
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas-phase synthesis of oxymethylene ethers over Si-rich zeolites†
Anna
Grünert
 ,
Pit
Losch
,
Cristina
Ochoa-Hernández
,
Wolfgang
Schmidt
and
Ferdi
Schüth
*
,
Pit
Losch
,
Cristina
Ochoa-Hernández
,
Wolfgang
Schmidt
and
Ferdi
Schüth
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: schueth@mpi-muelheim.mpg.de
First published on 27th September 2018
Abstract
Oxymethylene ethers are a class of synthetic fuels that allows significantly reduced levels of pollutant emissions from compression-ignition engines. Conventionally they are synthesized in liquid-phase. As a new approach for the production of oxymethylene ethers the continuous gas-phase synthesis from methanol and formaldehyde was studied. A broad range of zeolites has been studied as the catalysts for the reaction and a relationship between reactivity and silica-to-alumina ratio was established. Moderate acidity as found in silicon-rich zeolites proved to be advantageous. Even aluminum-free zeolite shows high selectivity and activity to OME indicating that silanol groups as found on the external surface or in defects provide sufficient acidity for the reaction. The zeolitic catalysts deactivate with time but can be fully regenerated with common regeneration protocols.
1 Introduction
As transportation is one of the main sectors contributing to global greenhouse gas emission,1 many efforts in politics as well as research are directed towards solutions for a more sustainable mobility. Synthetic fuels on the basis of CO2 and H2 are considered promising for reducing the overall life-cycle CO2 emissions of transportation via CO2 consumption during fuel production.2 In addition, such fuels allow shifting from fossil carbon to renewable feedstocks. CO2 preferentially originates from industrial exhaust gases, but could as well be provided from biomass or via direct air capture. In order to design a sustainable process, the source of H2 is also important. Considering such a scenario, H2 is preferably supplied from water electrolysis using electricity generated from sunlight and/or wind energy.The group of CO2-based fuels includes methane, methanol, dimethyl ether (DME) and Fischer–Tropsch (FT) fuels. Recently a new class of compounds, called oxymethylene ethers (OME), gained increasing attention due to its favorable combustion characteristics. OME have the chemical formula CH3O(CH2O)nCH3 with n denoting the length of the central ether chain in the abbreviation OMEn. Motor tests using different homologues of the chain ethers, either neat or in blends, show significant reduction of soot particle emission in compression-ignition engines as compared to conventional diesel fuel. Due to the absence of a soot-NOx trade-off, parameters such as exhaust gas recirculation can also be adjusted to minimize harmful NOx emissions.3–6
The physical properties of higher OME homologues, such as OME3–5, or OME/diesel blends allow their use as drop-in fuels in conventional motors (with only minor adjustments required) and their distribution via existing infrastructure and supply chains.7 The market introduction barrier for OME driven cars is hence lower than for vehicles driven by liquefied fuels such as methane or DME or electricity. An additional advantage is the non-toxic character of OMEs allowing their safe handling.
The main current challenge remains the development of a process for large-scale production. In literature, many studies report OME liquid-phase syntheses, where OME1 (also called dimethoxymethane (DMM) or methylal) is reacted with trioxane7–17 or paraformaldehyde.18–22 Current industrial synthesis routes are based on such systems.23 The major drawback of this route is the large number of process steps, yielding five main steps: (1) formation of MeOH, (2) production of aqueous formaldehyde, (3) + (4) synthesis of the intermediate OME1 and trioxane or paraformaldehyde and (5) OMEn formation. Another disadvantage is the need for a highly energy demanding separation of the intermediates.
In order to circumvent isolation of intermediates, the direct synthesis of OME from methanol (MeOH) and formaldehyde (FA) is of interest. First studies, including batch mode OME syntheses with acidic catalysts, such as ion exchange resins23–27 or various zeolites (H-BEA-25, H-MFI-90, H-MFI-27, H-FAU-30, H-MFI-400, H-MOR-30; the suffixes denoting SiO2/Al2O3 ratios),26 and continuous-flow OME synthesis over Zr-modified γ-Al2O3,28,29 have been reported with methanolic or mixed aqueous methanolic solutions of formaldehyde as reactants. Alternative approaches for avoiding isolation of intermediates are the direct synthesis of OME1 from carbon dioxide, hydrogen and methanol over homogeneous catalysts30,31 and the one-step oxidation of MeOH over bifunctional catalysts.32–42 OME1 as well as OME2 were described to form via DME oxidation, however in yields below 8%.43–45
Ouda et al. report a theoretical comparison of the conventional pathway (OME1 + trioxane) with the alternative pathway (via dehydrogenation of methanol to formaldehyde and subsequent OME synthesis) starting from syngas. They concluded that in addition to technological simplicity due to a reduced amount of synthesis steps, the alternative route shows a 20% higher thermodynamic theoretical efficiency due to a significantly reduced need for hydrogen.23
In this work, the gas-phase reaction of methanol and formaldehyde to OME was studied. In perspective of a future large-scale production of OME for supplying large enough quantities to use OME as a sustainable fuel, gas-phase technology has the advantage of scalability, improved process integration and the easy implementation of continuous processes. Potentially, OME can be produced in a complete continuous gas-phase process in only three process steps starting from CO2 and H2: (1) synthesis of methanol, (2) subsequent partial non-oxidative dehydrogenation to formaldehyde to yield the FA/MeOH reactant mixture, and (3) formation of OME. As reaction products and by-products from each step do not interfere with the succeeding reaction, energy intensive product separation can be minimized. While the first two reaction steps have extensively been studied,46,47 a catalytic study of the continuous gas-phase synthesis of OME from methanol and formaldehyde has, to our knowledge, not yet been described. In the present work, we aim to close the gap towards a gas-phase syngas-to-OME process.
