
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bio-based route to the carbon-5 chemical glutaric acid and to bionylon-6,5 using metabolically engineered Corynebacterium glutamicum†
Christina Maria
Rohles
a,
Lars
Gläser
a,
Michael
Kohlstedt
a,
Gideon
Gießelmann
a,
Samuel
Pearson
b,
Aránzazu
del Campo
 b,
Judith
Becker
a and
Christoph
Wittmann
*a
b,
Judith
Becker
a and
Christoph
Wittmann
*a
aInstitute of Systems Biotechnology, Saarland University, Saarbrücken, Germany. E-mail: christoph.wittmann@uni-saarland.de; Tel: +49-(0) 681 302 71970
bLeibniz Institut für Neue Materialien, Saarbrücken, Germany
First published on 24th September 2018
Abstract
In the present work, we established the bio-based production of glutarate, a carbon-5 dicarboxylic acid with recognized value for commercial plastics and other applications, using metabolically engineered Corynebacterium glutamicum. The mutant C. glutamicum AVA-2 served as a starting point for strain development, because it secreted small amounts of glutarate as a consequence of its engineered 5-aminovalerate pathway. Starting from AVA-2, we overexpressed 5-aminovalerate transaminase (gabT) and glutarate semialdehyde dehydrogenase (gabD) under the control of the constitutive tuf promoter to convert 5-aminovalerate further to glutarate. The created strain GTA-1 formed glutarate as a major product, but still secreted 5-aminovalerate as well. This bottleneck was tackled at the level of 5-aminovalerate re-import. The advanced strain GTA-4 overexpressed the newly discovered 5-aminovalerate importer NCgl0464 and formed glutarate from glucose in a yield of 0.27 mol mol−1. In a fed-batch process, GTA-4 produced more than 90 g L−1 glutarate from glucose and molasses based sugars in a yield of up to 0.70 mol mol−1 and a maximum productivity of 1.8 g L−1 h−1, while 5-aminovalerate was no longer secreted. The bio-based glutaric acid was purified to >99.9% purity. Interfacial polymerization and melt polymerization with hexamethylenediamine yielded bionylon-6,5, a polyamide with a unique structure.
Introduction
There is a rapid growth of interest to use renewables for the manufacture of sustainable chemicals and materials. In particular, bioplastics are promising products among the different bio-based sectors. At present, the bioplastics market grows between 20–100% per year and the production is expected to surpass six million tons in 2021. This trend explains the huge market potential assigned to bio-based chemicals, which can serve as building blocks for bioplastics, including dicarboxylic acids,1–9 diamines10–19 and diols.20–24Glutaric acid (1,5-pentanedioic acid) is such an attractive building block for polymers.25,26 It is used in the production of commercial polyesters and polyamides.27 Moreover, it is built into organometallics, leading to antimicrobial agents,28 transistors, and capacitors.29 The dimethyl ester of glutaric acid is regarded as a green solvent and used as an ingredient in cleaning products, paints and coating formulations.30 The hydrogenated form, 1,5-pentanediol,31 is a monomer for polyesters and polyurethanes.32 The costs and environmental concerns of traditional petrochemical routes to glutaric acid have stimulated research to produce this important chemical from renewable resources. Different approaches have demonstrated the production of glutarate and its derivative 5-aminovalerate.26,33–35 However, glutarate titers have remained unattractively low so far. The most promising concepts to derive glutarate by fermentation build on L-lysine overproducing microbes,25,26 previously engineered to accumulate the amino acid in high titers and yields.36 Introducing the so-called aminovalerate pathway, L-lysine is converted into 5-aminovalerate by L-lysine 2-monooxygenase (DavB) and δ-aminovaleramidase (DavA) (Fig. 1A). The downstream glutarate pathway involves the enzymes aminobutyrate/aminovalerate aminotransferase (GabT) and succinate/glutarate semialdehyde dehydrogenase (GabD) to finally form glutarate. In C. glutamicum, these are encoded by the operons NCgl0462 (gabT) and NCgl0463 (gabD).34 Using the L-lysine-mediated route, initial studies reported genetically modified E. coli, which produced glutarate at a level of 0.8 g L−1 (ref. 25) and 1.7 g L−1.26E. coli strains, engineered with a reverse adipate-degradation pathway, accumulated up to 4.8 g L−1 glutarate.37 Another approach to glutarate recently suggested a bio-reduction of glutaconate, but is greatly limited by the supply of the precursor in the milligram range.38 Likewise, glutarate biosynthesis via α-keto acid carbon chain extension and decarboxylation in recombinant E. coli has remained at titers of only 0.4 g L−1 until recently.27
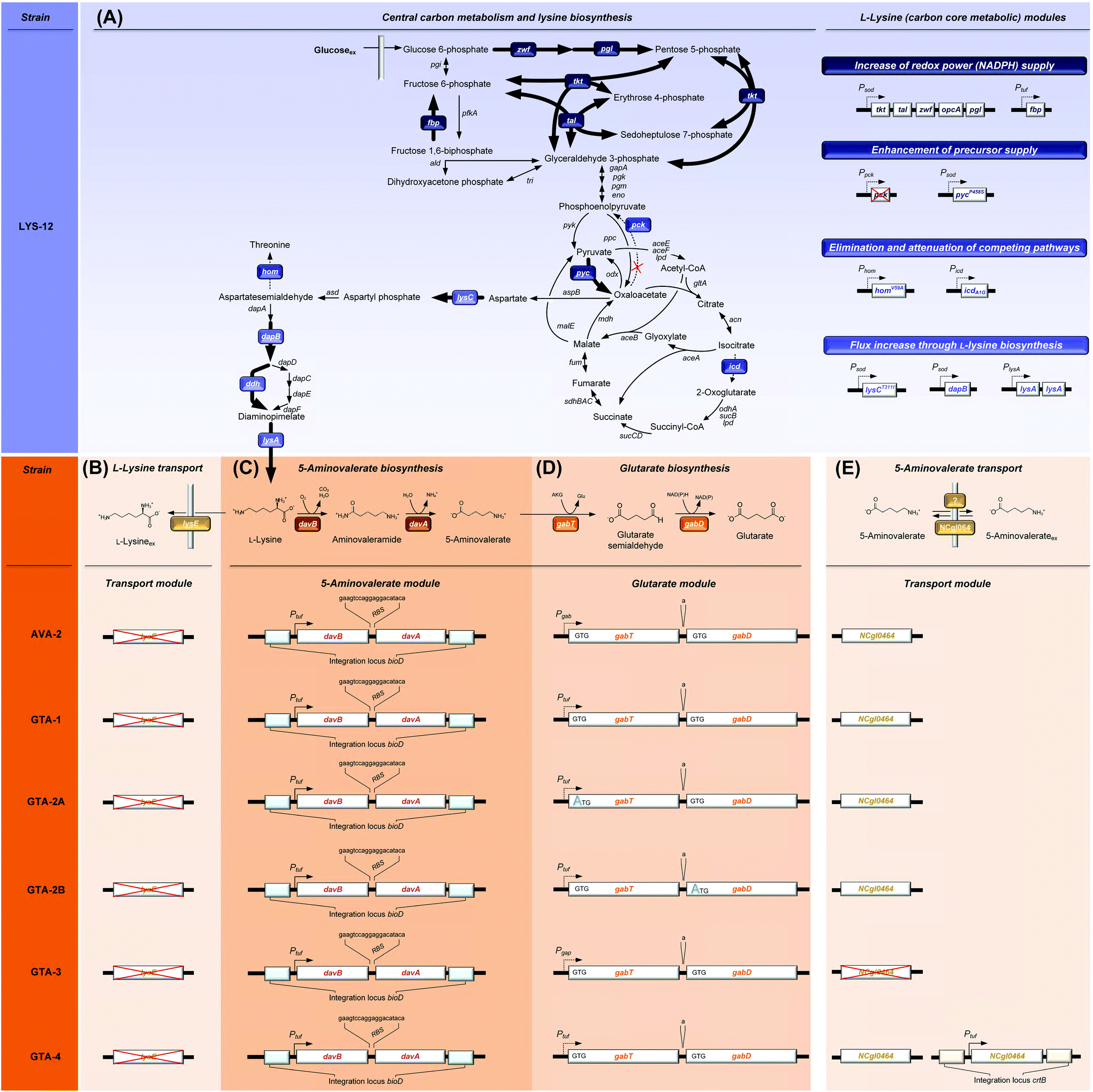 | ||
| Fig. 1 Metabolic pathway design for the production of glutarate in Corynebacterium glutamicum. The overview illustrates the genomic layout of each producer, created in this work, including core carbon metabolism and L-lysine synthesis (A) and secretion (B), 5-aminovalerate synthesis (C), glutarate synthesis (D), and the newly discovered re-import of 5-aminovalerate (E). All modifications were implemented into the genome. The genes (davBA) from P. putida KT2440, encoding L-lysine monooxygenase (DavB) and 5-aminovaleramidase (DavA), were used to establish the 5-aminovalerate module. The two genes were integrated under the control of the constitutive tuf-promoter into the bioD locus of C. glutamicum LYS-12. LYS-12 comprised twelve genome-based modifications for lysine hyper production.34 The glutarate module comprised 5-aminovalerate transaminase (GabT) and glutarate semialdehyde dehydrogenase (GabD), encoded by the native genes gabTD. Different variants of the modules were tested for optimization. The transport module comprised the L-lysine exporter lysE and the amino acid permease NCgl0464, expressed in its native form, deleted, and amplified via the genomic expression of a second gene copy in the crtB locus. The gene annotations for central metabolism and L-lysine synthesis are given in the ESI.† | ||
C. glutamicum seems to be a most straightforward host to produce glutarate, because it is the major workhorse for the industrial manufacture of L-lysine,39,40 a glutarate precursor. Different from other bacteria such as E. coli25 and P. putida,41,42C. glutamicum does not degrade L-lysine, but retains the amino acid inside the cell upon deletion of the lysine exporter lysE,34 preventing the loss of the precursor. From a metabolic viewpoint, the L-lysine-based pathway is the preferred route to make glutarate. It offers the highest theoretical yield (0.75 mol (mol glucose)−1), higher than that of the alternative α-ketoglutarate-based synthesis (0.67 mol (mol glucose)−1).27 Despite these advantages, C. glutamicum has not been metabolically engineered so far to produce glutarate. It is even claimed that C. glutamicum has “serious limitations for glutarate production”.37 The fact that 5-aminovalerate-producing C. glutamicum strains accumulate glutarate as a by-product,34,43 however, demonstrates the basic capability of the microbe to synthesize this interesting molecule.
