
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal analysis in polymers using tandem LA-ICP-MS/LIBS: eliminating matrix effects using multivariate calibration†
Maximilian
Bonta
 and
Andreas
Limbeck
*
and
Andreas
Limbeck
*
TU Wien, Institute of Chemical Technologies and Analytics, Getreidemarkt 9/164-I2AC, 1060 Vienna, Austria. E-mail: andreas.limbeck@tuwien.ac.at
First published on 25th June 2018
Abstract
Nowadays, metal analysis in polymers is experiencing growing interest due to increased environmental regulations and the need for sustainable polymer recycling strategies. Quick and reliable analyses are required to fulfill the demands of today's industry. Due to the high chemical inertness of most polymers, traditional solution-based analysis is often not an option and solid-sampling techniques such as Laser Ablation Inductively Coupled Plasma Mass Spectrometry (LA-ICP-MS) or Laser Induced Breakdown Spectroscopy (LIBS) have to be employed as an alternative. These, however, are typically prone to matrix effects and for each polymer type a separate reference material with known concentration may be required – an approach which is obviously not suitable if the polymer type is not even known. To overcome these difficulties, a tandem LA-ICP-MS/LIBS procedure coupled with statistical analysis has been used in this study. LIBS is known to be especially prone to matrix effects – which has been used as a benefit here. Complete broadband LIBS spectra with a wealth of information have been used as signatures for the investigated polymer types (polyimide, polymethylmethacrylate and polyvinylpyrrolidone) to serve the purpose of reducing matrix effects. While LIBS allowed the detection of alkali metals and alkali earth metals even at lower concentrations, LA-ICP-MS was used simultaneously for the analysis of other trace metals in the μg g−1 regime. Na, Sr, Co, In, and Pt were used as exemplary analytes at concentrations ranging from as low as 0.1 μg g−1 up to 300 μg g−1. Using the combined dataset of all three polymer types (in total 23 samples), multivariate calibration models could be constructed for all elements of interest. Validation was performed using a set of 22 external samples showing relative average deviations from their actual elemental content of 4.4%, but not more than 9.6%.
Introduction
Polymers are nowadays widely used in industrial applications, ranging from food packaging to the housing of electronic devices.1 With increasing complexity of the composition of these materials, also their trace element composition is gaining more and more interest. Factors to be assessed are for example toxicity,2 environmental concerns, or possible detrimental effects to the material properties. Traditionally, for this task, polymers are first brought into a solution and subsequently analyzed using liquid analysis techniques such as inductively coupled plasma-optical emission spectroscopy (ICP-OES) or inductively coupled plasma-mass spectrometry (ICP-MS).3,4 The problematic aspect of this strategy is the sample preparation step; due to the high chemical inertness of most polymers, harsh reaction conditions (e.g., oxidizing agents and microwave digestion) have to be used in order to produce a homogeneous sample solution. Not only have hazardous chemicals to be employed, but also the workload of the aforementioned sample preparation steps is a major point of concern. Alternately, direct solid sampling techniques can be applied to solve the problem of sample preparation. Here, the number of available techniques is limited by the usual absence of the electrical conductivity of polymers, which aggravates the analysis using OES with an arc/spark excitation source5 or glow discharge-optical emission spectroscopy/mass spectrometry (GD-OES/MS).6,7 Most commonly applied techniques are therefore X-ray based (XRF and EPMA);8,9 furthermore laser-assisted techniques such as LIBS10,11 or LA-ICP-MS12,13 have been used. Due to their non-destructive nature, XRF and EPMA are limited to the analysis of the surface near sample regions, whereas LA-ICP-MS and LIBS also offer the possibility of depth profiling.14,15 However, both laser based methods are inherently susceptible to so-called matrix effects meaning a variable absolute signal response based on the bulk composition of the sample. This is especially problematic in the case of polymers, where a large variety of matrix compositions can be encountered. Different properties of said materials, e.g., absorption, volatility, hardness, or other factors play a crucial role in the occurrence of such matrix effects.16–19The most straightforward way to overcome these problems is the preparation of matrix-matched standards where the composition of the calibrant (also referred to as the external standard) is matched to the closest possible extent with that of the sample. In the case of polymers, two problems arise with this strategy. Firstly, the preparation of bulk polymers homogeneously spiked with trace elements of interest is not necessarily a simple task. Secondly, the calibration strategy proves to be problematic when more than one such polymer type or mixture needs to be analyzed, or especially if the polymer composition of the sample of interest is not exactly known.
However, LIBS has already been shown to be an excellent technique for qualitative analysis, even when simultaneously used with ICP-MS detection.20 Especially, broadband LIBS analysis is useful if the elements to be analyzed are not known prior to analysis. In the field of polymer analysis, broadband LIBS spectra have already been used for the classification of different polymer types by exploiting the matrix effect which is in most cases rather considered problematic than useful.21,22
To overcome the above-mentioned difficulties, a novel procedure for standard preparation using spin-coating combined with multi-elemental analysis using tandem LA-ICP-MS/LIBS and multi-variate data evaluation for the matrix-independent quantification of trace elements in polymers is presented in this work. Spin-coating to form thin polymer layers can be used as a simple substitution procedure for the preparation of bulk polymer standards. A multi-variate chemometric treatment of the data has been shown to allow the reduction of matrix-effects by building a multi-matrix training set for obtaining calibration functions. The applicability of the approach is demonstrated using three polymer matrices and five elements of interest present at concentrations in the low to high μg g−1 range.
