled between ‘load’ and ‘inject’ to produce highly accurate timed injections of small sample volumes. The FI-ICP-MS measurements of the iron meteorites in this study incorporated an injection toggle time of 0.25 seconds. During this short period of time the solutions were pumped into the instrument (using an Elemental Scientific MP2 peristaltic pump, set to a speed of 9 and peristaltic pump tubing with an internal diameter of 0.38 mm) at a flow rate of 253 μL min−1, which introduced aliquots of 1.05 μL per injection into the plasma. These micro-volume sample aliquots contained TDS concentrations of 5000 μg g−1, much higher than the 2000 μg g−1 limit. However, due to their small volumes they provided minimal matrix loading onto the plasma and therefore plasma suppression and signal drift was negligible. The system that drives this flow injection process is known as prepFAST FIAS.The ICP-MS instrument used with this flow injection set-up was a PerkinElmer NexION 350D quadrupole ICP-MS (qICP-MS). Table 1 states the typical instrument operating parameters and components that were used to measure the micro-injections. The Elemental Scientific Pergo was an in-line addition to the nebuliser argon gas flow, to increase the humidity of this gas and thus reduce the likelihood of precipitates forming at the tip of the nebuliser, which could cause an eventual blockage.
| Parameter | PerkinElmer NexION 350D | Thermo Finnigan Element 2 |
|---|---|---|
| Nebuliser gas flow | 0.8–1.0 L min−1 | 1.05–1.25 L min−1 |
| Auxiliary gas flow | 1.2 L min−1 | 0.95 L min−1 |
| Cool gas flow | 18 L min−1 | 16 L min−1 |
| RF power | 1600 W | 1350 W |
| Nebuliser | Elemental Scientific PFA-ST | Elemental Scientific Microflow PFA |
| Nebuliser gas humidifier | Elemental Scientific Pergo | — |
| Peristaltic pump | Elemental Scientific MP2 | Thermo Finnigan |
| Peristaltic pump tubing | Orange-green (0.38 mm id) | Grey-grey (1.30 mm id) (drain use only) |
| Peristaltic pump speed | 9 | 8 rpm |
| Sample uptake flow rate | 253 μL min−1 | 100 μL min−1 (self aspiration) |
| Spray chamber | Elemental Scientific quartz cyclonic type | Elemental Scientific quartz cyclonic type |
| Peltier chiller for spray chamber | — | Elemental Scientific PC3 |
| Torch | PerkinElmer quartz demountable | Thermo Finnigan quartz demountable |
| Injector | PerkinElmer sapphire (1.8 mm id) | Thermo Finnigan quartz (1.75 mm id) |
| Sample and skimmer cones | PerkinElmer Nickel | Spectron Nickel (skimmer H type) |
Prior to undertaking the measurements the instrument was optimised using a 1 μg L−1 set-up solution to achieve the following specifications, as indicated by PerkinElmer: tuning of the torch position and nebuliser gas flow to obtain a maximum sensitivity for In (m/z 115), whilst maintaining CeO/Ce (m/z: 156/140) to be less than 0.025; tuning of the Quadrupole Ion Deflector (QID) lens plate voltages to obtain maximum transmission both with and without helium gas being purged into the cell; and finally a performance test from 5 replicate measurements to ensure the average sensitivities recorded for Be (m/z 9), In (m/z 115) and U (m/z 238) were greater than 2000 cps, 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cps and 30000 cps, respectively, in addition to the relative standard deviations from the replicates being less than 3%. Additionally, the ratio of Ce++ (m/z 70)/Ce+ (m/z 140) was also measured during the final performance test as a test for double-charged ion formations, which was required to be less than 0.03, and background measurements (from m/z 220), less than 1 cps. A typical measurement method was then developed using the PerkinElmer Syngistix software and is shown in Table S1 (in ESI†). These parameter settings, combined with the prepFAST-FIAS system allowed each sample digest to be measured in under 7 minutes. This includes a 70 second washout time, which was also used for the syringes to reset. During this washout sequence 4% HCl was rinsed around the entire sample introduction system by a vacuum pump at a flow of 1 mL per second. The exception to this was the sample line to the nebuliser, which was flushed with 4% HCl at a flow of 253 μL min−1.
000 cps and 30000 cps, respectively, in addition to the relative standard deviations from the replicates being less than 3%. Additionally, the ratio of Ce++ (m/z 70)/Ce+ (m/z 140) was also measured during the final performance test as a test for double-charged ion formations, which was required to be less than 0.03, and background measurements (from m/z 220), less than 1 cps. A typical measurement method was then developed using the PerkinElmer Syngistix software and is shown in Table S1 (in ESI†). These parameter settings, combined with the prepFAST-FIAS system allowed each sample digest to be measured in under 7 minutes. This includes a 70 second washout time, which was also used for the syringes to reset. During this washout sequence 4% HCl was rinsed around the entire sample introduction system by a vacuum pump at a flow of 1 mL per second. The exception to this was the sample line to the nebuliser, which was flushed with 4% HCl at a flow of 253 μL min−1.
Helium was selected as the cell gas to perform kinetic energy discrimination (KED), which is a powerful tool to reduce polyatomic interferences, especially those molecular ions with a large ionic radius. This gas is commonly used for KED as it is inert, light in mass and can fill the cell with high gas pressure. Bombardment of large molecular ions with the densely packed helium atoms will cause them to lose significant energy (over the smaller analyte single ions) and then a bias voltage at the cell exit excludes them from the ion beam. The loss of the polyatomics significantly reduces interference on analytes that have the same mass. In this study the KED cell mode was set with two different gas flow settings; a high flow (5.2 mL min−1) to ensure maximum recovery of analytes affected by classic polyatomic interferences, and a low flow (0.5 mL min−1) to maintain higher sensitivity for those analytes that should not suffer from polyatomic interference. Table S1† indicates which cell gas flow was employed for each element.
After the instrument was optimised, several blanks were analysed, using the method conditions stated in Table S1,† to ensure there was a low and steady baseline. This was then followed by an external calibration of ten calibrants, which utilised the multi-dilution feature of the prepFAST to prepare the series in-line from two sets of standard stock solutions.
After the calibration, each sample was measured five times and then a monitor standard (a mid-level calibrant) was tested, to ensure that each analyte signal was not drifting by more than 20% of its theoretical value. This sequence was repeated for each sample in addition to the measurement of a QC standard. Blanks were also measured periodically throughout the run to determine the background memory and to calculate the limit of detection.