2 Results and discussion
2.1 Catalyst screening
The catalytic tests were carried out in a set-up comprising an evaporator unit for the reactant mixture feed, a plug-flow reactor and a gas chromatograph for online analysis (Fig. S5†). An evaluation of suitable reaction temperatures in the range of 130–270 °C preceded the catalyst screening and was conducted with an exemplary solid acid catalyst (zeolite H-MOR-40). A strong influence of temperature on the OME selectivity was observed for the gas-phase reaction studied. OME1+2 selectivity decreases from 72% at 130 °C to below 3% at 220 °C and no OME is detected at 270 °C (Fig. S6†). Methyl formate (MeFo) formed via condensation-disproportionation of two formaldehyde molecules and dimethyl ether (DME) from methanol condensation were identified as by-products. In an attempt to account for both, the described positive impact of a low reaction temperature and the necessity to keep reactants in the gas-phase, 130 °C was chosen as reaction temperature and adopted for all further test runs.In the catalyst screening, four different zeolites with varying SiO2/Al2O3-ratios (H-Zeolite-SAR) were chosen. The selected zeolites were used in protonated form. Three samples of zeolite Y (H-FAU-12/129/340), two of zeolite Beta (H-BEA-35/150), three of ZSM-5 (H-MFI-27/90/∞, the latter will be referred to as Silicalite-1 throughout the study) and two of Mordenite (H-MOR-14/40) were tested under the chosen conditions. For further information on materials, analysis and evaluation routines, see Experimental section and Table S1.†
When evaluating the product distributions (Fig. 1), it is evident that OMEn yield is decreasing with increasing n – a typical feature of chain-growth reactions – and that products (OMEn) and by-products (MeFo, DME) are formed in varying ratios. While the latter observation might seem trivial, it has to be noted that in the analogous liquid phase reaction (batch mode) from formaldehyde (FA) and methanol (MeOH) equilibrium composition is reached irrespective of the employed catalyst. The time for reaching equilibrium is different for different catalysts, though.26
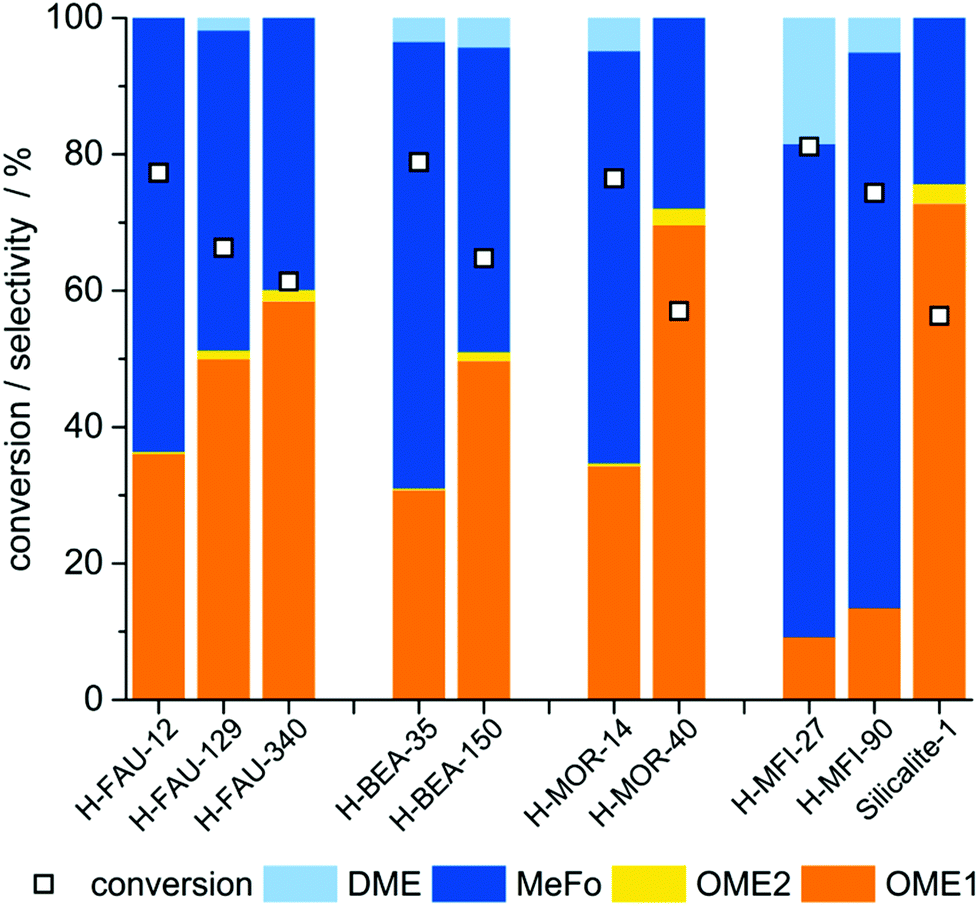 | ||
| Fig. 1 Catalyst screening: Initial selectivity and conversion of zeolitic catalysts determined in the interval of 40–70 min reaction time. Reaction conditions: 10 bar, 130 °C, 0.5 g of catalyst, 100 mL min−1 inert gas flow, 14 μL min−1 FA/MeOH solution feed (60% FA, 38% MeOH and 2% H2O obtained by dissolving paraformaldehyde and methanol). Weight hourly space velocity (WHSV) for formaldehyde: 1.1 g(FA) g(cat)−1 h−1. | ||
The reversibility of the competing reactions in the gas-phase was tested by feeding only MeFo or OME1 + H2O to the catalyst. It could be confirmed that MeFo formation is irreversible under reaction conditions while OME1 formation is reversible. Indeed, when OME1 + H2O were fed over H-MOR-40, MeOH and FA were detected resulting from back reaction of OME1 into its constituents. In addition, MeFo and DME were formed as by-products from the released FA and MeOH as well as OME2 from chain growth reaction of OME1 (Fig. S8†). It can therefore be assumed that the activity of the catalysts towards the irreversible formation of MeFo has a major influence on the final product ratio. Although reversibility of DME formation was not experimentally assessed, the same effect may also be expected in this case.
