
Direct conversion of shrimp shells to O-acylated chitin with antibacterial and anti-tumor effects by natural deep eutectic solvents†
Mi
Feng
ab,
Xingmei
Lu
 *ab,
Jie
Zhang
b,
Yi
Li
c,
Chunyan
Shi
b,
Lingling
Lu
c and
Suojiang
Zhang
*ab
*ab,
Jie
Zhang
b,
Yi
Li
c,
Chunyan
Shi
b,
Lingling
Lu
c and
Suojiang
Zhang
*ab
aCAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Multiphase Complex Systems, Beijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: xmlu@ipe.ac.cn; sjzhang@ipe.ac.cn; Fax: +86-010-82544800; Tel: +86-010-82544800
bSchool of Chemical and Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
cCenter of Parkinson's Disease, Beijing Institute for Brain Disorders, Department of Neurobiology, Beijing Center of Neural Regeneration and Repair, Key Laboratory for Neurodegenerative Diseases of the Ministry of Education, Capital Medical University, Beijing 100054, China
First published on 9th October 2018
Abstract
To obtain O-acylated chitin from shrimp shells directly, the used solvent should have multiple functions to remove calcium carbonate, protein, and acylated chitin. Herein, we use the acidic natural deep eutectic solvents (NADESs) with the ability to release H+ and various hydrogen bonding sites to achieve the above goal. The involved NADESs with three abilities of decalcification, deproteinization and acylation replaced acids, alkalis, catalysts, and acylation reagents in the conventional method. The experimental results revealed that the components of the NADESs, experiment temperature and time played key roles in the purity and degree of substitution (DS) of O-acylated chitin. Meanwhile, the ratio of shrimp shells to NADESs and a small amount of water had little effect on the preparation of O-acylated chitin. With the optimal NADES (choline chloride/DL-malic acid 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, ChCl 1–DL Mal 2) treatment under the optimal conditions, the purity of O-malate chitin reached 98.6% with a DS of 0.46, exhibiting antibacterial and anti-tumor effects. The experimental results showed that the removal of calcium carbonate and protein and acylation of chitin were carried out simultaneously. Mechanistic exploration using spectroscopy and experiments confirmed that H+ release from ChCl 1–DL Mal 2 was the main reason for the removal of calcium carbonate and initiation acylation reaction. The protein was degraded to amino acids because of the acidity of ChCl 1–DL Mal 2 and dissolved in the NADES due to hydrogen bonding formation with ChCl 1–DL Mal 2.
:2, ChCl 1–DL Mal 2) treatment under the optimal conditions, the purity of O-malate chitin reached 98.6% with a DS of 0.46, exhibiting antibacterial and anti-tumor effects. The experimental results showed that the removal of calcium carbonate and protein and acylation of chitin were carried out simultaneously. Mechanistic exploration using spectroscopy and experiments confirmed that H+ release from ChCl 1–DL Mal 2 was the main reason for the removal of calcium carbonate and initiation acylation reaction. The protein was degraded to amino acids because of the acidity of ChCl 1–DL Mal 2 and dissolved in the NADES due to hydrogen bonding formation with ChCl 1–DL Mal 2.
Introduction
The global production of shrimp is about 6–8 million metric tons annually generating a tremendous amount of waste shells.1 Continuous production of these waste shrimp shells without the development of utilization methods causes serious environment problems and large resource waste.2 Fortunately, the shrimp shells contain calcium carbonate (25–50%), protein (35–50%), and chitin (15–25%), which can be transformed to various useful substances or materials.1,3–8 Among these bioactive substances, chitin has attracted more attention due to its many beneficial properties such as biocompatibility, biodegradability, bioactivity, and non-toxicity,9,10 and can be applied in numerous fields such as biomedicine,4,11–13 agriculture,14–16 water treatment17–19 and cosmetics.20 Moreover, chitin derivatives including deacylated,21,22 acylated,23–25 thiolate,26 and grafted27–29 chitin have attracted more interest because of their high solubility and unique properties compared with chitin. In particular, the O-acylation of chitin, which usually occurs at the C3–OH and C6–OH sites of chitin (Fig. 1), gives a common but important derivative that exhibits good performance in wound healing,30,31 biomedical material preparation32–35 and drug carriers.36,37 Normally, acylation of chitin is easier as it occurs on C6–OH due to its lower steric hindrance.38,39 The O-acylated chitin shows huge potential for manufacturing biomedical materials and diverse industrial applications. Obviously, to obtain O-acylated chitin from shrimp shells, calcium carbonate and protein need to be removed and acyl groups need to be introduced.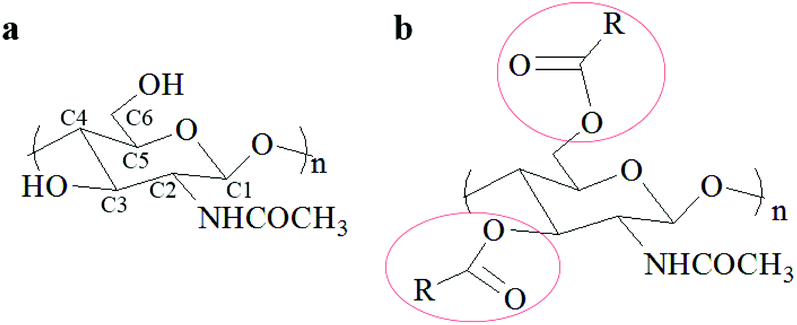 | ||
| Fig. 1 The structure of (a) chitin and (b) O-acylated chitin. | ||
However, the structure of shrimp shells is complex and compact. In the shrimp shells, the α-chitin nanofibrils are associated with protein to form long chitin protein fibers embedded in the calcium carbonate matrix. These chitin protein fibers are arranged parallelly to form horizontal planes, which are stacked and form a twisted plywood structure.40,41 It is difficult to use mild solvents for destroying the covalent chemical bonds between chitin and the protein to remove the protein and react with calcium carbonate to remove it. Additionally, the high purity O-acylated chitin is also hard to synthesize using shrimp shells as the starting material due to its complicated components and structure. Hence, the conventional approach for O-acylated chitin preparation needs three steps: firstly, the removal of calcium carbonate with dilute HCl aqueous solution; secondly, the removal of the protein with dilute NaOH aqueous solution at about 90 °C;42 thirdly, the reaction of the obtained chitin with carboxylic acid,43–45 anhydride23,46 or acyl chloride47,48 as the acylation reagent and methanesulfonic acid23,46 or trifluoroacetic anhydride (TFAA)/H3PO443,44,49 as the catalyst under homogeneous or heterogeneous conditions. The first and second steps were to extract chitin which has been considered harmful to the environment and to consume a huge amount of water. The third step was the acylation of chitin with an acid as the catalyst. Hence, to develop a one-step method for preparing O-acylated chitin using shrimp shells as the starting materials, we need to design a substance having the abilities of calcium carbonate removal, protein elimination, and acylation reaction initiation. Moreover, O-acylated chitin is usually used in biomaterials, so the designed substance is required to be nontoxic. This method is challenging but meaningful for achieving the goal of direct conversion of shrimp shells to O-acylated chitin, which will greatly reduce environmental pollution, and water and time consumption.
The use of NADESs provides a promising alternative to realize the direct preparation of O-acylated chitin from shrimp shells. NADESs from natural components, such as choline derivatives, organic acids, amino acids, or sugars, are considered as “readily biodegradable” solvents.50,51 They have been used in numerous fields, such as natural substance extraction,52–55 biotechnology,56–58 and organic reactions.59,60 Among the numerous NADESs, choline chloride (ChCl)-derived NADESs showed high solubility towards biopolymers, such as chitin,61,62 lignin,63–66 and proteins67,68 due to their hydrogen bonding network. Moreover, ChCl-organic acid NADESs could provide acidic conditions in acid-catalyzed reactions.69,70 Hence, ChCl-organic acid NADESs could be used as multifunctional solvents to remove calcium carbonate because of their acidity, and they dissolve protein through their hydrogen bonding network, and cause acylation reaction due to released H+, which would realize the direct conversion of shrimp shells to O-acylated chitin. What's more, NADESs are greener, lower cost and easier to synthesize on a large scale compared with ionic liquids,71 which is beneficial for industry application. It is obvious that the NADES-based O-acylated chitin preparation will provide a green, simple but effective approach for direct utilization of waste shrimp shells.
Herein, we used the acidic NADESs with the roles of decalcification, deproteinization and acylation to prepare O-acylated chitin from shrimp shells in one step. The acidic NADESs not only acted as solvents, but also as acylation reagents and catalysts in the process. The used acidic NADESs were synthesized by mixing choline chloride (ChCl) and organic acids including L-lactic acid, L-malic aicd, D-malic acid, DL-malic acid and citric acid in a fixed mole ratio. The impact of NADESs including the type and chirality of the organic acid, and the ratio between ChCl and malic acid, on the purity and DS of the O-acylated chitin was explored firstly. Subsequently, experimental conditions including temperature, time, ratio of NADES to shrimp shells, and the water content of NADES were investigated. Furthermore, the highest purity O-acylated chitin was characterized completely and tested against Gram-positive bacteria Bacillus subtilis and C6-ceramide tumor cells. Finally, the mechanism of the process was preliminarily investigated by spectroscopy methods and blank experiments. The results demonstrated that the released H+ reacted with calcium carbonate to remove it and caused the acylation reaction to produce the O-acylated chitin. Meanwhile, the protein was removed by dissolution and degradation due to the acidity and the hydrogen-bond interaction with ChCl 1–DL Mal 2.
Experimental
Materials
The shells of red shrimp (Solenocera crassicornis) from a Walmart Supermarket were cleaned with water, freeze-dried, ground and sieved, and they were composed of 56.1 wt% calcium carbonate, 8.1 wt% protein and 35.8 wt% chitin.The used choline chloride was from Xi Long Chemical Co. and dried before use. L-Lactic acid, D/L/DL-malic acid and citric acid were also purchased from Xi Long Chemical Co. and used without further purification. NADESs were prepared by mixing choline chloride (hydrogen-bond acceptor, HBA) and organic acids (hydrogen-bond donor, HBD) in a fixed mole ratio at 80 °C until a transparent and homogeneous liquid was formed. Then the NADESs were stored in a desiccator before use. The structures of HBA and HBD are shown in Fig. 2. The pH values of the NADESs were measured with a short range pH test paper. The list of NADESs is shown in Table 1. The NADESs were characterized by FT-IR (as shown in Fig. S1†).