2.4 ICP-SFMS analysis
Measurements by ICP-SFMS were undertaken with a conventional sample introduction system that continuously delivered much larger solution volumes to the plasma. Separate samples were analysed using this technique, which were dried down and re-suspended in 2% HNO3 to TDS concentrations of 100 μg g−1 (as described earlier in this section). This was conducted in order to negate chloride-based interferences, which would have a much greater impact in this operation mode.Although these aliquots were already diluted below the 2000 μg g−1 matrix threshold, they were diluted further by a factor of 10 to produce working solutions with TDS concentrations of 10 μg g−1. During this dilution step 1 ng mL−1 Pr was also added as the internal standard. The prepared samples were then ready for measurement by a Thermo Finnigan Element 2 ICP-SFMS and were subsequently placed onto its coupled Elemental Scientific (Omaha, U.S.A.) SC-μ DX autosampler. Table 1 lists the instrument components used for this analysis, together with the typical operating settings that were adopted to achieve a sensitive and steady state signal from this conventional sample introduction system format.
The instrument was optimised in all three resolutions (LR, MR and HR) prior to undertaking the sample measurements, using a 1 μg L−1 tune solution to achieve routine performance criteria indicated by Thermo Scientific. The measurement method (see Table S2 in ESI† for details) was developed using the Thermo Finnigan Element software and exploited the high resolution capability of this magnetic-sector type ICP-MS instrument. The washout times that are quoted in Table S2† are for the samples only and were sufficient for the levels that were introduced into the instrument. However, during the analysis those times increased to a maximum of 480 seconds, after the measurement of the high standard from the calibration standard series, in order to flush-out any significant memory.
The measurement running order was arranged in a similar way to the analysis conducted using the PerkinElmer NexION ICP-MS, except each sample was measured only once. Several blanks were initially measured to ensure that there was a steady baseline, prior to the standard addition calibration. The calibrants for this measurement procedure were prepared in four stages: (1) praseodymium, as the internal standard, was added at the same concentrations as those added to the samples and blanks; (2) aliquots of one sample were added to each of the seven calibrants at concentrations to match the TDS concentrations in the samples (i.e. 10 μg g−1); (3) calibrants were then doped with increasing concentrations of the analytes of interest (at a range from 0 to 10 ng g−1) to cover the likely concentrations in the diluted samples. These analytes were added from either a high concentration stock standard (100 ng g−1), for the more concentrated elements, or a low concentration stock standard (1 ng g−1), for those elements in the pg g−1 range; (4) 2% HNO3 was added at variable aliquots to dilute each standard to a final volume of 10 mL volume to complete the formation of each calibrant solution.
The samples were then measured in groups of 10 and a new calibration performed after 20 samples. Monitor standards and a quality control were routinely measured throughout the run in a similar fashion and purpose to those measurements made by the FI-ICP-MS, and were sandwiched between the sample groups when a re-calibration wasn't undertaken.
3. Results and discussion
3.1 Detection limits and signal strength
The method detection limits (MDLs) presented in Table 2 are calculated as three times the standard deviation of a series of instrument blanks (a blank containing the same acid matrix in the samples) and corrected for the sample preparation dilutions (circa 2 × 102 for FI-ICP-MS and circa 1 × 105 for ICP-SFMS). More than ten blanks were measured in both analytical techniques (n = 19 for FI-ICP-MS and n = 13 for ICP-SFMS). The blanks were periodically measured throughout both runs and were bracketed before and after a series of at least five samples or standards.| Isotope | Abundance (%) | Potential spectral interferences | MDL FI-ICP-MS (μg g−1) | MDL ICP-SFMS (μg g−1) |
|---|---|---|---|---|
| 52Cr | 83.8 | 36Ar16O, 35Cl16O1H, 40Ar12C, 37Cl14N1H | 0.081 | 0.088 |
| 63Cu | 30.8 | 31P(16O)2 | 0.020 | 0.367 |
| 69Ga | 60.1 | 37Cl(16O)2,53Cr16O, 32S37Cl | 0.329 | 0.068 |
| 99Ru | 12.8 | 59Co40Ar, 62Ni37Cl | 0.117 | 0.117 |
| 103Rh | 100 | 63Cu40Ar, 31P35Cl37Cl | 0.011 | 0.078 |
| 105Pd | 22.3 | 35Cl3,65Cu40Ar, 31P(37Cl)2 | 0.075 | 0.026 |
| 185Re | 37.4 | — | 0.002 | 0.001 |
| 193Ir | 62.7 | — | 0.017 | 0.495 |
| 195Pt | 33.8 | — | 0.003 | 0.114 |
| 197Au | 100 | — | 0.051 | 0.058 |
Preparation blanks were also measured, but their levels were all found to be below detection limit, except for Cu and Pt from FI-ICP-MS, which recorded slight elevations above detection. However, when compared to the working sample concentrations these concentrations were actually very similar to those found from the instrument blank. Also, as the number of replicates of the preparation blanks was constrained by their fixed 15 mL final volume, the number of allowable measurements would have been less than 10. The ability to measure many more blanks from the instrument blanks, and so to capture any deviation in instrument backgrounds, in our view was a better way to measure the detection limit for this study. Additionally, the blank measurements were carefully positioned at timed intervals throughout the analytical runs in order to capture any memory build-up (i.e. straight after a series of standards or samples). Therefore, the detection limit data, in Table 2, will also provide an insight into any contamination build-up, which can be a problem for this suite of elements.
Most qICP-MS instruments, including the PerkinElmer NexION 350, are widely considered to have lower performance characteristics than their high sensitivity and high resolution sector-field counter-parts (e.g. Thermo Finnigan Element 2). However, once coupled with the prepFAST-FIAS, the NexION is able to measure samples with TDS concentrations that exceed the 2000 μg g−1 limit, which opens up the possibility to analyse at significantly lower sample dilutions and therefore benefit from lower method detection limits. In Table 2 the MDLs for the FI-ICP-MS technique are lower for all elements in comparison to those measured by ICP-SFMS, with the exception of Ru, Ga, Pd and Re. In the case of Ga, the formation of 37Cl(16O)2 and 32S37Cl are likely candidates to have caused interference on m/z 69, which will account for the high recorded backgrounds and also the raised detection limit for this element, even after the use of He in the cell to achieve reductions in these molecular interferences. Instead of having the samples in a hydrochloric acid matrix, measurements by ICP-SFMS were made in 2% HNO3. In addition to this the ICP-SFMS was utilised with its high resolution capability to fully separate the analyte signal from the interference,28 and thus was able to record a much lower MDL for elements such as Ga.