The remarkable findings of the catalyst screening are the pronounced correlations between the silica-to-alumina ratio (SAR) on the one hand and conversion and selectivity on the other hand. For all four structural classes of zeolites, an increase in selectivity to OME is observed with increasing SAR. At increased SAR, the amount of Al and hence the amount of Brønsted-acidic protons is decreased.
An influence of SAR on catalyst performance has been reported for H-ZSM-5,12,48 H-MCM-22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 and Al-SBA-1516 when OME was synthesized in batch-mode from OME1 or MeOH and trioxane. In these cases however, a maximum in OME yield was generally observed with conversion drastically decreasing at higher SAR due to insufficient release of formaldehyde by acid catalyzed decomposition of trioxane. In our study, no constraints by trioxane decomposition exist and we have confirmed the trend over a wide range of SAR and for a broad range of samples.
49 and Al-SBA-1516 when OME was synthesized in batch-mode from OME1 or MeOH and trioxane. In these cases however, a maximum in OME yield was generally observed with conversion drastically decreasing at higher SAR due to insufficient release of formaldehyde by acid catalyzed decomposition of trioxane. In our study, no constraints by trioxane decomposition exist and we have confirmed the trend over a wide range of SAR and for a broad range of samples.
2.2 Investigation of correlation between acid site properties and catalyst performance
In order to study the suggested correlation of catalyst performance and the properties of its acid sites, temperature programmed desorption of ammonia (NH3-TPD) was carried out for all materials under investigation (Fig. S9–S12†). While it has been discussed for liquid-phase OME synthesis over different zeolites that moderately strong acid sites are best suited for OME syntheses,9,12 this cannot be confirmed in case of gas-phase synthesis from MeOH and FA.In Fig. 2, the conversion and the OME yield are presented as a function of the total amount of ammonia desorbed in the NH3-TPD measurement, the latter being related to the total amount of acid sites in the zeolite. In addition to the zeolitic catalysts, an amorphous siliceous reference material (fumed silica Aerosil 200) is included.
 | ||
| Fig. 2 Left: Conversion and right: OME yield as a function of total amount of ammonia desorbed. Filled symbols denote zeolitic catalysts; the hollow symbol indicates the siliceous reference sample. Suffixes at H-MOR-40 samples indicate calcination temperature as discussed below. | ||
In accordance with the above described correlation between SAR and conversion, an increased amount of acidic sites seems to correlate to higher conversion for the zeolitic samples (Fig. 2a). It is also evident that amorphous silica is not active.
The NH3-TPD curves show relatively broad and/or flat signals. Therefore, a deconvolution into low- and high-temperature contributions was not performed.
When the OME yield is related to the total amount of acid sites (Fig. 2b), zeolites with a low acid site concentration seem to perform best. The highest OME yields of 42% and 43% are achieved by H-MOR-40_350 and Silicalite-1, respectively. The latter is a siliceous zeolitic material that is characterized by the presence of only very weakly acidic silanol groups (not detected in NH3-TPD). The described high activity of Silicalite-1 is unexpected. Conventionally, classical Brønsted acid sites created by Si–OH–Al bridges or Lewis acid sites are thought to be responsible for the formation of OME. Since these are absent in Silicalite-1, another active site than hitherto thought must be responsible for the high activity of this catalyst. The amorphous silica used as a reference has no catalytic activity.
In order to substantiate the finding that Brønsted acid sites are not necessary to catalyze the formation of OME in the gas-phase, two Al-containing zeolite catalysts were transferred into their Na-exchanged form. In the catalytic tests, both materials showed a significantly improved performance (Fig. 3), resulting in an increase of OME yield of as high as 38% in case of the Na-MFI-27 zeolite.
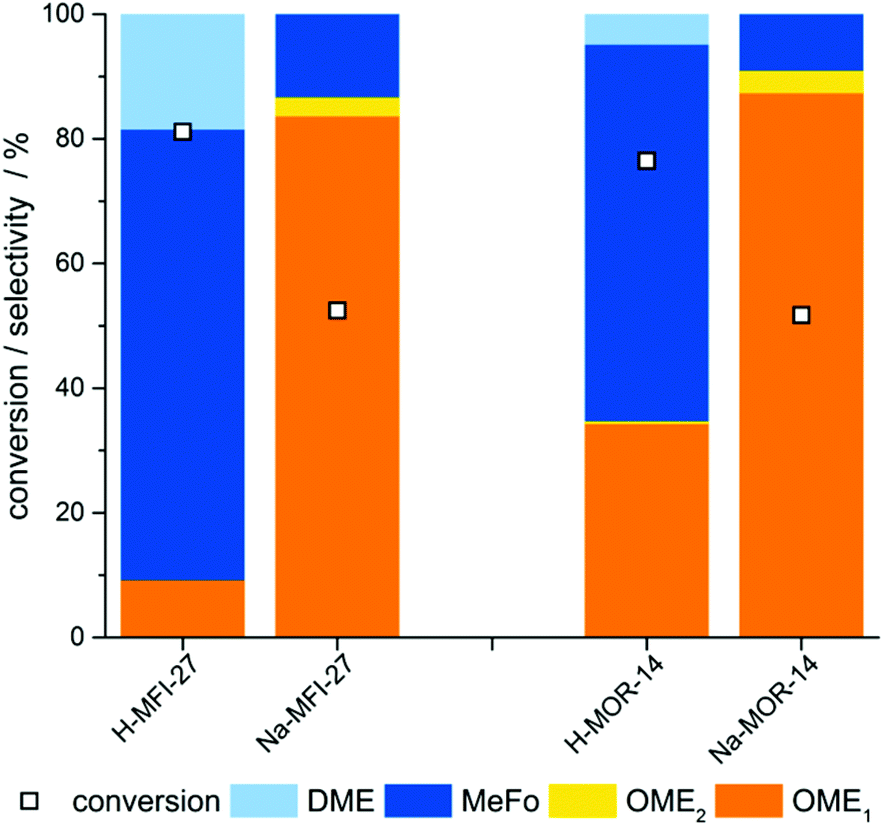 | ||
| Fig. 3 Initial selectivity and conversion of H- and Na-form zeolites determined in the interval of 40–70 min reaction time. Reaction conditions: 10 bar, 130 °C, 0.5 g of catalyst, 100 mL min−1 inert gas flow, 14 μL min−1 FA/MeOH solution feed, WHSV for formaldehyde: 1.1 g(FA) g(cat)−1 h−1. | ||
As mentioned above, the product ratio is influenced by the activity of the catalysts towards the irreversible formation of by-products (MeFo, DME). The observations that the formation of by-products is suppressed by Na-exchange in Al-containing zeolites and that Silicalite-1 shows high OME selectivity suggest that the presence of Brønsted acid sites may be related to by-product formation. Weakly acidic sites such as silanol-groups in framework defects or at pore mouths seem to provide sufficient acidity for the formation of OME. Besides, the presence of weakly Lewis acidic sodium ions in the framework does also not have an adverse impact on the OME selectivity.