Previously, our group has metabolically engineered C. glutamicum for the production of L-lysine,36,44 ectoine,45 diaminopentane,14 and 5-aminovalerate,34 all via the lysine route. Here, we describe systems metabolic engineering of C. glutamicum for high-level glutarate production. The recently developed producer C. glutamicum AVA-2 served as a starting point for strain engineering, because it exhibited an engineered aminovalerate pathway and excreted glutarate as a by-product in addition to 5-aminovalerate.34 The advanced producer C. glutamicum GTA-4, obtained after several rounds of strain optimization, was evaluated in a fed-batch process. Subsequently, a strategy for down-stream purification was developed and applied to purify glutaric acid from fermentation. The obtained glutaric acid was used to derive bio-nylon-6,5.
Results and discussion
C. glutamicum can grow in the presence of up to 60 g L−1 glutarate
C. glutamicum could grow in the presence of up to 60 g L−1 glutarate (ESI, Fig. S1†). Increasing concentrations caused reduced growth rates, leading to a 50% growth reduction at a rather high glutarate concentration (KI) of 46.0 ± 0.4 g L−1. No growth was observed at 80 g L−1 glutarate. Similarly, the microbe also grew on agar plates up to a glutarate level of 60 g L−1. C. glutamicum appeared significantly more tolerant than E. coli, already growth inhibited at 5 g L−1 glutarate and severely affected at 20 g L−1.25The overexpression of the 5-aminovalerate pathway module is not sufficient for selective glutarate production in C. glutamicum
During the growth of C. glutamicum AVA-2 on glucose as the sole source of carbon, 5-aminovalerate and glutarate were both secreted into the medium (Fig. 2A). The pathway precursor L-lysine was not secreted, resulting from the previously eliminated L-lysine exporter lysE in the genome of the AVA-2 strain.34 The production of 5-aminovalerate and glutarate was growth-associated and stopped after the depletion of the substrate. With regard to a selective production of glutarate, the formation of 5-aminovalerate was undesired. | ||
| Fig. 2 Growth and production characteristics of glutarate producing C. glutamicum strains. The strains AVA-2 (A, B), GTA-1 (C, D), and GTA-4 (E, F) were cultivated in shake flasks at 30 °C in a chemically defined glucose medium. The cultivation profiles show growth, product formation, and glucose consumption over time (A, C, E) and yields (B, D, F). Error bars represent standard deviations from three biological replicates. | ||
Overexpression of the native gabTD genes increases the glutarate production flux
To improve the glutarate production, we expressed the entire gabTD operon under the control of the strong constitutive tuf promoter (Fig. 1D). For this purpose, a 200 bp tuf promoter fragment was inserted into the genome upstream of the gabTD locus. Positive clones revealed a PCR fragment of 1.3 kb in contrast to the wild type (1.1 kB). After verification by sequencing, one clone was designated C. glutamicum GTA-1.Cultured on glucose, GTA-1 accumulated 12.5 mM glutarate (Fig. 2C): 50% more than its ancestor AVA-2. Furthermore, the new mutant exhibited a twofold increased glutarate yield (265 ± 8 mmol mol−1) and an eightfold reduced 5-aminovalerate production (Table 1 and Fig. 2D). The strong decrease in the 5-aminovalerate yield revealed that the major fraction of the precursor was successfully channeled to glutarate by the amplified glutarate module. As shown by enzymatic assays, the enhanced product yield benefited from a substantially enhanced activity of the pathway, which converted 5-aminovalerate to glutarate in two enzymatic steps (Fig. 1D). The GTA-1 strain revealed a 5 to 7-fold higher specific activity for 5-aminovalerate transaminase (159 mU mg−1) and glutarate semialdehyde dehydrogenase (222 mU mg−1) than the parent AVA-2 strain (GabT: 24 mU mg−1, GabD: 42 mU mg−1). The glutarate production was not further increased, when pyridoxal 5-phosphate (10 mg L−1), a known cofactor for transaminases, was supplied to the cultivation medium (data not shown). Obviously, the endogenous supply of the vitamin was sufficient. Overall, the glutarate formation in GTA-1 made up 96% of the total production flux (Fig. 3).
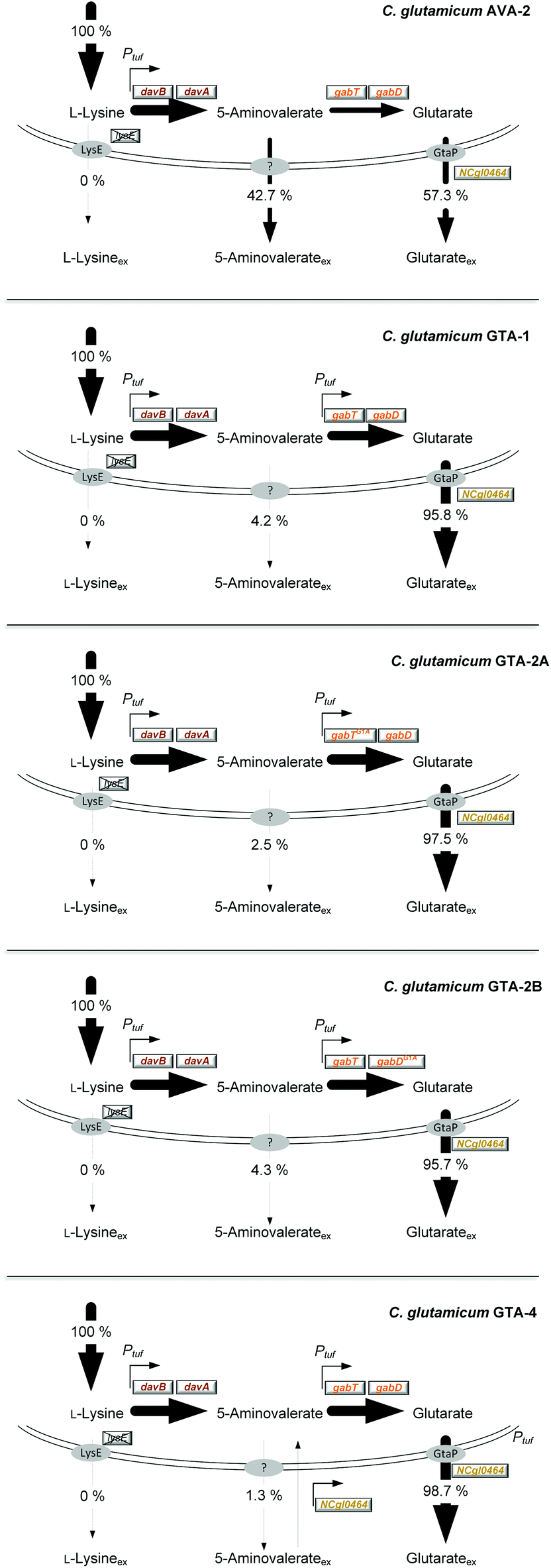 | ||
| Fig. 3 Relative contribution of L-lysine, 5-aminovalerate and glutarate secretion to the overall production flux in C. glutamicum AVA-2, GTA-1, GTA-2A, GTA-2B and GTA-4. The production is given as a relative flux related to the overall L-lysine-based production flux, which was set to 100%. | ||
| AVA-2 | GTA-1 | GTA-2A | GTA-2B | GTA-4 | |
|---|---|---|---|---|---|
| μ [h−1] | 0.22 ± 0.04 | 0.16 ± 0.00 | 0.14 ± 0.00 | 0.10 ± 0.00 | 0.18 ± 0.00 |
| q Glc [mmol g−1 h−1] | 4.6 ± 0.7 | 3.0 ± 0.1 | 2.8 ± 0.3 | 2.3 ± 0.1 | 3.3 ± 0.0 |
| q 5-Ava [mmol g−1 h−1] | 0.4 ± 0.1 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| q Glt [mmol g−1 h−1] | 0.6 ± 0.1 | 0.8 ± 0.0 | 0.8 ± 0.0 | 0.7 ± 0.0 | 0.9 ± 0.0 |
| Y X/Glc [g mol−1] | 47.6 ± 3.6 | 53.4 ± 4.0 | 49.4 ± 5.5 | 44.3 ± 1.6 | 54.3 ± 1.0 |
| Y 5-Ava/Glc [mmol mol−1] | 98 ± 2 | 12 ± 1 | 7 ± 3 | 14 ± 0 | 4 ± 0 |
| Y Glt/Glc [mmol mol−1] | 123 ± 2 | 265 ± 8 | 277 ± 6 | 316 ± 5 | 271 ± 3 |
Reinforcement of the glutarate pathway module at the level of translation upgrades the glutarate yield, but causes a decrease in cellular fitness
The genetic structure of the glutarate operon suggested a bicistronic transcript with individual translational start points for 5-aminovalerate transaminase (gabT) and glutarate semialdehyde dehydrogenase (gabD). The sequence further revealed that each of the genes contained the start codon GTG, previously found to be weak in translation efficiency.46,47 To enhance translation, the GTG codon of the gabT gene was replaced by the stronger variant ATG. The obtained mutant C. glutamicum GTA-1 gabTG1A, designated GTA-2A, revealed a shift of the product spectrum towards higher glutarate and lower 5-aminovalerate formation (Fig. 3), but suffered from 20% reduction of the specific growth rate (Table 1). In the same manner, the GTG codon in the gabD gene was replaced by ATG in the GTA-1 strain. The resulting mutant GTA-1 gabDG1A (GTA-2B) accumulated glutarate in a higher yield (Table 1 and Fig. 3), but also revealed an undesired decrease in fitness: its specific growth rate was reduced by almost 50% and the biomass yield was reduced by almost 20%, indicating undesired growth defects. This behavior was taken as an indication that the additional overexpression of only one of the two glutarate pathway genes was not the best approach to further enhance the pathway flux, so that the strains GTA-2A and GTA-2B were not considered further. Moreover, the remaining secretion of the by-product 5-aminovalerate was still an undesired feature.Re-uptake of the secreted intermediate 5-aminovalerate contributes to glutarate formation
An isotope experiment was designed to study the 5-aminovalerate/glutarate metabolism in C. glutamicum in more detail and to identify further possibilities to improve production. Briefly, C. glutamicum AVA-2 was grown on minimal medium with 99% [13C6] glucose, additionally amended with naturally labeled 5-aminovalerate. An incubation without 5-aminovalerate served as a control. GC/MS analysis of the 13C enrichment in glutarate, formed in the two set-ups, provided a direct readout, to which extent the external pathway intermediate was available for the metabolic conversion. When the non-labeled 5-aminovalerate had been added to the medium, the 13C enrichment of glutarate was substantially reduced, as compared to the control (Table 2 and ESI Fig. S2†). The analysis revealed that 53% of glutarate was synthesized from glucose, whereas the remaining fraction (47%) stemmed from the externally supplied 5-aminovalerate. This feature appeared to be beneficial for production from glucose: secreted 5-aminovalerate was still available for the cells to be taken up and channeled to glutarate.| Strain | Conditions | SFL |