Fig. 1 illustrates the differences found in the detection of the trace elements in the iron meteorites, between the FI-ICP-MS and ICP-SFMS techniques. The chart plots the range and average ratios of the sample concentrations to the method detection limit, which is an indicator for the sensitivity achieved by each method.
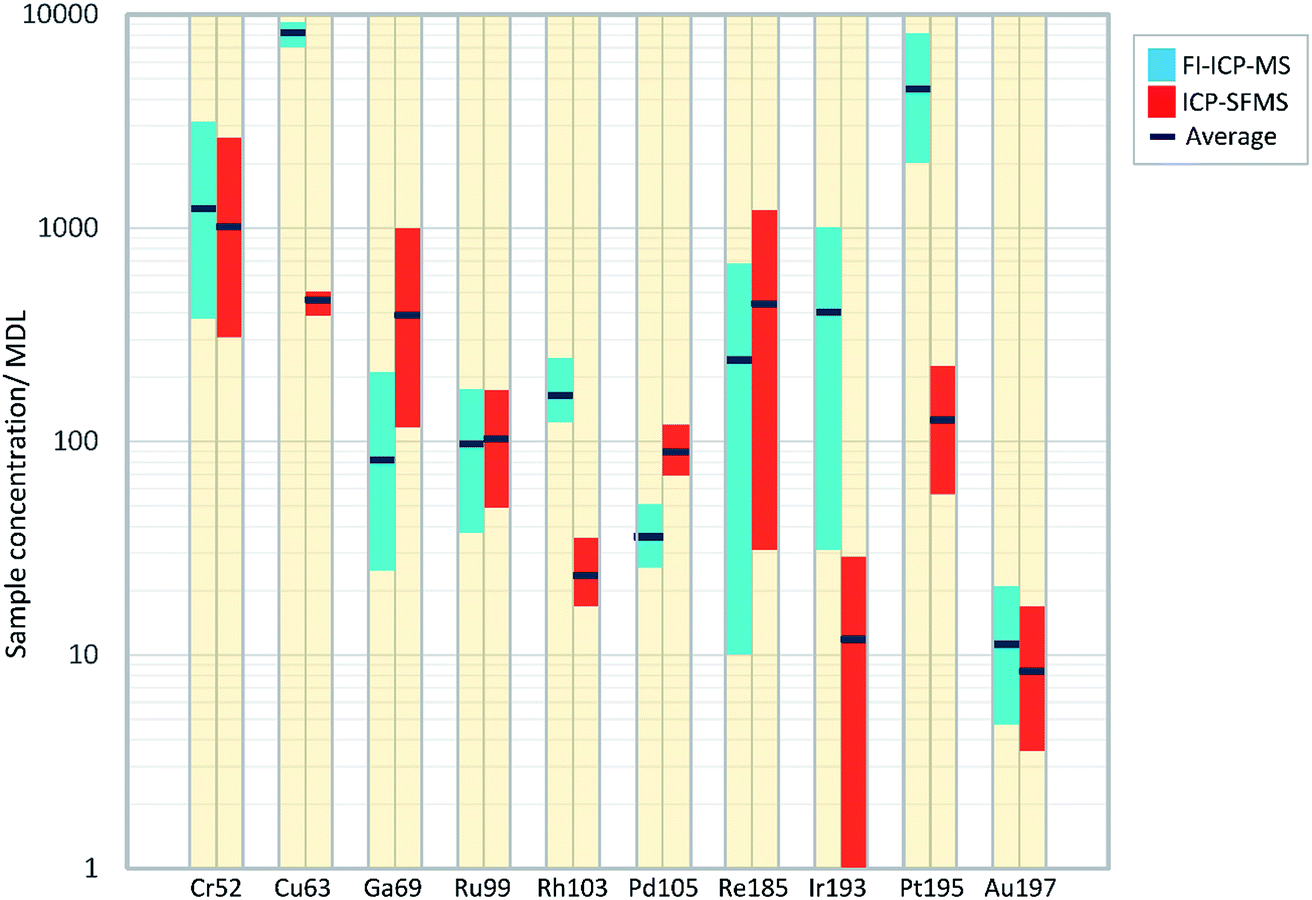 | ||
| Fig. 1 Plot of sample concentrations/method detection limit for both techniques, where the coloured bars illustrate the range found between the measured samples. For some elements the range is large, where it extends over an order of magnitude (e.g. Re and Ir). This reflects the broad, natural variations in the compositions of many trace elements between samples for this type of meteorite. The variability in the sample concentrations is also reflected with the position of the average value, which for several elements is skewed away from the middle of the bars. | ||
From an initial inspection it can be seen that six out of the ten elements (Cr, Cu, Rh, Ir, Pt and Au) have elevated ratios for FI-ICP-MS over ICP-SFMS, which indicates that the signal to background noise is better from measurements by the FI-ICP-MS technique for these elements. Indeed, for Cu, Rh, Ir and Pt the range bars for FI-ICP-MS exceed those reported by ICP-SFMS without any over-lap, thus indicating that there is a definite sensitivity advantage by using the FI-ICP-MS approach over ICP-SFMS for these elements. All of the ratios plot with values greater than 10 from measurements by FI-ICP-MS, with the analysis of Au in Henbury, Boxhole and Clark County being the only exceptions, which indicates that most of the data are recorded significantly above detection limit levels.
Values from the ICP-SFMS measurements were only found to better those conducted by FI-ICP-MS for Ga, Pd and Re, which in the case of the former two elements is probably due to the interferences encountered by the FI-ICP-MS (as has just been discussed). Elements that were reported only just above detection by ICP-SFMS include Ir and Au. Indeed, in the case of Ir the measurement for Orange River was actually below detection, where the range bar extends down to the minimum value in the graph to indicate this.
From the group of trace elements (Rh, Ir, Pd, Re and Au) all elements, with the exception of Pd and Re report higher ratios by FI-ICP-MS, and as previously discussed Rh and Ir are significantly better. The lowest average ratio from both instruments is found from Au. Although generally found at a higher concentration in iron meteorites than Re, the lower ratio values presented by Au may essentially be a result of its relatively high first ionisation energy (890 kJ mol−1) and the well documented memory problems for this element in the sample introduction system.29
From the group of intermediate concentration elements (Cr, Cu, Ga, Ru, Ir and Pt), improved sensitivities were recorded by all from the FI-ICP-MS with the exception of Ga and Ru. Significant advances were made for Ir, Cu and Pt, where average ratios greater than 30 times those made by ICP-SFMS were found for Ir and Pt.
