Use of an inline dilution method to eliminate species interconversion for LC-ICP-MS based applications: focus on arsenic in urine†
Received
9th February 2018
, Accepted 23rd March 2018
First published on 9th April 2018
Abstract
Measuring arsenic in urine provides important information for clinical and epidemiological studies. While many researchers have studied ways to improve sample storage and understand why arsenic species undergo species interconversion, none have investigated inline dilution as a solution for arsenic speciation sample stability. A fast inline dilution method for AsB, DMA, MMA, As III, and As V was demonstrated in this work. Inline dilution calibrations from a single stock standard were shown to have good linearity and resulted in LODs in the single digit ppt range. Inline dilutions of 30X, 50X, and 100X resulted in 0.1 s to 1.1 s variation in retention time. Manual sample preparation resulted in poor recovery (61%) for As III over a 24 h time period, which was a direct result of As III converting to As V. Inline dilution of urine spiked with As III resulted in good recovery (101%) and reduced the species interconversion of As III to As V to ∼1%. Accuracy to NIST SRM 2669 (arsenic in frozen urine) was found to be within reported values for the five arsenic species tested for in this method.
Introduction
Trace elemental analysis is vital for industrial, government, and academic communities for health and safety, industrial hygiene, and research purposes. Knowing the total concentration of an element in a given sample is important, but it cannot provide vital information for which species are present in the sample. The chemical species (e.g., arsenite, arsenate, or arsenobetaine) plays an important part in understanding how it can affect the environment, its bioavailability, or its toxicity.1,2 For example, inorganic arsenic is more toxic than organic arsenic, whereas organic mercury is more toxic than its inorganic form.3
Arsenic contamination comes from natural geological sources that leach into our water systems, industrial exposures, or mining.1–4 Typically, arsenate (As V) or arsenite (As III) are both the predominant forms found in contaminated drinking water or rice and are also the most toxic.1,5 Other common sources of arsenic intake can occur from the consumption of seafood, where the arsenic is generally in the organic form (e.g., monomethylarsonic acid (MMA), dimethylarsinic acid (DMA), or arsenobetaine (AsB)).6–9 Organic arsenic is considered to have low bio-availability and thus is less toxic than the inorganic form (As III (LD50 = 14 mg kg−1) or As V (LD50 = 20 mg kg−1)) which is considered highly bio-available.4,10 Exposure to arsenic has been linked to adverse health effects such as respiratory disease, lung cancer, and damage to kidneys and/or liver.4 Arsenic is excreted in the urine, thus monitoring human urine samples can reveal information regarding intake exposure over the previous 2 days.11,12
In general, speciation measurements are performed by coupling a separation instrument with that of an element specific detector. The most common technique is to couple liquid chromatography (LC) with an inductively coupled plasma-mass spectrometer (ICP-MS). A great deal of progress has been reported over the last few decades describing arsenic speciation for various LC-ICP-MS based methods.1,3–9,11–21 For large commercial based laboratories, having a single instrument dedicated for speciation by LC-ICP-MS is common. However, smaller laboratories that have to utilize one or two ICP-MS systems for all analyses require instrumentation that is interchangeable. The changing of front-end sample introduction equipment can be laborious and time consuming, thus instrumentation that is more flexible with multi-purpose capabilities is desired.
Another concern for elemental speciation analysis is the ability to keep the sample in its native or preserved state so the species of interest do not undergo unwanted interconversion. Sample collection and sample treatment are two of the main reasons for species interconversion; for example, changing pH, temperature, acidity, or addition of oxidants can cause the stability of the collected sample to be compromised.9,11,13 Ion exchange chromatography is the preferred method for arsenic speciation since it can remove the argon chloride interference, separate species based on charge, and utilize a buffer that is compatible with the arsenic species of interest.22,23 However, for most chromatographic columns the sample capacity is small, therefore the sample loaded has to be small volumes (μL's) and/or diluted to lower concentrations.
In this work, the evaluation of an arsenic speciation anion-exchange syringe-driven chromatography method coupled to an ICP-MS was performed. The prepFAST IC, is a new, state-of-the-art instrument that allows for chromatography (speciation) or total metals analysis (general ICP-MS analysis) sample introduction with one single instrument. The ability to perform arsenic speciation measurements with this instrument was evaluated for column reproducibility, method robustness, accuracy, precision, and limits of detection. Furthermore, arsenic spiked urine samples were analyzed by manual dilutions and inline dilutions to evaluate species stability.
Experimental
Reagents and standards
All reagents, diluents, and eluents were prepared using 18 MΩ cm water from an EMD Millipore high purity filtration system (MilliporeSigma, Burlington, MA, USA). Ammonia (23%, SEASTAR, Sidney, BC, CAN) and phosphoric acid (85%, ICL Performance Products, St. Louis, MO, USA) were used to prepare 0.4 mM ammonium phosphate (eluent 1, pH = 9.6) and 80 mM ammonium phosphate (eluent 2, pH = 8.6). Calibration standards were prepared from stock solutions of arsenobetaine (AsB, Chem Services, West Chester, PA, USA), dimethylarsinic acid (DMA, Chem Services), monomethylarsonic acid (MMA, Chem Services), arsenite (As III, High-Purity Standards, Charleston, SC, USA), and arsenate (As V, High-Purity Standards).