Experimental
Chemicals
Silicon wafers (n-doped) were obtained from Infineon Austria AG (Villach, Austria). Polyimide P84 in powder form (>98% purity) was kindly provided by HP Polymer GmbH (Lenzing, Austria). Polymethylmethacrylate (average MW > 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and polyvinylpyrrolidone (PVP 40) were of high purity grade (>99%) and were purchased from VWR International GmbH (Vienna, Austria).
000) and polyvinylpyrrolidone (PVP 40) were of high purity grade (>99%) and were purchased from VWR International GmbH (Vienna, Austria).
Acetylacetonate salts of Na, Sr, In, Co, and Pt were all of 99% grade or higher and were obtained from Merck (Darmstadt, Germany). 1-Methyl-pyrrolidone was of p.a. grade and was also purchased from Merck (Darmstadt, Germany).
Instrumental
Tandem LA-ICP-MS/LIBS experiments were carried out using a commercially available J200 tandem LA/LIBS system (Applied Spectra, Inc., Fremont, CA) equipped with a frequency quadrupled Nd:YAG laser operating at a wavelength of 266 nm. Radiation emitted from the laser induced plasma upon firing of the laser was collected by an optical fiber. Subsequently, analysis was performed using a Czerny–Turner type spectrometer with six-channel CCD detection, enabling the analysis of a wide spectral range between 190 and 1050 nm (spectral resolution approx. 0.1 nm) for every laser shot. Axiom software provided by the manufacturer of the instrument was used for the collection of the LIBS data. The system was connected to an iCAP Qc ICP-MS system (ThermoFisher Scientific, Bremen, Germany) using PTFE tubing with 3 mm inner diameter and a length of 1.2 m. A helium flow rate of 0.9 L min−1 provided optimal washout properties for the sample aerosol; 0.3 L min−1 of argon was mixed with the helium flow using a t-piece placed directly after the ablation chamber to be used as make-up gas. ICP-MS data were acquired in time resolved mode using Qtegra software provided by the manufacturer of the ICP-MS instrument.Instrumental parameters of the LIBS system were optimized in preliminary experiments. Polyimide (PI) samples containing all analytes of interest at the highest levels were ablated and the parameters (i.e., laser output energy and spectrometer gate delay) were selected in order to detect the maximum background corrected signal for Sr at an analytical wavelength of 460.733 nm. Emission lines of all other elements of interest also showed near to maximum signal intensities under the optimized conditions. Prior to every analysis, instrumental parameters of the ICP-MS system were optimized by ablating the very same PI sample with the optimized laser settings for the LIBS domain. Settings were tuned to achieve maximum signal intensity for 23Na, 115In, and 195Pt; typical instrumental parameters are shown in Table 1.
| Laser ablation system | |
| Laser output energy [mJ] | 11.7 |
| Laser ablation crater diameter [μm] | 100 |
| Laser repetition rate [Hz] | 10 |
| Carrier gas flow (He) [L min−1] | 0.9 |
| Make-up gas flow (Ar) [L min−1] | 0.3 |
| Laser beam geometry | Circular |
|
|
| Spectrometer system (Czerny–Turner type) | |
| Detection channels | 6 |
| Detector | CCD |
| Gate delay [μs] | 0.3 |
| Gate width [ms] | 1.05 |
|
|
| ICP-MS (Thermo iCAP Q) | |
| Cool gas flow (Ar) [L min−1] | 15.0 |
| Auxiliary gas flow (Ar) [L min−1] | 0.8 |
| RF power [W] | 1550 |
| Dwell time per isotope [ms] | 10 |
| Cone system | Ni |
| Measured isotopes | 13C, 23Na, 59Co, 113In, 115In, 195Pt, 196Pt |
| Measurement mode | Standard, no collision gas |
Production of polymer layers
Polymer layers were produced by spin-coating onto high purity grade silicon wafer pieces with dimensions of 1 × 1 cm2 and a thickness of 0.5 mm (Infineon Technologies AG, Villach, Austria). For the purpose of spin-coating, polymer solutions spiked with different amounts of the elements of interest were required. Solid polymers were dissolved in N-methylpyrrolidone (NMP) to obtain solutions each containing 10% by mass of the respective polymers. Standard solutions were prepared by dissolving acetylacetonate salts of the analytes of interest in NMP. Subsequently, salt solutions were mixed with the polymer solutions in a volume ratio of 1:10. Thus, 9% (w/w) polymer solutions were obtained containing analytes at different concentrations. Analyte mixtures and concentrations were chosen randomly to avoid collinearities during multivariate data evaluation. 15 mixtures were prepared for each of the three investigated polymer types yielding 45 different samples. A detailed summary of the element concentrations of the tested analytes Na, Sr, Co, In, and Pt in all samples is given in the ESI.† In short, samples contained between 0 and 300 μg g−1 of the five elements of interest.