|---|---|---|
| AVA-2 | [13C6]-glucose | 0.93 ± 0.00 |
| AVA-2 | [13C6]-glucose + 5-AVA | 0.53 ± 0.00 |
| GTA-3 | [13C6]-glucose + 5-AVA | 0.65 ± 0.00 |
The obviously existing 5-aminovalerate importer protein, however, was not known. On a first glance, the amino acid permease NCgl0464 (cgl0841) appeared as a promising candidate, given its functional assignment to transport γ-amino butyrate,50 which is structurally related to 5-aminovalerate. Moreover, the localization of NCgl0464 immediately downstream of the gabTD operon provided a reason to investigate its role experimentally. First, the deletion of the NCgl0464 gene was realized in the parent AVA-2 producer. A positive clone, which revealed the desired PCR fragment of 1.2 kb and the correctly modified sequence, in contrast to the wild type (2.4 kb), was studied for its production characteristics. The ΔNCgl0464 mutant, designated GTA-3, revealed a substantially reduced re-cycling of external 5-aminovalerate (Table 2 and Fig. S1†). Only about 35% of glutarate stemmed from the re-uptake of 5-aminovalerate, while 65% was synthesized from glucose. In addition, the overall glutarate production was reduced (6 mM), while the 5-aminovalerate level was substantially increased (9 mM), which indicated that the protein NCgl0464 had 5-aminovalerate permease activity. The isotope data also demonstrated that C. glutamicum possesses one or more additional proteins besides NCgl0464 to import 5-aminovalerate. The fortification of 5-aminovalerate re-import emerged as a relevant metabolic engineering target.
The overexpression of the permease NCgl0464 enables almost exclusive glutarate production
The overexpression of the permease NCgl0464 was tackled by inserting a second gene copy under the control of the tuf promoter into the genome of the GTA-1 strain, the best performing strain in the constructed genealogy (Table 1 and Fig. 1E). Positive clones, which carried the desired second gene copy, were identified on the basis of a 2.4 kb PCR fragment, which contained the inserted sequence in contrast to the wild type (1.1 kb). The successful modification was further confirmed by sequencing. The new strain GTA-1 PtufNCgl0464 was designated GTA-4. When tested in a shake flask in a batch culture, it exhibited improved performance and accumulated higher amounts of glutarate (Fig. 2E and F). In addition, GTA-4 formed the product in increased yield (271 mmol mol−1) and productivity (0.9 mmol g−1 h−1) (Table 1). The secretion of 5-aminovalerate was reduced by a factor of more than three, as compared to the GTA-1 strain, to a level of only about 1% of the glutarate formed (Fig. 3). Interestingly, the GTA-4 strain grew faster than its ancestor did. Regarding metabolic engineering, the transport of molecules across cell membranes is regarded crucial, although only relatively few successful cases are described.13 So far, efforts have been focussed on the transport of the final product itself, to drive its excretion out of the cell13,51 or block its re-uptake.52,53 In this regard our strategy, i.e. the amplification of a transport protein for enhanced re-assimilation of a pathway intermediate displays an interesting novel extension, which holds promising potential to be applied to other processes in biotechnology, often associated with the undesired secretion of valuable intermediates.The cell factory C. glutamicum GTA-4 performs well under fed-batch conditions and accumulates more than 90 g L−1 of glutarate within 60 hours
To assess performance under industrially relevant conditions, we benchmarked the GTA-4 strain in a fed-batch process on a glucose-molasses medium (Fig. 4). During the initial batch phase, the strain grew exponentially (Fig. 4A) and accumulated glutarate in a yield of 0.28 mol mol−1 (Fig. 5). After 15 h, the initially supplied sugar (84 g L−1) was depleted and the feed phase was started (Fig. 4A). Sucrose and fructose, contained at a lower level as molasses-based sugars (data not shown), were efficiently consumed during the first 8 hours and remained below the detection limit during the feed phase. In this way, C. glutamicum, equipped with efficient pathways to metabolize sucrose54 and fructose,55 handled the sugar mixture very well, including the high start concentration chosen. Pulses of the concentrated feed were automatically added, when the sugar was exhausted, which was nicely reflected by a sudden increase of the dissolved oxygen (DO) level (Fig. 4B). This correlation allowed a precise and robust control. Previous fermentations with C. glutamicum have shown that the microbe also performs well with other feed regimes such as linear ramps and pulse-wise additions, manually controlled using at-line monitoring of the sugar level.14 The DO-based control chosen here, however, appears particularly robust and does not need external monitoring.36,56 Further on, the glutarate level continuously increased from 15 g L−1 at the end of the batch phase to a final titer of more than 90 g L−1 after 60 hours. The cells produced glutarate exclusively, which provided a strong benefit for the subsequent down-stream processing (see below). The formation of 5-aminovalerate was negligible. A small amount, accumulating during the batch phase, was completely taken up again later (Fig. 4A). Other known by-products such as trehalose were not formed. The molar glutarate yield increased about 2.5-fold during the feed phase to 0.70 mol mol−1, based on consumed sugar (Fig. 5). This value is nearly as high as the theoretical optimum of the pathway (0.75 mol mol−1). Although a slight overestimation cannot be excluded, due to small amounts of yeast extract added (below 3/100 of sugar in feed) and certain levels of non-sugar compounds contained in the molasses, the created cell factory exhibited an enormous synthetic selectivity. The space–time yield for glutarate was maximal during the feed phase (1.8 g L−1 h−1). Averaged over the full process, production occurred at more than the half-maximum rate (1.4 g L−1 h−1).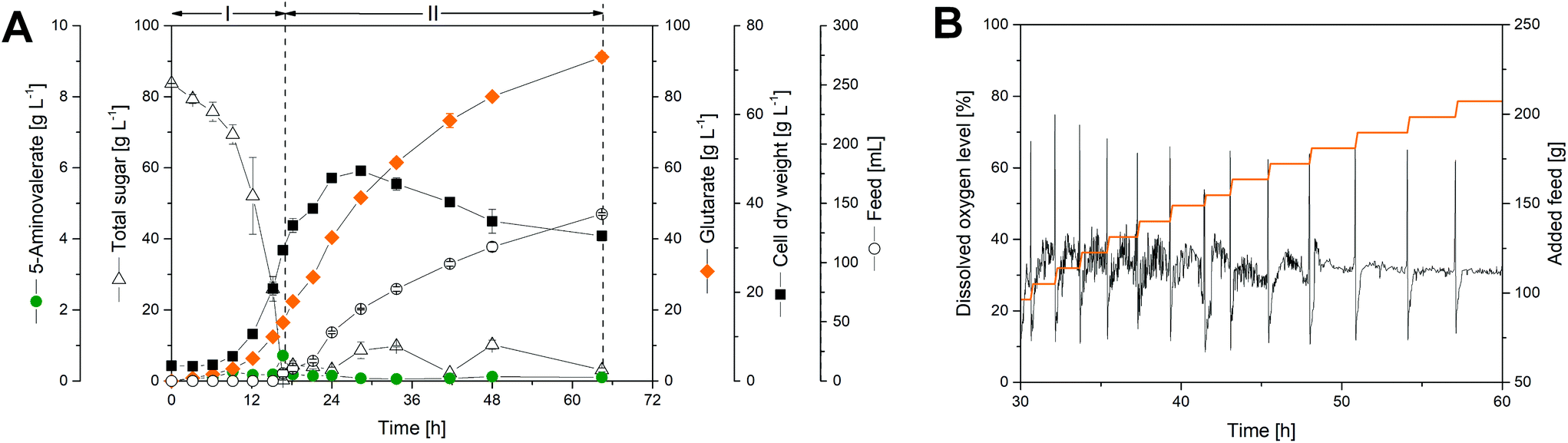 | ||
| Fig. 4 Fed-batch production of glutarate by metabolically engineered Corynebacterium glutamicum GTA-4: culture profile (A), visualization of the automatic process control for a period of 30 hours during the feed phase (B). The substrate is given as total sugar, i.e. the lumped concentration of glucose, sucrose, plus fructose, either added as pure glucose or as molasses-based sugar (sucrose, glucose, fructose). After depletion of the initial sugar at the end of the batch phase, pulses of feed were added automatically, using an increase of the dissolved oxygen (DO) above 45% as a trigger. The data represent mean values and deviations from two replicates. | ||
 | ||
| Fig. 5 Glutarate yield during fed-batch production by metabolically engineered Corynebacterium glutamicum GTA-4. The substrate is given as total sugar, i.e. the lumped concentration of glucose, sucrose, plus fructose, added as pure glucose or as molasses-based sugar (glucose, sucrose, fructose). The molar amount of the sugar reflects hexose units, i.e. one sucrose molecule in the mixture is considered as two hexoses (glucose plus fructose). The corresponding c-molar yields are obtained via multiplication of the molar yield values shown in the plot by a factor of 5/6: 0.24 c-mole c-mole−1 (phase I), and 0.58 c-mole c-mole−1 (phase II). The data represent mean values and deviations from two replicates. | ||
