
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported gold- and silver-based catalysts for the selective aerobic oxidation of 5-(hydroxymethyl)furfural to 2,5-furandicarboxylic acid and 5-hydroxymethyl-2-furancarboxylic acid†
Oliver R.
Schade
ab,
Kai F.
Kalz
ab,
Dominik
Neukum
b,
Wolfgang
Kleist
 c and
Jan-Dierk
Grunwaldt
*ab
c and
Jan-Dierk
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology, KIT, 76344 Eggenstein-Leopoldshafen, Germany
cIndustrial Chemistry – Nanostructured Catalyst Materials, Ruhr-University Bochum, 44801 Bochum, Germany
First published on 3rd July 2018
Abstract
The sustainable synthesis of two important intermediates relevant for the production of bio-based polymers, 2,5-furandicarboxylic acid (FDCA) and 5-hydroxymethyl-2-furancarboxylic acid (HFCA), via oxidation of 5-(hydroxymethyl)furfural (HMF) was investigated using supported gold- and silver-based catalysts in water with air as the oxidant. High yields and selectivities for the production of FDCA (89%) and HFCA (≥98%) were achieved under the optimized reaction conditions with Au/ZrO2 and Ag/ZrO2 catalysts, respectively. While FDCA was mainly formed in the presence of gold catalysts at a maximum productivity of 67 molFDCA h−1 molAu−1, silver catalysts showed a remarkably high activity in aldehyde oxidation producing HFCA in almost quantitative yields with a maximum productivity of 400 molHFCA h−1 molAg−1. By variation of the reaction parameters, the Au/ZrO2 catalyst could be tuned to produce also HFCA, whereas the Ag/ZrO2 catalyst exclusively produced HFCA in a wide range of reaction parameters. The observed differences in catalyst selectivities can be taken as a starting point for further mechanistic investigation on the oxidation of HMF, contributing to a fundamental understanding of this reaction which is particularly important for establishing the production of bio-based polymers.
Introduction
With diminishing fossil resources, efforts have to be taken for the future synthesis of chemicals. In this context, biomass can serve as an attractive renewable feedstock for chemical syntheses.1 One of the most versatile molecules that can be synthesized from C6 carbohydrates, which are e.g. present in biomass feedstocks containing cellulose and hemicellulose, is 5-(hydroxymethyl)furfural (HMF).2,3 HMF can be produced from hexoses by acid-catalyzed dehydration and can subsequently be converted into various other products.2,3 In numerous studies, the conversion of HMF into valuable secondary products has been investigated including e.g. via hydrogenation,4 dehydrogenation5 or hydrodeoxygenation.6 Products originating from HMF oxidation are 2,5-diformylfuran (DFF), 5-hydroxymethyl-2-furancarboxylic acid (HFCA) and 2,5-furandicarboxylic acid (FDCA). Among these, FDCA and HFCA are considered as very important secondary products from HMF, since they might substitute fossil monomers in bio-based polymers.7–10Besides the use of stoichiometric oxidants like KMnO4,11 numerous pathways for the synthesis of FDCA have been reported to produce FDCA in a sustainable way. These methods include bio-12,13 or electrocatalytic14,15 reactions as well as homogeneous and heterogeneous metal catalysis. A method which is industrially applied is the homogeneously catalyzed AMOCO mid-century process, which uses Co(OAc)2 and Mn(OAc)2 as catalysts in acetic acid solvent at 125 °C and 70 bar air pressure.16,17 More recent studies focused on the heterogeneously catalyzed oxidation of HMF for a more sustainable process. In these studies, mainly nanoparticles of noble metals like Pt,18–24 Pd,18,25–27 Ru28–31 or Au32–36 on various supports have been used for the oxidation of HMF to FDCA, often giving almost quantitative yields. For example, FDCA was produced with 96% yield after 5 h reaction time using a Au/CeO2 catalyst at 130 °C.32 If the reaction is carried out in basic aqueous solution, the oxidation proceeds via the oxidation of the aldehyde group with the oxidation of the alcohol functionality being the rate-limiting step.32,37 Therefore, the partially oxidized HFCA is an intermediate