
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical preparation and applications of copper(I) acetylides: a demonstration of how electrochemistry can be used to facilitate sustainability in homogeneous catalysis†
Peter W.
Seavill
 ,
Katherine B.
Holt
and
Jonathan D.
Wilden
*
,
Katherine B.
Holt
and
Jonathan D.
Wilden
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: j.wilden@ucl.ac.uk
First published on 13th November 2018
Abstract
Copper(I) acetylides are important intermediates for many syntheses and have been prepared here electrochemically in an energy efficient manner. These were subsequently employed in simple organic C–C bond forming reactions. We also demonstrate that application of Faraday's laws allows the charge to be calculated so that only the required amount of metal is used. In addition, the application of copper-coated graphite electrodes allows the maximum atom efficiency for this process and even offers a recovery strategy to extract the metal following completion of the reaction.
Introduction
Recent years have seen a renaissance in organocopper chemistry as our understanding of the mechanistic pathways of the catalytic systems involved has grown. The elegant work of Evano1a,b and Hwang1c–e has done much to popularise the chemistry of copper(I) acetylides as versatile reagents for a wealth of synthetic operations. The observation that the reagents can behave ‘classically’ by reacting with electrophiles to form C–C bonds as well as displaying ‘Umpolung’ behaviour to react with nucleophiles makes them attractive building blocks for the construction of complex molecular frameworks.1a Furthermore, they can be utilised cooperatively in a variety of noble metal-catalysed processes (most notably the palladium-catalysed Sonogashira reaction) to produce an even greater diversity of products.2 As shelf-stable yellow solids these species can be prepared in bulk and stored almost indefinitely. We here demonstrate that the preparation and utility of these species can be improved in terms of energy efficiency and waste management by the application of electrochemistry and by considering the practical issues that classical methods present.There are two main methods for the preparation of copper acetylides: (i) reaction of two equivalents of CuI with the parent acetylene in an alcohol solvent with aqueous ammonia or (ii) reaction of CuI with the parent alkyne in the presence of K2CO3 (Scheme 1). In terms of their ‘green’ credentials3 the solvent system of method (i) is reasonably good, however the method requires two equivalents of cuprous halide to combat the known propensity of CuI salts to undergo disproportionation in aqueous solvent mixtures and thus produces both excess copper and iodide waste that must be disposed of appropriately.
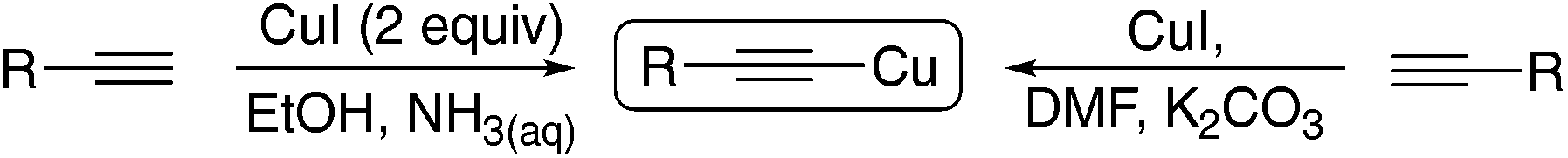 | ||
| Scheme 1 Classical methods of copper acetylide synthesis. | ||
Method (ii) requires the use of dimethylformamide:4 a dipolar aprotic solvent which has been identified as a major problem for industrial scale synthesis.5 Where dipolar aprotic solvents are required, acetonitrile is much preferred, particularly with new ‘green’ processes being identified for its preparation from benign feedstocks.6 Drawbacks aside, both methods allow easy access to the copper acetylide as they can simply be collected via filtration.
Our group has long been interested in the chemistry of acetylenes7–9 and has also developed a more recent interest in electro-organic synthesis and its application to ‘green’ and sustainable synthesis.10,11 As such, we wanted to explore the reactivity of acetylenes under electrochemical conditions and were attracted to the prospect of preparing the functionalised acetylides using a sacrificial copper anode where CuI ions could be released selectively by control of the applied potential. A schematic representation of our approach is outlined in Scheme 2.