The increase in the glutarate yield was largely enabled by the systematically streamlined metabolism, which included 18 genomics traits that covered the entire route from the substrate to the product: the L-lysine module (12 genomic changes in 4 different carbon core metabolic modules) to drive L-lysine formation from sugar, the 5-aminovalerate module (2 genomic changes) to convert L-lysine into 5-aminovalerate, the glutarate module (2 genomic changes) to drive 5-aminovalerate further to glutarate, and the transport module (2 genomic changes) to optimize inflow and outflow of the pathway intermediates L-lysine and 5-aminovalerate (Fig. 1). In this way the substrate carbon, which became available during the feed phase, when the growth of the cells slowed down and later even ceased and the anabolic demand diminished, could be fully driven to the desired product in a yield close to the theoretical maximum. This demonstrates the power of systems metabolic engineering.4,14,34,36,57–59 In addition, the performance benefited from a good robustness of C. glutamicum, which allowed cell growth up to about 60 g L−1 glutarate (ESI, Fig. S1†) and efficient product formation even at high glutarate levels, once growth slowed down or even stopped (Fig. 4A). Enabled by its systems-wide engineered metabolism and its native robustness, the GTA-4 strain accumulated twenty-fold more glutarate than any other microbe engineered so far.37 Taken together, the developed strain provides glutarate in an attractive yield, titer and productivity and places the developed process into the high-level range of industrial bio-processes, using C. glutamicum for chemical production.14,36,56
Downstream processing of the fermentation broth provides bio-based glutaric acid with more than 99.9% purity
Following fermentation, glutaric acid was recovered from the culture broth by two-step acidification, vacuum evaporation, and crystallization (Fig. 6). After cell separation and pH adjustment to 2.5, the filtrate was concentrated in a vacuum evaporator, thereby precipitating significant fractions of inorganic salts. Crude glutaric acid was obtained by acidification to pH 1.0 and further volume reduction. The different acidification and concentration steps were found crucial to enable glutaric acid crystallization, likely due to the excellent solubility of this organic acid.60 The crude crystals dissolved in acetone, while residual salts, insoluble in the solvent were separated by filtration. Recrystallization, washing and lyophilization, finally yielded a white crystalline powder. The product contained 98.2% pure glutaric acid with no organic impurities, but only traces of phosphate (ESI, Fig. S3A and B†). Alternative washing and recrystallization in chloroform provided the product at >99.9% purity (ESI, Fig. S3C and S4†). Nuclear magnetic resonance (NMR) spectroscopy showed no discernable difference to commercial glutaric acid (ESI, Fig. S5 and S6†). | ||
| Fig. 6 Downstream purification of bio-based glutaric acid and polymerization into nylon-6,5. Crude glutaric acid was recovered from the broth by a two-step acidification and vacuum concentration procedure. Purified glutaric acid was obtained by crystallization. The bio-based glutaric acid was activated with oxalyl chloride and was then used to perform interfacial polycondensation with 1,6-hexamethylene diamine into nylon-6,5. | ||
Bio-based glutaric acid gives access to bio-polyamide PA6.5
Nylons are important industrial polymers displaying good temperature resistance, relatively high moduli, and excellent strength and toughness, but are produced almost exclusively from non-renewable feedstocks. Nylon-6,6, formed by the poly-condensation of adipic acid and hexamethylene diamine, has the highest global production of all polyamides, and shows good all-round performance.7,61 A greener route to nylon-6,6 via bio-adipic acid was recently reported1,62–64 and reflects growing impetus to find renewable pathways to polyamides.14,15,18,64 To demonstrate the potential of (bio-based) glutaric acid as a polyamide precursor, we successfully synthesized nylon-6,5 from the novel bio-based glutaric acid and from the commercial petrochemical for comparison. The interfacial polymerization (the nylon “rope trick”) (Fig. 6) and the industrially relevant melt polymerization approaches (Fig. 5 and Fig. 7) were tested. The obtained polymers were analyzed by size exclusion chromatography (SEC) to determine their molar weight distribution. Thermal properties were characterized by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA).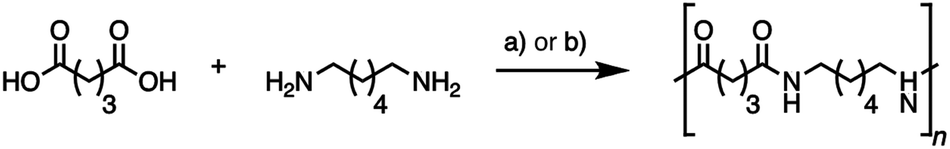 | ||
| Fig. 7 Synthesis of nylon-6,5 by nylon “rope trick” interfacial polymerization via glutaryl chloride (a) and melt polymerization for 9 h under an inert atmosphere (b). | ||
All polymers showed similar characteristics (Table 3): molecular weights around 10.000 g mol−1, polydispersity around 2.7 in interfacial polymerization and 1.8 in melt polymerization, melting temperature at 241–242 °C, crystallization enthalpy around 80 J g−1 and thermal stability up to 330 °C (ESI, Fig. S7, S8, and S9†). A small perturbation at the melting peak onset likely corresponded to the melting and recrystallization of a minor crystalline phase nylon-6,5. No significant differences were observed between the bio-based and the petrochemical based polymer. Bio-based glutaric acid is therefore a viable alternative to chemical glutaric acid for bioplastic synthesis, producing polymers of identical quality. The melt polymer molar masses are comparable to literature values for laboratory-scale melt-phase polyamide syntheses,65 with higher values expected by increasing the reaction time and chain mobility by plasticization with water vapor throughout polymerization.66 It seems that only one single study so far substantially studied nylon-6,5.67 While reporting similar values for viscosity-average molar mass and melting point for their nylon-6,5 made by interfacial polymerization, the authors encountered the same minor endotherm at around 230 °C and attributed this phenomenon to polymer reorganization during heating of smaller crystalline domains, also observed for other nylons.68 The unique structure of nylon-6,5 is a courtesy of its particular hydrogen bonding pattern, but other literature on nylon-6,5 is scarce, suggesting that its properties and potential applications remain largely unexplored. Nylons with fewer carbons per amide bond are more polar and therefore display higher water affinities. While increased water absorption can have unwanted plasticizing effects, more polar nylons can also offer advantages. Nylon-4 for example exhibits breathability, rivalling that of cotton due to its relatively high water affinity69 and nylon-4,6 shows a particularly high crystallinity and melting point (>280 °C) due to a high density of hydrogen bonds.70
| Nylon-6,5 properties | Glutaric acid chemical | Glutaric acid biological | Glutaric acid chemical | Glutaric acid biological |
|---|---|---|---|---|
| Synthesis method | Interfacial polymerization | Interfacial polymerization | Melt polymerization | Melt polymerization |
| Glass transition temperature Tg (°C) | 77 | 69 | 72 | 78 |
| Melting temperature Tm (°C) | 242 | 241 | 242 | 241 |
| Enthalpy of crystallization ΔHc (J g−1) | 79 | 82 | 88 | 79 |
| Number avg. molar mass Mn (g mol−1) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
9900 | 11300 |
10800 |
| Weight avg. molar mass Mw (g mol−1) | 28900 |
26300 |
20700 |
19800 |
| Dispersity | 2.8 | 2.6 | 1.8 | 1.8 |
For future research, nylons with lower carbon to amide ratios are interesting materials in their own right, and glutaric acid is a prime candidate as a precursor for such polymers. Moreover, the novel production route, described here, opens sustainable alternatives to many other products, accessible from glutaric acid.