Finally, it is presented in Table 2 that Re, Ir, Pt and Au are not affected by any potential spectral interferences, but as discussed in Section 2.3 were still measured under He KED conditions, rather than no gas (or standard mode). As has been described in Section 2.3, the interference removal capability provided by KED significantly improves the accuracy for many elements, with the only detrimental effect being a reduction in the analyte signal. However, as all of these elements still recorded good detection from the KED mode measurements (as described above and shown in Fig. 1), a sole cell gas mode was selected for this study. Also, an additional benefit from running only in KED mode was the reduced time required to purge the cell per sample, which ensured that a rapid measurement methodology was maintained.
3.2 Accuracy and precision
| Meteorite | Cr | (±2σ) | Cu | (±2σ) | Ga | (±2σ) | Ru | (±2σ) | Rh | (±2σ) | Pd | (±2σ) | Re | (±2σ) | Ir | (±2σ) | Pt | (±2σ) | Au | (±2σ) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a All uncertainties are calculated from the repeat measurement of a synthetically prepared monitor standard solution (n = 8 for FI-ICP-MS and n = 6 for ICP-SFMS). <DL indicates below detection limit. | ||||||||||||||||||||
| Boxhole (group IIIAB) | ||||||||||||||||||||
| FI-ICP-MS | 105 | (±20.3) | 165 | (±27.0) | 20.7 | (±4.35) | 12.2 | (±1.58) | 1.72 | (±0.19) | 2.29 | (±0.28) | 0.75 | (±0.11) | 9.73 | (±1.22) | 15.8 | (±1.67) | 0.48 | (±0.037) |
| ICP-SFMS | 92.9 | (±6.98) | 184 | (±16.0) | 20.5 | (±1.56) | 13.4 | (±0.26) | 1.86 | (±0.021) | 2.00 | (±0.023) | 0.80 | (±0.013) | 9.09 | (±0.19) | 15.7 | (±0.22) | 0.49 | (±0.066) |
|
||||||||||||||||||||
| Orange River (group IIIAB) | ||||||||||||||||||||
| FI-ICP-MS | 43.7 | (±8.45) | 148 | (±24.3) | 23.1 | (±4.86) | 4.38 | (±0.57) | 1.41 | (±0.16) | 3.81 | (±0.47) | 0.021 | (±0.003) | 0.52 | (±0.065) | 6.81 | (±0.72) | 1.08 | (±0.085) |
| ICP-SFMS | 40.1 | (±3.02) | 156 | (±13.6) | 21.6 | (±1.64) | 5.73 | (±0.11) | 1.47 | (±0.016) | 3.06 | (±0.035) | 0.037 | (±0.001) | <DL | — | 6.45 | (±0.090) | 0.98 | (±0.13) |
|
||||||||||||||||||||
| North Chile (group IIAB) | ||||||||||||||||||||
| FI-ICP-MS | 53.2 | (±10.3) | 138 | (±22.6) | 69.1 | (±14.5) | 20.6 | (±2.67) | 2.64 | (±0.29) | 1.93 | (±0.24) | 0.21 | (±0.030) | 4.19 | (±0.52) | 27.5 | (±2.89) | 0.54 | (±0.043) |
| ICP-SFMS | 46.9 | (±3.53) | 142 | (±12.4) | 67.2 | (±5.11) | 20.3 | (±0.40) | 2.74 | (±0.031) | 1.77 | (±0.020) | 0.22 | (±0.004) | 3.56 | (±0.076) | 25.7 | (±0.36) | 0.35 | (±0.047) |
|
||||||||||||||||||||
| Henbury (group IIIAB) | ||||||||||||||||||||
| FI-ICP-MS (A) | 130 | (±25.1) | 181 | (±29.7) | 21.4 | (±4.48) | 15.4 | (±2.00) | 1.87 | (±0.21) | 2.26 | (±0.28) | 1.44 | (±0.20) | 16.7 | (±2.10) | 18.6 | (±1.96) | 0.41 | (±0.033) |
| FI-ICP-MS (B) | 129 | (±24.8) | 179 | (±29.2) | 22.0 | (±4.62) | 15.5 | (±2.01) | 1.84 | (±0.20) | 2.27 | (±0.28) | 1.41 | (±0.20) | 16.5 | (±2.06) | 18.4 | (±1.94) | 0.41 | (±0.032) |
| ICP-SFMS | 110 | (±8.27) | 179 | (±15.6) | 20.5 | (±1.56) | 15.3 | (±0.30) | 1.87 | (±0.021) | 1.79 | (±0.020) | 1.44 | (±0.023) | 14.3 | (±0.31) | 17.1 | (±0.24) | 0.30 | (±0.040) |
|
||||||||||||||||||||
| Charcas (group IIIAB) | ||||||||||||||||||||
| FI-ICP-MS | 80.5 | (±15.5) | 167 | (±27.3) | 22.3 | (±4.68) | 9.59 | (±1.24) | 1.67 | (±0.18) | 2.72 | (±0.33) | 0.15 | (±0.021) | 2.68 | (±0.34) | 13.0 | (±1.37) | 0.58 | (±0.046) |
| ICP-SFMS | 69.9 | (±5.26) | 178 | (±15.5) | 22.8 | (±1.73) | 10.6 | (±0.21) | 1.78 | (±0.020) | 2.29 | (±0.026) | 0.16 | (±0.003) | 2.26 | (±0.048) | 12.6 | (±0.18) | 0.36 | (±0.049) |
|
||||||||||||||||||||
| Cumpas (group IIIAB) | ||||||||||||||||||||
| FI-ICP-MS | 30.2 | (±5.84) | 170 | (±27.9) | 23.8 | (±4.99) | 9.08 | (±1.17) | 1.71 | (±0.19) | 3.08 | (±0.38) | 0.24 | (±0.034) | 3.79 | (±0.47) | 12.9 | (±1.35) | 0.70 | (±0.055) |
| ICP-SFMS | 27.0 | (±2.03) | 175 | (±15.3) | 23.9 | (±1.82) | 10.1 | (±0.20) | 1.79 | (±0.020) | 2.73 | (±0.031) | 0.27 | (±0.004) | 3.44 | (±0.073) | 12.3 | (±0.17) | 0.72 | (±0.096) |
|
||||||||||||||||||||
| Clark County (group IIIF) | ||||||||||||||||||||
| FI-ICP-MS | 252 | (±48.6) | 162 | (±26.4) | 8.18 | (±1.72) | 8.17 | (±1.06) | 1.32 | (±0.15) | 2.84 | (±0.35) | 0.75 | (±0.11) | 9.28 | (±1.16) | 11.2 | (±1.18) | 0.24 | (±0.019) |
| ICP-SFMS | 231 | (±17.3) | 166 | (±14.5) | 7.91 | (±0.60) | 8.35 | (±0.16) | 1.32 | (±0.015) | 2.30 | (±0.026) | 0.76 | (±0.012) | 7.95 | (±0.17) | 10.5 | (±0.15) | 0.21 | (±0.028) |
Much of the data in the table show very good agreement between the two techniques, which can be seen from their quoted precision ranges (calculated as two times the standard deviation of repeat measurements of a synthetic standard [n = 8 for FI-ICP-MS and n = 6 for ICP-SFMS]). Together with the excellent agreement in the Henbury duplicates, this dataset offers significant confidence in support of the preparation method, as in addition to the measurements being conducted at different times, the samples were also prepared on two different occasions. The exception is for Au, which has high variance in values for North Chile and Charcas. As discussed in Section 3.1 this poor agreement may be a result of these values being only marginally above detection.