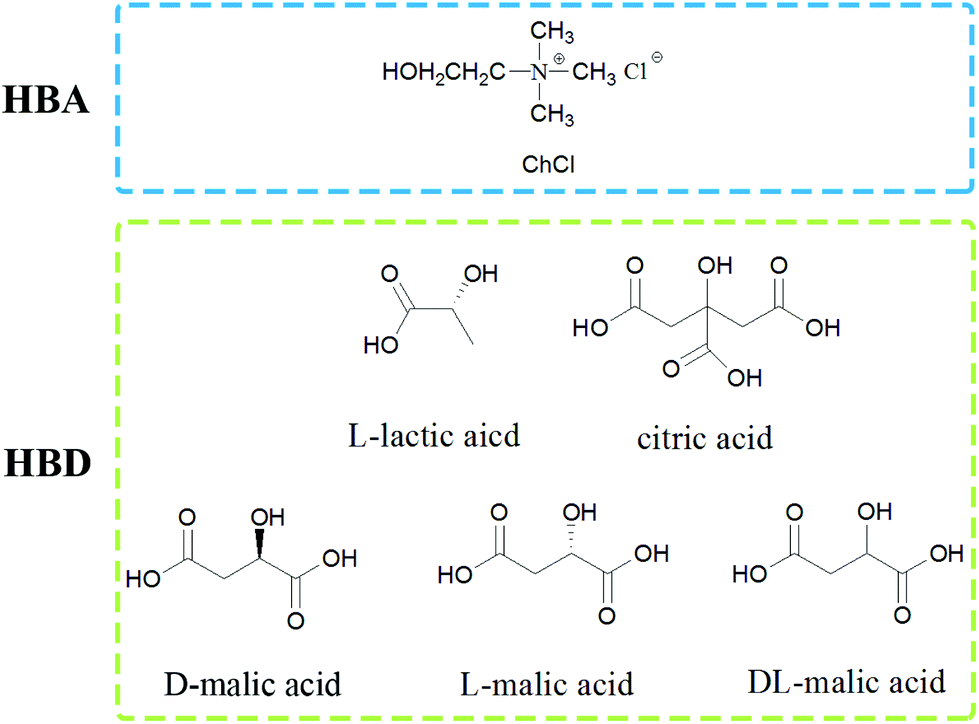 | ||
| Fig. 2 The structure of the used HBA and HBD. | ||
| Abbreviation | HBA | HBD | Mole ratio (HBA:HBD) |
|---|---|---|---|
| ChCl 1–L Lac 2 | Choline chloride | L-Lactic acid | 1:2 |
| ChCl 2–Citr 1 | Citric acid | 2:1 |
|
| ChCl 1–Citr 2 | Citric acid | 1:2 |
|
| ChCl 2–L Mal 1 | L-Malic acid | 2:1 |
|
| ChCl 1–D Mal 2 | L-Malic acid | 1:2 |
|
| ChCl 1–D Mal 2 | D-Malic acid | 1:2 |
|
| ChCl 1–DL Mal 1 | DL-Malic acid | 1:1 |
|
| ChCl 1–DL Mal 2 | DL-Malic acid | 1:2 |
|
| ChCl 1–DL Mal 3 | DL-Malic acid | 1:3 |
|
| ChCl 1–DL Mal 4 | DL-Malic acid | 1:4 |
|
| ChCl 1–DL Mal 5 | DL-Malic acid | 1:5 |
Preparation of O-acylated chitin from shrimp shells
The typical procedure of O-acylated chitin preparation from shrimp shells is as follows. The shrimp shells (0.25 g) and NADES (5 g) were added to a glass tube and mixed uniformly by magnetic stirring. The NADES/shrimp shell mixture was heated at 130 °C for 3 hours. After the experiment, dimethyl sulfoxide (DMSO) or water was added to the mixture to decrease the viscosity. Then the obtained mixture was separated by centrifugation to obtain a precipitate (P) and supernatant. The regenerated product (R) was obtained by adding water to the supernatant. The precipitate and the regenerated product were washed with water repeatedly and freeze dried. All the experiments in this work were carried out three times. To express briefly, the precipitates obtained from the NADES treatment were abbreviated as PChCl 1–L Lac 2, PChCl 2–Citr 1, PChCl 1–Citr 2, PChCl 2–L Mal 1, and PChCl 1–L Mal 2. Similarly, the regenerated products from different NADESs were denoted as RChCl 1–L Lac 2, RChCl 2–Citr 1, RChCl 1–Citr 2, PChCl 2–L Mal 1, and RChCl 1–L Mal 2.Recycling of ChCl 1–DL Mal 2
The recycling experiments were carried out as follows. After the experiment of O-malate chitin preparation, water was added to the ChCl 1–DL Mal 2/shrimp shell mixture. Then it was separated by centrifugation and the upper liquid was collected. The water in upper liquid was removed with rotary evaporation to obtain ChCl 1–DL Mal 2. However, a solid was formed during water removal, which was the dissolved protein and organic acid calcium in the ChCl 1–DL Mal 2. So, ethanol was added to the mixture to extract ChCl 1–DL Mal 2. The ethanol/ChCl 1–DL Mal 2 was obtained by centrifugation to remove the solid. Finally, the recycled ChCl 1–DL Mal 2 was obtained by rotary evaporation to removal of the ethanol. The recycled ChCl 1–DL Mal 2 was characterized by FT-IR. The recycled ChCl 1–DL Mal 2 was reused to prepare O-malate chitin with the same process as described above. ChCl 1–DL Mal 2 was used five times and the obtained O-malate chitin was analyzed. The recovery rate of ChCl 1–DL Mal 2 was calculated as follows,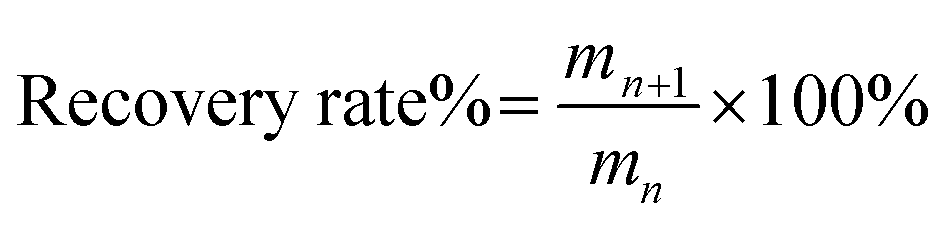 | (1) |
where mn is the weight of ChCl 1–DL Mal 2 used n times and mn+1 is the weight of ChCl 1–DL Mal 2 used n + 1 times. n = 1, 2, 3, 4, 5.
Content analysis of products
The content of CaCO3 in products was determined by using an inductively coupled plasma atomic emission spectrometer (ICPE-9000, Shimadzu, Japan). The samples were dissolved in HCl aqueous solution, and then the solution was diluted with water and analyzed with an ICPE-9000. The CaCO3 content of the sample was calculated as follows: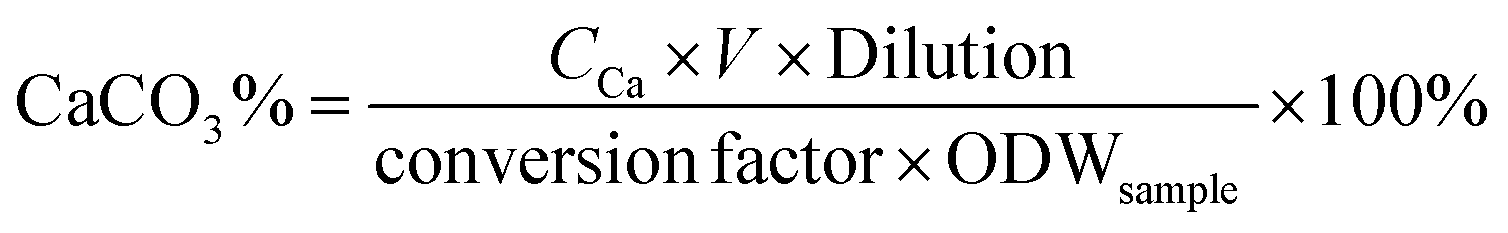 | (2) |
The protein content of the samples was measured by the Kieldahl method.72 Prior to analysis, 10 mg of sample was dissolved in 5 mL of 5 wt% NaOH for 1 h at 95 °C, followed by filtration to collect the liquid, which was subjected to protein determination. The amount of protein in the samples was calculated as follows:
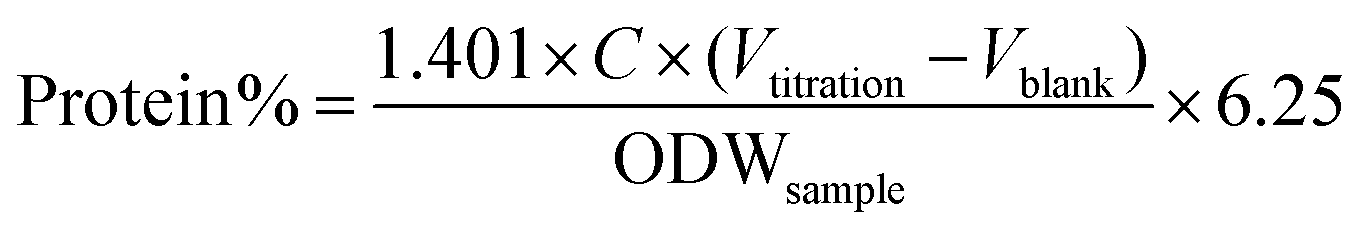 | (3) |
The obtained O-malate chitin under the optimal conditions was characterised by several methods as follows. Fourier transform-infrared (FT-IR) spectroscopy was conducted with a Nicolet 380 spectrometer (Thermo Fisher Scientific, America) using KBr as the blank in the range 4000–400 cm−1. Powder X-ray diffraction spectra (XRD) were recorded by using a SmartLab X-ray diffractometer (Rigaku Corporation, Japan) with Ni-filtered CuKα radiation (λ = 0.15418 nm) in the range 90–5° with a scanning rate of 10° min−1. Solid-state 13C nuclear magnetic resonance (13C NMR) was measured with an Avance III HD 500 (Bruker, Switzerland). X-ray photoelectron spectroscopy (XPS) measurements were conducted with an ESCALAB 250Xi (Thermo Fisher Scientific, America) with Al-K alpha as the radiation source (1486.6 eV). All spectra were calibrated with the binding energy of the C 1s peak at 284.8 eV. Survey and high-resolution spectra were acquired in CAE: pass energy mode with 100.0 and 20 eV, respectively. The assignments of each peak of the XPS spectrum resulted from the decomposition method used by the Thermo Avantage software based on the binding energies (BE) and atomic concentrations (AC, %). Ultraviolet (UV) spectrometry was carried out with a UV-2550 (SHIMADZU, Japan) in the range 400–800 nm. The content of amino acids was analyzed with a high performance liquid chromatograph (HPLC, Agilent, America, 1100) equipped with a UV detector (Pharmacia, Sweden, Ultrospec 2000). The chromatography conditions were as follows: the chromatography column was Zorbax 300 SB C18 [250 mm × 4.6 mm (I.D.), 5 μm]; mobile phase A (MP A) was water with 0.1% trifluoroacetic acid (TFA) and mobile phase B (MP B) was acetonitrile with 0.1% TFA; the eluent gradient was 5–60% MP B from 0 to 45 min and 60–90% MP B from 45 to 50 min; the rate of the mobile phase was 0.75 mL min−1; the wavelength of the UV detector was 280 nm. The pre-column derivatization was conducted with a NaHCO3 buffer solution and DNFB at 60 °C for 1 h, and then NaH2PO4 was added.