Conclusions
In this work, we established C. glutamicum as a production platform for the polymer building block glutaric acid through genome manipulation. As shown, we generated a strain with a highly attractive glutarate titer, yield and productivity. The created producer is fully genome based and offers genetic stability without the need for plasmids and antibiotic selection pressure, otherwise incompatible with industrial production. Recent claims that “C. glutamicum has serious limitations to produce glutarate”37 should be revised: the GTA-4 strain accumulates twenty-fold more product than any other microbe engineered so far and displays a milestone towards bio-based glutarate production. Further optimization can be expected by additional rounds of strain analysis, design and genetic engineering,71–74 which will leverage the performance further to industrial efficiency.36,75–77 In addition to molasses, it appears feasible to use other raw materials also. C. glutamicum can be engineered to consume xylose, arabinose, and cellobiose,78–81 which could open the use of lignocellulosic biomass to produce glutarate. Furthermore, the microbe has been recently upgraded to use mannitol, a major constituent of seaweed, which could fuel glutarate production with algal biomass from ocean farms in the future.44 The bionylon-6,5 provides a promising addition to the portfolio of bio-based polyamides for future industrial applications.Experimental
Microorganisms and plasmids
The ATCC 13032-derived C. glutamicum strain AVA-234 was used as a host to establish glutarate production. The wild type ATCC 13032 (American Type Culture Collection, Manassas, VA, USA) was used for tolerance testing. The amplification of transformation vectors on the basis of the integrative plasmid pClik int sacB82,83 was carried out in the Escherichia coli strains DH5α and NM522 (Invitrogen, Carlsbad, CA, USA).36 All strains and plasmids of this study are listed in Table 4.| Strain | Description | Ref. |
|---|---|---|
| AVA-2 | Metabolically engineered producer of 5-aminovalerate and glutarate | 34 |
| GTA-1 | AVA-2 + PtufgabTD encoding 5-aminovalerate transaminase (NCgl0462) and glutarate semialdehyde dehydrogenase (NCgl0463) | This work |
| GTA-2A | GTA-1 + gabTG1A | This work |
| GTA-2B | GTA-1 + gabDG1A | This work |
| GTA-3 | AVA-2 + deletion of the amino acid permease gene NCgl0464 | This work |
| GTA-4 | GTA-1 + genome-based expression of a second copy of NCgl0464 under the control of the tuf promoter | This work |
| Plasmids | Description | Ref. |
|---|---|---|
| pTC | Expression vector for DNA methylation with ORI for E. coli and tetracycline resistance. | 34 |
| pClik int sacB | Integrative transformation vector for C. glutamicum with MCS, ORI for E. coli, and KanR and sacB as selection markers | 82 |
| pClik int sacB tufp-gabTD | Integrative transformation vector for insertion of the tuf promoter upstream of the gabTD operon | This work |
| pClik int sacB gabTG1A | Integrative transformation vector for exchanging the start codon of gabT from GTG to ATG | This work |
| pClik int sacB gabDG1A | Integrative transformation vector for exchanging the start codon of gabD from GTG to ATG | This work |
| pClik int sacB ΔNCgl0464 | Integrative transformation vector for deletion of the amino acid permease NCgl0464 | This work |
| pClik int sacB tufp-NCgl0464 | Integrative vector for insertion of NCgl0464 under the control of the tuf promoter into the crtB locus | This work |
Molecular design and genetic engineering
For molecular design, Clone Manager Professional 9 (Sci-Ed Software, Denver, USA) was used. The genetic construct for genome-based integration of the tuf promoter from C. glutamicum, upstream of the gabT (NCgl0462)/gabD (NCgl0463) operon, comprised (i) 500 bp-sized flanking regions as homologous recombination sites, and (ii) a 200 bp-sized DNA fragment of the promoter of the structural tuf gene (NCgl0480) (Fig. 1). The genetic construct for genome-based expression of a second copy of the permease NCgl0464 comprised (i) 500 bp-sized flanking regions as homologous recombination sites for the crtB locus (NCgl0598), and (ii) a 200 bp-sized DNA fragment of the promoter of the structural tuf gene (NCgl0480) and the permease gene NCgl0464 (Fig. 1). For a replacement of the translational start codon GTG by ATG in the gabT and the gabD gene, respectively, DNA fragments of 550 bp size were amplified upstream and downstream of the start codon of the corresponding gene, whereby the start codon exchange was implemented using modified primers. The amplification and assembly of DNA fragments and the amplification, purification and transformation of plasmid vectors into E. coli and C. glutamicum strains were performed as described previously.34 PCR and sequence analysis (GATC Biotech AG, Konstanz, Germany) were used for plasmid and strain validation. Primer sequences are given in Table 5.| No. | Sequence | AT [°C] |
|---|---|---|
| PRtuf_gabTD_1 | CTGCGTTAATTAACAATTGGCGCTGGAGGTGATCGAGATAAATG | 59 |
| PRtuf_gabTD_2 | CATTCGCAGGGTAACGGCCAGGTTCCTCCTGTGAGGTGAGATAC | 60 |
| PRtuf_gabTD_3 | CTCACCTCACAGGAGGAACCTGGCCGTTACCCTGCGAATG | 62 |
| PRtuf_gabTD_4 | CGGTATGAGAGATCTTCCACTGTATGTCCTCCTGGACTTCGTG | 60 |
| PRtuf_gabTD_5 | GAAGTCCAGGAGGACATACAGTGGAAGATCTCTCATACCGC | 57 |
| PRtuf_gabTD_6 | AATCCCGGGTCTAGAGGATCCCGAATCCGGACTTGTATGG | 57 |
| PRgabT_G1A_1 | CTGCGTTAATTAACAATTGGTGCCAACGGATAAGACCACGCT | 63 |
| PRgabT_G1A_2 | ATCTTCCATTGTATGTCCTCCTGGACTTCGTGGT | 69 |
| PRgabT_G1A_3 | GACATACAATGGAAGATCTCTCATACCGCATCCC | 68 |
| PRgabT_G1A_4 | AATCCCGGGTCTAGAGGATCGGTTCTTCGCGGTCATCGCCAT | 64 |
| PRgabD_G1A_1 | CTGCGTTAATTAACAATTGGCCAGCGACGCAGAAGGTGTGAT | 64 |
| PRgabD_G1A_2 | GTCAAAGACATTTTAGCCCACCTTCTGGTGCG | 68 |
| PRgabD_G1A_3 | GTGGGCTAAAATGTCTTTGACCTTCCCAGTAATC | 68 |
| PRgabD_G1A_4 | AATCCCGGGTCTAGAGGATCTGAGACCAAGCCCTGCGGGATA | 64 |
| PRΔNCgl0464_1 | CTGCGTTAATTAACAATTGGTCGCAGCTAACCGTTTCTTG | 57 |
| PRΔNCgl0464_2 | GGAAGTGACTATGCCCAACCGTGCTGTACCTGCAGCATTG | 58 |
| PRΔNCgl0464_3 | CAATGCTGCAGGTACAGCACGGTTGGGCATAGTCACTTCC | 57 |
| PRΔNCgl0464_4 | AATCCCGGGTCTAGAGGATCAGGCGGTGAGCTGTACTTG | 59 |
| PRtuf_NCgl0464_1 | AATTGGGATCCTCTAGACCCGGTGGCTAGTCCTGCTAGTC | 63 |
| PRtuf_NCgl0464_2 | CATTCGCAGGGTAACGGCCACTCCTATGATCCGACTCAGTTG | 61 |
| PRtuf_NCgl0464_3 | CAACTGAGTCGGATCATAGGAGTGGCCGTTACCCTGCGAATG | 67 |
| PRtuf_NCgl0464_4 | GCAACTATTGATTCGGTAGTCATGTTGTATGTCCTCCTGGACTTC | 60 |
| PRtuf_NCgl0464_5 | GAAGTCCAGGAGGACATACAACATGACTACCGAATCAATAGTTGC | 63 |
| PRtuf_NCgl0464_6 | GAGTGGCCGATGAGGTTGTGGAAGTGACTATGCCCAACCC | 63 |
| PRtuf_NCgl0464_7 | GGGTTGGGCATAGTCACTTCCACAACCTCATCGGCCACTC | 65 |
| PRtuf_NCgl0464_8 | CCGCTAGCGATTTAAATCCCAGCACCAGTGACATTCCTTC | 62 |
Batch cultivation in shake flasks
C. glutamicum was grown in baffled shake flasks with 10% filling volume at 30 °C and 230 rpm on an orbital shaker (Multitron, Infors AG, Bottmingen, Switzerland). The cultivation procedure involved one pre-culture in complex medium (37 g L−1 BHI, Becton Dickinson, Franklin Lakes, NJ, USA) followed by another pre-cultivation in minimal medium.84 The main cultivation was carried out as triplicate in a chemically defined mineral salt medium with glucose as the carbon source.47 The medium contained: (A) 500 mL of a salt stock (1 g NaCl, 55 mg MgCl2·7H2O and 200 mg CaCl2), (B) 100 mL of a substrate stock (100 g L−1 glucose), (C) 100 mL of a buffer stock (2 M potassium phosphate, pH 7.8), (D) 100 mL of an ammonium stock (150 g L−1 (NH4)2SO4, pH 7.0), (E) 20 mL of a vitamin stock (25 mg L−1 biotin, 50 mg L−1 thiamine·HCl, and 50 mg L−1 pantothenic acid), (F) 10 mL of an iron stock (2 g L−1 FeSO4, pH 1.0), (G) 10 mL of a trace element stock,85 (H) 1 mL of a DHB stock (30 mg mL−1 3,4-dihydroxybenzoic acid in 0.3 M NaOH), and 159 mL deionized water. The stock solutions were sterilized by autoclaving (A–D), and by filtration (E–H), respectively, and combined at room temperature freshly before use.Fed-batch cultivation in stirred tank bioreactors