Sample preparation
Human urine from anonymous donation was collected and screened; the urine found to have no detectable arsenic was used as the base urine for all experiments. Spiked urine was prepared by spiking in 0.1 mL of water or arsenic standard to 9.9 mL of base urine (10 mL in total). NIST SRM 2669 was prepared based on the recommended protocol included in the certificate of analysis.11 Samples of 10 ppb As spiked urine (average adult in the United States has ∼8–10 ppb total As in urine) were prepared for the manual and inline dilution study. The inline dilution study was prepared as mentioned above, whereas the manual dilutions were prepared such that the final concentration reflected that of the 30X dilution factor (0.33 ppb As) that was applied for the inline study.
prepFAST IC
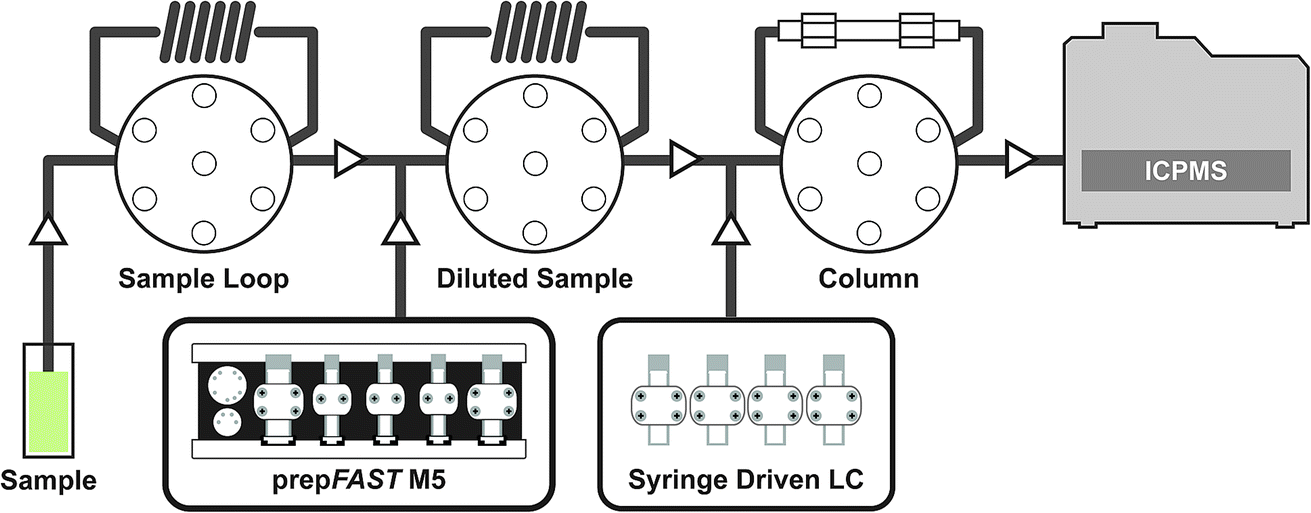
The syringe-driven liquid chromatography methods were performed using the prepFAST IC (Elemental Scientific Inc. (ESI), Omaha, NE, USA). The prepFAST IC is equipped with a speciation module (S400V2) that is capable of gradient elution and has a switching valve to allow the chromatographic column to be either on-line or off-line during analysis. The four syringes of the S400V2 are used for tuning, eluent 1, eluent 2, and post-column internal standard addition, dilution, or derivatization. An Elemental Scientific anion exchange column (50 × 4 mm) was used for the separation of AsB, DMA, MMA, As III, and As V species. The system is also equipped with a prepFAST M5 (S500V2) unit that allows for inline dilution or calibration of samples or standards. Fig. 1 displays a simplified flow diagram showing the sample path through the sample loop, dilutions sample loop, through the column, and introduction to the ICP-MS. The system is connected directly to a PolyPro-ST micro flow nebulizer (Elemental Scientific) and a quartz cyclonic spray chamber (Elemental Scientific). Two 500 μL sample loops are used: the first sample loop fills with original sample from autosampler and the second loop is used to dilute the sample and add base urine for matrix matching of standards (not used for samples) prior to injection onto the column. The diluted urine standards/samples are introduced onto the column through a switching valve for 7 seconds (optimized time based on 30X dilution) which is an equivalent to ∼23 μL injection.
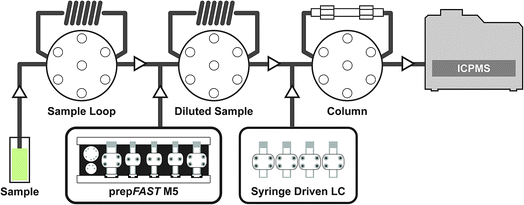 |
| | Fig. 1 Simplified flow path diagram for the prepFAST IC. | |
ICP-MS
A Perkin Elmer NexION 350 ICP-MS (Shelton, CT, USA) was employed for all experiments. The Ar flow rate for the ICP plasma, auxiliary, and nebulizer gases were set to 18, 1.2, and ∼1.0 L min−1, respectively, and the ICP was operated with an RF power of 1.60 kW. The analyses were performed in time resolved mode by monitoring 75As at a dwell time of 100 ms, with 5 sweeps, 720 readings, and 1 replicate equaling 6 minutes of measurement time (0.5 s per data point). The method was operated in kinetic energy discrimination (KED) mode with a helium flow rate of 2.0 mL min−1 and RPq of 0.25. The KED flow rate chosen was the minimum flow rate that resulted in no false positive from urine blanks spiked with Cl to mimic ArCl potential interferences. The sample flush and read delay times were set to 0 s and the wash time was set to 240 s. The wash time (ICP-MS software setting) of 240 s was required to allow for sufficient amount of time to reset the syringes for eluent 1 and 2 and to equilibrate the column prior to the next sample being prompted by the ICP-MS.