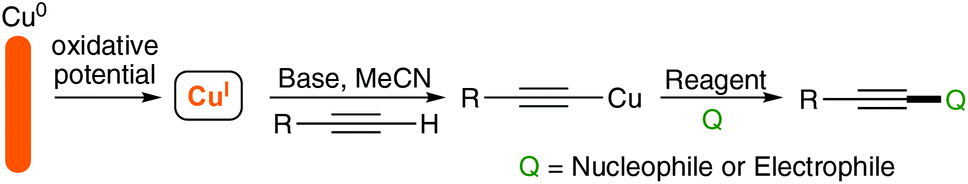 | ||
| Scheme 2 Proposed electrochemical generation of Cu acetylides. | ||
Some years ago, Mitsudo demonstrated that silver(I) acetylides could be prepared via a constant current protocol and successfully demonstrated their applicability in palladium catalysed coupling reactions.12 We were keen to know if the cheaper and potentially more versatile copper reagents would behave similarly, as such an approach would offer various advantages to the preparation of these species and their subsequent reactions. Firstly, with electrochemical techniques it is (theoretically at least) possible to calculate and measure the charge required to generate the desired amount of CuI. Therefore, only the energy required for the formation of the copper acetylide is consumed. Secondly, bulk copper metal is cheap and readily available in pure forms due to its almost ubiquitous applications in electrical wiring systems. Finally, the process is atom efficient as there is no iodide present in the starting copper species; for comparison only around 33% of a sample of CuI is copper, not to mention the issues surrounding the disposal of iodide waste.
Results and discussion
We therefore began by examining whether this approach would deliver copper acetylides in yields competitive with those known in the literature. We elected to use a divided cell (‘H-cell’) with a copper working-electrode, a Pt wire counter-electrode and a silver wire quasi reference-electrode. Initial cyclic voltammetry (CV) experiments (Fig. 1) indicated that copper ions (we hypothesised CuI ions in the form of stabilised Cu(MeCN)4PF6 complexes) could be released at oxidising potentials in acetonitrile so we were confident that including the alkyne and a suitable base in the mixture would yield the corresponding copper(I) acetylides.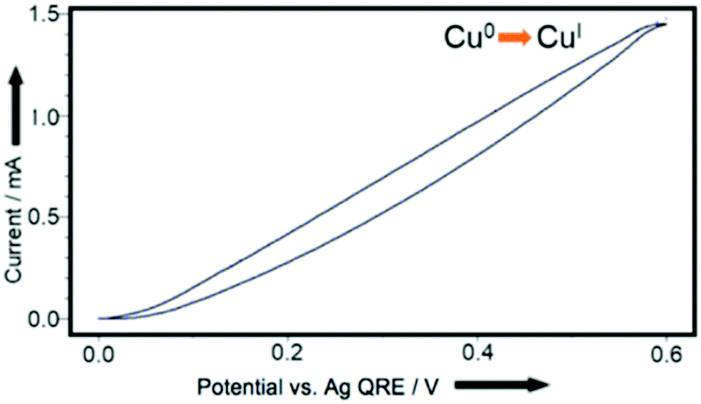 | ||
| Fig. 1 CV plot, using Cu0-coated glassy carbon working-electrode, Pt wire counter-electrode, Ag wire quasi reference-electrode in 0.1 M Bu4NPF6/MeCN. Axes redrawn for clarity. | ||
Pleasingly, application of an oxidative potential (constant potential, +0.5 V vs. Ag wire) to the copper electrode in a solution of the terminal alkyne in a 0.1 M Bu4NPF6/MeCN background electrolyte and the organic base 1,4-diazabicyclo[2.2.2]octane (DABCO) yielded the copper acetylides in excellent yields, when O2 was excluded, allowing isolation simply by filtration. Presumably, the current flow is maintained by the well-known electrochemically-mediated Hofmann elimination of the Bu4NPF6 electrolytic salt occurring in the cathodic chamber.13 Control and optimisation reactions are summarised in the ESI (Table S1†) and include a demonstration that commercial Cu(MeCN)4PF6 complex also successfully liberates 1 supporting our hypothesis regarding the identity of the active copper species released from the electrode. The yields and reaction times compare favourably with the classical methods outlined in the literature (Scheme 3).1a,b As a synthetic preparation of these species, this method is extremely efficient, however we wanted to develop a more sustainable and general method of generating and using these species without having to use a bulk metal plate as the electrode, which would be less viable if a similar approach were to be extended to other, potentially precious, metals. In addition, we wanted to develop a method where the amount of metal and charge passed could be more accurately measured to determine the efficiency of the process.