The production performance of the glutarate-producer C. glutamicum GTA-4 was evaluated in a fed-batch process. Fermentation was carried out in a glucose molasses medium using 1 L DASGIP bioreactors (Eppendorf, Jülich, Germany). The initial batch medium (300 mL), contained per liter: 72.4 g L−1 sugar cane molasses (Hansa Melasse, Bremen, Germany), 50 g L−1 glucose, 35 g L−1 yeast extract (Difco), 20 g L−1 (NH4)2SO4, 100 mg L−1 MgSO4, 60 mg L−1 Ca-pantothenate, 18 mg L−1 nicotinamide, 15 mg L−1 thiamine·HCl, 11 mg L−1 FeSO4·7H2O, 10 mg L−1 citrate, 9 mg L−1 biotin, 250 μL H3PO4 (85%) and 5 mL Antifoam 204 (Sigma-Aldrich). The process was inoculated with exponentially growing cells from pre-cultures on BHI medium. Dissolved oxygen-based feeding (500 g L−1 glucose, 162.5 g L−1 sugar cane molasses, 40 g L−1 (NH4)2SO4, 15 g L−1 yeast extract, 2 mL antifoam) was initiated, when the sugar concentration was depleted.Feed shots of 5 mL were automatically added, when the dissolved oxygen level (pO2) increased above 45%. Cultivation temperature and aeration rate were maintained constant at 30 °C and 1 vvm. The pH and the pO2 level were monitored online with a pH electrode (Mettler Toledo, Gießen, Germany) and a pO2 electrode (Hamilton, Höchst, Germany). The pH was kept constant at 7.0 ± 0.05 by automated addition of 10 M NaOH. The dissolved oxygen level was maintained above 30% of saturation by variation of stirrer speed and oxygen content in the gas inflow. Data acquisition and process operations were controlled by the DASGIP control software 4.0 (Eppendorf, Jülich, Germany).
Substrate and product quantification
Glucose, sucrose, fructose, trehalose, and organic acids including glutarate were separated by HPLC (Agilent 1260 Infinity Series, Agilent Technologies, Waldbronn, Germany) on an Aminex HPX-87H column (Bio-Rad, Hercules, California, USA) using 3.5 mM H2SO4 at 1 mL min−1 and 55 °C as the mobile phase. The analytes were quantified using refractive index detection (Agilent 1260 RID G1362A, Agilent Technologies) and external standards.Amino acids and 5-aminovalerate were quantified by HPLC as described before.34 The concentration of the cell dry mass (CDM) was calculated from the optical density, using a correlation factor of CDM [g L−1] = 0.32 × OD660.34
GC-MS analysis of glutaric acid
The labeling pattern of glutaric acid, obtained from the culture supernatant, was analyzed after derivatization into the t-butyl-dimethylsilyl derivative.4 For this purpose, 10 μL culture supernatant was dried under a nitrogen stream, followed by incubation with 50 μL dimethylformamide (0.1% pyrimidine) and 50 μL methyl-t-butyldimethylsilyl-trifluoroacetamide (Macherey and Nagel, Düren, Germany) for 30 minutes at 80 °C. The obtained t-butyldimethylsilyl derivate was analyzed by gas chromatography-mass spectrometry (GC-MS, 7890A, 5975C quadrupole detector, Agilent Technologies, Santa Clara, CA, USA).86 For comparison, commercial glutaric acid was analyzed as a reference standard.Determination of enzyme activities
Crude cell extracts were prepared from exponentially growing cells by mechanical cell disruption. A cultivation and harvest procedure was carried out as previously described.34 Aliquots of 1 mL cell suspension were transferred into FastPrep-24 vials (MP Biomedicals, Illkirch-Graffenstaden, France) containing silica beads (Ø 0.1 mm). Cell disruption was carried out in 2 × 30 s cycles at 5000 rpm (Precellys-24, PeqLab, Hannover, Germany) including 1 minute cooling pauses on ice. Removal of cell debris and protein quantification was performed as previously described.86 The activity of 5-aminovalerate transaminase was assayed in 50 mM Tris hydrochloride buffer (pH 8.5), additionally containing 2 mM 5-aminovalerate, 2 mM α-ketoglutarate, 50 mM KCl, 5 mM MgCl2 100 μM pyridoxal 5-phosphate, and crude cell extract (50 μL mL−1). The reaction mixture was incubated at 30 °C. Samples were regularly taken and thermally inactivated (5 min, 100 °C). The consumption of 5-aminovalerate was quantified by HPLC as given above and used to calculate the specific enzyme activity. The GabD activity was assayed in 100 mM Tris/HCl buffer (pH 8.5), additionally containing 100 mM KCl, 10 mM MgCl2, 2 mM glutarate semialdehyde, and 0.5 mM NAD. The formation of NADH was monitored online at 340 nm and used for activity calculation (ε340,NADH = 6.22 L mmol−1 cm−1). Negative controls were conducted without the addition of crude cell extract and substrate, respectively.Glutaric acid recovery and purification
After biomass separation via centrifugation (10 min, 8500g, 4 °C), the fermentation supernatant was vacuum-filtered (Whatman filter paper, Grade 3, Sigma-Aldrich). The pH of the filtrate was adjusted to pH 2.5, using 37% HCl, followed by volume reduction to about 30% in a vacuum concentrator (15 mbar, 40 °C, 1200 rpm, Christ, Germany). Precipitated inorganic salts were removed (Whatman filter paper, Grade 3, Sigma-Aldrich) and the obtained concentrate was incubated for 1 h with 5% (w/vol) activated carbon. After removal of the activated carbon (Whatman filter paper, Grade 3, Sigma-Aldrich), glutaric acid was crystallized by acidification to pH 1.0 with 85% H3PO4, a further volume reduction to about 10% of the initial volume, and incubation at 4 °C.The obtained crude crystals were dissolved in acetone, while insoluble salts were separated via vacuum filtration (regenerated cellulose, 0.45 μm, Sartorius). After evaporation of the acetone, the obtained crystals were washed with deionized water and were then lyophilized (Christ Gefriertrocknungsanlagen, Osterode am Harz, Germany), which yielded a white crystalline powder. Alternatively, re-crystallization was conducted using chloroform. The purity of glutaric acid was assessed by GC-MS, as described above.
Synthesis of nylon-6,5 by interfacial polymerization
A stock solution of glutaryl chloride was prepared by suspending glutaric acid (1.60 g, 12.1 mmol, 1 equiv.) in a commercial oxalyl chloride solution (2.0 M in dichloromethane (DCM), 13.3 mL, 26.6 mmol, 2.2 equiv.) under nitrogen and adding dry dimethylformamide (DMF, 50 μL) as a catalyst. A bubbler was fitted, and the suspension was stirred at r.t. for 1 h, at which point bubbling had ceased and the reaction was complete. The DCM and unreacted oxalyl chloride were removed under vacuum. The residue was re-dissolved in dry DCM (24.2 mL) under nitrogen to give a 0.5 M solution. 1,6-Hexamethylene diamine (HMDA, 0.352 g, 3.03 mmol, 1 equiv.) was dissolved in 0.5 M NaOH (6.1 mL) to give 0.5 M HMDA.For polymerization, a portion of glutaryl chloride stock solution (6.1 mL, 3.0 mmol, 1 equiv.) was transferred to a small beaker, and the HMDA solution was carefully added on top to avoid phase mixing. The polymer, formed at the interface of the two solutions, was taken with tweezers, and was wound around a glass stirring rod to generate a nylon rope. The resulting nylon-6,5 was washed with deionized water and vacuum dried before characterization.