When discussing the acidic properties of zeolites, it is also important to consider the influence of extra-framework aluminum (EFAl), which is typically characterized by Lewis acidity. The influence of the presence of EFAl on the formation of OME from MeOH and FA was exemplarily studied using H-MOR-40. In a series of H-MOR-40 material calcined at varying calcination temperatures, emergence of EFAl was induced at temperatures above 350 °C. This was evidenced by 27Al-MAS-NMR (Fig. S13†). The pristine H-MOR-40 shows mainly tetrahedrally-coordinated Al (signal centered at 57 ppm) and only little Al in octahedral environment (signal centered at 0 ppm). Upon temperature treatment, an increase in the asymmetric broadening of the signal related to tetrahedral framework indicates the formation of distorted tetrahedrally-coordinated and/or penta-coordinated Al. Furthermore, a rise in the peak at 0 ppm and the additional emergence of a broad peak centered at −5 ppm, assigned to various Al species in octahedral environment, indicate the removal of Al from the mordenite framework and the formation of Lewis acidic EFAl species.50
The change in the ratio of Brønsted- to Lewis-acidity as a result of EFAl formation was confirmed by Pyridine-FTIR measurements (Table 1 and Fig. S14†). As expected, a decrease in the ratio of Brønsted to Lewis acidity with increasing calcination temperature is observed at a constant level of Si/Al-ratio.
| Pyridine desorption temperature (°C) | C B (mmol g−1) |
C
La (mmol g−1) |
B/L | Si/Alb | |
|---|---|---|---|---|---|
| a Considering the band at 1455 cm−1. b Calculated at 150 °C. | |||||
| H-MOR-40_350 °C | 150 | 0.34 | 0.09 | 3.8 | 30 |
| 250 | 0.29 | 0.08 | 3.6 | ||
| 350 | 0.18 | 0.06 | 3.0 | ||
| H-MOR-40_450 °C | 150 | 0.30 | 0.11 | 2.7 | 30 |
| 250 | 0.25 | 0.09 | 2.8 | ||
| 350 | 0.14 | 0.07 | 2.0 | ||
| H-MOR-40_550 °C | 150 | 0.28 | 0.15 | 1.9 | 28 |
| 250 | 0.26 | 0.13 | 2.0 | ||
| 350 | 0.18 | 0.10 | 1.8 | ||
The effect of EFAl formation is reflected in the catalytic performance of H-MOR-40. A significant drop in OME selectivity was observed when calcination temperatures above 350 °C were employed (Fig. 4).
 | ||
| Fig. 4 Initial conversion and selectivity of H-MOR-40 as a function of calcination temperature. | ||
The H-MOR-40 samples were also characterized by NH3-TPD (Fig. S15†). An increased amount of ammonia desorbed in the high-temperature range of 500–700 °C is evident in the curves of samples calcined at 450 and 550 °C as compared to 350 °C suggesting upon temperature treatment, stronger acid sites were created. These could be due to strongly acidic EFAl sites and/or Brønsted acid sites with increased acidity due to interaction with EFAl.51 NH3-TPD analysis also supports that strongly acidic sites favor competing reactions leading to by-product formation.
In summary, one may conclude that three different acidic species in zeolites – namely Brønsted acid sites, Lewis acidic EFAl species as well as silanol groups – all affect the catalytic performance of the zeolites. This complex interplay of acidic sites along with the competition of OME formation with irreversible side-reactions render it difficult to exactly determine specific contributions of each type of acid site. However, the general conclusions can be drawn that catalysts characterized by a low number of Brønsted and/or EFAl acid sites show better performance and that weakly acidic species such as silanol groups are sufficient to catalyze the OME formation.
In order to rule out an effect of external surface area of the zeolites on the catalyst performance, external surface areas were determined from nitrogen sorption isotherms via t-plot analysis. When plotted against conversion (Fig. S16†) and OME yield (Fig. S17†), no clear correlation with the external surface area is evident.