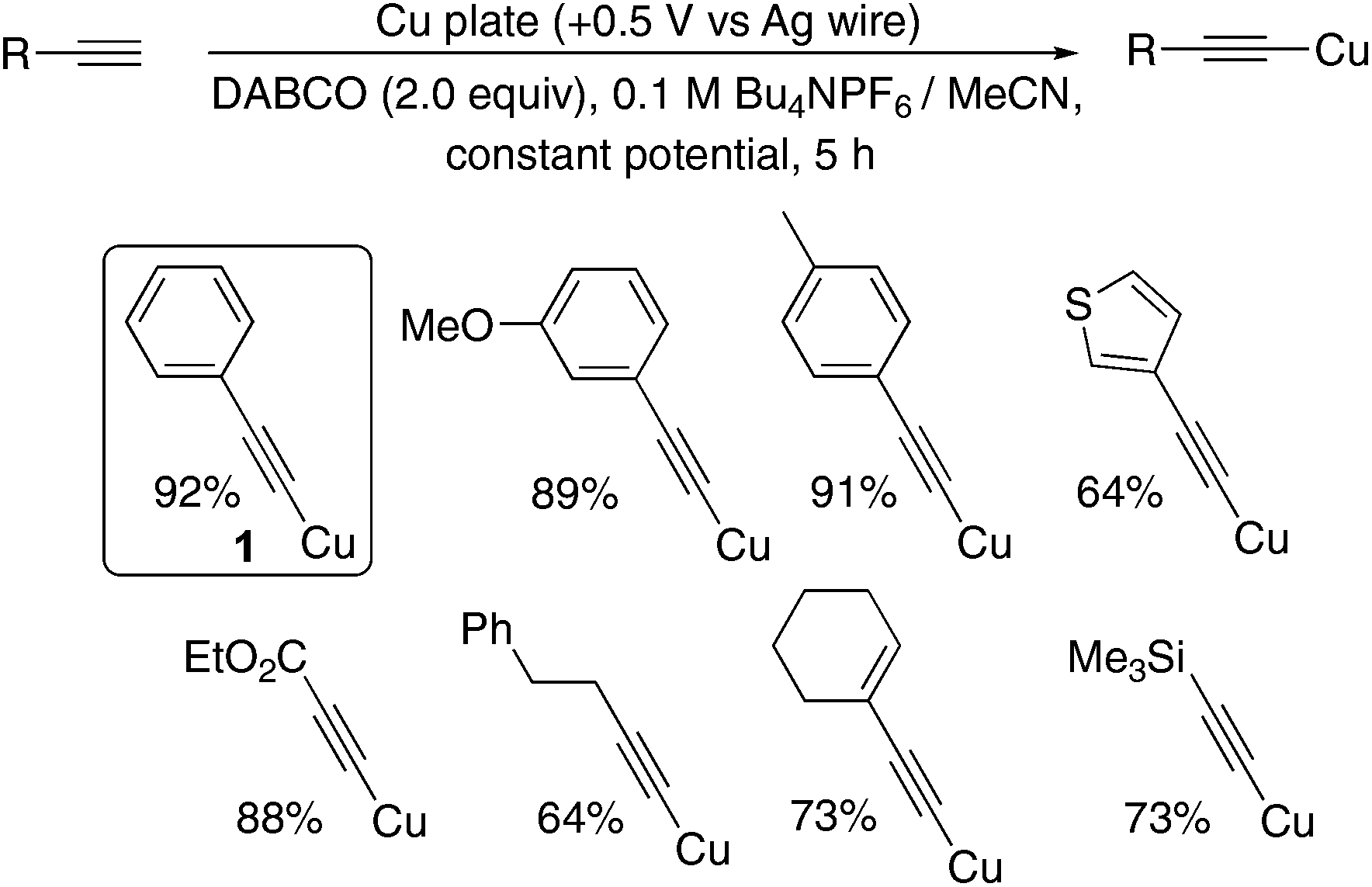 | ||
| Scheme 3 Electrochemical preparation of copper acetylides. | ||
We were attracted by the prospect of electroplating a thin layer of metal onto the surface of an inert electrode. In doing so we hoped to be able to: (i) measure the exact amount of metal deposited and used in reactions and (ii) use the metal catalytically to perform carbon–carbon bond forming reactions. We envisaged that the surface of a graphite rod could be coated with metal by electroplating from an aqueous solution of the metal salt. The metal ions could then be released by the application of an oxidative potential as required. This approach is outlined in Fig. 2.