Synthesis of nylon-6,5 by melt polymerization
Hexamethylene diamine (5.419 g, 46.6 mmol) was dissolved in water (1.35 mL) and a solution of glutaric acid (6.161 g, 46.6 mmol) in water (5.5 mL) was added dropwise with stirring in an ice bath. The resulting nylon-6,5 salt was precipitated in 2-propanol (200 mL) with vigorous stirring, isolated by vacuum filtration, and dried under vacuum. The salt was loaded into a 50 mL Schlenk flask, along with extra HMDA (0.02 equiv.) and a stir bar. The flask was heated to 170 °C in an oil bath under a gentle nitrogen flow for 1.5 h, by which time the reaction medium solidified. The flask was cooled to room temperature, and the solid was crushed with a spatula before heating at 210 °C under vacuum (20 mbar) for a further 7.5 h.Polymer characterization
Size exclusion chromatography was performed with an 1260 Infinity system (Agilent), equipped with a Polymer Standards Service mixed bed column (PSS SDV linear S + 5 μm, separation range 100–150000 g mol−1) as a stationary phase, THF at 24 °C and a flow rate of 1 mL min−1 as a mobile phase, and a RID detector. Calibration was conducted with PMMA standards at 1 mg mL−1 (Mp = 602, 2160, 5980, 12900, 22800, 41400, and 88500 g mol−1). Prior to the analysis, the nylon samples were functionalized overnight with trifluoroacetic anhydride in dry dichloromethane (DCM) to render them soluble. After drying, they were re-dissolved in dry THF at 2 mg mL−1 for analysis.
Thermogravimetric analysis was performed on an STA Jupiter 449 F 3 analyzer (Netzsch, Germany) under an argon atmosphere. A sample of about 5 mg was weighed into an aluminum crucible, and was then heated from room temperature to 600 °C at a rate of 5 °C min−1. Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC 1 (Gießen, Germany) under an argon atmosphere. A sample of about 5 mg was weighed into an aluminum crucible, and was then analyzed by consecutive heating and cooling cycles between 0 °C and 280 °C, with 5 min isotherms between each ramp. After an initial heating and cooling cycle at 20 °C min−1 to erase the thermal history, the glass transition temperature (Tg) and melting temperature (Tm) were determined by heating the sample at a rate of 20 °C min−1, and the enthalpy of crystallization (ΔHc) was determined using a cooling rate of 20 °C min−1. Thermograms were analyzed using STARe (Mettler Toledo) software.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the excellent assistance of Michel Fritz, Robert Drumm, and the INM analysis department for the analytics.References
- M. Kohlstedt, S. Starck, N. Barton, J. Stolzenberger, M. Selzer, K. Mehlmann, R. Schneider, D. Pleissner, J. Rinkel, J. S. Dickschat, J. Venus, J. B. J. H. van Duuren and C. Wittmann, Metab. Eng., 2018, 47, 279–293 CrossRef PubMed.
- N. Barton, L. Horbal, S. Starck, M. Kohlstedt, A. Luzhetskyy and C. Wittmann, Metab. Eng., 2018, 45, 200–2010 CrossRef PubMed.
- J. Becker, A. Lange, J. Fabarius and C. Wittmann, Curr. Opin. Biotechnol, 2015, 36, 168–175 CrossRef PubMed.
- J. Becker, J. Reinefeld, R. Stellmacher, R. Schäfer, A. Lange, H. Meyer, M. Lalk, O. Zelder, G. von Abendroth, H. Schröder, S. Haefner and C. Wittmann, Biotechnol. Bioeng., 2013, 110, 3013–3023 CrossRef PubMed.
- B. J. Harder, K. Bettenbrock and S. Klamt, Metab. Eng., 2016, 38, 29–37 CrossRef PubMed.
- S. Okino, R. Noburyu, M. Suda, T. Jojima, M. Inui and H. Yukawa, Appl. Microbiol. Biotechnol., 2008, 81, 459–464 CrossRef PubMed.
- D. R. Vardon, N. A. Rorrer, D. Salvachua, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- Y. Tsuge, T. Hasunuma and A. Kondo, J. Ind. Microbiol. Biotechnol., 2015, 42, 375–389 CrossRef PubMed.
- J. M. Otero, D. Cimini, K. R. Patil, S. G. Poulsen, L. Olsson and J. Nielsen, PLoS One, 2013, 8, e54144 CrossRef PubMed.
- J. Schneider and V. F. Wendisch, Appl. Microbiol. Biotechnol., 2010, 88, 859–868 CrossRef PubMed.
- S. Kind, W. K. Jeong, H. Schröder and C. Wittmann, Metab. Eng., 2010, 12, 341–351 CrossRef PubMed.
- S. Kind, W. K. Jeong, H. Schröder, O. Zelder and C. Wittmann, Appl. Environ. Microbiol., 2010, 76, 5175–5180 CrossRef PubMed.
- S. Kind, S. Kreye and C. Wittmann, Metab. Eng., 2011, 13, 617–627 CrossRef PubMed.
- S. Kind, S. Neubauer, J. Becker, M. Yamamoto, M. Völkert, G. V. Abendroth, O. Zelder and C. Wittmann, Metab. Eng., 2014, 25, 113–123 CrossRef PubMed.
- S. Kind and C. Wittmann, Appl. Microbiol. Biotechnol., 2011, 91, 1287–1296 CrossRef PubMed.
- Z. G. Qian, X. X. Xia and S. Y. Lee, Biotechnol. Bioeng., 2009, 104, 651–662 Search PubMed.
- Z. G. Qian, X. X. Xia and S. Y. Lee, Biotechnol. Bioeng., 2011, 108, 93–103 CrossRef PubMed.
- K. Imao, R. Konishi, M. Kishida, Y. Hirata, S. Segawa, N. Adachi, R. Matsuura, Y. Tsuge, T. Matsumoto, T. Tanaka and A. Kondo, Bioresour. Technol., 2017, 245, 1684–1691 CrossRef PubMed.
- N. Ikeda, M. Miyamoto, N. Adachi, M. Nakano, T. Tanaka and A. Kondo, AMB Express, 2013, 3, 67 CrossRef PubMed.
- N. E. Altaras and D. C. Cameron, Appl. Environ. Microbiol., 1999, 65, 1180–1185 Search PubMed.
- E. Celinska, Biotechnol. Adv., 2010, 28, 519–530 CrossRef PubMed.
- D. Rados, A. L. Carvalho, S. Wieschalka, A. R. Neves, B. Blombach, B. J. Eikmanns and H. Santos, Microb. Cell Fact., 2015, 14, 171 CrossRef PubMed.
- H. Yim, R. Haselbeck, W. Niu, C. Pujol-Baxley, A. Burgard, J. Boldt, J. Khandurina, J. D. Trawick, R. E. Osterhout, R. Stephen, J. Estadilla, S. Teisan, H. B. Schreyer, S. Andrae, T. H. Yang, S. Y. Lee, M. J. Burk and S. Van Dien, Nat. Chem. Biol., 2011, 7, 445–452 CrossRef PubMed.
- S. Niimi, N. Suzuki, M. Inui and H. Yukawa, Appl. Microbiol. Biotechnol., 2011, 90, 1721–1729 CrossRef PubMed.
- J. Adkins, J. Jordan and D. R. Nielsen, Biotechnol. Bioeng., 2013, 110, 1726–1734 CrossRef PubMed.
- S. J. Park, E. Y. Kim, W. Noh, H. M. Park, Y. H. Oh, S. H. Lee, B. K. Song, J. Jegal and S. Y. Lee, Metab. Eng., 2013, 16, 42–47 CrossRef PubMed.
- J. Wang, Y. Wu, X. Sun, Q. Yuan and Y. Yan, ACS Synth. Biol., 2017, 6, 1922–1930 CrossRef PubMed.
- S. W. Jaros, M. F. Guedes da Silva, M. Florek, P. Smolenski, A. J. Pombeiro and A. M. Kirillov, Inorg. Chem., 2016, 55, 5886–5894 CrossRef PubMed.
- A. F. Baldwin, R. Ma, A. Mannodi-Kanakkithodi, T. D. Huan, C. Wang, M. Tefferi, J. E. Marszalek, M. Cakmak, Y. Cao, R. Ramprasad and G. A. Sotzing, Adv. Mater., 2015, 27, 346–351 CrossRef PubMed.
- S. Trivedi, D. Fluck, A. Sehgal, A. Osborne, M. S. Dahanayake, R. Talingting-Pabalan, J. Ruiz and C. Aymes, US8222194B2, 2008 Search PubMed.
- H. Nagahara, M. Ono and K. Nakagawa, JPS6485937A, 1987 Search PubMed.
- J. Lu, L. Wu and B. G. Li, ACS Sustainable Chem. Eng., 2017, 5, 6159–6166 CrossRef.
- S. J. Park, Y. H. Oh, W. Noh, H. Y. Kim, J. H. Shin, E. G. Lee, S. Lee, Y. David, M. G. Baylon, B. K. Song, J. Jegal, S. Y. Lee and S. H. Lee, Biotechnol. J., 2014, 9, 1322–1328 CrossRef PubMed.