2.3. Optimization and catalyst deactivation and regeneration
In order to further investigate the catalyst performance for OME gas-phase formation, the two best performing zeolites from the screening were tested under optimized conditions and the deactivation and regeneration behavior was studied.For both materials, an improved OME yield was achieved when the weight hourly space velocity (WHSV) was increased from 1.1 to 6.4 g(FA) g(cat)−1 h−1 by adapting reactant mass flow as well as reactant partial pressure (Fig. 5, left). Experimentally, a further increase of WHSV was limited by the saturation pressure of reactants as well as a limit of gas flows that can be handled within the set-up.
 | ||
| Fig. 5 Left: Initial selectivity and conversion of H-MOR-40 and Silicalite-1 at increased WHSV and reactant partial pressure determined in the interval of 60–90 min reaction time. Reaction conditions: 10 bar, 130 °C, 1 g of catalyst, 400 mL min−1 inert gas flow, 168 μL min−1 FA/MeOH solution feed. WHSV for formaldehyde: 6.4 g(FA) g(cat)−1 h−1. Center: TG-MS curve of Silicalite-1 measured in argon. Right: Initial selectivity and conversion determined in the interval of 1–3 h reaction time of fresh samples and of regenerated samples. Reaction conditions: 10 bar, 130 °C, 0.5 g of catalyst, 100 mL min−1 inert gas flow, 14 μL min−1 FA/MeOH solution feed. WHSV for formaldehyde: 1.1 g(FA) g(cat)−1 h−1. | ||
Under the mentioned conditions, total OME selectivity reaches 95% at a conversion of 49% (H-MOR-40) or 47% (Silicalite-1) and, in contrast to screening conditions, OME3 was detected. Notably, trioxane is also observed as a by-product. However, the amount of trioxane formed decreases strongly within the first 60 min reaction time and subsequently shows a stable level. Initial conversion and selectivity under optimized conditions was therefore determined at 60–90 min reaction time.
Whereas catalytic properties of Silicalite-1 and H-MOR-40 with regards to conversion and product distribution are very similar, a difference is observed in deactivation behavior. In tests that were performed under the same reaction conditions as the catalyst screening, deactivation proceeded much slower for H-MOR-40 than for Sillicalite-1 (Fig. S18†). The deactivation onset was defined as the time at which conversion has decreased to 85% of the steady-state conversion level. Deactivation experiments were repeated three times and the average deactivation onset time was determined to be 38.3 h for H-MOR-40 and 11.1 h for Silicalite-1 with a broader spread of data in case of H-MOR-40 compared to Silicalite-1. It has to be noted that after the defined deactivation onset, the conversion drops with a smaller slope in case of H-MOR-40 as compared to Silicalite-1.
Several factors can affect the starting point of deactivation. For example, the deactivation mechanism will have a major impact on the deactivation behavior of the catalyst. As the formation of OME is a chain growth reaction, formation of higher, non-volatile OME homologues in small quantities is expected and could lead to a surface, pore or active site blocking of the catalyst. In the TG-MS curve of Silicalite-1 measured in an inert gas stream (Fig. 5, center), the release of FA and MeOH along with CO2 and H2O in the range of 170–350 °C is evident. Similar data is obtained when measured in a stream of air (Fig. S19†). For H-MOR-40, the mass loss occurs in several stages, but also in this case, the release of the starting materials FA and MeOH along with CO2 and H2O is observed (Fig. S20 and S21†).
The release of FA and MeOH can either be related to a release of monomeric FA and MeOH from the pores and/or active sites or to the presence and decomposition of non-volatile OME homologues or other non-volatile FA-containing species such as paraformaldehyde. When pore or surface blocking is discussed as possible deactivation mechanism, several factors can be considered effective to result in the differences in deactivation onset between Silicalite-1 and H-MOR-40. The two samples have a pronounced difference in crystallite sizes and size distribution (Silicalite-1: approx. 42 × 8 μm, H-MOR-40 large size distribution with an average of about 0.15 μm). The smaller external surface area of the Silicalite-1 could result in a faster blocking of the surface or pore entrances. A further parameter possibly influencing the deactivation behavior is the difference in diameters of the micropores (ring size of largest channel: 12 (MOR) vs. 10 (MFI); computed as 6.45 Å for MOR vs. 4.7 Å for MFI).52
Both catalysts could successfully be regenerated: Silicalite-1 was calcined in air at 550 °C to restore activity. For H-MOR-40, such a treatment would be too harsh and result in decreased OME selectivity (vide supra), and so the mordenite sample was regenerated in inert gas flow at 350 °C (Fig. 5, right). Whether such a treatment would also be sufficient for the Silicalite-1 was not explored.
2.4 Comparison of siliceous materials
As mentioned above, the amorphous silica reference material (Aerosil 200) was found to be inactive for OME synthesis, while Silicalite-1 (crystalline zeolite with MFI structure) is one of the best performing catalysts in this study. In order to study the difference between the two siliceous materials, a FTIR-DRIFTS adsorbate study was performed.For spectra of activated samples see Fig. S22 and S23.† The pristine Aerosil 200 shows only isolated silanol groups [3746 cm−1].53 Signals in the IR spectrum of Silicalite-1 can be attributed to unperturbed internal silanol groups [3723 and 3675 cm−1]54 and H-bonded internal silanol groups and silanol groups interacting with water [broad signal at 3000–3600 cm−1]. No isolated external silanols are observed, which can be attributed to the large dimensions of the Silicalite-1 crystals that feature a very low external surface area compared to the bulk volume. At the activation temperature, which is the maximal temperature achievable in the DRIFTS set-up, water is not completely removed as evident from the presence of a signal at 1634 cm−155 and the broadness of the peak at 3000–3600 cm−1. A harsher treatment to completely remove water was not applied as water being a by-product of OME formation will also always be present under reaction conditions.
