
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclic allylic carbonates as a renewable platform for protecting chemistry in water†
Peter
Olsén
 *a,
Jennifer
Morvan
a,
Supaporn
Sawadjoon
a,
Andrey
Shatskiy
a,
Eric V.
Johnston
*b and
Björn
Åkermark
*a
*a,
Jennifer
Morvan
a,
Supaporn
Sawadjoon
a,
Andrey
Shatskiy
a,
Eric V.
Johnston
*b and
Björn
Åkermark
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE 106 91, Stockholm, Sweden. E-mail: polsen@kth.se; bjorn.akermark@su.se
bSigrid Therapeutics AB, SE 111 52 Stockholm, Sweden. E-mail: eric@sigridthx.com
First published on 14th June 2018
Abstract
The present work explores different cyclic allylic carbonates as a potential class of allylcarbamate precursors. The 5-membered carbonate formed a carbamate with very good thermal and pH stability, which could be cleanly deprotected in aqueous solution, in just 30 min with 2 mol% Pd(OAc)2 as catalyst. The polar nature of the installed motif made it possible to deprotect highly unpolar substrates in water as solvent.
Introduction
The ability to veil and unveil the functional groups of molecules constitutes a fundamental strategy in organic synthesis.1 Different protecting groups, with strict and clear removal boundaries, make it possible to synthesize some of nature's macromolecules.2 Similarly, the creation of protecting groups with functional handles, more commonly referred to as cleavable linkers, permits the introduction of more versatile functions, ranging from support linkers to highly refined luminescent probes.3 Although protecting groups are indispensable in both chemistry and biology, there are fairly few green alternatives.4The question we raised was, whether it would be possible to develop a new class of amine protecting groups from a renewable origin, which has the potential to be readily cleaved off. Our hypothesis was that a specific alkene functionalized cyclic carbonate could be used to achieve this.
Cyclic carbonates exist in many different ring-sizes with a vast array of different functionalities. One useful feature of a ring opening reaction is that it leads to the formation of a chemically active chain end, which can be used in further chemical reactions. The ring-opening reaction between cyclic carbonates and amine nucleophiles has been shown to work effectively in a wide range of organic solvents and under neat conditions with many different catalytic systems.5 Our contribution is that we have shown that it is possible to ring-open cyclic carbonates selectively with unprotected amino acids in water.5h This feature and the diversity in accessible cyclic carbonate motifs provide ample opportunities to specifically tailor a protecting group with the desired properties.6 The work presented takes a holistic view of the role and properties of cyclic carbonates, both as precursors and in the product carbamate.
Results and discussion
Equilibrium behaviour and its connection to synthesis
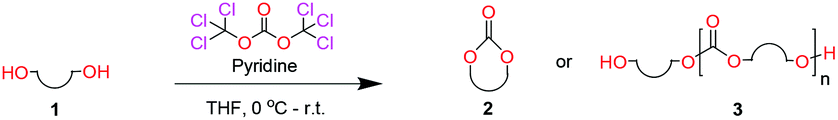
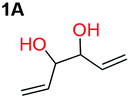
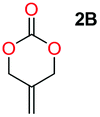
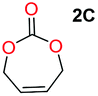
When installing a protection group originating from a cyclic precursor it is important that the system leads to mono-addition only and avoids oligomerization. The ideal cyclic substrate should be thermodynamically stable under standard conditions and form a new thermodynamically stable product upon ring opening. Generally, five membered cyclic carbonates will be the most thermodynamically stable. However, the equilibrium between open and closed rings may change with different ring substituents.7That the ring size is the most important factor is nicely demonstrated by the properties of the cyclic carbonates 2A–2C (Table 1). The equilibria were measured under the same ring closing conditions, at 2 M concentration of diol with 0.5 equiv. of triphosgene at an initial temperature of 0 °C for 2 h, followed by r.t. for 6 h (Table 1). Under these conditions, 4,5-divinyl-1,3-dioxolan-2-one (Table 1, compound 2A) was obtained in a quantitative yield, in contrast to 4,7-dihydro-1,3-dioxepin-2-one (Table 1 compound 2C) where only oligomers were formed (for more details see ESI Fig. S1 and S2†).8 These results suggest that only the cyclic carbonate 2A is a potential protecting group precursor because of its stability towards oligomerisation. Accordingly, the equilibrium features of 2A made it possible to develop a robust, green and scalable protocol for its synthesis (Scheme 1). Thermodynamics makes it possible to ring-close the diol under quite harsh conditions, using what is referred to as either ring-closing depolymerization or the Braun method.9 The diol can be synthesized through the pinacol coupling of acrolein, which in turn may be derived from glycerol, providing an overall green route to this building block.10 The target compound 2A was obtained by a one-pot, two step synthetic operation (using diethyl carbonate as the carbonyl donor) at an overall yield of 70% after purification by simple distillation (Scheme 1).