Before spin coating, the silicon wafers were etched in conc. HF to activate the SiO surface for allowing better friction of the polymer solutions. After washing with ultrapure water and drying under ambient conditions, the wafer pieces could be readily used for spin coating. The process was performed using a conventional lab spin coating device with variable spinning speed and programmable control. The parameters of the spin coating device were optimized in preliminary experiments to obtain polymer films with reproducible appearance and visual homogeneity. 200 μL of the polymer solution was manually deposited onto the Si wafer spinning at 3000 rpm. Samples were spun for another 180 seconds after the application of the polymer solution before removing the sample from the apparatus. The complete procedure was performed under a dry nitrogen atmosphere to avoid influences of humidity on the drying process of the polymer layer. To completely remove the solvent fraction from the polymers, samples were cured at 180 °C for three hours. Preliminary experiments showed that no further solvent could be evaporated after the curing step. Thus, element concentrations in the thin films could be calculated based on the assumption that the solvent was completely removed from the polymers. Until analysis, samples were stored in gas tight boxes under ambient conditions.
Polymer layers prepared by such means had thicknesses in the low μm range. However, due to irrelevance for the further analyses, the sample thickness was not further investigated in the frame of this study. Most importantly, samples were not penetrated during tandem LA-ICP-MS/LIBS analysis which enabled an independent analysis of the polymer material from the silicon carrier.
Tandem LA-ICP-MS/LIBS measurements
Tandem LA-ICP-MS/LIBS measurements were run without ablating polymer layers covering the Si wafers in their complete thickness. Thus, initial experiments were performed to evaluate the experimental conditions at which no signal for silicon at a wavelength of 288.2 nm above a signal-to-noise ratio of 3 was obtained.All samples were analyzed using 5 parallel linescans, each linescan being 5 mm in length and placed in the center of the samples to avoid wall effects originating from the spin coating process. Using a 100 μm laser beam diameter at a stage scan speed of 100 μm s−1 and a repetition rate of 10 Hz, 500 laser shots were fired on each of the five lines ablated on every sample piece. For LIBS analysis, the emission spectra from these 500 laser shots were accumulated. When intregrating the emission intensities of specific analytical wavelengths, averages with respective standard deviations could be calculated based on the five replicate measurements of each sample. For LA-ICP-MS analysis, the dry sample aerosol generated during laser ablation was transported from the ablation cell towards the ICP-MS device allowing a simultaneous ICP-MS detection alongside with LIBS analysis. Based on the experimental parameters chosen, each ablated linescan would create a transient signal 50 s in length. Signals acquired during the complete time were integrated for each element and used for further data evaluation. Again, values obtained from the five replicate measurements could be used to calculate averages and standard deviations.
Signals of Sr (Sr(II) at 407.771 nm) and Na (Na(I) at 588.995 nm) could be well detected in all samples using LIBS while Co and In were only detectable at higher concentrations. LA-ICP-MS was able to detect all elements except Na in the lower concentration range (LOD for Na using LA-ICP-MS was between 42 and 85 μg g−1 depending on the polymer type). Thus, Sr and Na were selected to be analyzed using LIBS while Co, In and Pt were investigated using the ICP-MS domain of the tandem LA-ICP-MS/LIBS setup.
Results and discussion
Homogeneity of the spin-coated polymer layers