in the oxidation reaction from HMF to FDCA. Although a number of active catalysts have been reported in the literature, stability and, consequently, recyclability are still challenging.32 Further strategies to increase the activity and stability of the catalysts utilize alloy formation of different noble metals,38–41 innovative support materials, such as carbonized MgO,23 carbon nanotubes24 or N-doped carbon42 or a combination of both approaches.43 In addition, the base-free conversion of HMF to produce FDCA has attracted attention, often using basic hydrotalcites (HT) as the catalyst support.27,36,44 For example, a Au/HT catalyst tested by Gupta et al.36 was able to produce FDCA in almost quantitative yield at 95 °C. However, when HT is used as the catalyst support, leaching of Mg remains a key issue.27,36,44
Besides its use as a monomer,8–10 HFCA shows antitumor activity and can be used in the synthesis of an interleukin inhibitor.45,46 In contrast to the synthesis of FDCA, the targeted synthesis of HFCA has only rarely been pursued. For example, biocatalytic oxidation reactions using Serratia liquefaciens LF14 whole cells47 or xanthine oxidase48 were reported. Kang et al.49 used a Cannizzaro reaction in ionic liquids for the synthesis of HFCA with a yield of 46%. In this reaction, the dialcohol was produced in equimolar amounts limiting the maximum yield of HFCA to 50%. As mentioned above (cf.Scheme 1), HFCA can also be generated as an intermediate in the FDCA synthesis using noble metal catalysts, but in most cases reaction conditions are optimized for FDCA production. For example, HFCA was produced in almost quantitative yield over a Au/CeO2 catalyst at 65 °C and 10 bar air pressure in less than one hour and subsequently oxidized to FDCA.32 One of the first methods for the targeted synthesis of HFCA was reported by Reichstein50 in the 1920s using stoichiometric amounts of silver oxide as the oxidant in basic solution. The pure product was separated with 84% yield. Also supported silver oxide on copper oxide was used for the oxidation of HMF to HFCA.51 Zhang et al.52 studied the synthesis of HFCA using a molybdenum acetylacetonate complex immobilized on montmorillonite K-10 clay as a heterogeneous catalyst in HMF oxidation. This catalyst was able to produce HFCA in 87% yield in toluene at 110 °C within three hours.52 However, for the sustainable production of HFCA, the oxidation reaction should preferably be performed in aqueous medium. In a recent review article, Ventura et al.9 pointed out that the search for highly active systems to produce HFCA as an alternative green monomer is still highly desired.
 | ||
| Scheme 1 Oxidation products of HMF using gold and silver catalysts. | ||
Herein, we report on supported gold and silver catalysts, which are able to produce FDCA and HFCA, respectively, in high yields from HMF in basic aqueous solution (Scheme 1). The reactions have been performed in water as the solvent and using air as the oxidant. Different preparation methods for silver-based catalysts have been compared and the reaction conditions for Ag and Au catalysts have been optimized for the targeted synthesis of HFCA and FDCA, respectively. In addition, first studies towards the identification of the active sites of the silver catalysts have been carried out.
Experimental
Materials
All chemicals were of analytical grade and have been used without further purification: HMF, FDCA, AgNO3, ZrO(NO3)2 (Sigma-Aldrich), HFCA, NaOH, silver benzoate (Merck), 5-formyl-2-furoic acid, DFF (TCI Chemicals), zirconium(IV) 2-ethylhexanoate, HAuCl4·3H2O, ZrO2 1/8′′ pellets, TiO2 1/8′′ pellets, MgO (Alfa Aesar), CeO2 (MEL chemicals) and synthetic air (Air Liquide).Catalyst preparation
In a variation of the dp method, the support was suspended in water, the pH was adjusted to a value of 9 and the metal precursor (HAuCl4·3H2O or AgNO3) was added dropwise at constant pH. No differences in activity were observed for the catalysts obtained from the two different methods.‡
After preparation, the catalyst was calcined and reduced as described above.