 | ||
| Scheme 1 Synthetic route towards 4,5-divinyl-1,3-dioxolan-2-one. | ||


Thermal and pH stability window
An important aspect of protecting groups and cleavable linkers is that they exhibit both great stability under a wide range of conditions and clearly defined cleavage boundaries. To determine the stability, ring opened product 4 of 4,5-divinyl-1,3-dioxolan-2-one was studied at different temperatures and pH values (for more synthetic details see the ESI†).Glycine was selected as a suitable nucleophile for the main reason that the formed carbamate 4 is highly soluble in water. The degradation occurred mainly by two pathways, direct hydrolysis or ring closure, processes which are both increased in polar environments.11 Pleasingly, 4 displayed high thermal stability in DMSO-d6 at both 50 °C and 100 °C, with only 20% degradation after 20 h at 100 °C (Fig. 1a). For comparison, the ring-opened product 5, from the reaction between glycine and ethylene carbonate, was also studied. The thermal stability of 5 was even higher, with 10% degradation after 20 h at 100° (Fig. 1b). The difference is interpreted as a combination of the pKa of the alkoxy next to the carbamate and the Thorpe–Ingold effect exerted by the external substituents.12 The high thermal stability of 4 is in sharp contrast to the classical Fmoc-protection group that is completely removed in just 15 minutes at 120 °C in DMSO.13
 | ||
| Fig. 1 Thermal stability at 50 °C and 100 °C in DMSO-d6 at a concentration of 0.16 M of both 4 (a) and 5 (b). | ||
To get a clearer picture of pH stability, the degradation of 4 and 5 was analysed as a function of both pH and time (Fig. 2). It was found that 4 is quite stable between pH 1 to 10, with a moderate degradation rate between pH 10 to 12.5; however, at pH 14, fast degradation occurred and only 20% remained after 6 h (Fig. 2b). Compound 5 was slightly more stable than 4 under the same conditions.
 | ||
| Fig. 2 pH stability as a function of time and pH in D2O at a concentration of 0.16 M of both 4 (a) and 5 (b). | ||
Deprotection behavior
We envisioned the creation of a robust protocol for the selective removal of our protecting group, without the necessity of inert or dry conditions. Chemical motifs similar to 4 are susceptible to transition metal catalysed nucleophilic substitution.8a,14 In the initial experiments we used triethylamine, TEA, 6 as a nucleophile. At room temperature, using 2% palladium catalyst, the deprotection was very slow but at 100° C deprotection was complete after 2 h; Table 2, entries 1–3. Under these conditions the simple carbamate 5 was completely stable. It was found that addition of a more nucleophilic amine, piperazine (7), led to a considerable increase in the rate to give complete deprotection, 2 h at 75° or 0.5 h at 100° (Fig. 4b and Table 2, entries 5 and 6). By contrast, the addition of less nucleophilic bases, such as pyridine and 4-hydroxyaniline, had no effect (Fig. 4 and ESI, Table S2†). | ||
| Fig. 3 Initial rate dependence as a function of mol% Pd(OAc)2 in the reaction mixture (a), revealing a second order dependence in [Pd] (b). | ||
 | ||
| Fig. 4 Deprotection conversion of 4 in the presence of different allyl scavengers after 2 h at r.t., (b) deprotection conversion as a function of temperature between alloc-GLY-OH and 3 under identical condition after 2 h. All deprotections were performed in 0.16 M concentration of substrate in D2O with 2 mol% of Pd(OAC)2 and 6 equiv. of TEA. In the case of (b) additional 3 equiv. of piperazine was added. | ||