Antibacterial and antitumor activity test in vitro
The antibacterial activity test of O-malate chitin was evaluated against Escherichia coli (CCTCC AB 2012883) using a liquid culture method. Escherichia coli was pre-cultured in LB liquid medium at 37 °C with 180 rpm for 12 h and diluted with normal saline to 103 colony forming units (CFU) mL−1. Then O-malate chitin (0.025 g) was added to the bacterial suspensions (20 mL) and incubated on a HYG-A double-deck TAT rotary shaker at 180 rpm, 37 °C for 12 h. The control and blank groups were treated by adding chitin or nothing under the same conditions. The bacterial concentration of the suspension was detected by using a UV-1800 spectrophotometer (Aoe Instruments) at 600 nm. The inhibition rate (IR%) was calculated as follows: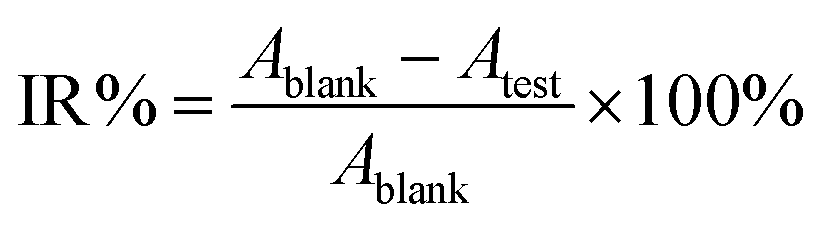 | (4) |
Rat C6 glioma cells (generously provided by Yi He, Anding Hospital, China, Beijing) were grown in DMEM/F12 medium containing 10% fetal bovine serum (FBS), 1 × GlutaMAX, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (all from Gibco, Carlsbad, CA, USA) at 37 °C with 5% CO2. Cell viability was determined with the MTT assay as previously described.73 Briefly, the cells were seeded in a 96-well microplate (1 × 104 cells per well) and cultured with or without O-malate chitin or chitin for 24 h. The medium was replaced by MTT (Sigma-Aldrich, M2128) at a final concentration of 0.5 mg ml−1 and incubated for 4 h subsequently. The cells were washed twice with PBS and the formazan crystals were dissolved in 100 ml DMSO. The absorbance of the above samples was read at 490 nm with a micro-plate reader (PerkinElmer, Waltham, MA, USA). The C6 glioma cell viability (V%) was calculated as follows,
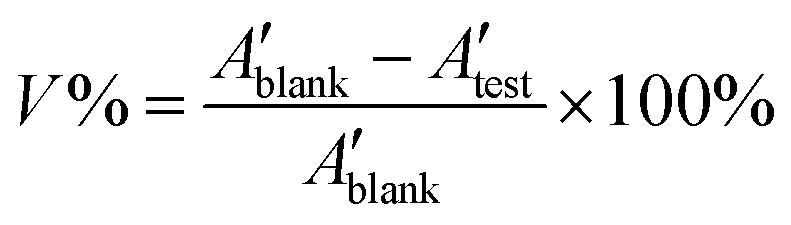 | (5) |
Results and discussion
The influence of NADES components on the O-acylated chitin preparation
To directly obtain the O-acylated chitin from shrimp shells, the used NADESs need to play three roles, namely decalcification, deproteinization and acylation. Hence, an organic acid was chosen as the HBD since calcium carbonate removal and acylation reaction were easy to carry out under acidic conditions. Meanwhile, the organic acid also acted as an acylation reagent in the acylation process. Moreover, the ChCl-based NADESs displayed high solubility towards proteins.67,68 Hence, five types of ChCl-organic acid NADESs were synthesized with ChCl and HBDs, including L-lactic acid, L-malic acid and citric acid. The five NADESs contained different amounts of COOH and OH groups leading to different pH values and molecular sizes (details in Table S1†), which may have different performances in the O-acylated chitin preparation.The O-acylated chitin preparation was carried out at 130 °C for 3 h with a mass ratio of 1:20 (shrimp shells:NADESs). DMSO was used as the solvent to dilute the NADES/shrimp shell mixture before centrifugation. Previously, the acidic ChCl-based NADESs were used to extract chitin from shrimp shells by selective dissolution of chitin or removal of protein and calcium carbonate.74 So, the main content of the precipitate and regenerated product was preliminarily determined by FT-IR spectroscopy, which could reveal the different performances of the five NADESs on calcium carbonate, protein and chitin in the shrimp shells.
As shown in Fig. 3a, the characteristic peaks of PChCl 2–Citr 1 and PChCl 2–L Mal 1 were basically the same with α-chitin except for peaks at 1665 and 1614 cm−1 ascribed to amide I, indicating that they were raw chitin due to the residual protein content. However, an additional peak at 1738 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the ester appeared in the spectra of PChCl 1–L Lac 2, PChCl 1–L Mal 2 and PChCl 1–Citr 2, indicating that the products were O-acylated chitin. The rest of the peaks of the three precipitates were well in accordance with α-chitin, proving that the functional groups and hydrogen network were not appreciably broken. The results revealed that the five NADESs could remove the calcium carbonate and protein, simultaneously. However, the heterogeneous acylation of chitin was only carried out when the mole ratio of the organic acid and ChCl was up to 2:1. Importantly, the spectra of RChCl 1–L Lac 2, RChCl 2–L Mal 1, RChCl 1–L Mal 2, RChCl 2–Citr 1, and RChCl 1–Citr 2 (Fig. 3b) all contained the characteristic peak of CO. All the dissolved chitin in the five NADESs was acylated since homogeneous acylation was easier to carry out than the heterogeneous reaction.
O group of the ester appeared in the spectra of PChCl 1–L Lac 2, PChCl 1–L Mal 2 and PChCl 1–Citr 2, indicating that the products were O-acylated chitin. The rest of the peaks of the three precipitates were well in accordance with α-chitin, proving that the functional groups and hydrogen network were not appreciably broken. The results revealed that the five NADESs could remove the calcium carbonate and protein, simultaneously. However, the heterogeneous acylation of chitin was only carried out when the mole ratio of the organic acid and ChCl was up to 2:1. Importantly, the spectra of RChCl 1–L Lac 2, RChCl 2–L Mal 1, RChCl 1–L Mal 2, RChCl 2–Citr 1, and RChCl 1–Citr 2 (Fig. 3b) all contained the characteristic peak of CO. All the dissolved chitin in the five NADESs was acylated since homogeneous acylation was easier to carry out than the heterogeneous reaction.
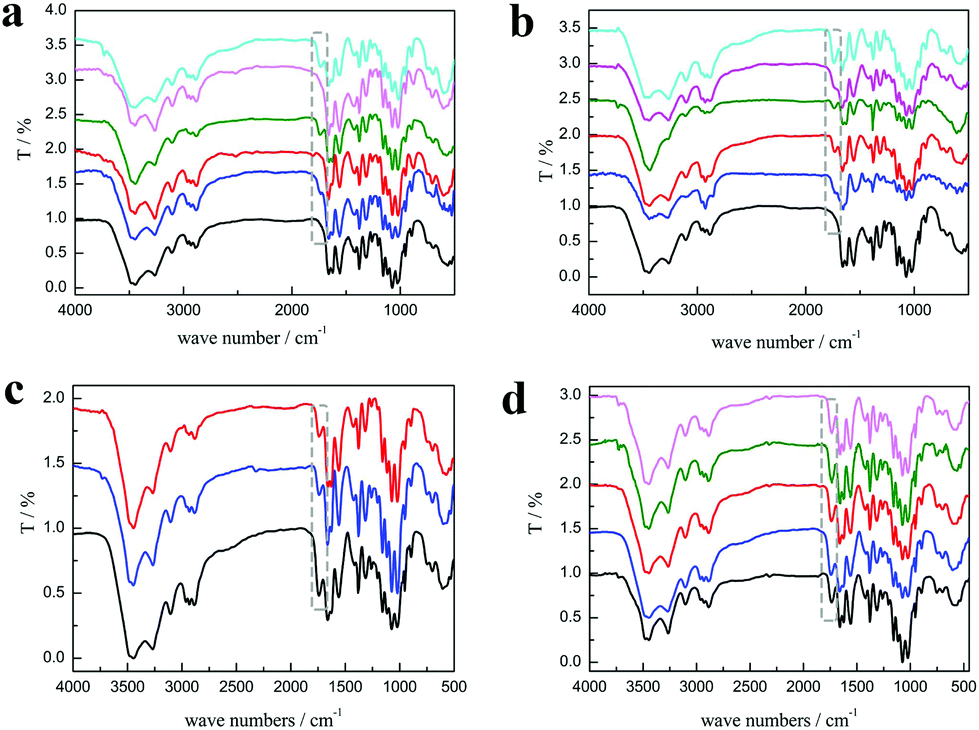 | ||
| Fig. 3 The FT-IR spectra of (a) α-chitin (black), PChCl 1–L Lac 2 (blue), PChCl 2–L Mal 1 (red), PChCl 1–L Mal 2 (olive), PChCl 2–Citr 1 (LT magenta), and PChCl 1–Citr 2 (cyan) (from down to up). (b) α-chitin (black), RChCl 1–L Lac 2 (blue), RChCl 2–L Mal 1 (red), RChCl 1–L Mal 2 (olive), RChCl 2–Citr 1 (LT magenta), and RChCl 1–Citr 2 (cyan) (from down to up). (c) PChCl 1–L Mal 2 (black), PChCl 1–D Mal 2 (blue), and PChCl 1–DL Mal 2 (red) (from down to up). (d)PChCl 1–DL Mal 1 (black), PChCl 1–DL Mal 2 (blue), PChCl 1–DL Mal 3 (red), PChCl 1–DL Mal 4 (olive), and PChCl 1–DL Mal 5 (LT magenta) (from down to up). Experimental conditions: 0.25 g shrimp shells; 5 g NADES; 130 °C; 3 h. | ||
Although the regenerated product held a higher DS, its yield was less than 1 wt%, which was not meaningful on the industrial scale (Table 2). So, the following quantitative study was focused on the precipitates, including the calcium carbonate content (CaCO3%) and protein content (protein%). As shown in Fig. 4a, the CaCO3% of the precipitate obtained from the five NADES treatments was about 3 wt% because the pH values of the five NADESs were very similar. However, the ability of protein removal of the five NADESs was affected by the components and the mole ratio of the organic acid. The NADES with L-malic acid as the HBD had the highest capacity for protein removal in the three organic acids. When the HBD was the same, the protein content of the products decreased with the increase of the HBD mole ratio. The protein removal ability of the five NADESs was in accordance with their pH values (details in Table S1†), showing that the acidity of the NADES partly affected the protein removal. It was obvious that ChCl 1–L Mal 2 exhibited the best performance in the protein removal.