After exposure of samples to FA and MeOH vapor, no additional signals could be observed in case of Aerosil 200 (Fig. 6). The signal related to external silanol groups shows decreased intensity indicating that there is interaction with adsorbed species. As the reactant molecules do not seem to adsorb on the Aerosil 200 surface, this decrease might be assigned to adsorption of additional water molecules. In case of Silicalite-1, a distinct pattern of signals in the range 2770–3000 cm−1 and signals at 1449, 1465 and 1475 cm−1 appear upon adsorption of the vapor containing FA and MeOH. Notably, the spectrum after adsorption of OME1 shows the same features. The OME1 features agree well with literature data (cf. liquid OME1 spectrum56). As a reference, pure MeOH was adsorbed on Silicalite-1. In the considered range, signals at 2950 and 2846 cm−1 are present in the difference spectrum after adsorption. Considering IR data of formaldehyde from literature [NIST database: 2785, 2850 and 2995 cm−1],60 the pattern arising after exposure to FA and MeOH vapour cannot be explained by a superposition of FA and MeOH signals. We assume that the reactants FA and MeOH have already reacted to OME1 at 40 °C. This is in good agreement with reports from literature describing liquid phase OME synthesis at temperatures as low as 50 °C.8
 | ||
| Fig. 6 Difference spectra of Aerosil 200 and Silicalite-1 after adsorption of probe molecules. | ||
At this point, a clear assignment of activity to certain silanol species in Silicalite-1 is difficult. In case of the Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam, for which Silicalite-1 is also highly active and selective, internal silanol nests as well as external silanol groups are discussed to be the active species.57,58 The IR-data obtained in this study does not allow such a straightforward interpretation as IR signals for silanol nests are not well resolved due to the presence of water and as a decrease in signal intensity upon adsorption could only be assigned to unperturbed internal silanol groups.
From the FTIR-DRIFTS adsorbate study, a clear difference in the adsorption behavior of Silicalite-1 compared to amorphous silica was shown. We assume that the high adsorption potential as present in micropores of the crystalline zeolite may be a key factor for activity in OME synthesis.
3. Conclusions
In summary, a broad range of zeolites were tested in the gas-phase synthesis of OME from methanol and formaldehyde. It was demonstrated that catalysts characterized by a low number of Brønsted acid sites and/or EFAl show a better performance and that very weakly acidic species such as silanol groups can catalyze OME formation with a lower tendency for by-product formation than strong acid sites.With respect to catalytic activity, Silicalite-1 and H-MOR-40 showed the best performance. Both catalysts allow producing OME with selectivity as high as 95%. A deactivation study showed that H-MOR-40 features increased long-term stability compared to the all-silica material Silicalite-1, while both catalysts could be fully regenerated by thermal treatment.
This study provides insights into OME gas-phase synthesis from formaldehyde and methanol without any further solvents and shows that OME can be produced in high selectivity over zeolites. For application of OME as diesel additives, the need to increase the yield of the oligomers OME3–5 in the presented gas-phase reaction can be addressed by advanced process-technology such as additional units for water removal to shift the acetalization equilibrium and recycling of OME with undesired chain length. On a longer perspective, the presented process step could be coupled with well-established methanol technology to form a complete gas-phase route towards OME fuels.
4 Experimental
4.1 Materials
4.2 Set-up
The catalytic tests were carried out in a set-up comprising an evaporator unit for the reactant mixture feed, a plug-flow reactor and a gas chromatograph for online-analysis (Fig. S5†). For catalytic tests, a packed bed of catalyst pellets (300–400 μm) diluted with silicon carbide powder (Alpha Aesar, 46 grit, mass ratio of catalyst to SiC: 1:6) was prepared between plugs of quartz wool inside the reactor tube (5.8 mm inner diameter).
The reactant mixture was obtained by refluxing 120 g paraformaldehyde (prilled, Sigma) in 100 mL methanol (Honeywell Riedel-de-Haën) for 24 h. Subsequently, the solution was cooled to room temperature and filtered. A solution with a composition of 60 wt% FA, 38 wt% MeOH and 2 wt% H2O was obtained.
4.3 Analysis
Gas-phase samples were analyzed using an Agilent 6890N gas chromatograph with a DB-WAX capillary column equipped with a TCD and FID detector. Methane was used as an internal standard and was fed into the set-up in form of a 5% CH4/N2 mixture used as carrier gas. Gases were supplied by Air Liquide.For calibration, response factors of pure components OME1, OME3, OME4, trioxane (TRI) and methyl formate (MeFo) with respect to methanol were identified by manually injecting pure components. In order to obtain response factors for OME2 and OME>4, the area/mole ratio was extrapolated. In the gas-phase, response factors of MeOH, OME1 and OME3 with respect to the internal standard CH4 were determined by evaporation and analysis of a known liquid feed supplied by a calibrated HPLC pump. Ratios of the response factors of MeOH, OME1 and OME3 in the gas-phase coincided with ratios determined via liquid phase injections, which allowed translating response factors of further OME oligomers, trioxane and MeFo to methane-based response factors. DME was calibrated using a 5% DME/N2 calibration gas.
Formaldehyde was calibrated by evaporation and analysis of a known liquid feed of a methanolic formaldehyde solution supplied by a calibrated HPLC pump. The formaldehyde content of the FA/MeOH solution was determined by iodometry and the water content by Karl-Fischer titration. In the obtained chromatograms, FA, MeOH and water could be well resolved. In addition, a peak assigned to the hemiacetal of methanol and formaldehyde is observed. Due to difficulty in calibration of the hemiacetal and the marginal amount detected, it was not considered in the evaluation of GC results. This may result in a minor systematic undervaluation of reactant concentrations, which is, however, not expected to significantly influence conversion and selectivity data. In contrast to liquid-phase systems, methylene glycol and longer chain hemiformals are not detected.