Selective oxidation of HMF
The catalytic tests were performed in home-built autoclaves (max. temperature 350 °C at a max. pressure of 200 bar, 52 mL with Teflon® inlets). In a typical reaction, 1 mmol (126 mg) HMF was dissolved in water and the desired amount of NaOH was added using a 2.5 M solution resulting in a total volume of 10 mL. Then, the catalyst was added, the reactor was closed, purged three times with synthetic air and the desired pressure was adjusted. The autoclaves were heated with heating sleeves and heating plates and stirred magnetically. The time when the desired reaction temperature was first reached was defined as the starting point (t = 0). After the reaction, the reactors were cooled to room temperature using an ice bath, depressurized and the solutions were separated from the catalysts by decantation. The samples were taken, filtered with a 0.45 μm Pall Teflon filter and diluted for HPLC analysis (Hitachi Primaide, Bio-Rad Aminex HPX-87H column, solvent 5 mM H2SO4, temperature 50 °C, 50 bar). After the reactions, HFCA and FDCA could be separated from the reaction mixtures, in case the reactions gave the desired products in high yields. FDCA was separated by acidification with HCl, whereas HFCA was separated by acidification with HCl and subsequent extraction using EtOAc and evaporation of the solvent. The amount of catalyst needed per reaction was calculated using the nominal metal loading of 2 wt%.Characterization
XRD measurements of the powder catalysts were performed on a PANalytical X'Pert Pro instrument using Cu Kα radiation (wavelength = 1.54060 Å) at diffraction angles 2θ from 20° to 80° and a step size of 0.017° (0.53 s acquisition time). Support materials were measured in the calcined state, and catalysts in the calcined and reduced state. Measurements were performed on rotating sample holders.N2 physisorption was used to determine the specific surface areas using the Brunauer–Emmet–Teller method on a Rubotherm BELSORP-mini II instrument. Catalysts were heated to 300 °C for 2 h under reduced pressure prior to the experiment.
The metal loading of the catalysts and the concentrations of metals in the reaction solutions were determined by ICP-OES measurements (Agilent 720/725-ES instrument) and XRF of powder catalysts (Bruker S4 Pioneer spectrometer). For the analysis of the solid catalysts via ICP-OES, the samples were digested in a mixture of sulfuric and nitric acid.
Electron microscopy was performed on carbon covered copper grids using reduced catalyst samples. For TEM measurements, the catalyst powders were mounted onto a carbon coated Cu TEM grid covered with a holey carbon film in the dry state. Subsequently, the samples were examined under an FEI Titan 80-300 aberration corrected electron microscope operated at 300 kV. High angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) images were acquired with a Fischione model 3000 HAADF-STEM detector.
The separated products, HFCA and FDCA, were characterized using NMR spectroscopy. 1H and 13C NMR spectra were obtained in DMSO-d6 at room temperature with a Bruker Avance 250 and a Bruker Avance 400 spectrometer, respectively.