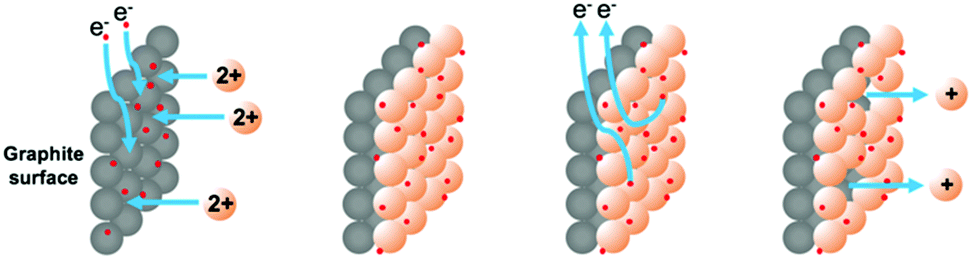 | ||
| Fig. 2 Selective coating and release of copper ions for organic catalysis. | ||
As expected, a simple graphite rod was easily plated with a fine layer of copper metal by application of a reducing potential (−0.5 V vs. Ag wire) to a 0.5 M solution of CuSO4(aq.). In an example where this potential was applied for 600 s, a total charge of 11.99 C was passed, and copper was clearly visible on the graphite surface (Fig. 3). By applying Faraday's laws (eqn (1)), this corresponds to a maximum of 6.21 × 10−5 moles of copper metal deposited (or 3.95 mg). To place this in context, the mass of cuprous iodide containing the same amount of copper would be 11.83 mg.
 | ||
| Fig. 3 Graphite rod electrode coated with metallic copper and Faraday's Laws of Electrolysis represented in eqn (1). | ||
We found that this method proved effective at confirming our suspicion that CuI was released into solution rather than CuII. A simple experiment was devised in which a graphite rod was coated with Cu0 from CuSO4(aq.) (13.40 C passed during coating, equating to a maximum of 6.94 × 10−5 moles of Cu0) and used to produce copper acetylide 1 from phenylacetylene (0.5 mmol) with the conditions seen in Scheme 3 (Pt wire counter electrode), with the exception that all of the Cu present on the graphite rod was released within 30 min and in this experiment the alkyne was the reagent that was in excess rather than the Cu. Our maximum theoretical yield for this experiment was 11.43 mg of 1 if all available oxidised Cu reacted. We isolated 8.10 mg (4.92 × 10−5 moles) of 1, which pertains to an excellent 71% efficiency of Cu atom integration into the product. Furthermore, the charge passed during the release of Cu ions was measured to be 6.17 C. Substituting this figure back into eqn (1) along with the number of moles of isolated 1 gives a good approximation of the oxidation state of the copper released into solution, z, as being 1.30 (1) or in other words, CuI rather than CuII. When employing a graphite counter electrode of the same dimensions as the working electrode, a value of z of 1.12 was obtained and an efficiency of 77% was recorded (more details are available in the ESI†). This indicates our method is robust even when asymmetric electrodes are employed.
Graphs showing charge passed as a function of time for the Cu coating (A) and release (B) in this experiment are shown in Fig. 4. These plots demonstrate that the charge employed to coat the electrode via reduction of CuII (A) is approximately twice that required to release it (B), consistent with the release of CuI into the solution.