The reproducibility of catalytic tests was evaluated by 5-fold repetition of a test run with following reaction conditions: 10 bar, 130 °C, 0.5 g H-MOR-40, 100 mL min−1 inert gas flow, 14 μL min−1 FA/MeOH solution feed. The deviation of the arithmetic mean of obtained conversion and selectivity results was below 3%.
In all test screening test runs, a quasi-plateau of product streams was reached after 40 minutes (Fig. S7†). Initial conversion and initial selectivity presented in the publication are averaged over data from four consecutive GC runs in this regime (ca. 40–70 minutes reaction time). Selectivity comprises only C-containing products as water is not quantified. The conversion is calculated as an average of the conversion of both reactants, FA and MeOH. This value is used as a simple, lumped indicator for catalytic activity. Due to the spectrum of possible products with correspondingly different consumption of both reagents, calculation of conversion normalized to one of the reagents could give a misleading impression. For a full assessment of catalyst performance, conversion and product selectivities should be considered together.
Within the range of reaction conditions that could be applied in the test set-up, conversion levels of different catalysts could not be adapted to a sufficient extent in order to compare all catalysts at the same conversion level. Therefore, catalyst performance was chosen to only be compared at identical reaction conditions.
4.4 Catalyst characterization
For the H-MOR-40 samples, a milder activation procedure was applied: 100 mg of catalyst were activated at 350 °C for 5 h (heating ramp of 2 °C min−1) and then cooled to 150 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Max Planck Society and the Fonds der Chemischen Industrie (FCI) for financial support and B. Zibrowius for Al-MAS-NMR spectroscopy.References
- I. P. o. C. C. (IPCC), Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, New York, 2014 Search PubMed.
- S. Deutz, D. Bongartz, B. Heuser, A. Kätelhön, L. Schulze Langenhorst, A. Omari, M. Walters, J. Klankermayer, W. Leitner, A. Mitsos, S. Pischinger and A. Bardow, Energy Environ. Sci., 2018, 11, 331–343 RSC.
- D. Pélerin, K. Gaukel, M. Härtl and G. Wachtmeister, in Internationaler Motorenkongress 2017, ed. J. Liebl and C. Beidl, Springer Fachmedien Wiesbaden, 2017, pp. 439–456 Search PubMed.
- M. Härtl, P. Seidenspinner, E. Jacob and G. Wachtmeister, Fuel, 2015, 153, 328–335 CrossRef.
- L. Pellegrini, M. Marchionna, R. Patrini and S. Florio, Emission Performance of Neat and Blended Polyoxymethylene Dimethyl Ethers in an Old Light-Duty Diesel Car, SAE Technical Paper 2013-01-1035, 2013, DOI:10.4271/2013-01-1035.
- B. Lumpp, MTZ worldwide, 2011, 72, 34–38 CrossRef.
- L. Lautenschütz, D. Oestreich, P. Haltenort, U. Arnold, E. Dinjus and J. Sauer, Fuel Process. Technol., 2017, 165, 27–33 CrossRef.
- J. Burger, E. Ströfer and H. Hasse, Ind. Eng. Chem. Res., 2012, 51, 12751–12761 CrossRef CAS.
- J. Cao, H. Zhu, H. Wang, L. Huang, Z. Qin, W. Fan and J. Wang, Ranliao Huaxue Xuebao, 2014, 42, 986–993 CAS.
- W. H. Fu, X. M. Liang, H. Zhang, Y. M. Wang and M. Y. He, Chem. Commun., 2015, 51, 1449–1452 RSC.
- S. Heiner, S. Eckhard, P. Rolf, H. Andrea, T. Gerd-Dieter, H. Hans and B. Sergej, Method for producing polyoxymethylene dimethyl ethers from methylal and trioxane in the presence of an acidic catalyst, Basf Aktiengesellschaft, Germany, WO2006045506A1, 2006 Search PubMed.
- J. Wu, H. Zhu, Z. Wu, Z. Qin, L. Yan, B. Du, W. Fan and J. Wang, Green Chem., 2015, 17, 2353–2357 RSC.
- Q. Wu, M. Wang, Y. Hao, H. Li, Y. Zhao and Q. Jiao, Ind. Eng. Chem. Res., 2014, 53, 16254–16260 CrossRef CAS.
- Y. Wu, Z. Li and C. Xia, Ind. Eng. Chem. Res., 2016, 55, 1859–1865 CrossRef CAS.
- Z. Xue, H. Shang, C. Xiong, C. Lu, G. An, Z. Zhang, C. Cui and M. Xu, RSC Adv., 2017, 7, 20300–20308 RSC.
- Z. Xue, H. Shang, Z. Zhang, C. Xiong, C. Lu and G. An, Energy Fuels, 2017, 31, 279–286 CrossRef CAS.
- Z. Yang, Y. Hu, W. Ma, J. Qi and X. Zhang, Chem. Eng. Technol., 2017, 40, 1784–1791 CrossRef CAS.
- F. Liu, T. Wang, Y. Zheng and J. Wang, J. Catal., 2017, 355, 17–25 CrossRef CAS.
- Y. Liu, Y. Wang and W. Cai, Trans. Tianjin Univ., 2018 DOI:10.1007/s12209-018-0131-0.
- G.-F. Shi, J. Miao, G.-Y. Wang, J.-M. Su and H.-X. Liu, Asian – J. Chem., 2015, 27, 2149–2153 CrossRef CAS.
- Y. Zhao, Z. Xu, H. Chen, Y. Fu and J. Shen, J. Energy Chem., 2013, 22, 833–836 CrossRef CAS.
- Y. Zheng, Q. Tang, T. Wang, Y. Liao and J. Wang, Chem. Eng. Technol., 2013, 36, 1951–1956 CrossRef CAS.