|
|
||||
|---|---|---|---|---|
| Entry | 1 (M) | Temp. (°C) |
4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:7:8 :6:7:8 |
Yield, 9 [%] |
| Reaction conditions: 1 equiv. of 4 with 2 mol% of [Pd] with 6 equiv. of 6 and 3 equiv. of 7 in D2O at different temperatures.a After 0.5 h.b After 2 h.c Pd/C basic support.d Major deprotection found was the corresponding diol.e After 24.5 h. | ||||
| 1 | 0.16 | 50 | [1.0]:[6.0]:[0.02]:[0.0] |
3a(13)b |
| 2 | 0.16 | 75 | [1.0]:[6.0]:[0.02]:[0.0] |
28(40) |
| 3 | 0.16 | 100 | [1.0]:[6.0]:[0.02]:[0.0] |
32(100) |
| 4 | 0.16 | 50 | [1.0]:[6.0]:[0.02]:[3.0] |
13(36) |
| 5 | 0.16 | 75 | [1.0]:[6.0]:[0.02]:[3.0] |
67(100) |
| 6 | 0.16 | 100 | [1.0]:[6.0]:[0.02]:[3.0] |
100(100) |
| 7 | 0.16 | 50 | [1.0]:[6.0]:[0.02]c:[3.0] |
4(13)d |
| 8 | 0.16 | 75 | [1.0]:[6.0]:[0.02]c:[3.0] |
10(60)d |
| 9 | 0.16 | 100 | [1.0]:[6.0]:[0.02]c:[3.0] |
14(68)d |
| 10 | 0.16 | 50 | [1.0]:[6.0]:[0.50]c:[3.0] |
9(91e)d |
| 11 | 0.16 | 50 | [1.0]:[6.0]:[1.0]c:[3.0] |
18(92e)d |

In order to study the influence of the catalyst concentration, a series of kinetic experiments were performed. It was found that at very high catalyst concentration, 13 mol% at ambient temperature in water under air, with 5.4 equiv. of TEA, complete deprotection was observed after 24 h, whereas the carbamate 5 was stable, as might be expected (see the ESI, Table S1†). The major species formed, besides free glycine, was the positively charged TEA adduct (for more details see the ESI Fig. S20, S23–S25†).15
In an extensive kinetic study, the highest loading of Pd(OAc)2, 38 mol% with 5.4 equiv. of TEA, resulted in complete deprotection after 1.5 hours (for more details see the ESI Table S1† entry 11). When the initial loading was decreased from 38 mol% to 6 mol% under identical conditions, the initial rate of removal was decreased by two orders of magnitude but useful rates were still obtained with only 2 mol% catalyst by increasing the temperature (Fig. 3 and Table 2). It was found that deprotection followed a second-order dependence on Pd(OAc)2 (Fig. 3b). This suggests that the active catalyst here is a dinuclear palladium complex.
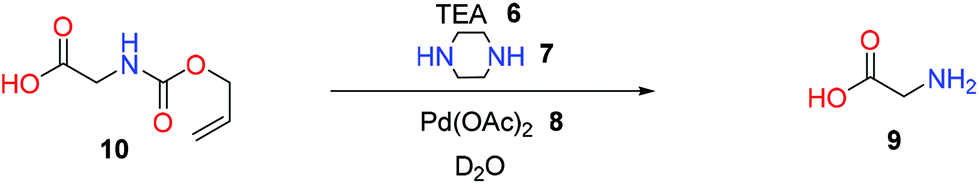
Clear differences in removal behaviour were seen between 4 and the parent allyl carbamate 10 (Table 3 entries 1–3). Specifically, reaction at 50 °C yielded only 2% removal for compound 10 compared to 36% for 4 under identical conditions. Also commercial Pd/C could be used for efficient deprotection; see Table 2 entries 7–11.
|
|
||||
|---|---|---|---|---|
| Entry | 1 (M) | Temp. (°C) |
10:6:7:8 |
Yield. 9[%] |
| Reaction conditions: 1 equiv. of 10 with 2 mol% of [Pd] with 6 equiv. of 6 and 3 equiv. of 7 in D2O at different temperature.a After 0.5 h.b After 2 h. | ||||
| 1 | 0.16 | 25 | [1.0]:[6.0]:[0.02]:[3.0] |
0a(2)b |
| 2 | 0.16 | 75 | [1.0]:[6.0]:[0.02]:[3.0] |
7(33) |
| 3 | 0.16 | 100 | [1.0]:[6.0]:[0.02]:[3.0] |
16(56) |
The generality of divinyl carbonate (DVC) (2A) as a precursor to the protected amino-group and the unmasking behaviour in water was studied with four different substrates (Fig. 5). The substrates were selected on the basis that they would have very different solubilities in water. Poor solubility in an aqueous environment was observed for compounds 12 and 13.