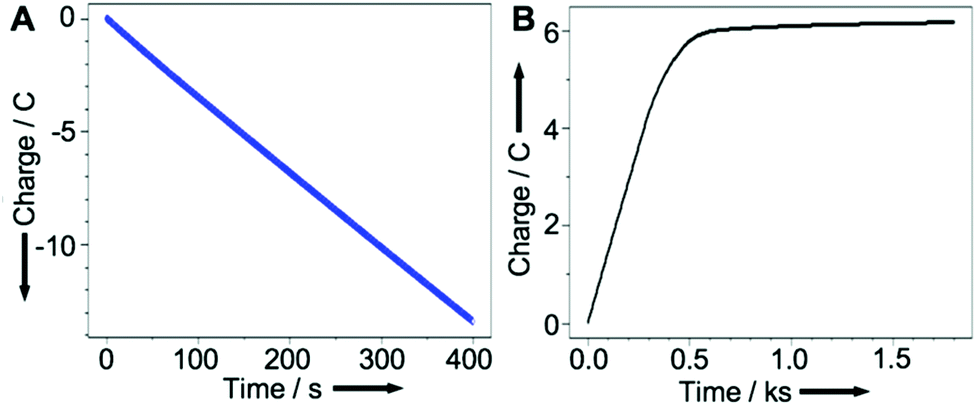 | ||
| Fig. 4 Charge passed as a function of time when: (A) coating the graphite rod from a CuSO4(aq.) solution and (B) for the release of CuI. Axes redrawn for clarity. | ||
In addition to preparing copper acetylides for isolation we wished to examine the possibility of using the metal-coated rod to generate copper acetylides in situ which would then undergo a catalytic reaction requiring only a sub-stoichiometric amount of copper. It also occurred to us that if an oxidising potential of +0.5 V could release CuI into the solution, then it should also be possible to recover at least some of the copper at the end of the reaction by the application of a reducing potential as a method of ‘cleaning up’ the reaction mixture. In order to test this ‘inverting potential’ approach, we chose to examine the simple Glaser–Hay14 oxidative dimerization of phenylacetylene to see if the copper-coated graphite could serve as a catalytic CuI source for this C–C bond forming reaction. We hoped that the graphite electrode would then serve as a vehicle for the removal of the copper from the solution at the end of the reaction by switching to a reducing potential (−1.0 V). For this reaction we found that dichloromethane proved to be a more effective solvent than MeCN in that the intermediary copper acetylide did not precipitate out of solution upon formation as it did in MeCN, providing better yields of the diyne product when exposed to aerial O2. Work is ongoing to find more sustainable conditions for this reaction. We found that when 1 mmol phenylacetylene was used with a Cu-coated graphite electrode (14.14 C passed when coating, equating to a maximum of 7.33 × 10−5 moles of Cu0) we had a maximum catalytic loading of 7.33 mol%. Pleasingly, when we applied an oxidative potential of +0.5 V to the coated graphite electrode in this coupling reaction, the corresponding diyne, 2, was isolated in a good yield of 68%, in spite of a small portion of the copper acetylide intermediate precipitating out of solution (Scheme 4). We also found that while the yield of the reaction could be improved by adding a tetramethylethylenediamine (TMEDA) ligand, interestingly, the TMEDA proved capable of oxidising the electroplated Cu, without requiring an applied potential. The ligand-free conditions are shown in Scheme 4.
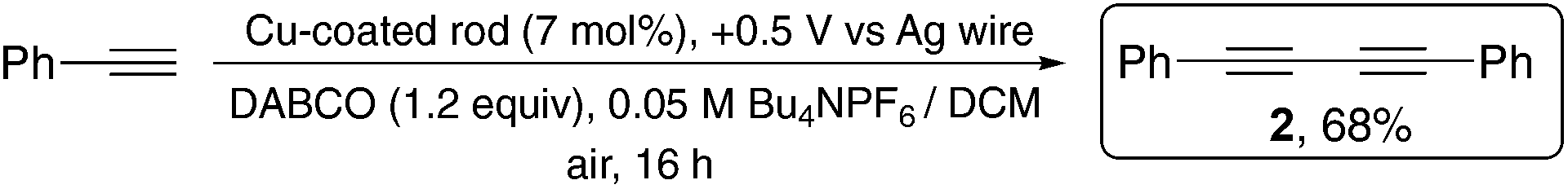 | ||
| Scheme 4 Electrochemically-controlled ligand-free Glaser–Hay coupling. | ||
Emboldened by this result, we attempted the ambitious concept of recovering the copper after the reaction had been completed by switching the polarity of the potential of the working electrode. Such an approach would represent a major advantage in terms of reusing the catalyst and cleaning up the reaction mixture which are major considerations in sustainable chemistry, especially when undertaking large scale reactions where the catalyst is often a precious metal. The approach is conceptualised in Fig. 5.