- M. Ouda, G. Yarce, R. J. White, M. Hadrich, D. Himmel, A. Schaadt, H. Klein, E. Jacob and I. Krossing, React. Chem. Eng., 2017, 2, 50–59 RSC.
- N. Schmitz, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2015, 54, 12553–12560 CrossRef CAS.
- N. Schmitz, F. Homberg, J. Berje, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2015, 54, 6409–6417 CrossRef CAS.
- D. Oestreich, L. Lautenschütz, U. Arnold and J. Sauer, Chem. Eng. Sci., 2017, 163, 92–104 CrossRef CAS.
- M. Shi, X. Yu, L. Wang, F. Dai, G. He and Q. Li, Kinet. Catal., 2018, 59, 255–261 CrossRef CAS.
- J. Zhang, D. Fang and D. Liu, Ind. Eng. Chem. Res., 2014, 53, 13589–13597 CrossRef CAS.
- J. Zhang and D. Liu, Int. J. Energy Res., 2018, 42, 1237–1246 CrossRef CAS.
- B. G. Schieweck and J. Klankermayer, Angew. Chem., Int. Ed., 2017, 56, 10854–10857 CrossRef CAS PubMed.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed.
- S. Chen, S. Wang, X. Ma and J. Gong, Chem. Commun., 2011, 47, 9345–9347 RSC.
- Y. Fu and J. Shen, Chem. Commun., 2007, 2172–2174, 10.1039/b618898b.
- H. Guo, D. Li, D. Jiang, W. Li and Y. Sun, Catal. Commun., 2010, 11, 396–400 CrossRef CAS.
- X. Lu, Z. Qin, M. Dong, H. Zhu, G. Wang, Y. Zhao, W. Fan and J. Wang, Fuel, 2011, 90, 1335–1339 CrossRef CAS.
- O. A. Nikonova, M. Capron, G. Fang, J. Faye, A.-S. Mamede, L. Jalowiecki-Duhamel, F. Dumeignil and G. A. Seisenbaeva, J. Catal., 2011, 279, 310–318 CrossRef CAS.
- N. T. Prado, F. G. E. Nogueria, A. E. Nogueira, C. A. Nunes, R. Diniz and L. C. A. Oliveira, Energy Fuels, 2010, 24, 4793–4796 CrossRef CAS.
- J.-M. Tatibouët and H. Lauron-Pernot, J. Mol. Catal. A: Chem., 2001, 171, 205–216 CrossRef.
- K. Thavornprasert, M. Capron, L. Jalowiecki-Duhamel, O. Gardoll, M. Trentesaux, A.-S. Mamede, G. Fang, J. Faye, N. Touati, H. Vezin, J.-L. Dubois, J.-L. Couturier and F. Dumeignil, Appl. Catal., B, 2014, 145, 126–135 CrossRef CAS.
- E. Zhan, Y. Li, J. Liu, X. Huang and W. Shen, Catal. Commun., 2009, 10, 2051–2055 CrossRef CAS.
- M. Li, Y. Long, Z. Deng, H. Zhang, X. Yang and G. Wang, Catal. Commun., 2015, 68, 46–48 CrossRef CAS.
- H. Zhao, S. Bennici, J. Shen and A. Auroux, J. Catal., 2010, 272, 176–189 CrossRef CAS.
- X.-J. Gao, W.-F. Wang, Y.-Y. Gu, Z.-Z. Zhang, J.-F. Zhang, Q.-D. Zhang, N. Tsubaki, Y.-Z. Han and Y.-S. Tan, ChemCatChem, 2018, 10, 273–279 CrossRef CAS.
- Q. Zhang, W. Wang, Z. Zhang, Y. Han and Y. Tan, Catalysts, 2016, 6, 43 CrossRef.
- Q. Zhang, Y. Tan, G. Liu, C. Yang and Y. Han, J. Ind. Eng. Chem., 2014, 20, 1869–1874 CrossRef CAS.
- G. Bozzano and F. Manenti, Prog. Energy Combust. Sci., 2016, 56, 71–105 CrossRef.
- N. Y. Usachev, I. M. Krukovskii and S. A. Kanaev, Pet. Chem., 2004, 44, 379–394 Search PubMed.
- X. Gao, W. Yang, Z. Liu and H. Gao, Cuihua Xuebao, 2012, 33, 1389–1394 CrossRef CAS.
- Q. Zhao, H. Wang, Z. Qin, Z. Wu, J. Wu, W. Fan and J. Wang, J. Fuel Chem. Technol., 2011, 39, 918–923 CrossRef CAS.
- T.-H. Chen, K. Houthoofd and P. J. Grobet, Microporous Mesoporous Mater., 2005, 86, 31–37 CrossRef CAS.
- A. Corma and H. García, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS PubMed.
- http://www.iza-structure.org/databases/ , (accessed 22.06.2018).
- R. S. McDonald, J. Am. Chem. Soc., 1957, 79, 850–854 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. Spoto, L. Marchese, G. Petrini, G. Leofanti and M. Padovan, J. Phys. Chem., 1992, 96, 4985–4990 CrossRef CAS.
- K. Vikulov, G. Martra, S. Coluccia, D. Miceli, F. Arena, A. Parmaliana and E. Paukshtis, Catal. Lett., 1996, 37, 235–239 CrossRef CAS.
- J. K. Wilmhurst, Can. J. Chem., 1958, 36, 285–289 CrossRef.
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev.: Sci. Eng., 2001, 43, 381–441 CrossRef CAS.
- C. Flego and L. Dalloro, Microporous Mesoporous Mater., 2003, 60, 263–271 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- https://webbook.nist.gov/cgi/cbook.cgi?ID=C50000&Type=IR-SPEC&Index=1#IR-SPEC , accessed on 22.06.2018.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc02617c |
| This journal is © The Royal Society of Chemistry 2018 |