Results and discussion
Column chemistry
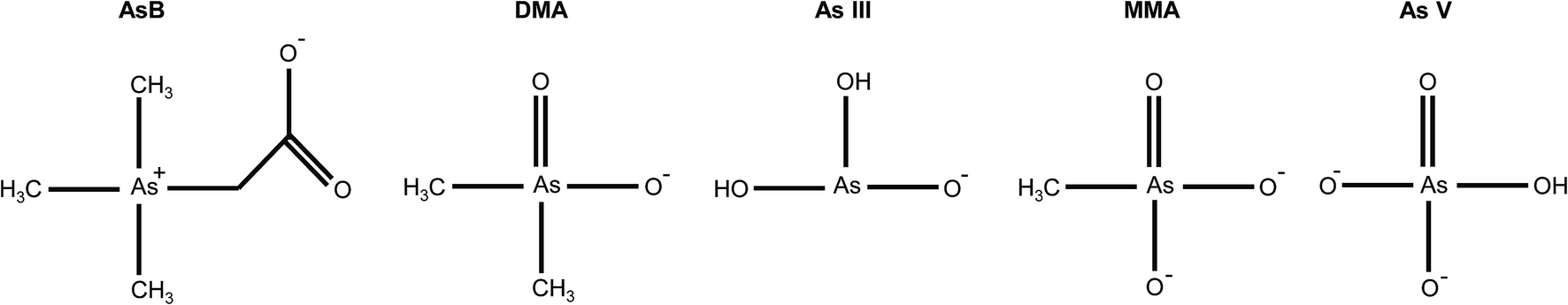
The optimal eluent conditions used with the Elemental Scientific anion exchange column were determined to be 0.4 mM ammonium phosphate buffer (pH = 9.6) and 80 mM ammonium phosphate buffer (pH = 8.6) for eluent 1 and eluent 2, respectively. The two step gradient was 100% eluent 1 at a flow rate of 200 μL min−1 for t = 0–200 s and 100% eluent 2 at a flow rate of 300 μL min−1 for t = 200–325 s. At the aforementioned conditions, the five arsenic species of interest eluted at different times. Table 1 displays the pKa's and net charge of each species under the conditions for eluent 1. AsB was the first species to elute from the column and had no interaction with the stationary phase of the column. DMA elutes before arsenite, due to the delocalization of the negative charge on the oxygen with the carbonyl group (Fig. 2). MMA and arsenate both stick to the stationary phase under eluent 1 conditions, therefore an increase in buffer concentration and lowering of the pH were needed to elute the species. Both species have a net charge of −2, however the difference in elution can be attributed to the methyl group on MMA and the hydroxyl group on As V (Fig. 2). The hydroxyl group on As V causes the pKa to be lower than that of MMA, meaning MMA will elute before As V.
Table 1 Arsenic species investigated in this study
| Arsenic species |
Abr. |
pKa (ref. 12) |
Net charge@pH = 9.6 |
| Arsenobetaine |
AsB |
2.2 |
0 |
| Dimethylarsinic acid |
DMA |
6.2 |
−1 |
| Arsenite |
As III |
9.2, 12.1, 13.4 |
−1 |
| Monomethylarsonic acid |
MMA |
3.6, 8.2 |
−2 |
| Arsenate |
As V |
2.3, 6.8, 11.6 |
−2 |
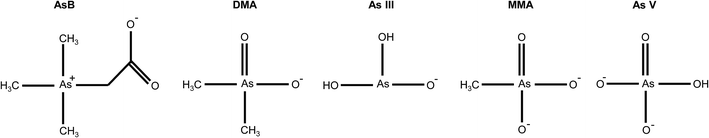 |
| | Fig. 2 Arsenic species at pH = 9.6 (eluent 1 conditions). | |
Column reproducibility
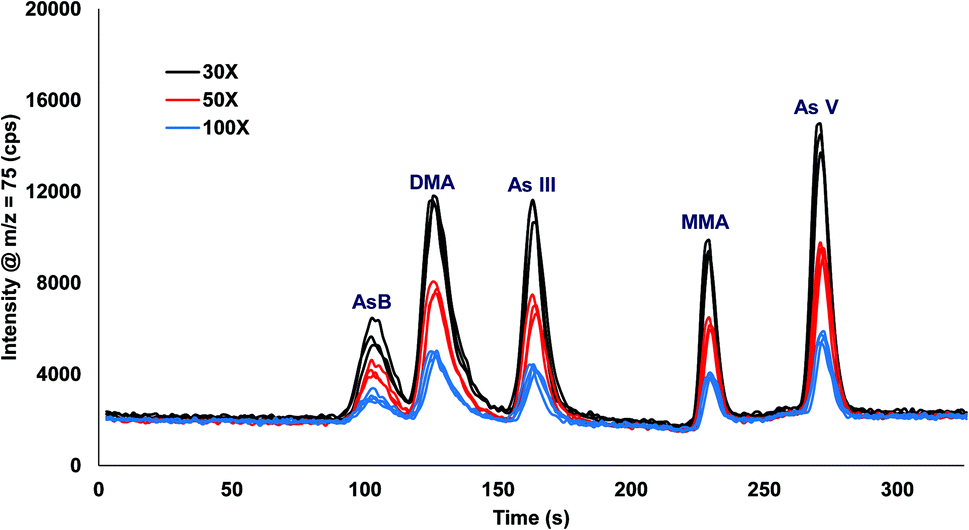
Urine was spiked with 10 ppb arsenic species and injected onto the column with varying inline dilution factors ranging from 1X up to 100X. For dilution factors ≤20X the peak shapes were distorted and the retention times were shifted earlier in time for AsB, DMA, and As III as compared to the 30X, 50X, and 100X (Fig. 3). The most likely reason for the poor results is that the dilution factors ≤20X were overloading the column. Table 2 shows the comparison of 30X, 50X, and 100X dilutions of urine for two columns prepared identically. The retention times for the different dilution factors varied by 0.1 to 1.1 s for a single column, with the largest variation in the As III peak coming off column A. There was a small variation in retention times when comparing two different columns, with the largest variation detected for As III. A 30X dilution factor was chosen for all subsequent experiments.