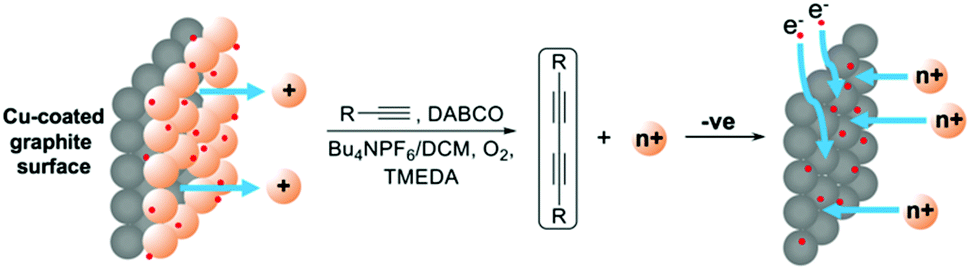 | ||
| Fig. 5 Conceptualized approach to Glaser–Hay coupling from a Cu-coated electrode and recovery of the metal catalyst by switching to a reducing potential. | ||
Our first attempts at recovering the metal post reaction proved troublesome, since the amine additives (e.g. TMEDA, amine bases, etc.) commonly added in such reactions to enhance their efficiency coordinate the copper and hold it in the organic solvent making it resistant to reduction at the electrode surface. As stated previously, TMEDA facilitates oxidation of the coated Cu making the use of an oxidative potential for release redundant, however, we opted to include the ligand in these reactions as it lead to a smoother reaction in which none of the copper acetylide intermediate precipitated out of solution (thereby maximising the amount of copper accessible for recovery) and allowed the use of a slightly lower catalyst loading (6 mol%) than that seen in ligand-free attempts (Scheme 4). Acidification allowed the copper to be liberated from the TMEDA, however also reduced the reductive potential window for recovering the copper. As such, we acidified the reaction medium with a minimum amount of 2 M HCl and water to drive all the available copper into the aqueous phase. The movement of copper ions from the organic to aqueous phase is clearly observable as displayed in Fig. 6.
 | ||
| Fig. 6 Location of copper species in: (A) organic phase (just after HCl(aq.) added) and (B) aqueous phase (after 30 min vigorous stirring). | ||
Essentially, the TMEDA-coordinated copper is converted to CuCl2 which is much more easily reduced and recovered. Pleasingly, application of a reducing potential to the solution then allowed us to re-coat the electrode with copper, again clearly visible as shown in Scheme 5.
 | ||
| Scheme 5 Glaser–Hay coupling and recovery of copper post reaction. | ||
We then undertook experiments to accurately determine the amount of copper redeposited on the electrode. Using the charge passed when recovering copper from the HCl(aq.) solution proved unreliable as a large component of the charge must be attributed to the electrolysis of the aqueous solution itself. Therefore, we transferred the electrode to a fresh solution of acetonitrile with 0.1 M Bu4NPF6 as background electrolyte to release the copper into solution as Cu(MeCN)4PF6 while monitoring the current and charge passed. As the metallic copper is exhausted, the current falls to zero and the charge approaches a maximum as can be seen from the plots of current (A) and charge (B) vs. time in Fig. 7.