Table 4 evaluates the repeatability of the measurement of the iron meteorites by the FI-ICP-MS technique. The individual five replicates from the measurement of North Chile are presented, together with a statistical analysis of the replicate data and a comparison to literature values. North Chile was chosen as it is a relatively homogenous meteorite and also has a large quantity of high quality literature data available.20
| Cr | Cu | Ga | Ru | Rh | Pd | Re | Ir | Pt | Au | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Those data from D'Orazio et al., 2004 (ref. 20) were measured by ICP-MS. Those data from Lovering et al., 1967 (ref. 30) and Moore et al., 1969 (ref. 31) were determined spectrographically. | ||||||||||
| Replicate 1 | 52.0 | 134 | 71.4 | 20.4 | 2.64 | 1.79 | 0.205 | 4.00 | 26.5 | 0.557 |
| Replicate 2 | 52.9 | 138 | 65.3 | 20.5 | 2.55 | 2.09 | 0.215 | 4.21 | 27.2 | 0.508 |
| Replicate 3 | 55.9 | 145 | 71.3 | 21.3 | 2.68 | 2.01 | 0.219 | 4.37 | 29.1 | 0.552 |
| Replicate 4 | 53.1 | 138 | 68.5 | 21.3 | 2.66 | 1.98 | 0.207 | 4.17 | 27.5 | 0.542 |
| Replicate 5 | 52.2 | 134 | 69.1 | 19.7 | 2.65 | 1.80 | 0.226 | 4.18 | 27.2 | 0.544 |
| Average | 53.2 | 138 | 69.1 | 20.6 | 2.64 | 1.93 | 0.214 | 4.19 | 27.5 | 0.540 |
| 2σ | 3.13 | 8.58 | 4.97 | 1.35 | 0.10 | 0.26 | 0.017 | 0.26 | 1.97 | 0.038 |
| % RSD | 2.94 | 3.11 | 3.60 | 3.27 | 1.87 | 6.81 | 4.05 | 3.14 | 3.58 | 3.53 |
|
||||||||||
| Literature values | ||||||||||
| D'Orazio et al., 2004 (ref. 20) | — | — | 65.7 | 17.6 | 2.50 | 1.75 | 0.207 | 3.20 | 24.2 | 0.557 |
| Lovering et al., 1957 (ref. 30) | 48 | — | — | — | — | — | — | — | — | — |
| Moore et al., 1969 (ref. 31) | — | 140 | — | — | — | — | — | — | — | — |
The repeatability is generally very good, where values of the standard deviation are less than 5% for all elements, with the exception of Pd. There is also very good agreement to the literature values for most of the data, especially for Au, Rh, Pd, Ga, Cu and Re, which are all found within 2σ precision. The good comparisons found to the literature are re-assuring for this study, especially as most of the cited values were also measured by ICP-MS.20
Also, after contrasting data for Au was found from the comparison of the two ICP-MS techniques (as described earlier and shown in Table 3), it is very promising to discover that an excellent agreement was found between the literature data and the FI-ICP-MS measurements for this element (see Table 4).20 Indeed, although the concentration of Au found in North Chile from the ICP-SFMS is shown to be nearly 50% lower than the FI-ICP-MS value at 0.35 μg g−1 (see Table 3), the procedure for measurement by this technique and the extra dilutions that ensued, where the samples were diluted by a further two orders of magnitude more than those measured by D'Orazio et al., 2004,20 make it likely that this measurement is compromised by over-dilution.
Of the elements that do not fall within error of the literature values, Ru and Pt still actually fall within a wider reported literature range, taken from sources cited in D'Orazio et al., 2004.20 The range is not reported here as, apart from the Cr and Cu data values, it is of value for this study to compare data also obtained from ICP-MS. In addition to this, the variance in the reported trace element concentrations of iron meteorites may reflect their heterogenous nature and not necessarily poor performance by the analytical technique.32 Although North Chile is considered to be more homogenous than many counter-part iron meteorites,20 it still doesn't provide the ideal assessment of accuracy that a certified reference material would provide.
| Synthetic solution 1 | Synthetic solution 2 | Synthetic solution 3 | |
|---|---|---|---|
| No dope (just matrix metals) | Low dope | High dope | |
| Fe | 5000 μg g−1 | 5000 μg g−1 | 5000 μg g−1 |
| Ni | 400 μg g−1 | 400 μg g−1 | 400 μg g−1 |
| Co | 25 μg g−1 | 25 μg g−1 | 25 μg g−1 |
| P | 10 μg g−1 | 10 μg g−1 | 10 μg g−1 |
| S | 35 μg g−1 | 35 μg g−1 | 35 μg g−1 |
| Cr | — | 5 ng g−1 | 1000 ng g−1 |
| Cu | — | 5 ng g−1 | 1000 ng g−1 |
| Ga | — | 5 ng g−1 | 1000 ng g−1 |
| Ru | — | 10 ng g−1 | 100 ng g−1 |
| Rh | — | 10 ng g−1 | 100 ng g−1 |
| Pd | — | 10 ng g−1 | 100 ng g−1 |
| Re | — | 5 ng g−1 | 100 ng g−1 |
| Ir | — | 10 ng g−1 | 100 ng g−1 |
| Pt | — | 10 ng g−1 | 100 ng g−1 |
| Au | — | 10 ng g−1 | 100 ng g−1 |
The additions of the matrix and trace elements into these solutions were made from Merck Certipur 1000 ppm ICP standards, with the exception of Fe, which was dissolved and added from an iron-hydrate nitrate solid (Alpha Aesar, Puratronic Iron III Nitrate Hydrate, 99.999%, lot. number 10191822). The added reagents were evaporated to dryness on a hotplate and then re-dissolved into 4% HCl, to mimic the matrix acid of the samples.