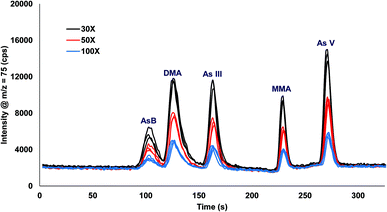 |
| | Fig. 3 Inline dilution (30X, 50X, and 100X dilution factors) of urine samples spiked with 10 ppb arsenic species. | |
Table 2 Comparison of retention times (s) for the five arsenic species studied based on dilution factors (30, 50, or 100X) and columns. Retention times were determined based on peak maximum. Replicates were determined on two different days (n = 5 for each day)
|
|
Dilution factor |
Retention times (s) |
| AsB |
DMA |
As III |
MMA |
As V |
| Column A |
30X |
103.3 ± 0.8 |
125.4 ± 0.8 |
160.8 ± 2.1 |
228.9 ± 0.6 |
270.7 ± 0.8 |
| 50X |
103.1 ± 0.5 |
125.6 ± 0.8 |
161.9 ± 1.5 |
229.2 ± 0.7 |
271.4 ± 0.6 |
| 100X |
103.2 ± 0.6 |
126.0 ± 0.9 |
162.9 ± 0.9 |
229.5 ± 0.6 |
271.6 ± 0.6 |
|
All
|
103.2 ± 0.1
|
125.7 ± 0.3
|
161.9 ± 1.1
|
229.2 ± 0.3
|
271.2 ± 0.5
|
|
|
|
|
|
|
|
|
| Column B |
30X |
103.0 ± 0.9 |
123.8 ± 0.8 |
156.6 ± 1.4 |
228.1 ± 0.5 |
268.9 ± 0.7 |
| 50X |
102.5 ± 0.4 |
124.3 ± 0.3 |
157.1 ± 0.9 |
228.5 ± 0.7 |
269.3 ± 0.6 |
| 100X |
102.7 ± 0.6 |
123.6 ± 0.4 |
157.5 ± 0.8 |
228.4 ± 0.7 |
269.5 ± 0.9 |
|
All
|
102.7 ± 0.3
|
123.9 ± 0.4
|
157.1 ± 0.5
|
228.3 ± 0.2
|
269.2 ± 0.3
|
|
|
|
|
|
|
|
|
| Column A & B |
All
|
103.0 ± 0.3
|
124.8 ± 1.0
|
159.5 ± 2.7
|
228.8 ± 0.5
|
270.2 ± 1.2
|
Calibration
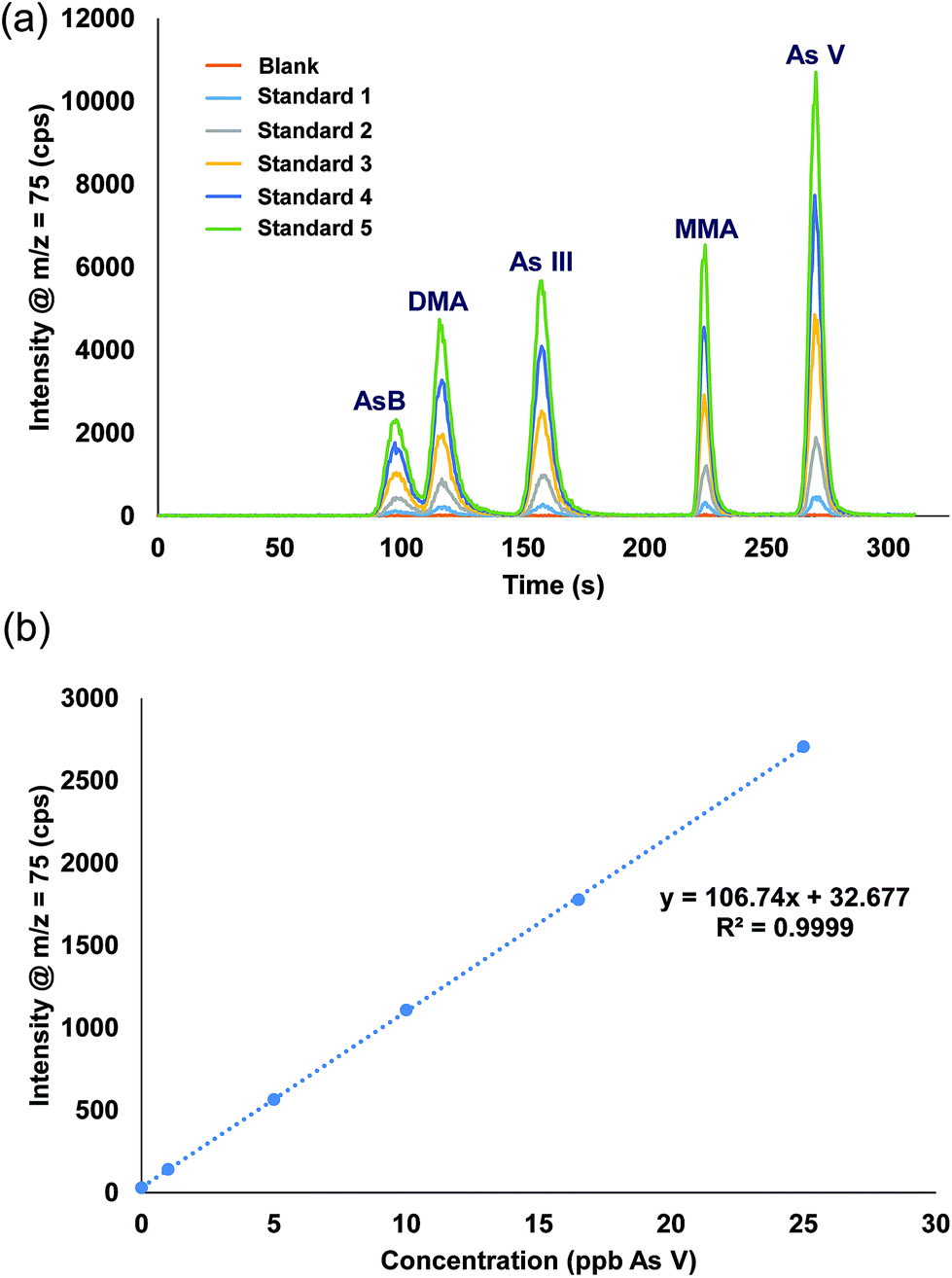
A single stock standard containing each of the arsenic species was prepared and used to calibrate the method. Utilizing the prepFAST feature, the 500 μL sample loop was filled with the stock solution and diluted inline with water to create each single calibration standard (e.g. 50 ppb stock standard with a 10X dilution factor results in 5 ppb being introduced onto the column, a 2X dilution factor results in 25 ppb). Rather than using the internal standard syringe of the prepFAST, which is normally used to spike constant amounts of an internal standard, blank urine was added (1 part urine: 29 part sample) into the 500 μL dilution sample loop prior to injection onto the column. Verdon et al., reported interconversion of As III to As V in urine samples that have a pH > 7.24 However, with the prepFAST IC configuration, the urine is spiked into the standard seconds before being injected onto the column, thus reducing/eliminating the time in which the interconversion from As III to As V could take place. Fig. 4 shows the chromatograms and the calibration curve for As V (only one curve shown for brevity) that were produced using the inline calibration function (prepFAST).