 | ||
| Fig. 5 Deprotection behaviors of different substrates in D2O at 75 °C with 2 mol% of Pd(OAC)2, 3 equiv. of piperazine and 6 equiv. of TEA. | ||
The reactions were performed in D2O at 75 °C at a substrate concentration of 0.16 M and 2 mol% Pd(OAc)2 with 6 equiv. of TEA and 3 equiv. of piperazine. To our delight all substrates were unmasked under these conditions (Fig. 5).
In the case of 4, 11 and 12 nearly complete removal was achieved within 2 h; however, in the case of substrate 13, 6 h were required. The ease of unmasking these substrates in water is attributed to the polar nature of the ring-opened product of 2A. In all cases the same major cationically charged TEA-adduct was also formed.
Conclusions
Ring size dictates the equilibrium between cyclic carbonates and oligomers. Under the same reaction conditions, the five membered cyclic carbonate ring, 4,5-divinyl-1,3-dioxolan-2-one, was formed in quantitative yield, whereas the attempt to prepare the seven membered analogue, 4,7-dihydro-1,3-dioxepin-2-one, gave only oligomers. The stability of 4,5-divinyl-1,3-dioxolan-2-one enabled the design of a one-pot, two step synthetic protocol, which gave 70% overall yield, based on abundant and green reactants, with only distillation as a final purification step.The installed protecting group was found to have excellent thermal and pH stability. In the pH range of 1 to 10 the carbamates were almost inert, even after three days. However, at more basic pH moderate to high degradation was observed. Additionally, even at 100 °C for 20 h only 20% loss of the protecting group was observed.
The unmasking was performed under air, in water, with catalytic amounts of Pd(OAc)2. Detailed kinetic evaluation revealed a second-order dependence in Pd(OAc)2. It was found that addition of the allyl scavenger piperazine increased the rate of removal. This, together with increase in temperature, permitted full deprotection in just 0.5 h with 2 mol% of Pd(OAc)2 as catalyst.
The generality of DVC as a protecting group was shown with four different substrates with different polarities. Even though two of the substrates were poorly soluble under the investigated reaction conditions, clean removal of the protecting group was achieved within 6 h with a substrate concentration of 0.16 M and 2 mol% Pd(OAc)2 as catalyst.
The results presented here are believed to provide a starting point for a renewable and inexpensive protection building-block with potential applications in many facets of chemistry and biology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swedish Research Council (621-2013-4872 and 621-2013-4868) and the Knut and Alice Wallenberg foundation is gratefully acknowledged. We gratefully acknowledge Dr Oscar Verho for fruitful discussions.Notes and references
- (a) P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed; (b) A. Isidro-Llobet, M. Alvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef PubMed.
- For seminal contributions and applications of protection chemistry, see: (a) R. B. Merrifield, Angew. Chem., Int. Ed. Engl., 1985, 24, 799–892 CrossRef; (b) R. N. Zuckermann, J. M. Kerr, W. H. Moosf and S. B. H. Kent, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef; (c) Z. J. Kamiński, P. Paneth and J. Rudziński, J. Org. Chem., 1998, 63, 4248–4255 CrossRef; (d) O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef PubMed; (e) C. M. Stevens and R. Watanabe, J. Am. Chem. Soc., 1950, 72, 725–727 CrossRef; (f) L. A. Carpino, J. Am. Chem. Soc., 1957, 79, 4427–4431 CrossRef; (g) G. W. Anderson and A. C. McGregor, J. Am. Chem. Soc., 1957, 79, 6180–6183 CrossRef; (h) L. A. Carpino and G. Y. Han, J. Am. Chem. Soc., 1970, 92, 5748–5749 CrossRef; (i) L. A. Carpino and G. Y. Han, J. Org. Chem., 1972, 37, 3404–3409 CrossRef; (j) H. Kunz and C. Unverzagt, Angew. Chem., Int. Ed. Engl., 1984, 23, 436–437 CrossRef; (k) F. S. Gibson, S. C. Bergmeier and H. Rapoport, J. Org. Chem., 1994, 59, 3216–3218 CrossRef.