The results gained from the measurement of these solutions are shown in Fig. 2, which presents scatter plots of the recorded concentrations (those that are similar to an average iron meteorite) versus the theoretical values. Table 6 presents the results for each replicate that were measured as part of the investigation, together with statistical data of the precision (2× RSD% from the seven replicates) and accuracy for the entire dataset.
 | ||
| Fig. 2 Measured versus theoretical trace element concentrations from the synthetic meteorites (a). Total plot: especially examines intermediate dope transition elements (b). Zoomed image of blue shaded square in (a), to highlight the intermediate dope PGEs (c). Zoomed image of red shaded square in (b), to highlight low dope PGEs and Re. | ||
| Cr | Cu | Ga | Ru | Rh | Pd | Re | Ir | Pt | Au | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Values in italic are data recorded in excess of those reported for the synthetic meteorite analysis and also plotted in Fig. 2. | ||||||||||
| Low spike | ||||||||||
| Replicate 1 | 5.44 | 6.24 | 4.66 | 9.02 | 8.83 | 8.23 | 4.62 | 9.47 | 8.88 | 9.02 |
| Replicate 2 | 4.81 | 4.52 | 3.46 | 9.32 | 8.79 | 8.07 | 4.81 | 9.44 | 8.52 | 8.04 |
| Replicate 3 | 6.10 | 6.03 | 3.91 | 9.89 | 9.17 | 7.92 | 4.83 | 9.32 | 9.02 | 8.27 |
| Replicate 4 | 5.24 | 5.54 | 3.65 | 8.64 | 8.51 | 8.11 | 4.69 | 9.14 | 8.95 | 8.24 |
| Replicate 5 | 4.16 | 6.66 | 3.40 | 8.94 | 8.55 | 7.66 | 4.85 | 9.36 | 8.53 | 8.28 |
| Replicate 6 | 5.94 | 6.15 | 3.93 | 9.32 | 8.87 | 8.49 | 4.59 | 9.51 | 8.55 | 8.76 |
| Replicate 7 | 5.60 | 5.12 | 3.81 | 8.93 | 8.75 | 8.00 | 4.88 | 9.46 | 8.68 | 8.59 |
| Average | 5.33 | 5.75 | 3.83 | 9.15 | 8.78 | 8.07 | 4.75 | 9.38 | 8.73 | 8.46 |
| Theoretical value | 4.86 | 4.82 | 4.84 | 9.71 | 9.58 | 9.69 | 4.72 | 9.70 | 9.73 | 9.78 |
| 2 × RSD % | 25.2 | 25.6 | 21.9 | 8.82 | 4.99 | 6.40 | 5.04 | 2.70 | 4.87 | 8.14 |
| Accuracy% | 9.67 | 19.2 | 20.8 | 5.75 | 8.32 | 16.7 | 0.77 | 3.24 | 10.21 | 13.5 |
|
||||||||||
| High spike | ||||||||||
| Replicate 1 | 941 | 855 | 920 | 95.1 | 0.95 | 85.1 | 91.8 | 94.1 | 90.1 | 85.2 |
| Replicate 2 | 959 | 861 | 954 | 95.4 | 0.97 | 86.8 | 95.0 | 94.8 | 90.2 | 88.3 |
| Replicate 3 | 929 | 843 | 906 | 87.3 | 0.93 | 85.5 | 91.4 | 93.4 | 87.4 | 84.6 |
| Replicate 4 | 965 | 874 | 970 | 95.5 | 1.19 | 89.5 | 94.6 | 96.6 | 91.9 | 88.9 |
| Replicate 5 | 1000 | 898 | 1005 | 91.1 | 1.00 | 90.1 | 96.5 | 99.7 | 92.5 | 93.0 |
| Replicate 6 | 928 | 834 | 921 | 90.7 | 1.02 | 82.3 | 88.9 | 91.2 | 86.2 | 84.9 |
| Replicate 7 | 933 | 856 | 938 | 91.5 | 0.93 | 87.3 | 91.9 | 95.1 | 88.7 | 86.6 |
| Average | 951 | 860 | 945 | 92.4 | 1.00 | 86.7 | 92.9 | 95.0 | 89.6 | 87.4 |
| Theoretical value | 969 | 963 | 966 | 96.6 | 97.1 | 96.4 | 94.2 | 98.0 | 96.8 | 97.3 |
| 2 × RSD % | 5.53 | 4.86 | 7.28 | 6.72 | 18.45 | 6.18 | 5.62 | 5.60 | 5.12 | 6.84 |
| Accuracy% | 1.89 | 10.68 | 2.16 | 4.38 | 98.97 | 10.11 | 1.36 | 3.08 | 7.44 | 10.23 |
In Fig. 2, 1:1 linears with propagated ±5%, ±10% and ±20% absolute error lines are included. Error bars are also shown for each data point, which denote 2σ precision calculated from the seven measurements of each doped solution. Fig. 2a highlights the entire data plot but due to their significant concentrations only the results for the high concentration transition elements (Cu, Cr and Ga) are clearly visible. Each of these elements are found with precision values less than 10% (values shown in Table 6), and for Cr and Ga the average values are well within ±5% absolute error. Copper also has a similar precision to Cr, with a value that is just below 5% (see Table 6), but records a higher uncertainty, which can be seen in Fig. 2, where the average value just plots outside ±10% error. Fig. 2b is a zoomed plot from the lower blue region of Fig. 2a, which especially highlights the intermediate PGEs. In the plot Ru and Ir form a pair which present excellent accuracy, where their average values plot within the 5% margin. The replicate data in Table 6 also illustrates the high levels of precision that were achieved for these elements, where all values were found to be less than 10%. Fig. 2c is a zoomed plot from the red region in Fig. 2b and focusses on the low concentration doped PGEs. As Ir shows variable concentrations in iron meteorites it has been duplicated at this level and again shows excellent agreement. Also, its precision value, of 2.70% (see Table 6) is actually better than that recorded from the high doped solution. Additionally, Rh also performs well where good precision and accuracy values were found. These performance values are reassuring and provide confidence that the system has the capability to provide accurate and precise data for elements, such as Ir, that have variable concentrations in iron meteorites. However, in contrast to the Ir and Rh data, Au and Pd record higher error values, where the average values for both elements, although lower than expected, plot in-between ±10% and ±20% absolute error, which is in contrast to their precision values which report values of less than 10% for both elements (see Table 6). Indeed, Au and Pd were also measured in the intermediate test (Fig. 2b), and although still lower than expected, are this time found just outside the ±10% absolute error line. Thus, this experiment indicates that these elements may induce higher error readings in comparison to others at the trace level in this type of sample, and is likely to be a result of the lower detection capability (Au) and chloride molecular interference (Pd), as discussed in Section 3.1. However, in contrast to these data, comparisons to real sample values found in the literature (e.g. the results from North Chile in Table 4) instead highlight excellent agreements for both elements. Finally, also presented in Fig. 2c is the result for Re, which unlike Au and Pd demonstrates excellent accuracy. Similarly, the replicate data for this element, shown in Table 6 also presents a good degree of precision, where values of just over 5% were obtained, both from the low doped and high doped solutions. Additionally, the Re results from both solutions also dictate similar attributes to those reported by Ir, where good performance is exhibited at low concentrations as well as high. Coupled to these findings, and similarly to the results found from many of the other trace siderophiles, Re also presents a good agreement to the literature value from the North Chile meteorite (see Table 4).