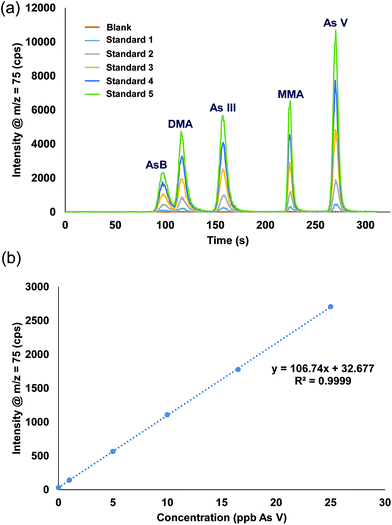 |
| | Fig. 4 (a) Chromatograms from the inline autocalibration of arsenic species, calibration standards range from 0 ppb (blank urine) up to 27 ppb. (b) Calibration curve for arsenic V. | |
Manual sample preparation vs. inline dilution sample preparation
Issues with the stability of arsenic species have been reported for matrices such as water, plant extracts, apple juice, and urine.12,24–27 Recent work from this laboratory has shown examples of inorganic arsenic interconverting in apple juice and urine after sample preparation. Initial experiments demonstrated that some of the urine samples tested showed interconversion of arsenic species (most likely due to the right combination of pH, oxides, and salt). Therefore, these anonymous urine samples (showing species interconversion) were spiked with all five arsenic species at a concentration of 1 ppb. The urine samples were analyzed at t = 0 h and 24 h, with one of the samples resulting in ∼40% interconversion from As III to As V. This urine sample was chosen for the subsequent experiments.
To study the effects of species interconversion, manual sample preparation was compared to inline dilution sample preparation to determine if it is advantageous to dilute samples mere seconds before injection onto the analytical column. The manual sample preparation involved spiking the urine sample with 10 ppb AsB, DMA, MMA, As III, or As V and then diluting the sample 30X. For each sample, the dilution occurred immediately before the 0 h analysis started, and each sample was only spiked with one arsenic species in order to easily track any possible interconversion. The study lasted for 24 h with analyses occurring at t = 0, 1, 3, 6, 12, and 24 h from the same sample vial. The analyses for the t = 1–24 h measurements were taken from the vial that was diluted at the 0 h time period. For the inline sample dilution study, the urine samples were spiked and placed undiluted onto the autosampler and analyzed with the same time intervals as mentioned above except that the dilution occurred in the valve just before the column for each measurement.