| Type of NADESs | DS of the precipitateb | DS of the regenerated product |
|---|---|---|
| a Experimental conditions: 0.25 g shrimp shells; 5 g NADES; 130 °C; 3 h. b DS = degree of substitution, which was calculated by comparing the absorption ratios of amide I at 1658 cm−1 and the ester at 1732 cm−1 according to the FT-IR spectra.25 | ||
| ChCl 1–L Lac 2 | 0.19 | 0.20 |
| ChCl 2–L Mal 1 | 0.00 | 0.19 |
| ChCl 1–L Mal 2 | 0.45 | 0.47 |
| ChCl 2–Citr 1 | 0.00 | 0.29 |
| ChCl 1–Citr 2 | 0.67 | 0.87 |
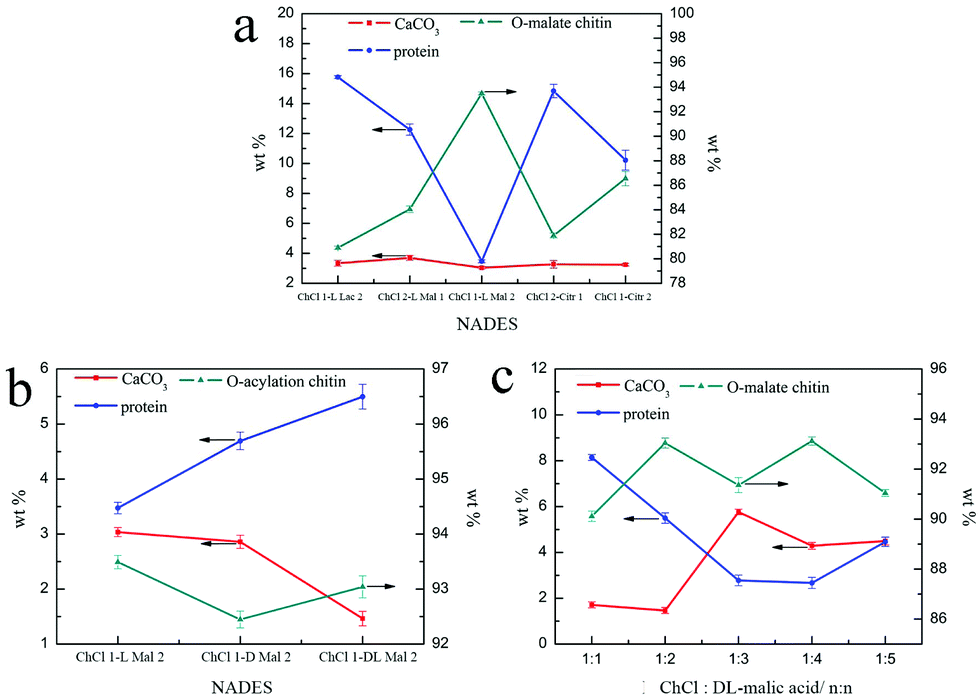 | ||
| Fig. 4 The CaCO3 (red), protein (blue), and O-acylated chitin (dark cyan) content of the precipitate treated with (a) ChCl 1–L Lac 2, ChCl 2–L Mal 1, ChCl 1–L Mal 2, ChCl 2–Citr 1, and ChCl 1–Citr 2; (b) ChCl 1–L Mal 2, ChCl 1–D Mal 2, and ChCl 1–DL–Mal 2. (c) ChCl 1–DL Mal 1, ChCl 1–DL Mal 2, ChCl 1–DL Mal 3, ChCl 1–DL Mal 4, and ChCl 1–DL Mal 5. Experimental conditions: 0.25 g shrimp shells; 5 g NADES; 130 °C; 3 h. | ||
Based on the above results, L-malic acid was chosen as the optimal HBD to explore the following influences on the O-acylated chitin. Hence, the product obtained would be O-malate chitin because the acylation reagent was malic acid. Malic acid exists in nature in three forms i.e.L, D and DL, the price gap of which is huge. Meanwhile, the protein of the shrimp shells consists of L-amino acids. Considering the cost and the rule of “like dissolves like”, ChCl/L-malic acid (1:2, ChCl 1–L Mal 2), ChCl/D-malic acid (1:2, ChCl 1–D Mal 2) and ChCl/DL-malic acid (1:2, ChCl 1–DL Mal 2) were involved to study the effect of chirality on the O-acylated chitin preparation. The experiments were carried out at 130 °C for 3 h with a mass ratio of 1:20 (shrimp shells:NADES). Considering the regenerated product with water as anti-solvent from water was still O-acylated chitin and too little to collect, DMSO was replaced by water to dilute the NADES/shrimp shell mixture in the following experiments. This would further reduce environmental pollution and improve the yield of O-malate chitin. The products consisted of precipitates and regenerated products.
As shown in Fig. 3c, the peak at 1738 cm−1 attributed to the CO group was included in the FT-IR spectra of the products, indicating that the O-acylation reaction of chitin can be carried out under the three NADES treatments. The DS of the three products, mainly O-malate chitin, were all about 0.4 (Table 3), indicating that the chirality of malic acid had no significant effect on the degree of acylation. According to Fig. 4b, ChCl 1–L Mal 2 has the highest ability to remove proteins, which may be attributed to the same chirality as the L-form structure of natural amino acids in shrimp shells. Additionally, the CaCO3% of the precipitate from ChCl 1–DL Mal 2 treatment was the lowest, and the yield was the highest due to its lowest solubility of chitin. Although the chirality of the organic acid exerted an influence on the calcium carbonate and protein removal, the difference is small. Based on the above results, DL-malic acid was chosen as the HBD for performing the following study because of its lowest cost, highest yield and purity of the O-acylated chitin.
| Sample | Purityb/% | Yieldc/% | DS |
|---|---|---|---|
| a Experimental conditions: 0.25 g shrimp shells; 5 g NADES; 130 °C; 3 h. b Purity% = 100 − CaCO3% − protein%. c Yield% = (mprecipitate × Purity%)/mo-malate chitin, where mo-malate chitin is a theoretical value without the consideration of the DS. | |||
| PChCl 1–L Mal 2 | 91.9 ± 0.3 | 17.3 ± 0.1 | 0.45 |
| PChCl 1–D Mal 2 | 92.6 ± 0.5 | 19.2 ± 0.3 | 0.41 |
| PChCl 1–DL Mal 1 | 89.9 ± 0.8 | 56.6 ± 2.7 | 0.40 |
| PChCl 1–DL Mal 2 | 92.6 ± 0.1 | 27.2 ± 1.0 | 0.43 |
| PChCl 1–DL Mal 3 | 91.3 ± 0.9 | 17.9 ± 1.8 | 0.51 |
| PChCl 1–DL Mal 4 | 93.4 ± 0.9 | 13.1 ± 0.8 | 0.55 |
| PChCl 1–D Mal 5 | 91.1 ± 0.4 | 21.0 ± 0.5 | 0.59 |
The alteration of the mole ratio of HBA and HBD can lead to a change of the pH and the molecular size of the NADESs, which may have an influence on the purity and DS of the products. So, DL-malic acid based NADESs, including ChCl/DL-malic acid (1:1, ChCl 1–DL Mal 1), ChCl/DL-malic acid (1:2, ChCl 1–DL Mal 2), ChCl/DL-malic acid (1:3, ChCl 1–DL Mal 3), ChCl/DL-malic acid (1:4, ChCl 1–DL Mal 4) and ChCl/DL-malic acid (1:5, ChCl 1–DL Mal 5), were employed to determine their effect. The shrimp shells were treated with the NADESs at 130 °C for 3 h with a mass ratio of 1:20 (shrimp shells:NADES). The products were collected and analyzed. Initially, the FT-IR spectra (Fig. 3d) showed that the main content of the products was O-malate due to the existence of peaks at 1738 cm−1 attributed to the CO group. Meanwhile, the addition of DL-malic acid in the NADESs showed a positive effect on the chitin acylation. As shown in Table 3, the DS of the precipitates slightly increased with the increase of the mole ratio of DL-malic acid. Subsequently, the precipitates were analyzed to determine their content (Fig. 4c). Interestingly, we found that the CaCO3% of the precipitates showed a wave type rather than a decrease trend with the increase of the mole ratio of malic acid, indicating that the amount of COOH is not the only factor affecting the calcium carbonate removal. The lowest CaCO3% of the precipitate was obtained with ChCl 1–DL Mal 2 treatment. However, the protein% trend of the precipitates decreased firstly and then increased with the lowest point at PChCl 1–DL Mal 4. The results revealed that the calcium carbonate and protein removal of the five NADESs depended on their different properties. Moreover, the yield of the O-malate decreased firstly and then increased slightly due to the more chitin degradation with the acidity increase. To determine the acidolysis of chitin in the NADES, the following experiments were carried out. ChCl 1–DL Mal 1, ChCl 1–DL Mal 2, ChCl 1–DL Mal 3 and ChCl 1–DL Mal 4 were reacted with pure chitin at 150 °C for 3 h, respectively. After the experiment, the supernatant was collected and analyzed with HPLC. The HPLC results (Fig. S2†) showed that with the increase in the moles of organic acid in the NADES, the area of the peak at 23 min increased. Compared with the standard chitosan oligosaccharide, the peak at 23 min was from a disaccharide. The results proved that part of the chitin was degraded to a disaccharide during NADES treatment, and its degradation rate increased with the increase in the acidity of NADES.
Considering the DS, purity, yield, and the cost, ChCl 1–DL Mal 2 was selected as the optimal NADES to perform the subsequent study.
Influence of experimental conditions on the O-malate chitin preparation
In the above study, attention was paid to the influence of NADESs on decalcification, deproteinization and acylation. ChCl 1–DL Mal 2 was chosen as the optimal NADES to explore the effect of experimental conditions on the O-malate preparation, including temperature, time, ratio of shrimp shells to NADESs, and the water content of NADES.Firstly, the influence of experiment temperature was studied due to its important role in acylation and deproteinization of chitin.75 The experiments were carried out with a ratio of 1:20 (shrimp shells:ChCl 1–DL Mal 2, m:m) for 3 h by changing the temperature from 90 to 150 °C. According to Fig. 5a, the FT-IR spectra reveal that the characteristic peaks of the products treated at 90 and 110 °C are the same as that of α-chitin and there is no characteristic peak of CO, indicating that the acylation of chitin was not carried out. When the temperature was changed from 130 to 150 °C, the characteristic peak of CO at 1732 cm−1 appeared and continued to increase. By calculating from the FT-IR spectra, it was found that the DS of the precipitates increased from 0 to 0.46.
 | ||
| Fig. 5 The FT-IR of the products treated with ChCl 1–DL Mal 2 under different conditions. (a) The experiment temperature was 90, 110, 130, 140, and 150 °C (from down to up), respectively. Other conditions: Pure ChCl 1–DL Mal 2, shrimp shells:ChCl 1–DL Mal 2 = 1:20 (m:m), 3 h. (b) The experiment time was 0.5, 1, 3, and 5 h (from down to up), respectively. Other conditions: Pure ChCl 1–DL Mal 2, shrimp shells:ChCl 1–DL Mal 2 = 1:20 (m:m), 150 °C. (c) The ratio of shrimp shells to ChCl 1–DL Mal 2 was from 1:10, 1:20, 1:30, 1:40, and 1:50 (from down to up), respectively. Other conditions: Pure ChCl 1–DL Mal 2, 3 h, 150 °C. (d) The water content of ChCl 1–DL Mal 2 was 0, 1, 4, 10, and 20 wt% (from down to up). Other conditions: 3 h, 150 °C, shrimp shells:ChCl 1–DL Mal 2 = 1:20 (m:m). | ||
Subsequently, attention was paid to the influence of temperature on the calcium carbonate and protein removal. As shown in Fig. 6a, the protein and calcium carbonate content of the products gradually decreased with the experiment temperature varying from 90 °C to 150 °C. When the temperature was increased up to 150 °C, the CaCO3% and protein% of the products were all about 0.6 wt%, which was close to chitin from Aladdin with 0.2% calcium carbonate and 1.2% protein. Additionally, the trend of yield was not an easy linear variation and all below 30 wt%, suggesting that the part of the chitin was also degraded in CLDM-DL2 even when the temperature was down to 90 °C. The highest yield of O-malate chitin was obtained at 130 °C, because at lower temperature it was not easy to remove calcium carbonate and protein but a higher temperature was beneficial for chitin degradation. Hence, the results revealed that temperature was the key factor in determining whether the acylation reaction can occur. However, a high DS was not obtained by a limited increase in the temperature (Table 4).