A concluding observation from this experiment is an outlier that was found from the data received for Rh from the high doped solution. Although good data was received from the low doped solution, which is the relevant concentration for the measurement of iron meteorites in this study, very poor recoveries and precision were received in the high doped solution. It is unclear why this occurred, but it is likely to be a preparation error rather than an instrumental issue. However, taking this outlier aside, these measurements have successfully been used to test the accuracy and precision of this technique, and in evaluation promote the capability of this FI-ICP-MS technique to measure elements with very low concentrations in a challenging, high matrix sample type to a high degree of performance.
3.3 Robustness

 | ||
| Fig. 3 Plot of internal standard recoveries for both techniques as the analytical runs progressed. (a) ICP-SFMS, (b) FI-ICP-MS. The symbols correspond to recoveries gained (as a percentage), relative to first blank. Samples were measured in groups (n = 10 for ICP-SFMS and n = 5 for FI-ICP-MS) that were bracketed with blanks (n = 2 for ICP-SFMS and n = 4 for FI-ICP-MS) and monitor standards (n = 2 for ICP-SFMS and FI-ICP-MS). TDS corresponds to ‘Total Dissolved Solids’ concentrations in the samples. Two standard addition calibrations were conducted for the ICP-SFMS analysis and were undertaken between acquisition numbers 6–12 and 43–49. The calibration standards for FI-ICP-MS were analysed at the beginning of the run and were undertaken between acquisition numbers 4–28. | ||
Even though the samples and standards from the ICP-SFMS analysis contained a TDS of only 10 μg g−1, a clear reduction in the internal standard intensity is seen (see Fig. 3a). As this analysis was calibrated by the method of standard additions, the blanks were the only solutions that were measured with a negligible TDS. Intriguingly, the internal standard intensities from the blanks actually have lower values relative to their neighbouring samples or standards. An important interpretation from this observation, and more especially as the signal does not return to values at or near to 100%, is that the instrument sample introduction system has suffered from significant matrix deposition onto its components. Fig. 3a illustrates this clearly, where the instrument response is shown to decrease by more than 20% over the course of the run. Additionally this highlights the extra impact that iron, as the near sole TDS contributor, has on the instrument system, given that the TDS levels in the samples measured by ICP-SFMS are 200 times lower than the generally accepted TDS limit.
In contrast to the ICP-SFMS profile, the FI-ICP-MS internal standard recovery profile (see Fig. 3b) has a clear definition between the blanks and the high matrix sample solutions. As illustrated by the blue markers in the plot, all of the blank (or standard) measurements, apart from a couple of excursions, have internal standard signals that return back to values recorded from the first blank. This clearly highlights that the instrument system is robust to the sample matrix and that it was never over-loaded with the high TDS concentrations. The divergence in the internal standard recoveries between the samples and the blanks indicates that the sample matrix is causing suppression on the plasma, however each measured element has been carefully paired with an internal standard that best fits with their mass and first ionisation potential, in order that this suppression is normalised effectively. This has been proved from the good accuracy and precision data gained from the tests on the synthetic meteorites, as discussed previously in Section 3.2.2.
The standards used for calibration of the FI-ICP-MS were prepared inline by the Elemental Scientific prepFAST system from two stock solutions and were diluted in only the 4% HCl diluent, together with an addition of the internal standard. The excellent agreement of the data collated from the FI-ICP-MS and ICP-SFMS measurements (presented in Table 3) highlights the capability of the flow injection system to operate robustly without the need to matrix match standards or to apply matrix ‘conditioning’ to the sample introduction system. This is unlike interpretations made by Wang et al., 2016,17 who also measured iron meteorites by ICP-MS using external standard calibrations, but instead found that correct matrix-matching of the standards was key to obtaining results of high accuracy.
4. Conclusions
The data compiled from the measurements of the iron meteorites by both FI-ICP-MS and ICP-SFMS provide very good agreement for most elements. Although the agreement is not as strong for Au, which generally has a lower concentration than many of the other elements studied, the FI-ICP-MS replicate data from North Chile present excellent agreement to the literature for this element. This indicates the excellent capability of the FI-ICP-MS technique to detect and measure trace elements with a high degree of accuracy. In addition to this the precision and agreement of all of the other elements from the North Chile replicates to literature values were also very good, where relative standard deviations for most elements were less than 5%.To the best of our knowledge the measurement of trace elements by ICP-MS has not been conducted before on iron meteorite sample solutions at TDS concentrations that exceed the generally accepted 2000 μg g−1 limit. In this study the FI-ICP-MS technique was tested for its capability to measure the prepared samples at a TDS concentration of 5000 μg g−1. The lower dilutions enabled the FI-ICP-MS to record lower MDLs for all but four elements measured in this study, in comparison to those gained by the highly sensitive ICP-SFMS, and also gain much higher signal to detection limit ratios (for those elements with a lower MDL). In addition to this, data from the measurement of the synthetic iron meteorites confirm the ability of the FI-ICP-MS technique to measure all of the elements studied to a good degree of accuracy and precision, and without the matrix effects described by Wang et al., 2016 (ref. 17) and D'Orazio and Folco, 2003.19 This further confirms the powerful capability of this technique to measure trace elements accurately and precisely in challenging, high matrix-containing samples.