- For examples on linker chemistry, see: (a) K. S. Lam, M. Lebl and V. Krchňák, Chem. Rev., 1997, 97, 411–444 CrossRef PubMed; (b) F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 3859 CrossRef PubMed; (c) E. E. Swayze, Tetrahedron Lett., 1997, 38, 8465–8468 CrossRef; (d) B. Sauerbrei, V. Jungmann and H. Waldmann, Angew. Chem., Int. Ed., 1998, 37, 1143–1146 CrossRef; (e) Y. Kwon, M. A. Coleman and J. A. Camarero, Angew. Chem., Int. Ed., 2006, 45, 1726–1729 CrossRef PubMed; (f) N. Kotagiri, D. M. Niedzwiedzki, K. Ohara and S. Achilefu, Angew. Chem., Int. Ed., 2013, 52, 7756–7760 CrossRef PubMed; (g) H.-Z. He, K.-H. Leung, W. Wang, D. S.-H. Chan, C.-H. Leung and D.-L. Ma, Chem. Commun., 2014, 50, 5313–5315 RSC.
- (a) J. V. Staros, R. W. Wright and D. M. Swingle, Anal. Biochem., 1986, 156, 220–222 CrossRef PubMed; (b) A. S. Galanis, F. Albericio, M. Grøtli and M. Grotli, Org. Lett., 2009, 11, 4488–4491 CrossRef PubMed; (c) C. M. Gabriel, M. Keener, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 3968–3971 CrossRef PubMed; (d) B. H. Lipshutz, J. Org. Chem., 2017, 82, 2806–2816 CrossRef PubMed; (e) S. B. Lawrenson, R. Arav and M. North, Green Chem., 2017, 19, 1685–1691 RSC; (f) M. Cortes-Clerget, J.-Y. Berthon, I. Krolikiewicz-Renimel, L. Chaisemartin and B. H. Lipshutz, Green Chem., 2017, 19, 4263–4267 RSC.
- (a) M. Blain, L. Jean-Gérard, R. Auvergne, D. Benazet, S. Caillol and B. Andrioletti, Green Chem., 2014, 16, 4286 RSC; (b) V. M. Lombardo, E. A. Dhulst, E. K. Leitsch, N. Wilmot, W. H. Heath, A. P. Gies, M. D. Miller, J. M. Torkelson and K. A. Scheidt, Eur. J. Org. Chem., 2015, 2015, 2791–2795 CrossRef; (c) W. Guo, J. Gõnzalez-Fabra, N. A. G. Bandeira, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed., 2015, 54, 11686–11690 CrossRef PubMed; (d) S. Sopeña, V. Laserna, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 2172–2178 CrossRef; (e) M. Blain, H. Yau, L. Jean-Gérard, R. Auvergne, D. Benazet, P. R. Schreiner, S. Caillol and B. Andrioletti, ChemSusChem, 2016, 9, 2269–2272 CrossRef PubMed; (f) M. Alves, R. Méreau, B. Grignard, C. Detrembleur, C. Jérôme and T. Tassaing, RSC Adv., 2017, 7, 18993–19001 RSC; (g) A. Cornille, M. Blain, R. Auvergne, B. Andrioletti, B. Boutevin and S. Caillol, Polym. Chem., 2017, 8, 592–604 RSC; (h) P. Olsén, M. Oschmann, E. Johnston and B. Åkermark, Green Chem., 2018, 20, 469–475 RSC.
- (a) S. H. Pyo and R. Hatti-Kaul, Adv. Synth. Catal., 2016, 358, 834–839 CrossRef; (b) M. Selva, A. Caretto, M. Noè and A. Perosa, Org. Biomol. Chem., 2014, 12, 4143 RSC; (c) P. Olsén, K. Odelius and A.-C. Albertsson, Macromolecules, 2014, 47, 6189–6195 CrossRef; (d) D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef.