To prove the suitability of the polymer layers for calibration purposes, the homogeneity of the analyte distributions had to be investigated. Thus, one sample of each polymer type (PI, PMMA, and PVP) was randomly selected and elemental mapping using tandem LA-ICP-MS/LIBS was performed. To avoid image blurring effects and increase lateral resolution in the resulting images, slightly different experimental parameters compared to typical sample analysis were selected; a laser beam diameter of 100 μm at a 1 Hz repetition rate and a stage scan speed of 100 μm s−1 was used when scanning the complete Si wafer area of 1 × 1 cm2. This procedure allowed creating lateral distribution images with sizes of 100 × 100 pixels. Fig. 1a shows a photographic image of the investigated sample coated with PI. The distributions of Sr measured by LIBS (analytical wavelength 407.771 nm; 248 μg g−1 in this sample) and Pt measured by LA-ICP-MS (m/z 195; 192 μg g−1 in this sample) simultaneously during one run of analysis are shown in Fig. 1b and c, respectively. An approx. 300 μm broad border on the edge of the coated wafers can be seen in the visual image as well as in the elemental distribution images. This results from thickening of the polymer layer during spin coating. Apart from that, no indication of heterogeneity of the analyte could be found using visual inspection. This finding applies for all elements in all three analyzed samples (not shown here). Due to thicker polymer layers on the wafer edges, only an area of 0.8 × 0.8 cm2 in the center of each wafer was analyzed.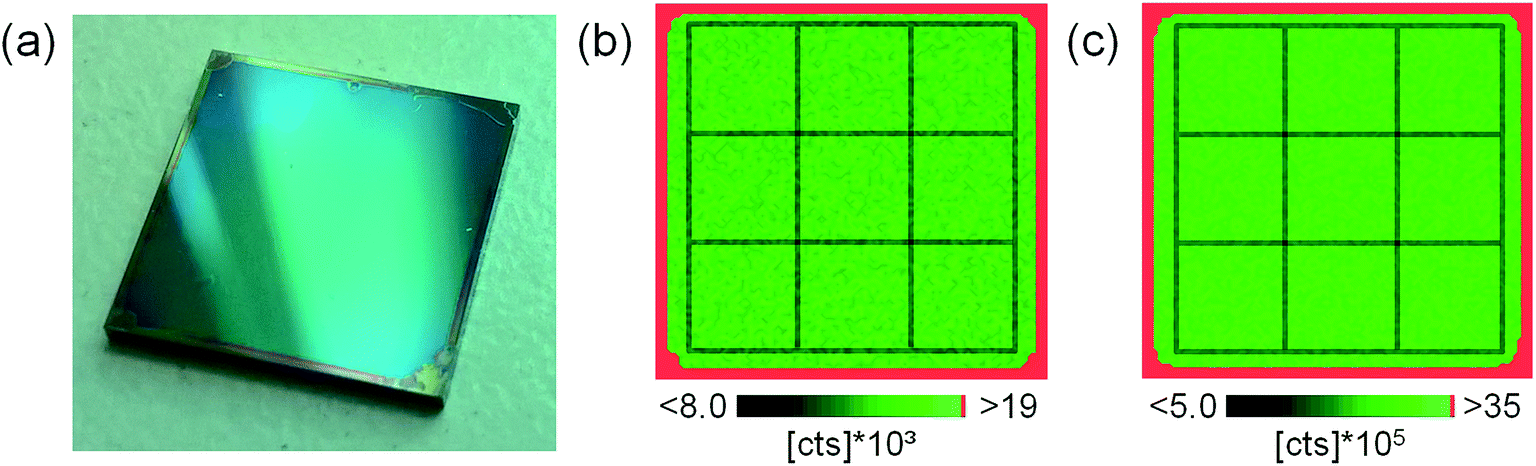 | ||
| Fig. 1 Photographic image of the investigated sample (a), Sr distribution determined using LIBS (Sr(II) 407.771 nm) (b), and Pt distribution measured by LA-ICP-MS (195Pt) (c). | ||
To underline the results with statistical figures rather than the visual appearance of the distribution images, the obtained images were sub-divided into 9 equally sized squares using a 3 × 3 grid, as indicated in Fig. 1b and c. Values of pixels from each area were averaged into 9 averages and compared using one-way ANOVA. The results do not indicate any significant difference between the 9 averages for any element in any of the three investigated samples (p = 0.99).
Univariate calibration procedure
The most straightforward way for external calibration is establishing univariate calibration functions for the elements of interest. Usually, a couple of external standards are sufficient for this type of quantification strategy. The said method was applied to the data collected within this study to evaluate the suitability of the polymer thin layers for external calibration. Elements detectable with a signal-to-noise ratio of 3 or above for atleast one analytical wavelength in atleast some samples using LIBS were Sr and Na. Background corrected signal intensities varied between 100 and 30000 cts. LA-ICP-MS was able to detect all elements of interest with signal intensities well above the background level. Signal intensities varied between 3000 and 17000000 cts. Relative standard deviations (RSDs) of the five replicate measurements of each standard were usually around 5% for LIBS and LA-ICP-MS, and never above 9%. Exemplary univariate calibration functions determined for Sr (LIBS) and Pt (LA-ICP-MS) are shown in Fig. 2 for the three mentioned polymer types.
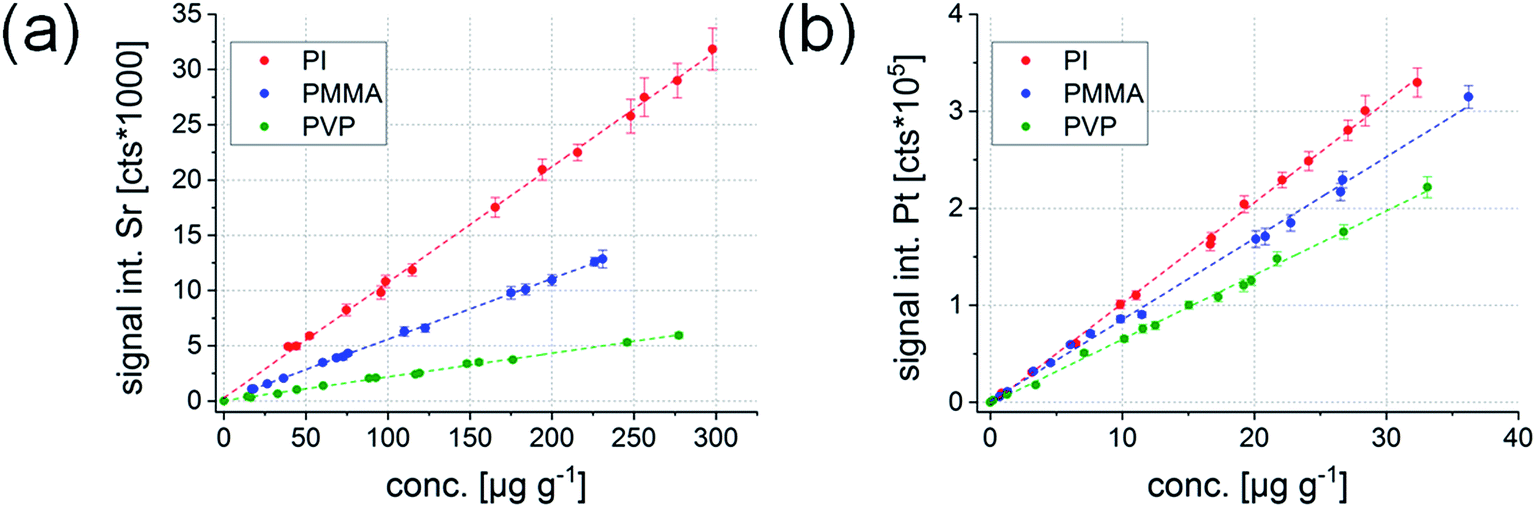 | ||
| Fig. 2 Univariate calibration functions for Sr determined by LIBS (Sr(II) 407.771 nm) (a) and Pt determined by LA-ICP-MS (m/z 195) (b). | ||
All elements show a very good linear correlation between the signal intensity and concentration of the respective element in the sample for all three polymer types, making the standards suitable for the quantification of an element in a known polymer type. Regression coefficients of the univariate calibrations were always above 0.9980. However, when comparing the signal response (i.e., the slope) of the calibration functions for the different polymers, it can be seen that they are significantly different from each other in the LIBS as well as the LA-ICP-MS results. This matrix effect seems to be larger for LIBS analysis, which becomes