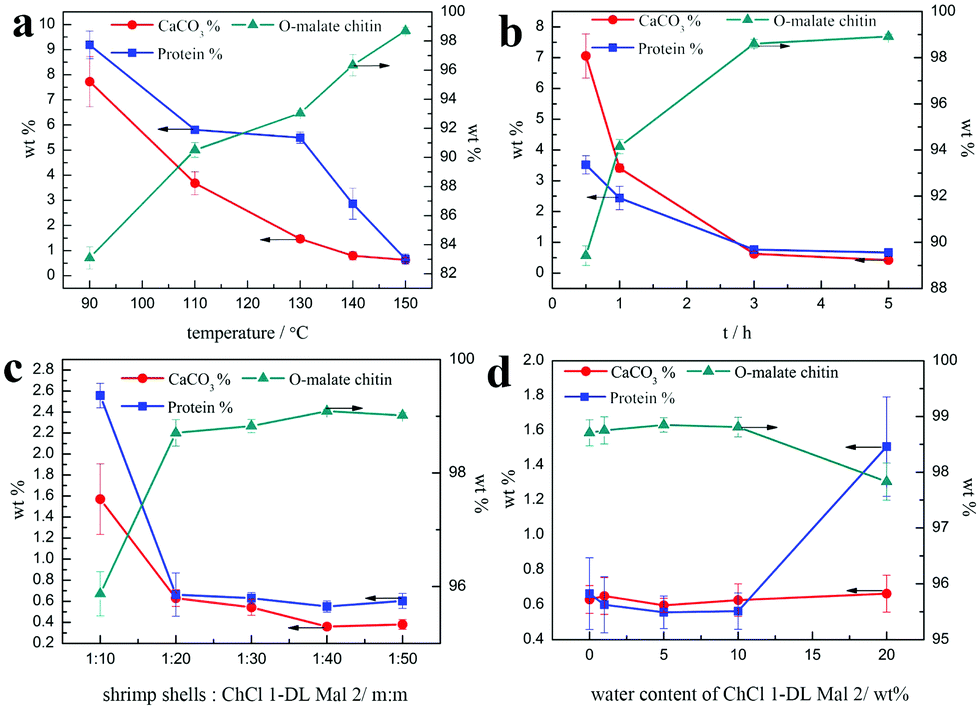 | ||
| Fig. 6 Different experiment effects on the CaCO3% (red), protein % (blue), and O-malate chitin (dark cyan) of products (a) temperature. Conditions: Shrimp shells:ChCl 1–DL Mal 2 = 1:20 (m:m), 3 h. (b) Time. Conditions: Shrimp shells:ChCl 1–DL Mal 2 = 1:20 (m:m), 150 °C. (c) Solid/liquid ratio. Conditions: 150 °C, 3 h. (d) Water content of ChCl 1–DL Mal 2. Conditions: Shrimp shells:ChCl 1–DL Mal 2/water mixture = 1:20 (m:m), 150 °C, 3 h. | ||
| T (°C) | Purity (wt%) | Yield (wt%) | DS |
|---|---|---|---|
| a Experimental conditions: 0.25 g shrimp shells; 5 g ChCl 1–DL Mal 2; 3 h. | |||
| 90 | 83.1 ± 0.8 | 21.0 ± 1.1 | 0 |
| 110 | 90.5 ± 0.5 | 19.8 ± 0.5 | 0 |
| 130 | 92.8 ± 0.3 | 27.2 ± 1.0 | 0.39 |
| 140 | 96.3 ± 0.7 | 15.9 ± 1.0 | 0.43 |
| 150 | 98.6 ± 0.2 | 13.2 ± 1.1 | 0.46 |
According to the above results, the optimal experiment temperature was selected as 150 °C to explore the impact of time on the preparation of O-malate chitin. The experiments were conducted at 150 °C with a ratio of 1:20 (shrimp shells:ChCl 1–DL Mal 2, m:m) for different experiment time, including 0.5, 1, 3, and 5 h. According to Fig. 5b, the characteristic peak of CO at 1732 cm−1 was observed for the precipitate treated only for 0.5 h. Meanwhile, the CaCO3% and protein% of the same product was 7.1 wt% and 3.5 wt% (Fig. 6b), respectively, which was significantly lower than raw shrimp shells with 56.1 wt% calcium carbonate and 8.1 wt% protein. The results indicated that the acylation of chitin, decalcification and deproteinization in the shrimp shells were carried out, simultaneously. Moreover, 87.4% calcium carbonate and 56.5% protein were removed in 0.5 h, because most of the calcium carbonate existed on the surface of the shrimp shells and all the chitin wrapped with protein was embedded in the mineral matrix.40,41
Obviously, the DS of the products increased (Table 5) and CaCO3% and protein% of the precipitates decreased with the increase in time. However, when the time was 5 h, the amount of precipitate increased possibly as a result of the decomposition of ChCl 1–DL Mal 2. The blank experiment, which involved heating pure ChCl 1–DL Mal 2 for 5 h at 150 °C, further showed white precipitate formation. Meanwhile, the yield of the O-malate chitin decreased due to the increase in the solubility of chitin.
| t (h) | Purity (wt%) | Yield (wt%) | DS |
|---|---|---|---|
| a Experimental conditions: 0.25 g shrimp shells; 5 g ChCl 1–DL Mal 2; 150 °C. b The purity and yield were not calculated because it is difficult to determine the impurity in the precipitate attributed to the decomposition of ChCl 1–DL Mal 2. | |||
| 0.5 | 89.4 ± 0.4 | 32.3 ± 0.9 | 0.13 |
| 1 | 94.1 ± 0.3 | 32.8 ± 0.1 | 0.28 |
| 3 | 98.6 ± 0.2 | 13.2 ± 1.1 | 0.46 |
| 5 | —b | —b | 0.52 |
In the subsequent experiments we evaluated the influence of the ratio of shrimp shells to ChCl 1–DL Mal 2 on the O-malate chitin preparation. The experiments were conducted with ChCl 1–DL Mal 2 at 150 °C for 3 h with different ratios of shrimp shells to ChCl 1–DL Mal 2, including 1:10, 1:20, 1:30, 1:40, and 1:50. The FT-IR spectra of the products in these experiments all have peaks at 1732 cm−1 attributed to the CO group. However, in the spectrum of products treated with shrimp shells with a mass ratio of 1:50 (shrimp shells:ChCl 1–DL Mal 2), the intensity of the peaks at 3259 cm−1 ascribed to the NH group and at 1312 cm−1 attributed to amine III weakened compared with other precipitates, and the peaks of amine I including 1658 and 1622 cm−1 changed from a doublet to singlet at 1634 cm−1. The results demonstrated that the increased ChCl 1–DL Mal 2 damaged the hydrogen bonding network of the amino group instead of increasing the DS of the O-malate chitin. Moreover, the results revealed that the ratio of shrimp shells to ChCl 1–DL Mal 2 had a slight effect on the DS of the O-malate chitin. Similarly, when the ratio was above 1:10, the change of the ratio had almost no effect on the decalcification and deproteinization. However, the yield of the O-malate chitin decreased obviously indicating that the increased amount of ChCl 1–DL Mal 2 destroyed chitin instead of leading to acylation of chitin (Table 6).
| Shrimp shells:ChCl 1–DL Mal 2 (m:m) |
Purity (%) | Yield (%) | DS |
|---|---|---|---|
| a Experimental conditions: 150 °C; 3 h. | |||
| 1:10 |
95.8 ± 0.4 | 27.3 ± 1.5 | 0.44 |
| 1:20 |
98.6 ± 0.2 | 13.2 ± 1.1 | 0.46 |
| 1:30 |
98.8 ± 0.1 | 9.8 ± 1.9 | 0.50 |
| 1:40 |
99.1 ± 0.1 | 4.9 ± 1.0 | 0.61 |
| 1:50 |
99.0 ± 0.1 | 4.6 ± 1.8 | 0.20 |
The water content can affect the structure of the NADES,76 which may change its ability of decalcification, deproteinization and acylation. Hence, experiments were carried out at 150 °C for 3 h with varied water content of ChCl 1–DL Mal 2 from 1 to 20 wt%. However, the results proved that the water content of ChCl 1–DL Mal 2 had a little influence on the purity and DS of chitin. Only ChCl 1–DL Mal 2 with 20 wt% water content exhibited a lower protein removal ability (Fig. 6d). Hence, pure ChCl 1–DL Mal 2 can be replaced by ChCl 1–DL Mal 2 with 10 wt% water content to reduce its cost on a large scale (Table 7).
| Water content/wt% | Purity (%) | Yield (%) | DS |
|---|---|---|---|
|
a Experimental conditions: Shrimp shells:ChCl 1–DL Mal 2 = 1:20, 150 °C; 3 h.
|
|||
| 0 | 98.6 ± 0.2 | 13.2 ± 1.1 | 0.46 |
| 1 | 98.8 ± 0.2 | 11.8 ± 2.6 | 0.46 |
| 5 | 98.8 ± 0.1 | 11.1 ± 2.8 | 0.43 |
| 10 | 98.8 ± 0.2 | 15.9 ± 1.6 | 0.49 |
| 20 | 97.8 ± 0.3 | 12.1 ± 1.3 | 0.49 |
Hence, O-malate chitin with a purity of 98.6% and a DS of 0.46 was obtained from shrimp shells by treatment with pure ChCl 1–DL Mal 2 (shrimp shells:ChCl 1–DL Mal 2 = 1:20) for 3 h at 150 °C, which was further characterized and tested in the following experiments.
The effect of ChCl 1–DL Mal 2 recycling on the O-malate chitin
Efforts were made to recycle and reuse ChCl 1–DL Mal 2. Obviously, the hydrogen bonding structure of NADESs could be broken at high temperature or in aqueous solution. Hence, it is difficult to achieve complete circulation of NADESs. In our work, we found that the used ChCl 1–DL Mal 2 could be reused and recycled.As shown in Fig. 7(a), the recycling rate of ChCl 1–DL Mal 2 was about 70 wt% in the first and second recycling experiments. However, the recycling rate of ChCl 1–DL Mal 2 was up to 97.6% in the third experiment, and decreased smoothly with the increase in recycling. The change in the ChCl 1–DL Mal 2 recycling rate was due to the destruction of the relatively unstable hydrogen bond structure. The destroyed ChCl 1–DL Mal 2 existed as a yellow solid. When the recycling time increased, the remaining ChCl 1–DL Mal 2 became stable for recycling. It was obvious that the components of the recycled ChCl 1–DL Mal 2 were changed. However, the FT-IR spectra of the recycled ChCl 1–DL Mal 2 were almost the same, which showed that the main functional groups were the same in ChCl 1–DL Mal 2.