- C. M. Rohles, G. Giesselmann, M. Kohlstedt, C. Wittmann and J. Becker, Microb. Cell Fact., 2016, 15, 154 CrossRef PubMed.
- P. Liu, H. Zhang, M. Lv, M. Hu, Z. Li, C. Gao, P. Xu and C. Ma, Sci. Rep., 2014, 4, 5657 CrossRef PubMed.
- J. Becker, O. Zelder, S. Haefner, H. Schröder and C. Wittmann, Metab. Eng., 2011, 13, 159–168 CrossRef PubMed.
- M. Zhao, G. Li and Y. Deng, Appl. Environ. Microbiol., 2018, 84(16), e00814-18 CrossRef PubMed.
- I. Djurdjevic, O. Zelder and W. Buckel, Appl. Environ. Microbiol., 2011, 77, 320–322 CrossRef PubMed.
- J. Becker and C. Wittmann, Curr. Opin. Biotechnol., 2012, 23, 718–726 CrossRef PubMed.
- L. Eggeling and M. Bott, Appl. Microbiol. Biotechnol., 2015, 99, 3387–3394 CrossRef PubMed.
- O. Revelles, M. Espinosa-Urgel, T. Fuhrer, U. Sauer and J. L. Ramos, J Bacteriol., 2005, 87, 7500–7510 CrossRef PubMed.
- J. P. Vandecasteele and M. Hermann, Eur. J. Biochem., 1972, 31, 80–85 CrossRef PubMed.
- J. H. Shin, S. H. Park, Y. H. Oh, J. W. Choi, M. H. Lee, J. S. Cho, K. J. Jeong, J. C. Joo, J. Yu, S. J. Park and S. Y. Lee, Microb. Cell Fact., 2016, 15, 174 CrossRef PubMed.
- S. L. Hoffmann, L. Jungmann, S. Schiefelbein, L. Peyriga, E. Cahoreau, J. C. Portais, J. Becker and C. Wittmann, Metab. Eng., 2018, 47, 475–487 CrossRef PubMed.
- J. Becker, R. Schäfer, M. Kohlstedt, B. J. Harder, N. S. Borchert, N. Stöveken, E. Bremer and C. Wittmann, Microb. Cell Fact., 2013, 12, 110 CrossRef PubMed.
- J. Becker, N. Buschke, R. Bücker and C. Wittmann, Eng. Life Sci., 2010, 10, 430–438 CrossRef.
- J. Becker, C. Klopprogge, H. Schröder and C. Wittmann, Appl. Environ. Microbiol., 2009, 75, 7866–7869 CrossRef PubMed.
- W. A. van Winden, C. Wittmann, E. Heinzle and J. J. Heijnen, Biotechnol. Bioeng., 2002, 80, 477–479 CrossRef PubMed.
- C. Wittmann, Microb. Cell Fact., 2007, 6, 6 CrossRef PubMed.
- Z. Zhao, J. Y. Ding, W. H. Ma, N. Y. Zhou and S. J. Liu, Appl. Environ. Microbiol., 2012, 78, 2596–2601 CrossRef PubMed.
- M. Li, D. Li, Y. Huang, M. Liu, H. Wang, Q. Tang and F. Lu, J. Ind. Microbiol. Biotechnol., 2014, 41, 701–709 CrossRef PubMed.
- X. Xie, L. Xu, J. Shi, Q. Xu and N. Chen, J. Ind. Microbiol. Biotechnol., 2012, 39, 1549–1556 CrossRef PubMed.
- Y. Li, H. Cong, B. Liu, J. Song, X. Sun, J. Zhang and Q. Yang, Antonie van Leeuwenhoek, 2016, 109, 1185–1197 CrossRef PubMed.
- C. Wittmann, P. Kiefer and O. Zelder, Appl. Environ. Microbiol., 2004, 70, 7277–7287 CrossRef PubMed.
- P. Kiefer, E. Heinzle, O. Zelder and C. Wittmann, Appl. Environ. Microbiol., 2004, 70, 229–239 CrossRef PubMed.
- J. Becker, M. Kuhl, M. Kohlstedt, S. Starck and C. Wittmann, Microb. Cell Fact., 2018, 17, 115 CrossRef PubMed.
- A. L. Rodrigues, J. Becker, A. O. de Souza Lima, L. M. Porto and C. Wittmann, Biotechnol. Bioeng., 2014, 111, 2280–2289 CrossRef PubMed.
- I. Poblete-Castro, D. Binger, A. Rodrigues, J. Becker, V. A. Martins Dos Santos and C. Wittmann, Metab. Eng., 2013, 15, 113–123 CrossRef PubMed.
- A. L. Rodrigues, N. Trachtmann, J. Becker, A. F. Lohanatha, J. Blotenberg, C. J. Bolten, C. Korneli, A. O. de Souza Lima, L. M. Porto, G. A. Sprenger and C. Wittmann, Metab. Eng., 2013, 20, 29–41 CrossRef PubMed.
- M. Z. H. Rozaini and P. Brimblecombe, J. Chem. Thermodyn., 2009, 41, 980–983 CrossRef.
- C. W. Johnson, D. Salvachua, P. Khanna, H. Smith, D. J. Peterson and G. T. Beckham, Metab. Eng. Commun., 2016, 3, 111–119 CrossRef PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- C. W. Johnson, P. E. Abraham, J. G. Linger, P. Khanna, R. L. Hettich and G. T. Beckham, Metab. Eng. Commun., 2017, 5, 19–25 CrossRef PubMed.
- T. Polen, M. Spelberg and M. Bott, J. Biotechnol., 2013, 167, 75–84 CrossRef PubMed.
- O. Turunc, M. Firdaus, G. Klein and M. A. R. Meier, Green Chem., 2012, 14, 2577–2583 RSC.
- E. Roerdink and J. M. M. Warnier, Polymer, 1985, 26, 1582–1588 CrossRef.
- E. Navarro, L. Franco, J. A. Subirana and J. Puiggali, Macromolecules, 1995, 28, 8742–8750 CrossRef.
- C. Millot, L. A. Fillot, O. Lame, P. Sotta and R. Seguela, J. Therm. Anal. Calorim., 2015, 122, 307–314 CrossRef.
- K.-S. Kang, Y.-K. Hong, Y. J. Kim and J. H. Kim, Fibers Polym., 2014, 15, 1343–1348 CrossRef.
- E. Roerdink and J. M. M. Warnier, Polymer, 1985, 26(10), 1582–1588 CrossRef.
- J. Nielsen, Annu. Rev. Biochem., 2017, 86, 245–275 CrossRef PubMed.
- K. Campbell, J. Xia and J. Nielsen, Trends Biotechnol., 2017, 35, 1156–1168 CrossRef PubMed.
- M. D. Lynch, Curr. Opin. Biotechnol., 2016, 38, 106–111 CrossRef PubMed.
- A. Lange, J. Becker, D. Schulze, E. Cahoreau, J.-C. Portais, S. Haefner, H. Schröder, J. Krawczyk, O. Zelder and C. Wittmann, Metab. Eng., 2017, 44, 198–212 CrossRef PubMed.
- S. Y. Lee and J. H. Park, Adv. Biochem. Eng./Biotechnol., 2010, 120, 1–19 Search PubMed.
- J. Becker and C. Wittmann, Curr. Opin. Microbiol., 2018, 45, 180–188 CrossRef PubMed.
- J. Ohnishi, S. Mitsuhashi, M. Hayashi, S. Ando, H. Yokoi, K. Ochiai and M. Ikeda, Appl. Microbiol. Biotechnol., 2002, 58, 217–223 CrossRef PubMed.
- H. Kawaguchi, A. A. Vertes, S. Okino, M. Inui and H. Yukawa, Appl. Environ. Microbiol., 2006, 72, 3418–3428 CrossRef PubMed.
- S. S. Yim, J. W. Choi, S. H. Lee and K. J. Jeong, ACS Synth. Biol., 2016, 5, 334–343 CrossRef PubMed.
- N. Buschke, H. Schröder and C. Wittmann, Biotechnol. J., 2011, 6, 306–317 CrossRef PubMed.
- M. Sasaki, T. Jojima, H. Kawaguchi, M. Inui and H. Yukawa, Appl. Microbiol. Biotechnol., 2009, 85, 105–115 CrossRef PubMed.
- J. Becker, C. Klopprogge, O. Zelder, E. Heinzle and C. Wittmann, Appl. Environ. Microbiol., 2005, 71, 8587–8596 CrossRef PubMed.
- S. Kind, J. Becker and C. Wittmann, Metab. Eng., 2013, 15, 184–195 CrossRef PubMed.
- J. Becker, C. Klopprogge and C. Wittmann, Microb. Cell Fact., 2008, 7, 8 CrossRef PubMed.
- J. J. Vallino and G. Stephanopoulos, Biotechnol. Bioeng., 1993, 41, 633–646 CrossRef PubMed.
- N. Buschke, J. Becker, R. Schäfer, P. Kiefer, R. Biedendieck and C. Wittmann, Biotechnol. J., 2013, 8, 557–570 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc01901k |
| This journal is © The Royal Society of Chemistry 2018 |