apparent when looking at Sr (a similar trend could be seen for Na). While the difference between the lowest absolute value for the slope (PVP) and the highest absolute value (PI) ranged between 75 and 79% for LIBS, it was only between 34 and 37% for LA-ICP-MS. It is well known that LIBS is very prone to matrix effects and even the smallest variations in physical sample properties may influence the formation of the laser induced plasma – a statement which is well supported by the current finding. In contrast, signal generation in LA-ICP-MS is much less dependent on the local plasma formation on the sample surface, whereby generally lower laser wavelengths lead to reduced elemental fractionation and less matrix effects.23
When matrix effects occur, one often applied approach to overcome them is to apply an internal standard. Besides the option of artificially adding an internal standard to the sample – which is usually not possible in the case of a real-world sample – sample inherent elements can be used. Such internal standards do not only allow the reduction of matrix effects, but usually their use also leads to an improvement of the relative standard deviation (i.e., precision). In the case of polymers, (in most cases) only carbon, hydrogen, oxygen, and nitrogen remain for this purpose, whereof only carbon can be detected by LIBS and LA-ICP-MS. Normalizing absolute signal intensities to the carbon signal improved the situation slightly for LIBS analysis (31 to 35% difference between the slopes for the calibration functions of the three polymers) but worsened it for LA-ICP-MS (45 to 48%). Two facts might contribute to this unsatisfactory result: on the one hand, carbon is surely not the prime choice for an internal standard due to significantly different transport and ionization properties compared to metals.24 However, most importantly, carbon in polymers does not meet the main prerequisite for an internal standard: equal (or known) concentration in all samples. The carbon content varies in different polymer types with often unknown concentrations (especially in industrial polymers with often undisclosed exact composition), making it an inacceptable choice when unknown polymer types are to be analyzed.
Principal component analysis for polymer classification
As mentioned above, LIBS is very prone to matrix effects. While this fact does not necessarily improve the possibilities for quantification at first sight, LIBS spectra can be used to visualize the appearing matrix effect quite well. Already an overlay of LIBS spectra from three polymer types shows a significant difference, as demonstrated in Fig. 3. Aside the atomic emission lines, a large number of features remain which cannot be assigned to one specific element present in the sample. Interestingly, these features make up the most apparent differences between the three polymer types. Only the most prominent peaks are shown in Fig. 3. Especially, signals of the weakly excitable elements such as In or Co cannot be seen in the overview spectra.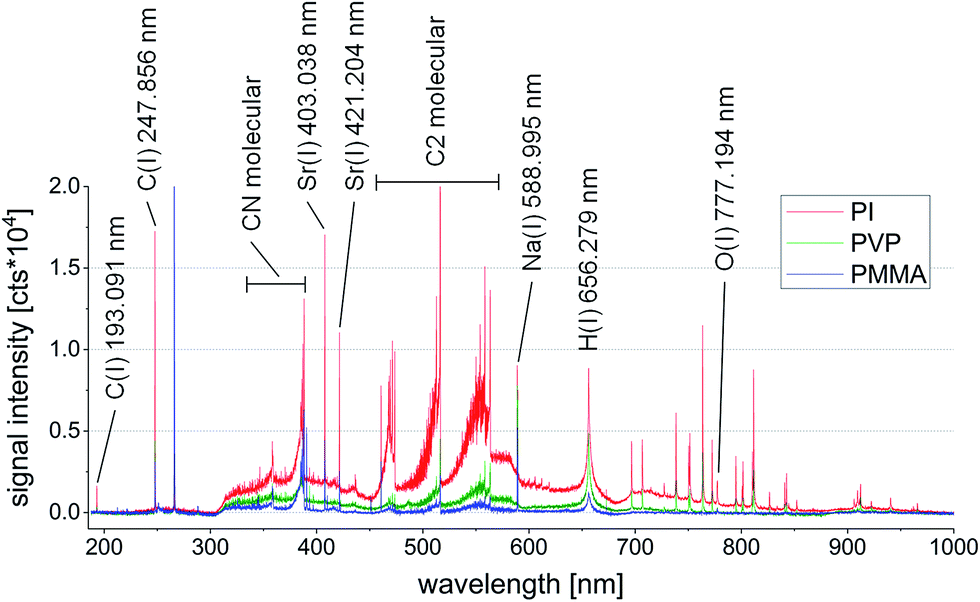 | ||
| Fig. 3 Overlay of three representative LIBS spectra from PI (red), PVP (green), and PMMA (blue) measured on samples having the same trace element profile. | ||
All three analyzed samples have the same trace metal profile at similar concentrations. Still, as mentioned above, the broadband LIBS emission spectra differ heavily from one another. Background emission, ratios between atomic and ionic emission lines, molecular emission intensity and other factors make up those differences. Especially, the occurrence of molecular emission lines (mostly originating from C–C and C–N species in the wavelength range between 350 and 575 nm) indicates distinct differences in atomization and molecule formation in the laser induced plasma when comparing the three polymer types. These variations may be attributed to the different physical properties of the polymeric materials, such as hardness, absorption behavior and also their different chemical compositions.