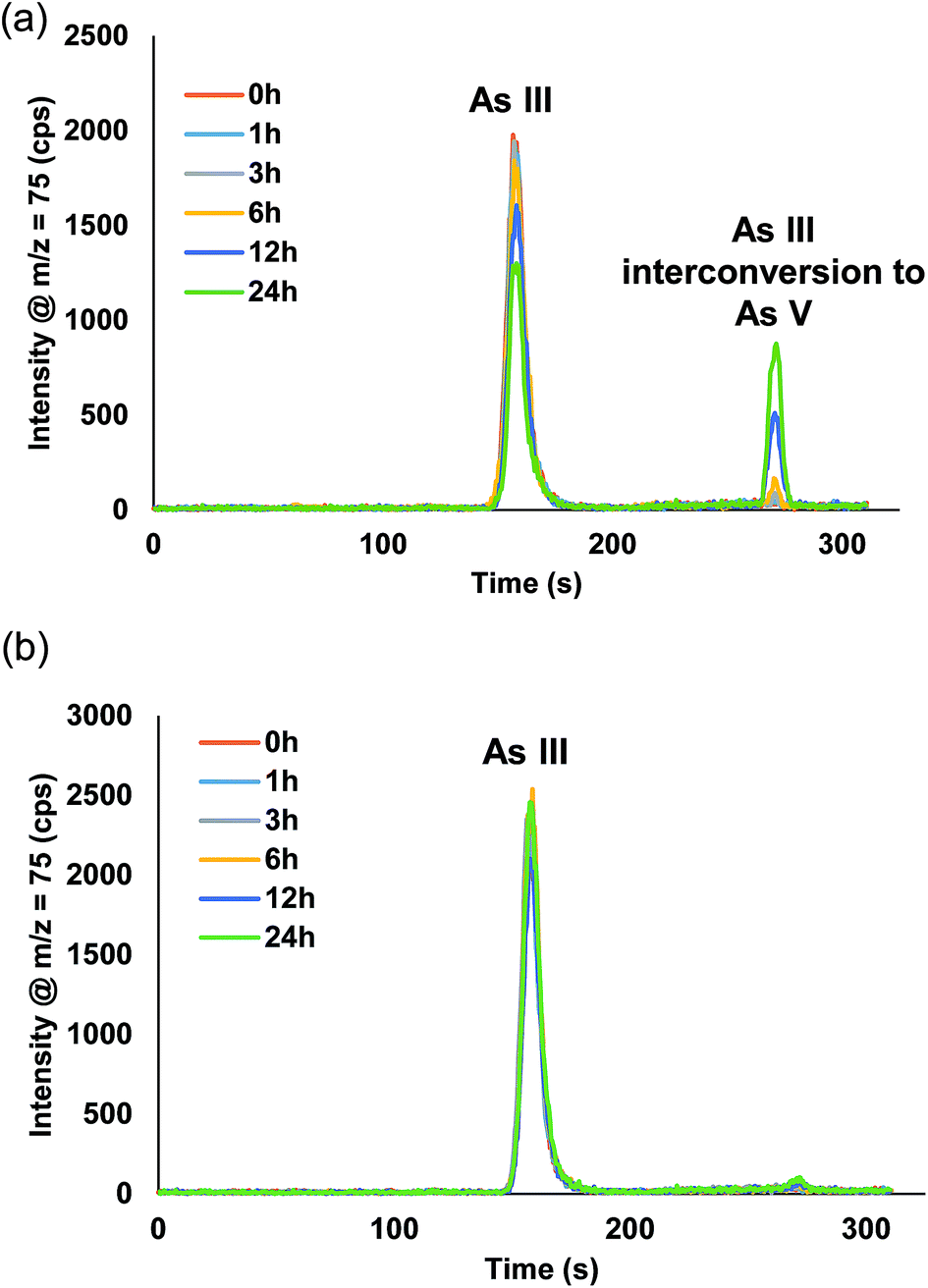
Fig. S1† displays the chromatograms from the manual dilution study. For AsB, DMA, MMA, and As V no changes were detected from t = 0 h up to t = 24 h. Fig. 5a, shows there was a distinct peak detected for As V during the As III study, indicative of As III interconverting to As V. The As III was detected at a level of 9.9 ppb at t = 0 h and after t = 24 h it had dropped to 6.1 ppb (Table 3). Table 4 displays the % of As III that converted to As V during the study. After 24 h, the As III was 38% less than the initial measured concentration at t = 0 h, whereas, 21% of the As III had converted to As V. There was ∼17% As III lost during the 24 h time period, which is similar to the reported loss in the work by Verdon et al.24
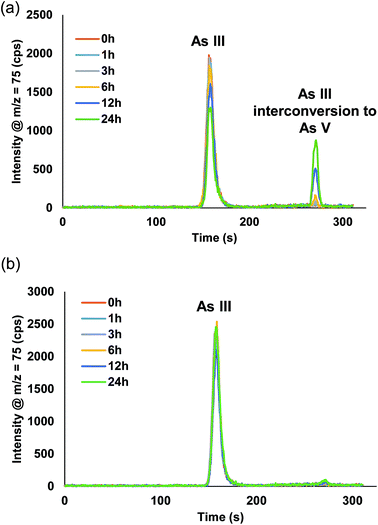 |
| | Fig. 5 Chromatograms for the 10 ppb As III time study (0–24 h) from the (a) manual dilution method and (b) inline dilution method. | |
Table 3 Manual dilution vs. inline autodilution for samples prepared and analyzed from 0 h up to 24 h after sample preparation
| Time (h) |
Measured concentration (ppb) |
| AsB |
DMA |
As III |
MMA |
As V |
|
Manual dilution
|
| 0 |
9.5 |
10.3 |
9.9 |
8.8 |
9.6 |
| 1 |
9.5 |
7.9 |
9.5 |
9.5 |
9.5 |
| 3 |
9.0 |
9.1 |
9.2 |
9.4 |
9.4 |
| 6 |
8.7 |
8.7 |
9.1 |
9.3 |
9.3 |
| 12 |
8.7 |
9.0 |
7.8 |
9.1 |
9.4 |
| 24 |
9.2 |
8.9 |
6.1 |
9.4 |
9.4 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
Inline autodilution
|
| 0 |
10.9 |
10.2 |
9.7 |
11.0 |
11.0 |
| 1 |
10.2 |
10.0 |
10.0 |
10.6 |
10.1 |
| 3 |
9.0 |
9.1 |
10.6 |
9.7 |
11.3 |
| 6 |
9.5 |
8.4 |
10.8 |
10.6 |
10.2 |
| 12 |
9.1 |
8.8 |
9.2 |
9.4 |
10.0 |
| 24 |
9.9 |
9.8 |
10.1 |
9.5 |
10.4 |
Table 4 Manual dilution vs. inline autodilution for As III measured over 24 ha
| Time (h) |
10 ppb As III spiked urine samples |
| As III % |
As V % |
As III + As V |
|
As III % = (x h As III/0 h As III) × 100, As V % = (x h As V/0 h As III) × 100, As III + As V = As III % + As V %.