- (a) K. J. Ivin, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2137–2146 CrossRef; (b) A. Duda and S. Penczek, ACS Symp. Ser., 2000, 160–198 Search PubMed; (c) P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- Examples in the literature on applications of the investigated cyclic carbonates; (a) B. M. Trost and A. Aponick, J. Am. Chem. Soc., 2006, 128, 3931–3933 CrossRef PubMed; (b) B. M. Trost, A. Aponick and B. N. Stanzl, Chem. – Eur. J., 2007, 13, 9547–9560 CrossRef PubMed; (c) B. M. Trost, B. M. O'Boyle, W. Torres and M. K. Ameriks, Chem. – Eur. J., 2011, 17, 7890–7903 CrossRef PubMed; (d) B. M. Trost, A. C. Burns and T. Tautz, Org. Lett., 2011, 13, 4566–4569 CrossRef PubMed; (e) T. Takata, M. Igarashi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 781–784 CrossRef; (f) F. Sanda, T. Fueki and T. Endo, Macromolecules, 1999, 32, 4220–4224 CrossRef.
- (a) W. Carothers and F. Natta, J. Am. Chem. Soc., 1930, 52, 314–326 CrossRef; (b) R. A. Braun, J. Org. Chem., 1963, 28, 1383–1384 CrossRef; (c) P. Olsén, K. Odelius, H. Keul and A.-C. Albertsson, Macromolecules, 2015, 48, 1703–1710 CrossRef PubMed; (d) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef; (e) K. Tomishige, H. Yasuda, Y. Yoshida, M. Nurunnabi, B. Li and K. Kunimori, Green Chem., 2004, 6, 206 RSC; (f) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC; (g) J. Ma, J. Song, H. Liu, J. Liu, Z. Zhang, T. Jiang, H. Fan and B. Han, Green Chem., 2012, 14, 1743 RSC; (h) M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef.
- (a) R. Hekmatshoar, I. Yavari, Y. S. Beheshtiha and M. M. Heravi, Monatsh. Chem., 2001, 691, 689–669 CrossRef; (b) A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC; (c) N. Sotto, C. Cazorla, C. Villette, M. Billamboz and C. Len, ACS Sustainable Chem. Eng., 2016, 4, 6996–7003 CrossRef.
- (a) M. L. Bender and W. A. Glasson, J. Am. Chem. Soc., 1959, 81, 1590–1597 CrossRef; (b) P. Olsén, J. Undin, K. Odelius, H. Keul and A. C. Albertsson, Biomacromolecules, 2016, 17, 3995–4002 CrossRef PubMed.
- (a) S. Takahashi, L. A. Cohen, H. K. Miller and E. G. Peake, J. Org. Chem., 1971, 36, 1205–1209 CrossRef; (b) M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef PubMed.
- S. Höck, R. Marti, R. Riedl and M. Simeunovic, Chimia Int. J. Chem., 2010, 64, 200–202 CrossRef.
- (a) F. Guibe and Y. Saint M′leux, Tetrahedron Lett., 1981, 22, 3591–3594 CrossRef; (b) J. E. Gómez, W. Guo and A. W. Kleij, Org. Lett., 2016, 18, 6042–6045 CrossRef PubMed; (c) A. Behr and J. Leschinski, Green Chem., 2009, 11, 609 RSC; (d) T. Nishikata and B. H. Lipshutz, Org. Lett., 2009, 11, 2377–2379 CrossRef PubMed; (e) S. C. Zheng, M. Zhang and X. M. Zhao, Chem. – Eur. J., 2014, 20, 7216–7221 CrossRef PubMed.
- (a) G. Tao, L. He, W. Liu, L. Xu, W. Xiong, T. Wang and Y. Kou, Green Chem., 2006, 8, 639 RSC; (b) M. Zawadzki, F. A. e Silva, U. Domańska, J. A. P. Coutinho and S. P. M. Ventura, Green Chem., 2016, 18, 3527–3536 RSC; (c) S. Bhattacharyya and F. U. Shah, ACS Sustainable Chem. Eng., 2016, 4, 5441–5449 CrossRef; (d) T. A. Faßbach, N. Gösser, F. O. Sommer, A. Behr, X. Guo, S. Romanski, D. Leinweber and A. J. Vorholt, Appl. Catal., A, 2017, 543, 173–179 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional kinetic data, experimental details, NMR spectra and mass data. See DOI: 10.1039/c8gc01622d |
| This journal is © The Royal Society of Chemistry 2018 |