Due to these very distinct patterns, such spectra could be used for the determination of the statistical correlation of the polymer types. Principal component analysis (PCA) was used for this purpose in the present case. A score plot of only two principal components (PCs) showed to be sufficient to clearly distinguish the polymer types based on their spectral signatures (see Fig. 4). PCs 2 and 3 have been used in this graph, as they depict grouping of the polymer classes in a two-dimensional graph in the best way.
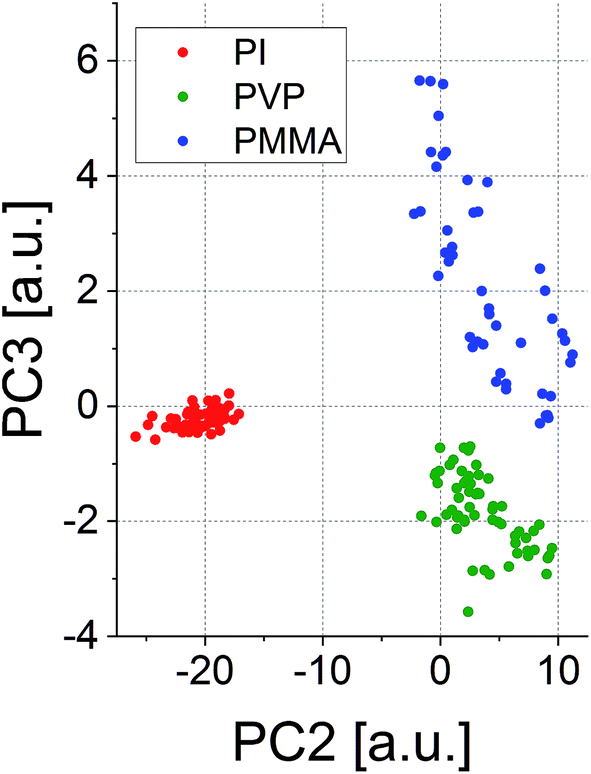 | ||
| Fig. 4 Score plot of PC2 and PC3 for the visualization of similarities between the LIBS spectra obtained from different polymers. | ||
Classification can be further improved by taking more PCs into account, but visualization would be hard for a multidimensional plot. In fact, eight PCs showed to describe the differences between the polymers in the most satisfactory way based on the eigenvalues of the principal components (90.5% accumulated variance). The results from the PCA already indicate that the broadband LIBS emission spectra may be able to serve as a kind of ‘classificatory tool’ to determine and quantify the extent of the matrix effect during LA-ICP-MS and LIBS analyses. The possibility of the classification of polymers is well in line with the results reported earlier by Grégoire et al.21 and Unnikrishnan et al.22
Principal component regression for matrix-independent calibration
Knowing the potential of the LIBS dataset, it could be further used in the data evaluation process. Combining the known element concentrations with the results from the PCA, building a linear regression model is possible – so-called principal component regression (PCR). Thus, data from ICP-MS and LIBS had to be merged into one combined dataset by adding six more variables (carbon and the five elements of interest) to the LIBS data cube, wherein each variable corresponds to the isotopes measured by ICP-MS and their respective intensities. In the derived model, PCA results are used to minimize the matrix differences, while building a regression model combining different polymer types. The number of PCs describing the maximum variance of the dataset while keeping the noise level at a minimum was 8 for all elements of interest.To construct a multivariate regression model, 23 samples were selected randomly out of the 45 available different polymer layers. Correlation plots between the actual concentrations and those calculated using the regression model show excellent comparability. As an example, the correlation plot for Co is shown in Fig. 5.
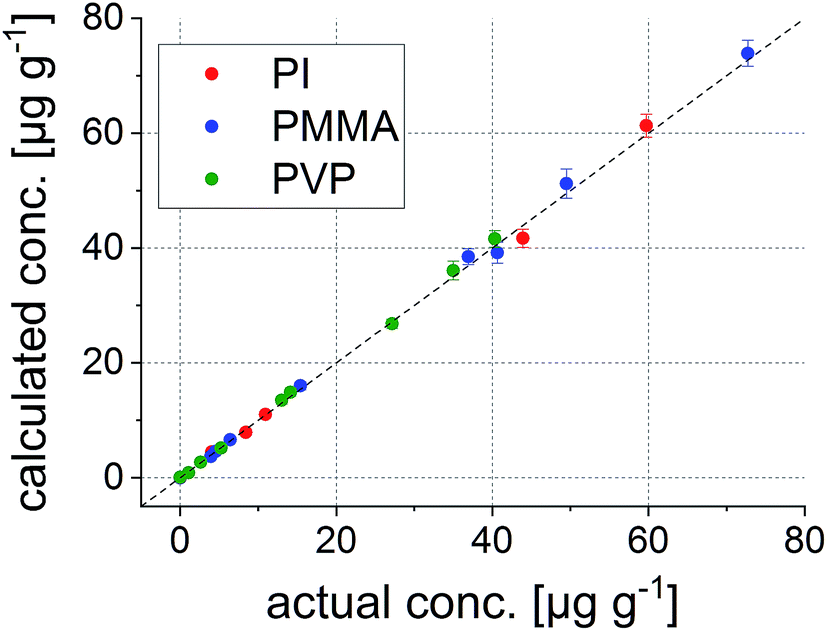 | ||
| Fig. 5 Correlation between the actual and the calculated concentrations of Co in the 23 polymer samples used for external calibration. | ||
This internal validation procedure already provides promising results. Concentrations predicted using the model fit excellently with the actual ones. However, it is not guaranteed that also samples which are not part of the initial model deliver correct values. Thus, the much more interesting external validation using the remaining 22 polymer samples was carried out. The results for all five elements are shown in Fig. 6.