|
|
Manual dilution
|
| 0 |
100.0 |
0.0 |
100.0 |
| 1 |
95.7 |
0.0 |
95.7 |
| 3 |
92.8 |
0.2 |
93.0 |
| 6 |
91.3 |
1.6 |
92.9 |
| 12 |
78.5 |
12.0 |
90.6 |
| 24 |
61.9 |
21.4 |
83.4 |
|
|
Inline autodilution
|
| 0 |
100.0 |
0.0 |
100.0 |
| 1 |
102.8 |
0.0 |
102.8 |
| 3 |
105.2 |
0.0 |
105.2 |
| 6 |
106.4 |
0.0 |
106.4 |
| 12 |
95.1 |
0.5 |
95.6 |
| 24 |
102.9 |
1.3 |
104.2 |
Fig. S2† displays the chromatograms for the inline dilution study. AsB, DMA, MMA, As III (Fig. 5b), and As V appeared to have no visual interconversion, but there was a 1.3% interconversion of As III to As V after 24 h (Table 4). There was no loss of total inorganic arsenic for the inline sample preparation study; conversely, the manual sample preparation study had a loss of ∼17% after 24 h. These studies suggest that inline dilutions offer a better solution when samples need to be diluted prior to introduction onto the column.
Analytical performance
To evaluate the method accuracy, NIST SRM 2669 (arsenic species in frozen human urine) levels I and II were analyzed using the calibration and inline dilution scheme described in the previous sections. All results obtained are shown in Table 5. The NIST certified value and the minimum/maximum reported values are listed for comparison purposes.11 All level I results were ≤10 %BIAS (%BIAS = (measured value − reference value)/(reference value × 100)) except for MMA which was −17.6 %BIAS, however the measured results fall within the reported values of 1.5–2.55 ppb MMA. NIST SRM 2669 level II includes arsenocholine (AC) and trimethylarsine oxide (TMAO) which were not part of the five species method developed in this study. The AsB results for level II were higher than the certified or reported values which is due to TMAO co-eluting with AsB.28 All other results for level II that were reported are within ≤10 %BIAS.
Table 5 Accuracy to NIST SRM 2669 for the five arsenic species measured (n = 3)a
|
|
NIST level I certified value (ppb) |
All methods reported11 (ppb) |
Measured values (ppb) |
| Min |
Max |
Avg ± 1 SD |
|
Min = minimum reported value in the original SRM manuscript. Max = maximum reported value in the original SRM manuscript.
AsB (1.43) + TMAO (1.94) = 3.37 ppb (2.44 ppb (min.) − 4.86 ppb (max.)).
|
| AsB |
12.4 ± 1.9 |
10.48 |
14.68 |
12.3 ± 1.0 |
| DMA |
3.47 ± 0.41 |
2.98 |
4.09 |
3.39 ± 0.35 |
| As III |
1.47 ± 0.10 |
1.14 |
1.86 |
1.34 ± 0.37 |
| MMA |
1.87 ± 0.39 |
1.5 |
2.55 |
1.54 ± 0.17 |
| As V |
2.41 ± 0.30 |
1.9 |
2.88 |
2.57 ± 0.53 |
| Total |
22.2 ± 4.8 |
18 |
26.06 |
20.85 |
|
|
NIST level II certified value (ppb) |
All methods reported11 (ppb) |
Measured values (ppb) |
| Min |
Max |
Avg ± 1 SD |
| AsB |
1.43 ± 0.08 |
1.26 |
1.84 |
2.45 ± 0.54b |
| DMA |
25.3 ± 0.7 |
24.1 |
27.1 |
26.0 ± 3.6 |
| As III |
5.03 ± 0.31 |
4 |
6.2 |
5.58 ± 0.24 |
| MMA |
7.18 ± 0.56 |
6.51 |
8.24 |
6.96 ± 0.79 |
| As V |
6.16 ± 0.95 |
4.99 |
7.48 |
6.68 ± 0.85 |
| AC |
3.74 ± 0.35 |
3.14 |
4.27 |
n/a |
| TMAO |
1.94 ± 0.27 |
1.18 |
3.02 |
n/a |
| Total |
50.7 ± 6.3 |
45.18 |
58.15 |
47.62 |
The limits of detection (LOD) and quantification (LOQ) were calculated for the five arsenic species using the 30X inline dilution method (Table 6). The LOD and LOQ values were determined using the criteria of 3σ and 10σ, respectively (urine blanks, n = 10). The LODs obtained were in the single digit ppt range (0.3–1.7 ppt) which are slightly better than what was reported by Carioni et al. (LODs = 4–25 ppt) and Leese et al. (LODs = 3–51 ppt).28,29
Table 6 Limits of detection and limits of quantificationa
|
|
AsB |
DMA |
As III |
MMA |
As V |
|
LOD = (3 × σblank)/m, LOQ = (10 × σblank)/m.
|
| LOD (ppt) |
1.7 |
0.7 |
0.6 |
1.0 |
0.3 |
| LOQ (ppt) |
5.8 |
2.5 |
1.9 |
3.2 |
1.0 |
Conclusion
The prepFAST IC, in conjunction with an ICP-MS, has been shown to be a reliable syringe-driven liquid chromatography system, capable of accurately measuring arsenobetaine, dimethylarsinic acid, arsenite, monomethylarsonic acid, and arsenate. Using the inline dilution function, the calibration curves were created from a single stock standard with excellent linearity. The ability to prepare a calibration curve from a single stock standard saves analyst time and reduces the amount of waste produced (e.g., plastic vials/consumables). Additionally, the use of inline dilution as compared to traditional manual sample preparation provided a solution to reduce/eliminate the interconversion detected when spiking urine with As III. The overall recovery was improved when samples were diluted directly before injection onto the column. Method accuracy for the inline dilution scheme was in agreement with the reported values for NIST SRM 2669.