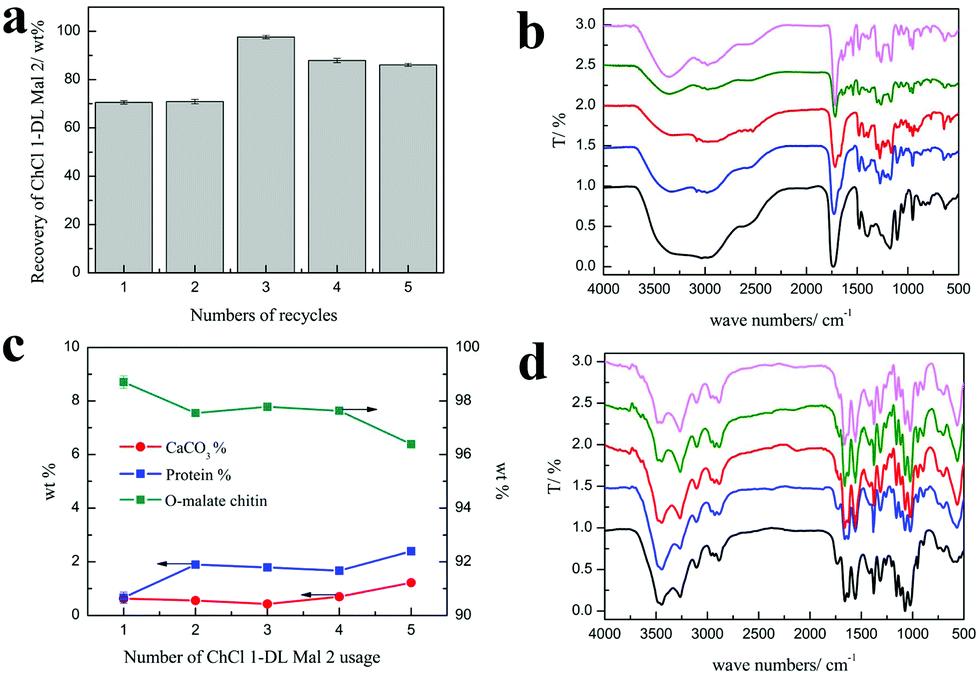 | ||
| Fig. 7 (a) The recovery rate of ChCl 1–DL Mal 2. (b) The FT-IR spectra of recycled ChCl 1–DL Mal 2. The usage time of ChCl 1–DL Mal 2 was 1 (black), 2 (blue), 3 (red), 4 (olive), and 5 (LT magenta). (c) The content of the obtained O-malate chitin under recycled ChCl 1–DL Mal 2 treatment. (d) The FT-IR spectra of the obtained O-malate chitin under reused ChCl 1–DL Mal 2 treatment. The usage time of ChCl 1–DL Mal 2 was 1 (black), 2 (blue), 3 (red), 4 (olive), and 5 (LT magenta). Experimental conditions: 150 °C, 3 h, shrimp shells:ChCl 1–DL Mal 2 = 1:20. | ||
On the other hand, the purity of the obtained O-malate chitin with the recycled ChCl 1–DL Mal 2 was maintained at about 97%, while the DS of the O-malate chitin decreased from 0.46 to 0.18 with the usage count increase from 1 to 5 times (Table 8). Meanwhile, a new peak at 1716 cm−1 ascribed to the carbonyl group of –COOH was detected in the O-malate chitin when the usage count was up to 3 times. This indicated that the carboxyl groups in ChCl 1–DL Mal 2 did not fully react due to the change of its components. Moreover, the yields of the O-malate chitin were higher than that of the previously obtained O-malate chitin. The structure of ChCl 1–DL Mal 2 also helped in chitin degradation. Overall, the results showed that the effect of recycling times on the acylation was greater than that of decalcification and deproteinization.
| Water content/wt% | Purity (%) | Yield (%) | DS |
|---|---|---|---|
|
a Experimental conditions: Shrimp shells:ChCl 1–DL Mal 2 = 1:20, 150 °C; 3 h.
|
|||
| 0 | 98.6 ± 0.2 | 13.2 ± 1.1 | 0.46 |
| 1 | 97.5 ± 0.1 | 34.6 ± 2.2 | 0.35 |
| 5 | 97.7 ± 0.1 | 28.6 ± 1.2 | 0.20 |
| 10 | 97.6 ± 0.1 | 40.1 ± 1.7 | 0.20 |
| 20 | 96.4 ± 0.1 | 36.6 ± 1.7 | 0.18 |
Characterization and properties of the obtained O-malate chitin
The product was determined as O-malate chitin according to the FT-IR spectrum of the product. To further prove its structure, the obtained O-malate chitin was characterized by XRD, solid state 13C NMR, and XPS. As shown in the XRD spectra (Fig. 8a), the O-malate chitin maintained the α-form as the main diffraction peaks were consistent with α-chitin. However, the crystallization index (CI%) of the O-malate chitin was 83.8%, which was lower than that of chitin from Sigma with 91.5%, demonstrating that ChCl 1–DL Mal 2 and the acylation reaction reduced the crystallinity of the chitin compared with the raw material. In the solid 13C NMR spectra (Fig. 8b) of the O-malate chitin, an additional signal was observed at 195.05 ppm due to CO from the ester or carboxyl group, proving that the acylation reaction was carried out successfully. Furthermore, additional peaks at 140.45 and 135.21 ppm were ascribed to C8 and C7 of the inserted group. The rest of the peaks were the same as that of chitin, proving that ChCl 1–DL Mal 2 did not break the carbon skeleton of chitin during the process.
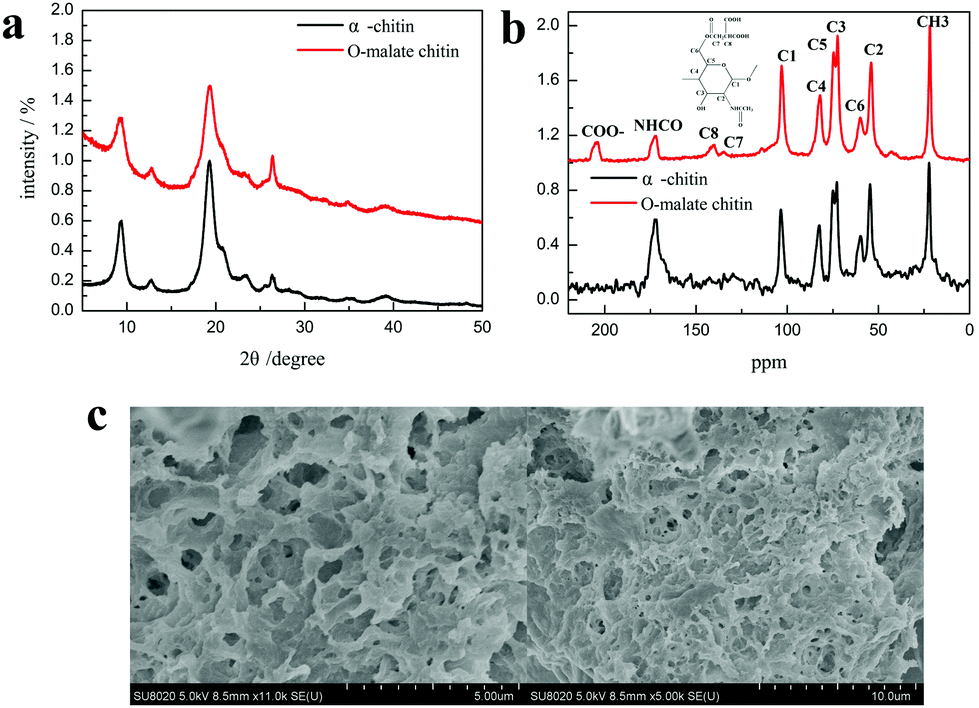 | ||
| Fig. 8 (a) The XRD spectra of α-chitin and O-malate chitin. (b) The solid 13C NMR spectra of α-chitin and O-malate chitin. (c) The SEM figures of O-malate chitin. | ||
The XPS analysis also showed that the precipitate was O-malate chitin. As shown in the XPS spectrum of chitin C 1s (Fig. 9a), three peaks are observed, including a peak at 284.8 eV (27.0 at%) due to the aliphatic carbon, a peak at 286.3 eV (46.6 at%) ascribed to the C–OH, C–N, and C–O–C moieties and linkage (C4), and a peak at 287.9 eV (26.4 at%) assigned to the CO and C–O–C (C1) moieties of the chitin rings.77,78 Obviously, a new peak at 288.9 eV (4.1 at%) attributed to the ester group was observed in the O-malate chitin C 1s XPS spectrum (Fig. 9b),79 which further proved that the acylation of chitin was successfully carried out. In the XPS O 1s spectra (Fig. 9c and d), the chitin and O-malate all contained peaks at 532.8 eV and 531.1 eV due to N–CO and OH/C–O, respectively.77 Moreover, the O 1s spectra of O-malate chitin had an additional peak at 532.0 eV ascribed to the O–CO group,80 which reconfirms that the acylation reaction was conducted.
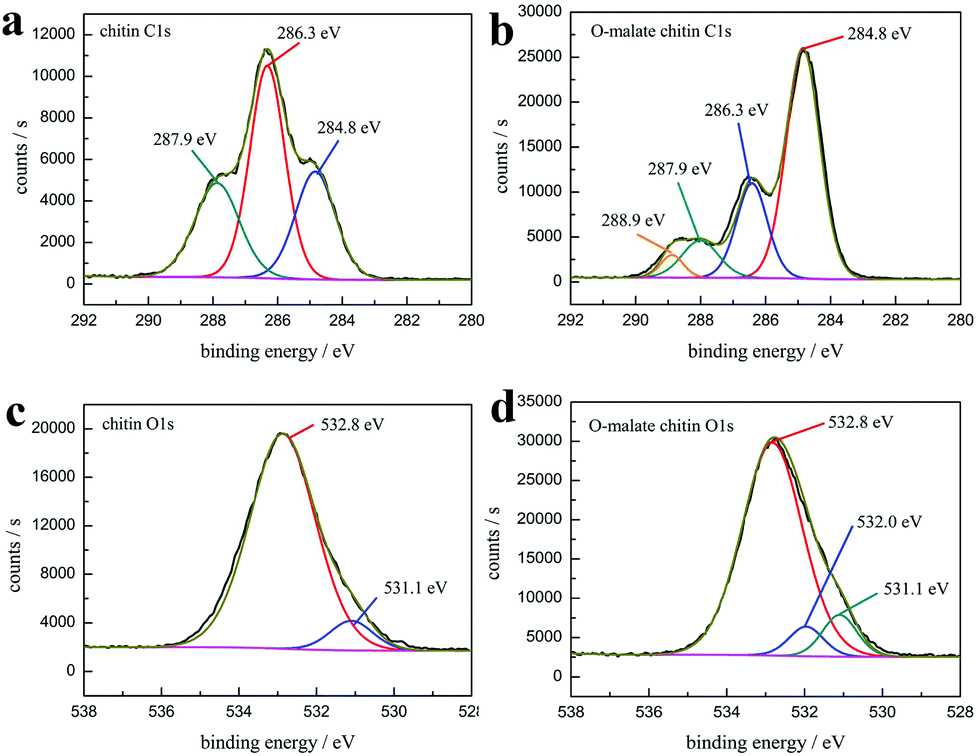 | ||
| Fig. 9 The high-resolution XPS spectra of (a) chitin C 1s (b) O-malate chitin C 1s (c) chitin O 1s and (d) O-malate chitin O 1s. | ||
Additionally, SEM was performed to detect the morphology of the O-malate chitin. Compared with the shrimp shells (Fig. S3†), the raw honeycomb-like structure was destroyed due to calcium carbonate and protein removal. Moreover, the surface morphology (Fig. 8c) of the O-malate chitin was porous and smooth without fibers, showing that the raw status of the chitin is broken. Furthermore, the experiments showed that the molecular weight of the O-malate chitin (DS 0.46) was 311725 g mol−1. Meanwhile, we also found that the molecular weight of chitin from Aladdin was 122492 g mol−1. By calculation, the molecular weight of O-malate chitin (DS 0.46) was 163047 g mol−1. The results showed that the NADES treatment was milder than the hydrochloric acid and sodium hydroxide treatment.