Also, the robustness of both measurement techniques were investigated thoroughly. Intra-run robustness, plotted from the internal standard recoveries illustrates very different profiles between the analytical techniques. Even though the TDS concentrations were significantly lower than the generally accepted limit for ICP-MS, the profile from the ICP-SFMS analysis shows that the instrument suffered from the negative impacts of analytical drift, caused by contamination build-up in the sample introduction system. In contrast the profile for the FI-ICP-MS results does not present any sign of analytical drift and the consistently high recovery values for the blank solutions indicate that the plasma was never over-loaded. Also, quality control data collated from the FI-ICP-MS technique over four separate sessions, using the same sample introduction components, show a high degree of repeatability. As this ICP-MS instrument never became over-loaded with the high matrix concentrations, these measured values provide confidence that the instrument system can be operated for a number of sessions without the concern of analytical drift.
Finally, the successful development of the flow injection ICP-MS method in this study to measure trace elements at high TDS concentrations in iron meteorites now provides the opportunity to employ this technique for other applications. Indeed, the same technique has also recently, successfully reported multi-elements for a bio-geochemistry study,34 but, its capabilities could also be demonstrated with other sample types for other research or industrial applications, where trace element analyses in high TDS concentrations or challenging sample matrices are required.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Jane Barling and Naomi Saunders for their help with the dissolution of the samples at the Department of Earth Sciences, University of Oxford. This research was supported by STFC grants (ST/M001318/1 and ST/G00272X/1). The research materials supporting this publication can be accessed by contacting PB. The Natural History Museum (London) and the Smithsonian are thanked for access to the samples. Finally, we thank Ms Harriet Brewerton and the two anonymous reviewers for their comments on this paper.Notes and references
- W. M. White, Geochemistry, Wiley-Blackwell, Chichester, 2013 Search PubMed.
- B. A. Haley, G. P. Klinkhammer and A. C. Mix, Earth Planet. Sci. Lett., 2005, 239, 79–97 CrossRef.
- Y. Huh, L.-H. Chan and O. A. Chadwick, Geochem., Geophys., Geosyst., 2004, 5, 1–22 CrossRef.
- G. A. Jenner, H. P. Longerich, S. E. Jackson and B. J. Fryer, Chem. Geol., 1990, 83, 133–148 CrossRef.
- R. C. Hutton and A. N. Eaton, J. Anal. At. Spectrom., 1988, 3, 547–550 RSC.
- E. McCurdy and W. Proper, Spectroscopy, 2014, 29, 14 Search PubMed.
- N. Praphairaksit and R. S. Houk, Anal. Chem., 2000, 72, 2356–2361 CrossRef PubMed.
- M. Rehkämper and A. N. Halliday, Talanta, 1997, 44, 663–672 CrossRef.
- M. Fischer-Gödde, H. Becker and F. Wombacher, Geochim. Cosmochim. Acta, 2010, 74, 356–379 CrossRef.
- X. Duan and M. Regelous, J. Anal. At. Spectrom., 2014, 29, 2379–2387 RSC.
- D. J. Malvin, D. Wang and J. T. Wasson, Geochim. Cosmochim. Acta, 1984, 48, 785–804 CrossRef.
- J. T. Wasson and J. W. Richardson, Geochim. Cosmochim. Acta, 2001, 65, 951–970 CrossRef.
- D. E. Ryan, J. Holzbecher and R. R. Brooks, Chem. Geol., 1990, 85, 295–303 CrossRef.
- E. R. D. Scott and J. T. Wasson, Rev. Geophys. Space Phys., 1975, 13, 527–546 CrossRef.
- E. Chajduk and H. Polkowska-Motrenko, J. Radioanal. Nucl. Chem., 2017, 311, 1347–1353 CrossRef PubMed.
- Sk. A. Latif, Y. Oura, M. Ebihara and H. Nakahara, Anal. Bioanal. Chem., 2013, 405, 8749–8759 CrossRef PubMed.
- G. Wang, Y. Wu and Y. Lin, Rapid Commun. Mass Spectrom., 2016, 30, 543–551 CrossRef PubMed.
- P. Bonnand and A. N. Halliday, Nat. Geosci., 2018, 11, 401–404 CrossRef.
- M. D'Orazio and L. Folco, Geostand. Geoanal. Res., 2003, 27, 215–225 CrossRef.
- M. D'Orazio, L. Folco and N. Perchiazzi, Meteorit. Planet. Sci., 2004, 39, A133–A138 CrossRef.
- J. Ruzicka and E. H. Hansen, Anal. Chim. Acta, 1975, 78, 145–157 CrossRef.
- W. Van Bussel, PerkinElmer Technical Report, An Introduction and Overview of FAST-FIAS Coupled to the NexION ICP-MS, Perkin Elmer, Waltham, 2016 Search PubMed.
- T. Wiesenhofer, K. A. Francesconi and D. Kuehnelt, Microchem. J., 2017, 130, 310–315 CrossRef.
- M. Hollenbach, J. Grohs, S. Marnich, M. Kroft and E. R. Denoyer, J. Anal. At. Spectrom., 1994, 9, 927–933 RSC.
- Z. Zhu and A. Zheng, Spectroscopy, 2017, 32, 50–59 Search PubMed.
- M. Azaroual, B. Romand, P. Freyssinet and J.-R. Disnar, Geochim. Cosmochim. Acta, 2001, 65, 4453–4466 CrossRef.
- I. Jarvis, M. M. Totland and K. E. Jarvis, Chem. Geol., 1997, 143, 27–42 CrossRef.
- M. D. Axelsson, I. Rodushkin, J. Ingri and B. Öhlander, Analyst, 2002, 127, 76–82 RSC.
- S. E. Jackson, B. J. Fryer, W. Gosse, D. C. Healey, H. P. Longerich and D. F. Strong, Chem. Geol., 1990, 83, 119–132 CrossRef.
- J. F. Lovering, W. Nichiporuk, A. Chodos and H. Brown, Geochim. Cosmochim. Acta, 1957, 11, 263–278 CrossRef.
- C. B. Moore, C. F. Lewis and D. Nava, Meteorite Research, 1969, pp. 738–748 Search PubMed.
- G. K. Benedix, T. J. McCoy and K. Keil, Meteorit. Planet. Sci., 1997, 32, A11–A12 Search PubMed.
- G. Faure, Principles and Applications of Geochemistry, Prentice Hall, New Jersey, 2nd edn, 1998 Search PubMed.
- Q. Zhang, J. Snow, P. Holdship, D. Price, P. Watson and R. E. M. Rickaby, J. Anal. At. Spectrom., 2018, 33, 1196–1208 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ja00162f |
| This journal is © The Royal Society of Chemistry 2018 |