Conflicts of interest
There are no conflicts of interest to declare.
References
- M. M. Nearing, I. Koch and K. J. Reimer, Spectrochim. Acta, Part B, 2014, 99, 150–162 CrossRef CAS.
- P. Apostoli, Fresenius. J. Anal. Chem., 1999, 363, 499–504 CrossRef CAS.
- T. Wang, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 807–831 CrossRef CAS.
- V. W. M. Lai, Y. Sun, E. Ting, W. R. Cullen and K. J. Reimer, Toxicol. Appl. Pharmacol., 2004, 198, 297–306 CrossRef CAS PubMed.
- I. Sen, W. Zou, J. Alvaran, L. Nguyen, R. Gajek and J. She, J. AOAC Int., 2015, 98, 517–523 CrossRef CAS PubMed.
- T. Narukawa, T. Suzuki, K. Inagaki and A. Hioki, Talanta, 2014, 130, 213–220 CrossRef CAS PubMed.
- N. S. Horner and D. Beauchemin, Anal. Chim. Acta, 2012, 717, 1–6 CrossRef CAS PubMed.
- A. V. Zmozinski, T. Llorente-Mirandes, J. F. Lopez-Sanchez and M. M. da Silva, Food Chem., 2015, 173, 1073–1082 CrossRef CAS PubMed.
- L. Schmidt, J. A. Landero, R. F. Santos, M. F. Mesko, P. A. Mello, E. M. M. Flores and J. A. Caruso, J. Anal. At. Spectrom., 2017, 32, 1490–1499 RSC.
- L. Benramdane, M. Accominotti, L. Fanton, D. Malicier and J.-J. Vallon, Clin. Chem., 1999, 45, 301–306 CAS.
- L. L. Yu, C. P. Verdon, W. C. Davis, G. C. Turk, K. L. Caldwell, R. L. Jones, B. Buckley and R. Xie, Anal. Methods, 2011, 3, 1107–1115 RSC.
- L. Munoz, M. Meneses, P. Pismante, O. Andonie, F. Queirolo and S. Stegen, J. Chil. Chem. Soc., 2014, 59, 2432–2436 CrossRef.
- M. Montes-Bayon, K. DeNicola and J. A. Caruso, J. Chromatogr. A, 2003, 1000, 457–476 CrossRef CAS PubMed.
- V. M. O. Carioni, J. A. McElroy, J. M. Guthrie, R. A. Ngwenyama and J. D. Brockman, Talanta, 2017, 165, 76–83 CrossRef CAS PubMed.
- T. Wang, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 807–831 CrossRef CAS.
- J. Delafiori, G. Ring and A. Furey, Talanta, 2016, 153, 306–331 CrossRef CAS PubMed.
- W. W. Buchberger and P. R. Haddad, J. Chromatogr. A, 1997, 789, 67–83 CrossRef CAS.
- R. Xie, W. Johnson, S. Spayd, G. S. Hall and B. Buckley, Anal. Chim. Acta, 2006, 578, 186–194 CrossRef CAS PubMed.
- K. Wrobel, K. Wrobel, B. Parker, S. S. Kannamkumarath and J. A. Caruso, Talanta, 2002, 58, 899–907 CrossRef CAS PubMed.
- T. I. Todorov, J. W. Ejnik, F. G. Mullick and J. A. Centeno, Microchim. Acta, 2005, 151, 263–268 CrossRef CAS.
- Z. Gong, X. Lu, M. Ma, C. Watt and X. C. Le, Talanta, 2002, 58, 77–96 CrossRef CAS PubMed.
- B. S. Sheppard, W. Shen, J. A. Caruso, D. T. Heitkemper and F. L. Fricke, J. Anal. At. Spectrom., 1990, 5, 431–435 RSC.
- B. S. Sheppard, J. A. Caruso, D. T. Heitkemper and K. A. Wolnik, Analyst, 1992, 117, 971–975 RSC.
- C. P. Verdon, K. L. Caldwell, M. R. Fresquez and R. L. Jones, Anal. Bioanal. Chem., 2009, 393, 939–947 CrossRef CAS PubMed.
- R. E. Wolf, S. A. Morman, P. L. Hageman, T. M. Hoefen and G. S. Plumlee, Anal. Bioanal. Chem., 2011, 401, 2733–2745 CrossRef CAS PubMed.
- S. D. Conklin and P. E. Chen, Food Addit. Contam., 2012, 29, 1272–1279 CrossRef CAS PubMed.
- C. D. B. Amaral, J. A. Nobrega and A. R. A. Nogueira, Talanta, 2013, 115, 291–299 CrossRef CAS PubMed.
- V. M. O. Carioni, J. A. McElroy, J. M. Guthrie, R. A. Ngwenyama and J. D. Brockman, Talanta, 2017, 165, 76–83 CrossRef CAS PubMed.
- E. Leese, J. Morton, E. Tan, P. H. E. Gardiner and V. A. Carolan, J. Anal. Toxicol., 2014, 38, 24–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ja00038g |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Patrick
Sullivan
,
M. Paul
Field
,
Scott
Smith
and
Daniel R.
Wiederin
*,
Patrick
Sullivan
,
M. Paul
Field
,
Scott
Smith
and
Daniel R.
Wiederin