The obtained O-malate chitin was evaluated for the antibacterial and antitumor activities with Gram-negative bacterium Escherichia coli and C6 glioma, respectively. As shown in Table 9, the O-malate chitin exhibited 14.2 ± 0.5% and 36.4 ± 0.4% against Escherichia coli and C6 glioma, respectively. Obviously, the acylation of α-chitin provided additional antibacterial activity and a better antitumor effect compared with α-chitin, which enhance its application.
| Sample | Antibacterial activity/IR% | Antitumor activity/MR% |
|---|---|---|
| α-Chitin | −43.3 ± 0.4 | 29.8 ± 1.1 |
| O-Malate chitin | 14.2 ± 0.5 | 36.4 ± 0.4 |
The mechanism of decalcification, deproteinization and acylation of chitin
According to previous reports, the chitin nanofibrils are individually wrapped with protein to form chitin–protein fibers embedded in the calcium carbonate matrix in the shrimp shells.80,81 When ChCl 1–DL Mal 2 was distributed around the shrimp shells, it invaded the inner membrane of the shrimp shell structure, and then came into contact with the calcium carbonate, protein and chitin at the same time leading to decalcification, deproteinization and acylation reaction simultaneously. The above experimental results on the time factor elucidated this conjecture. We speculate that the acidity and the hydrogen bond formation ability of ChCl 1–DL Mal 2 were the key factors in the O-acylation chitin preparation. Hence, several efforts have been made to explore the interaction between shrimp shells and ChCl 1–DL Mal 2 to prove this speculation.The pH of ChCl 1–DL Mal 2 was about 1.5, indicating that H+ can be released from ChCl 1–DL Mal 2. The released H+ reacted with calcium carbonate to produce CO2 and water-soluble calcium salts resulting in calcium carbonate removal. The amounts of bubbles produced during the experiments (shown in Fig. S4†) and the supernatant collected after experiments containing Ca2+ proved the above inference.
We proposed that the protein in the shrimp shells was changed to amino acids or a water-soluble protein, which can be dissolved in water to remove it. To prove this speculation, the supernatant after experiments was collected and analyzed directly by HPLC to determine the content of amino acids. The results showed that it contained 18 types of amino acids including glycine, histidine, serine and so on (details are given in Table S2†). In addition, the HPLC analysis of the supernatant after acidolysis revealed that the content of six amino acids increased compared with the untreated sample ascribed to the undegraded protein included in the supernatant (details are given in Table S2†).
Furthermore, a white precipitate was formed after adding ethanol to the supernatant, which was easy to dissolve in water. The UV spectrum of the white precipitate aqueous solution stained with Coomassie Brilliant Blue exhibited a peak at 595 nm proving that it was a protein (Fig. 10a). The results greatly proved our conjecture that the protein was degraded and dissolved in ChCl 1–DL Mal 2 to remove it. Additionally, 1H NMR spectra were recorded to explore the interaction between the protein and ChCl 1–DL Mal 2 to explain the reason for protein dissolution. ChCl 1–DL Mal 2/regenerated protein due to the –COOH group in the spectrum of ChCl 1–DL Mal 2 disappeared in the spectrum of ChCl 1–DL Mal 2/regenerated product. It partly proved that hydrogen bonding was formed between –COOH of ChCl 1–DL Mal 2 and the protein. However, the precise interaction sites and mode between the protein and NADES are complicated, hence further work on this is still in progress.
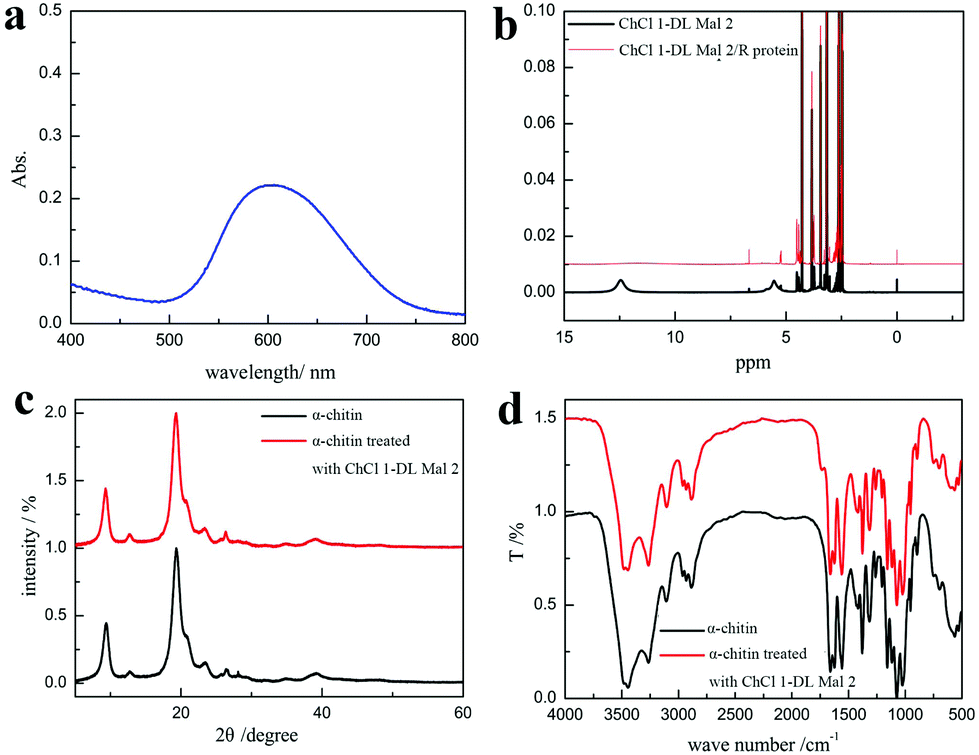 | ||
| Fig. 10 (a) The UV spectrum of the regenerated aqueous solution stained with Coomassie Brilliant Blue. (b) 1H NMR of ChCl 1–DL Mal 2 and ChCl 1–DL Mal 2/regenerated product. (c) XRD spectra of α-chitin and α-chitin treated with ChCl 1–DL Mal 2. (d) FT-IR spectra of α-chitin and α-chitin treated with ChCl 1–DL Mal 2. | ||
In the acylation of the chitin, ChCl 1–DL Mal 2 caused the chitin to swell and reacted with it simultaneously. To prove this speculation, pure chitin was treated with ChCl 1–DL Mal 2 at 110 °C for 0.5 h. Upon calculation using XRD results, it was found that chitin's CI% decreased from 96.6% to 94.1% (Fig. 10c). However, the FT-TR spectrum showed that the acylation of chitin was also conducted (Fig. 10d) at 110 °C, indicating that it was easier to acylate compared with the existing chitin in the shrimp shells. The findings were consistent with this speculation. Normally, the acylation of chitin in ChCl 1–DL Mal 2 was a nucleophilic reaction catalyzed by H+ released from ChCl 1–DL Mal 2, which was similar to the well-recognized aldol condensation mechanism.
Conclusion
For the first time, direct preparation of O-acylated chitin from shrimp shells with acidic NADESs was systematically investigated, providing a simple, effective and environmentally benign approach to produce value-added chitin derivatives from waste shrimp shells. The used NADESs not only acted as solvents to remove calcium carbonate and protein, but also acted as reagents and catalysts to react with chitin to produce O-acylated chitin. The types and mole ratios of organic acids in the NADESs and the experiment temperature were the key factors for the purity and DS of the O-acylation. O-Malate chitin with 98.6% purity and a DS of 0.46 was obtained from shrimp shells with choline chloride/DL-malic acid (1:2) treatment at 150 °C for 3 h. The structure of the obtained O-malate chitin was confirmed with XRD, solid 13C NMR and XPS, which exhibited antibacterial and anti-tumor effects. Herein, we point out that H+ was the main reason for the removal of calcium carbonate and caused acylation reaction. About the protein removal, we found that it was dissolved and degraded in the NADESs due to hydrogen bond formation and acidity respectively. A further study to reveal the exact interaction between protein/chitin and NAEDSs is still in progress. Importantly, we provide a green, simple but effective alternative to the shrimp shell utilization strategy based on multifunctional NADESs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Basic Research Program of China (973 Program, 2015CB251401), the Natural Scientific Fund of China (No. 21476234, 21506231), the National CAS/SAFEA International Partnership Program for Creative Research Teams (No. 20140491518), and the Key Research Program of Frontier Sciences CAS (QYZDY-SSW-JSC011).References
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- M. S. Islam, S. Khan and M. Tanaka, Mar. Pollut. Bull., 2004, 49, 103–110 CrossRef CAS PubMed.
- M. Borić, H. Puliyalil, U. Novak and B. Likozar, Green Chem., 2018, 20, 1199–1204 RSC.
- M. Feng, X. Lu, K. Jiang, J. Zhang, J. Xin, C. Shi, K. Wang and S. Zhang, Green Chem., 2018, 20, 2212–2217 RSC.
- D. Gómez-Ríos, R. Barrera-Zapata and R. Ríos-Estepa, Food Bioprod. Process., 2017, 103, 49–57 CrossRef.
- C. King, J. L. Shamshina, G. Gurau, P. Berton, N. F. A. F. Khan and R. D. Rogers, Green Chem., 2017, 19, 117–126 RSC.
- A. K. Mondal, K. Kretschmer, Y. Zhao, H. Liu, H. Fan and G. Wang, Microporous Mesoporous Mater., 2017, 246, 72–80 CrossRef CAS.
- L. Yan, J. Yu, J. Houston, N. Flores and H. Luo, Green Energy Environ., 2017, 2, 84–99 CrossRef.
- B. Duan, X. Zheng, Z. Xia, X. Fan, L. Guo, J. Liu, Y. Wang, Q. Ye and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5152–5156 CrossRef CAS PubMed.
- I. Younes and M. Rinaudo, Mar. Drugs, 2015, 13, 1133–1174 CrossRef CAS PubMed.
- F. Ding, H. Deng, Y. Du, X. Shi and Q. Wang, Nanoscale, 2014, 6, 9477–9493 RSC.
- S. Wu, B. Duan, X. Zeng, A. Lu, X. Xu, Y. Wang, Q. Ye and L. Zhang, J. Mater. Chem. B, 2017, 5, 2952–2963 RSC.
- M. Yuan, B. Bi, J. Huang, R. Zhuo and X. Jiang, Carbohydr. Polym., 2018, 192, 10–18 CrossRef CAS PubMed.
- Y. F. Aklog, M. Egusa, H. Kaminaka, H. Izawa, M. Morimoto, H. Saimoto and S. Ifuku, Int. J. Mol. Sci., 2016, 17, 1600 CrossRef PubMed.
- G. Ilangumaran, G. Stratton, S. Ravichandran, P. S. Shukla, P. Potin, S. Asiedu and B. Prithiviraj, Front. Microbiol., 2017, 8, 781 CrossRef PubMed.