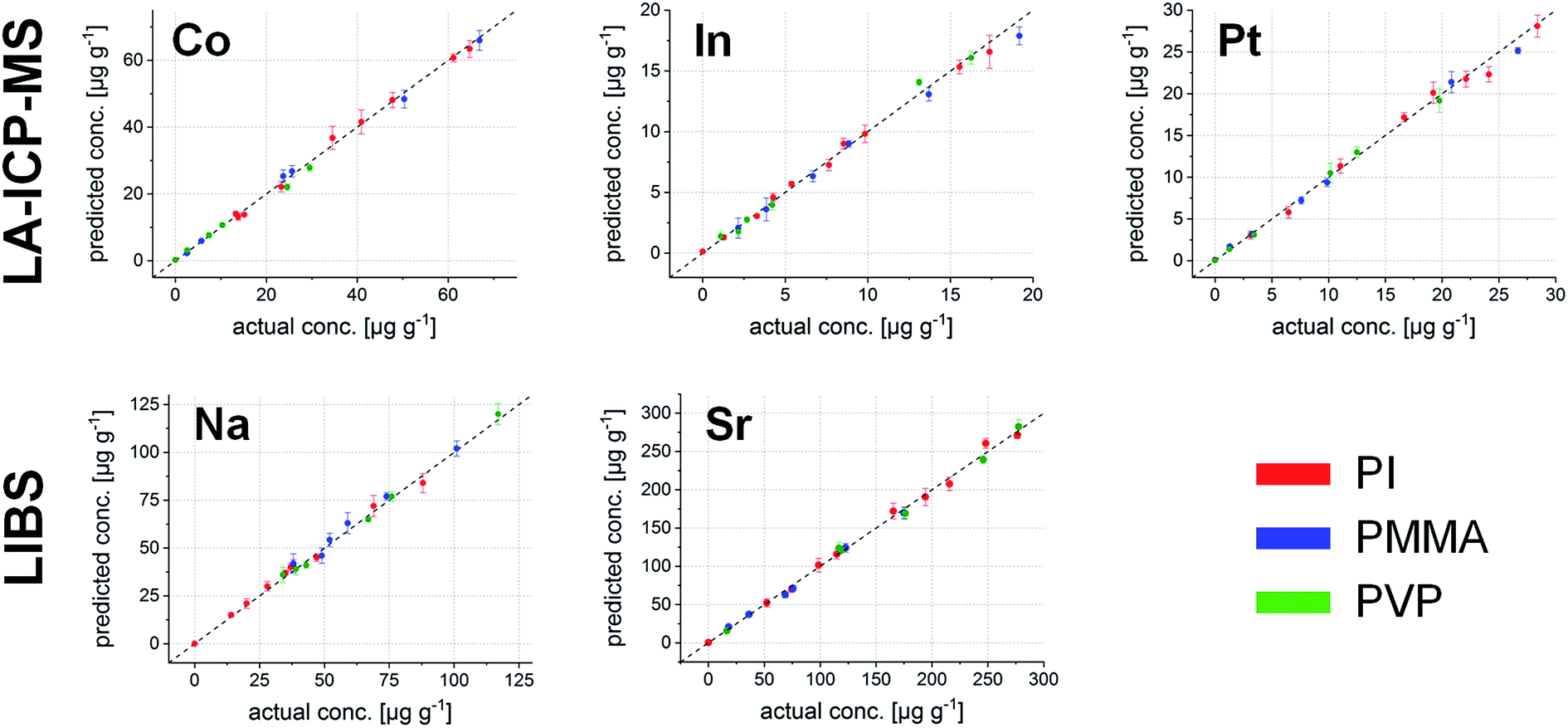 | ||
| Fig. 6 External validation of the regression model using 22 samples not being included in the model; first row: elements measured by LA-ICP-MS (Co, In, and Pt) and second row: elements measured by LIBS (Na and Sr). | ||
The average deviation from the actual concentrations is 4.4% throughout the complete external validation pool; the sample with the highest deviation between the found and actual value shows a difference of 9.6% (Co in the PVP sample at a nominal concentration of 7.4 μg g−1). These figures state excellent applicability for the performed calibration, even across different polymer types.
Conclusion
Matrix effects are a serious problem when performing polymer analysis using laser-based analysis methods. Aside from demonstrating this fact in a systematic way, a solution to this often encountered problem could be proposed in this work. Performing LIBS and LA-ICP-MS in one run of analysis (tandem LA-ICP-MS/LIBS) gives the opportunity of combining the very matrix-sensitive LIBS method with LA-ICP-MS offering a very high detection power. While matrix-sensitivity is usually considered as a negative aspect hampering accurate analysis, it could be shown that using statistical methods it is possible to filter the matrix effect from the LIBS data and use full LIBS spectra as a type of internal standard for accurate quantification. Further studies will focus on the applicability of this approach for the analysis of bulk polymer samples; a polymer database will be built up to include as many polymer types into a calibration model as possible. In the future, this may give rise to the possibility of analyzing polymers in a matrix-independent manner. Moreover, the possibility of spatially resolved analysis and depth profile analysis will be exploited, providing information about the distribution of major, minor and trace constituents within the polymer sample.Conflicts of interest
There are no conflicts of interest to declare.References
- H. Namazi, BioImpacts, 2017, 7, 73–74, DOI:10.15171/bi.2017.09.