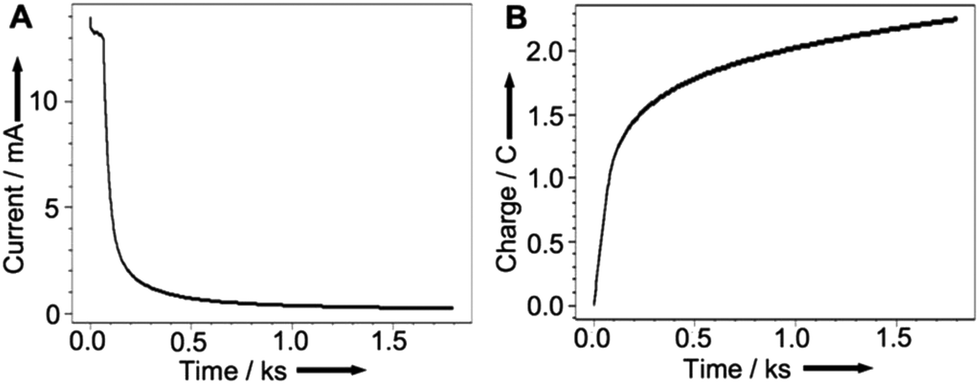 | ||
| Fig. 7 (A) Current and (B) Charge passed as a function of time for Cu recovery efficiency determination. Axes redrawn for clarity. | ||
At the point where the current approaches zero (i.e. when all of the copper metal had been stripped from the graphite surface) a charge of 2.34 C had passed. This corresponds to 24.26 μmol (1.54 mg of copper) and a corresponding recovery efficiency of 39%, as the graphite rod was initially coated with 3.95 mg of copper. In light of the difficulty of recovering metal catalysts from reaction media this is a remarkably good initial proof of concept. This preliminary study is highly promising, and we are confident that future refinements of the technology will only see the efficiency improve.
Conclusions
In conclusion, we have demonstrated how the combination of simple electrochemical techniques can be employed to improve the efficiency and sustainability of a copper-mediated organic reaction. The novel application of a metal-coated carbon electrode has also enabled us to perform simple calculations to consider the energy and atom efficiency of the process. We have also demonstrated that the technique could, with further refinements, offer opportunities to recover transition metals from reaction media. This offers an attractive and industrially-relevant approach for the future of transition metal-mediated organic synthesis, in particular where the metals involved are toxic and/or expensive.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge financial support from the Engineering and Physical Sciences Research Council (grant no. EP/M02220X/1), University College London and UCL Business. The contributions of the UCL Mass Spectrometry service (Dr K. Karu) and NMR Service (Dr A. Aliev) are also gratefully acknowledged.Notes and references
- (a) G. Evano, K. Jouvin, C. Theunissen, C. Guissart, C. Tresse, J. Heimburger, Y. Bouhoute, R. Veillard, M. Lecomte, A. Nitelet, S. Schweizer, N. Blanchard, C. Alayrac and A.-C. Gaumont, Chem. Commun., 2014, 50, 10008 RSC; (b) K. Jouvin, J. Heimburger and G. Evano, Chem. Sci., 2012, 3, 756 RSC; (c) A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896 CrossRef CAS PubMed; (d) A. Sagadevan, V. P. Charpe, A. Ragupathi and K. C. Hwang, J. Am. Chem. Soc., 2017, 139, 2896 CrossRef CAS PubMed; (e) D. K. Das, V. K. K. Pampana and K. C. Hwang, Chem. Sci., 2018, 9, 7318–7326 RSC.
- S. Díez-González, Adv. Organomet. Chem., 2016, 66, 93 CrossRef.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed.
- N. A. Bumagin, A. B. Ponomarev, A. N. Ryabtsev and I. P. Beletskaya, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1988, 37, 507 CrossRef.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchy, J. C. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- E. C. Corker, U. V. Mentzel, J. Mielby, A. Riisager and R. Fehrmann, Green Chem., 2013, 15, 928 RSC.
- V. J. Gray, J. Cuthbertson and J. D. Wilden, J. Org. Chem., 2014, 79, 5869 CrossRef CAS PubMed.
- J. D. Wilden and V. J. Gray, Org. Biomol. Chem., 2016, 14, 9695 RSC.
- R. M. Chowdhury and J. D. Wilden, Org. Biomol. Chem., 2015, 13, 5859 RSC.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC.
- K. Mitsudo, T. Shiraga, J.-I. Mizukawa, S. Suga and H. Tanaka, Chem. Commun., 2010, 46, 9256 RSC.
- C. E. Dahm and D. G. Peters, J. Electroanal. Chem., 1996, 402, 91 CrossRef.
- (a) C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422 CrossRef; (b) A. S. Hay, J. Org. Chem., 1960, 25, 1275 CrossRef CAS; (c) A. S. Hay, J. Org. Chem., 1962, 27, 3320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental detail, control experiments and characterisation data. See DOI: 10.1039/c8gc03262a |
| This journal is © The Royal Society of Chemistry 2018 |