- A. J. Winkler, J. A. Dominguez-Nunez, I. Aranaz, C. Poza-Carrion, K. Ramonell, S. Somerville and M. Berrocal-Lobo, Mar. Drugs, 2017, 15, 40 CrossRef PubMed.
- A. Bhatnagar and M. Sillanpaa, Adv. Colloid Interface Sci., 2009, 152, 26–38 CrossRef CAS PubMed.
- X. Liang, X. Fan, R. Li, S. Li, S. Shen and D. Hu, Bioresour. Technol., 2018, 250, 178–184 CrossRef CAS PubMed.
- L. Sellaoui, D. S. P. Franco, G. L. Dotto, É. C. Lima and A. B. Lamine, J. Mol. Liq., 2017, 233, 543–550 CrossRef CAS.
- I. Aranaz, N. Acosta, C. Civera, B. Elorza, J. Mingo, C. Castro, M. Gandía and A. Heras Caballero, Polymers, 2018, 10, 213 CrossRef.
- D. Kafetzopoulos, A. Martinou and V. Bouriotis, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2564–2568 CrossRef CAS.
- A. Tolaimate, J. Desbrieres, M. Rhazi, A. Alagui, M. Vincendon and P. Vottero, Polymer, 2000, 41, 2463–2469 CrossRef CAS.
- S. Mine, H. Izawa, Y. Kaneko and J. Kadokawa, Carbohydr. Res., 2009, 344, 2263–2265 CrossRef CAS PubMed.
- Y. Teramoto, T. Miyata and Y. Nishio, Biomacromolecules, 2006, 7, 190–198 CrossRef CAS PubMed.
- Y. Zou and E. Khor, Biomacromolecules, 2005, 6, 80–87 CrossRef CAS PubMed.
- A. Lutzke, A. Pegalajar-Jurado, B. H. Neufeld and M. M. Reynolds, J. Mater. Chem. B, 2014, 2, 7449–7458 RSC.
- S. Liang, Q. Dang, C. Liu, Y. Zhang, Y. Wang, W. Zhu, G. Chang, H. Sun, D. Cha and B. Fan, Carbohydr. Polym., 2018, 195, 275–287 CrossRef CAS PubMed.
- A. T. Ramaprasad, D. Latha and V. Rao, J. Phys. Chem. Solids, 2017, 104, 169–174 CrossRef CAS.
- S. S. Silva, J. F. Mano and R. L. Reis, Green Chem., 2017, 19, 1208–1220 RSC.
- A. Chilarski, I. Krucinska, P. Kiekens, A. Blasinska, G. Schoukens, R. Cislo and J. Szumilewicz, Fibres Text. East. Eur., 2007, 15, 77–81 CAS.
- A. Blasinska and J. Drobnik, Biomacromolecules, 2008, 9, 776–782 CrossRef CAS PubMed.
- I. P. Dobrovolskaya, V. E. Yudin, P. V. Popryadukhin, E. M. Ivan'kova, A. S. Shabunin, I. A. Kasatkin and P. Morgantie, Carbohydr. Polym., 2018, 194, 260–266 CrossRef CAS PubMed.
- B. Blanco-Fernandez, S. Chakravarty, M. K. Nkansah and E. M. Shapiro, Acta Biomater., 2016, 45, 276–285 CrossRef CAS PubMed.
- J. Drobnik, I. Krucinska, A. Komisarczyk, S. Sporny, A. Szczepanowska and J. Ciosek, Can. J. Surg., 2017, 60, 162–171 CrossRef PubMed.
- R. A. Muzzarelli, M. Guerrieri, G. Goteri, C. Muzzarelli, T. Armeni, R. Ghiselli and M. Cornelissen, Biomaterials, 2005, 26, 5844–5854 CrossRef CAS PubMed.
- L. Casettari, M. Cespi and E. Castagnino, Drug Dev. Ind. Pharm., 2012, 38, 979–984 CrossRef CAS PubMed.
- T. Jain, S. Kumar and P. K. Dutta, Int. J. Biol. Macromol., 2016, 82, 1011–1017 CrossRef CAS PubMed.
- F. I. Khan, S. Rahman, A. Queen, S. Ahamad, S. Ali, J. Kim and M. I. Hassan, Appl. Microbiol. Biotechnol., 2017, 101, 3513–3536 CrossRef CAS PubMed.
- H. Liu, Q. Yang, L. Zhang, R. Zhuo and X. Jiang, Carbohydr. Polym., 2016, 137, 600–607 CrossRef CAS PubMed.
- S. Nikolov, M. Petrov, L. Lymperakis, M. Friak, C. Sachs, H. O. Fabritius, D. Raabe and J. Neugebauer, Adv. Mater., 2010, 22, 519–526 CrossRef CAS PubMed.
- P. Romano, H. Fabritius and D. Raabe, Acta Biomater., 2007, 3, 301–309 CrossRef CAS PubMed.
- A. Percot, C. Viton and A. Domard, Biomacromolecules, 2003, 4, 12–18 CrossRef CAS PubMed.
- L. R. Bhatt, B. M. Kim, K. Hyun, K. H. Kang, C. Lu and K. Y. Chai, Carbohydr. Res., 2011, 346, 691–694 CrossRef CAS PubMed.
- B. Y. Yang, Q. Ding and R. Montgomery, Carbohydr. Res., 2009, 344, 336–342 CrossRef CAS PubMed.
- K. Kaifu, N. Nishi, T. Komai, S. Tokura and O. Somorin, Polym. J., 1981, 13, 241 CrossRef CAS.
- N. Nishi, J. Noguchi, S. Tokura and H. Shiota, Polym. J., 1979, 11, 27 CrossRef CAS.
- K. Kaifu, N. Nishi and T. Komai, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 2361–2363 CrossRef CAS.
- M. Sugimoto, M. Kawahara, Y. Teramoto and Y. Nishio, Carbohydr. Polym., 2010, 79, 948–954 CrossRef CAS.
- L. R. Bhatt, B. M. Kim, K. Hyun, G. B. Kwak, C. H. Lee and K. Y. Chai, Molecules, 2011, 16, 3029–3036 CrossRef CAS PubMed.
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- T. Bosiljkov, F. Dujmić, M. Cvjetko Bubalo, J. Hribar, R. Vidrih, M. Brnčić, E. Zlatic, I. Radojčić Redovniković and S. Jokić, Food Bioprod. Process., 2017, 102, 195–203 CrossRef CAS.
- Y. Huang, F. Feng, J. Jiang, Y. Qiao, T. Wu, J. Voglmeir and Z. G. Chen, Food Chem., 2017, 221, 1400–1405 CrossRef CAS PubMed.
- M. Ivanovic, M. E. Alanon, D. Arraez-Roman and A. Segura-Carretero, Food Res. Int., 2018, 111, 67–76 CrossRef CAS PubMed.
- B.-Y. Zhao, P. Xu, F.-X. Yang, H. Wu, M.-H. Zong and W.-Y. Lou, ACS Sustainable Chem. Eng., 2015, 3, 2746–2755 CrossRef CAS.
- S. Daneshjou, S. Khodaverdian, B. Dabirmanesh, F. Rahimi, S. Daneshjoo, F. Ghazi and K. Khajeh, J. Mol. Liq., 2017, 227, 21–25 CrossRef CAS.
- Y. Qiao, H. L. Cai, X. Yang, Y. Y. Zang and Z. G. Chen, Appl. Microbiol. Biotechnol., 2018, 102, 5695–5705 CrossRef CAS PubMed.
- T.-X. Yang, L.-Q. Zhao, J. Wang, G.-L. Song, H.-M. Liu, H. Cheng and Z. Yang, ACS Sustainable Chem. Eng., 2017, 5, 5713–5722 CrossRef CAS.
- L. Cicco, N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, F. M. Perna, V. Capriati, J. García-Álvarez and J. González-Sabín, Green Chem., 2018, 20, 3468–3475 RSC.
- F. Sebest, L. Casarrubios, H. S. Rzepa, A. J. P. White and S. Díez-González, Green Chem., 2018, 20, 4023–4035 RSC.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149–18155 RSC.
- C. Mukesh, D. Mondal, M. Sharma and K. Prasad, Carbohydr. Polym., 2014, 103, 466–471 CrossRef CAS PubMed.
- Y. Liu, W. Chen, Q. Xia, B. Guo, Q. Wang, S. Liu, Y. Liu, J. Li and H. Yu, ChemSusChem, 2017, 10, 1692–1700 CrossRef CAS PubMed.
- J. G. Lynam, N. Kumar and M. J. Wong, Bioresour. Technol., 2017, 238, 684–689 CrossRef CAS PubMed.
- C. Alvarez-Vasco, R. Ma, M. Quintero, M. Guo, S. Geleynse, K. K. Ramasamy, M. Wolcott and X. Zhang, Green Chem., 2016, 18, 5133–5141 RSC.
- Q. Xia, Y. Liu, J. Meng, W. Cheng, W. Chen, S. Liu, Y. Liu, J. Li and H. Yu, Green Chem., 2018, 20, 2711–2721 RSC.
- A. Sanchez-Fernandez, K. J. Edler, T. Arnold, D. Alba Venero and A. J. Jackson, Phys. Chem. Chem. Phys., 2017, 19, 8667–8670 RSC.
- Q. Zeng, Y. Wang, Y. Huang, X. Ding, J. Chen and K. Xu, Analyst, 2014, 139, 2565–2573 RSC.
- H.-C. Hu, Y.-H. Liu, B.-L. Li, Z.-S. Cui and Z.-H. Zhang, RSC Adv., 2015, 5, 7720–7728 RSC.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280 RSC.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. Sila, M. Nasri and A. Bougatef, Int. J. Biol. Macromol., 2012, 51, 953–959 CrossRef CAS PubMed.
- D. Gerlier and N. Thomasset, J. Immunol. Methods, 1986, 94, 57–63 CrossRef CAS PubMed.
- P. Zhu, Z. Gu, S. Hong and H. Lian, Carbohydr. Polym., 2017, 177, 217–223 CrossRef CAS PubMed.
- L. R. Bhatt, B. M. Kim, C. Y. An, C. Lu, Y. S. Chung, M. G. Soung, S. H. Park and K. Y. Chai, Carbohydr. Res., 2010, 345, 2102–2106 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- E. Ieva, A. Trapani, N. Cioffi, N. Ditaranto, A. Monopoli and L. Sabbatini, Anal. Bioanal. Chem., 2009, 393, 207–215 CrossRef CAS PubMed.
- T. G. Liu, Y. T. Wang, B. Li, H. B. Deng, Z. L. Huang, L. W. Qian and X. Wang, Carbohydr. Polym., 2017, 174, 464–473 CrossRef CAS PubMed.
- A. Manakhov, P. Kiryukhantsev-Korneev, M. Michlíček, E. Permyakova, E. Dvořáková, J. Polčák, Z. Popov, M. Visotin and D. V. Shtansky, Appl. Surf. Sci., 2018, 435, 1220–1227 CrossRef CAS.
- A. Al-Sawalmih, C. Li, S. Siegel, H. Fabritius, S. Yi, D. Raabe, P. Fratzl and O. Paris, Adv. Funct. Mater., 2008, 18, 3307–3314 CrossRef CAS.
- P. Romano, H. Fabritius and D. Raabe, Acta Biomater., 2007, 3, 301–309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc02506a |
| This journal is © The Royal Society of Chemistry 2019 |