- M. Jaishankar, T. Tseten, N. Anbalagan, B. Mathew Blessy and N. Beeregowda Krishnamurthy, in Interdisciplinary Toxicology, 2014, vol. 7, p. 60 Search PubMed.
- J. S. F. Pereira, C. L. Knorr, L. S. F. Pereira, D. P. Moraes, J. N. G. Paniz, E. M. M. Flores and G. Knapp, J. Anal. At. Spectrom., 2011, 26, 1849–1857, 10.1039/c1ja10050e.
- E. M. M. Flores, E. I. Muller, F. A. Duarte, P. Grinberg and R. E. Sturgeon, Anal. Chem., 2013, 85, 374–380, DOI:10.1021/ac3029213.
- Z. Zhou, K. Zhou, X. Hou and H. Luo, Appl. Spectrosc. Rev., 2005, 40, 165–185, DOI:10.1081/asr-200052001.
- J. Pisonero, N. Bordel, C. Gonzalez de Vega, B. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Bioanal. Chem., 2013, 405, 5655–5662, DOI:10.1007/s00216-013-6914-1.
- N. Tuccitto, L. Lobo, A. Tempez, I. Delfanti, P. Chapon, S. Canulescu, N. Bordel, J. Michler and A. Licciardello, Rapid Commun. Mass Spectrom., 2009, 23, 549–556, DOI:10.1002/rcm.3906.
- A. Turner and K. R. Solman, Talanta, 2016, 159, 262–271, DOI:10.1016/j.talanta.2016.06.026.
- A. Massos and A. Turner, Environ. Pollut., 2017, 227, 139–145, DOI:10.1016/j.envpol.2017.04.034.
- F. O. Leme, Q. Godoi, P. H. M. Kiyataka, D. Santos, J. A. M. Agnelli and F. J. Krug, Appl. Surf. Sci., 2012, 258, 3598–3603, DOI:10.1016/j.apsusc.2011.11.122.
- F. W. B. Aquino, C. M. Paranhos and E. R. Pereira-Filho, J. Anal. At. Spectrom., 2016, 31, 1228–1233, 10.1039/c6ja00038j.
- M. Voss, M. A. G. Nunes, G. Corazza, E. M. M. Flores, E. I. Müller and V. L. Dressler, Talanta, 2017, 170, 488–495, DOI:10.1016/j.talanta.2017.04.048.
- T. Stehrer, J. Heitz, J. D. Pedarnig, N. Huber, B. Aeschlimann, D. Günther, H. Scherndl, T. Linsmeyer, H. Wolfmeir and E. Arenholz, Anal. Bioanal. Chem., 2010, 398, 415–424, DOI:10.1007/s00216-010-3963-6.
- P. Jorge and G. Detlef, Mass Spectrom. Rev., 2008, 27, 609–623, DOI:10.1002/mas.20180.
- G. Galbács, Anal. Bioanal. Chem., 2015, 407, 7537–7562, DOI:10.1007/s00216-015-8855-3.
- A. Limbeck, M. Bonta and W. Nischkauer, J. Anal. At. Spectrom., 2017, 32, 212–232, 10.1039/c6ja00335d.
- F. A. Orellana, C. G. Gálvez, F. A. Orellana, C. G. Gálvez, M. T. Roldán, C. García-Ruiz, M. T. Roldán and C. García-Ruiz, TrAC, Trends Anal. Chem., 2013, 42, 1–34, DOI:10.1016/j.trac.2012.09.015.
- D. Günther and B. Hattendorf, TrAC, Trends Anal. Chem., 2005, 24, 255–265, DOI:10.1016/j.trac.2004.11.017.
- R. E. Russo, X. Mao, H. Liu, J. Gonzalez and S. S. Mao, Talanta, 2002, 57, 425–451, DOI:10.1016/s0039-9140(02)00053-x.
- M. Bonta, J. J. Gonzalez, C. D. Quarles, R. E. Russo, B. Hegedus and A. Limbeck, J. Anal. At. Spectrom., 2016, 31, 252–258, 10.1039/c5ja00287g.
- S. Grégoire, M. Boudinet, F. Pelascini, F. Surma, V. Detalle and Y. Holl, Anal. Bioanal. Chem., 2011, 400, 3331–3340, DOI:10.1007/s00216-011-4898-2.
- V. K. Unnikrishnan, K. S. Choudhari, S. D. Kulkarni, R. Nayak, V. B. Kartha and C. Santhosh, RSC Adv., 2013, 3, 25872–25880, 10.1039/c3ra44946g.
- S. Zhang, M. He, Z. Yin, E. Zhu, W. Hang and B. Huang, J. Anal. At. Spectrom., 2016, 31, 358–382, 10.1039/c5ja00273g.
- D. A. Frick and D. Gunther, J. Anal. At. Spectrom., 2012, 27, 1294–1303, 10.1039/c2ja30072a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ja00161h |
| This journal is © The Royal Society of Chemistry 2018 |