Results and discussion
Catalyst preparation and characterization
The metal loading and the specific surface area of the catalysts and the corresponding support materials are listed in Table 1. No change in the specific surface area of the supports was observed upon deposition–precipitation or impregnation of the noble metals (Table 1, entries 5 to 13). The ZrO2 support used for the preparation of catalysts via deposition–precipitation and impregnation had a specific surface area of ca. 90 m2 g−1. For the silver catalysts prepared by co-precipitation and flame-spray pyrolysis (Table 1, entries 14–16), higher specific surface areas were obtained. A maximum specific surface area of 147 m2 g−1 was observed for the Ag/ZrO2_cp catalyst calcined at 350 °C (Table 1, entry 14), and the specific surface area decreased upon calcination at 500 °C (Table 1, entry 15).| Entry | Support/catalyst | Metal loading/wt% | Specific surface area/m2 g−1 |
|---|---|---|---|
| a Determination of metal loading by ICP-OES. b Determination of metal loading by XRF. | |||
| 1 | ZrO2 | — | 88 |
| 2 | TiO2 | — | 42 |
| 3 | CeO2 | — | 40 |
| 4 | MgO | — | 6 |
| 5 | Au/ZrO2_dp | 1.6a | 90 |
| 6 | Ag/ZrO2_dp | 1.0b | 89 |
| 7 | Au/TiO2_dp | 1.1b | 38 |
| 8 | Ag/TiO2_dp | 1.7b | 39 |
| 9 | Au/CeO2_dp | 1.7b | 40 |
| 10 | Ag/CeO2_dp | 1.7b | 40 |
| 11 | Au/MgO_dp | 2.0b | 5 |
| 12 | Ag/MgO_dp | 2.0b | 4 |
| 13 | Ag/ZrO2_imp | 2.0b | 92 |
| 14 | Ag/ZrO2_cp_350 | 1.9b | 147 |
| 15 | Ag/ZrO2_cp_500 | 1.9b | 82 |
| 16 | Ag/ZrO2_fsp | 1.7b | 108 |
The efficiency of noble metal deposition was strongly dependent on the support material and the respective metal. For gold catalysts, a precipitation at lower pH values led to higher loadings, however larger gold particles were obtained.55 An optimal pH value for deposition–precipitation of gold seemed to be in the range of pH = 8–9.55–57 A low silver content of 1 wt% was observed for the Ag/ZrO2_dp catalyst (Table 1, entry 6), whereas higher silver loadings were achieved on the other support materials. Changing the preparation method allowed to receive an increased silver content on ZrO2 (Table 1, entries 13–16).
Most catalysts did not show reflections of noble metals or noble metal oxides in the XRD patterns besides the reflections of the corresponding support materials. This can be explained by the low noble metal loadings and the desired presence of small particles with a size below the detection limit of XRD (<5 nm).58 The presence of small particles with a mean size of 3.7 nm was confirmed for the Au/ZrO2_dp catalyst by TEM (Fig. S1†). No distinct Ag particles were found in the TEM images of the Ag/ZrO2_dp catalyst probably due to the low metal loading and the low contrast between silver and the support material. However, HRTEM showed the presence of some elliptical silver particles with a size of about 5 nm (Fig. S2b†). In addition, there were no detectable changes in the XRD patterns of commercial support materials before and after the deposition–precipitation or impregnation of noble metals. The only differences were those observed for the catalysts supported on MgO. For the Au/MgO_dp catalyst, reflections of Mg(OH)2 were observed after calcination, which disappeared after reduction (Fig. S4†). The calcined Ag/MgO_dp catalyst showed reflections of reduced silver and Ag2O (Fig. S5†). However, the reflections of Ag2O disappeared upon reduction. Due to the high basicity of MgO, the deposition–precipitation method was modified for MgO-supported catalysts (cf. catalyst preparation). This high pH value might have led to a rapid precipitation of large metal particles. Also note the low surface area of the MgO support (Table 1), which most likely also led to the formation of larger particles, as indicated by reflections from metallic phases for the Ag/MgO_dp catalyst. Although no reflections of Au were observed in the calcined or in the reduced Au/MgO_dp catalyst, large particle sizes are expected for this catalyst36 as stated above.
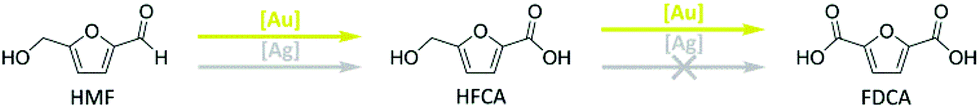
Different XRD patterns were observed for the Ag/ZrO2_cp and Ag/ZrO2_fsp catalysts, where the ZrO2 support was prepared together with the silver particles (Fig. 1). The Ag/ZrO2_cp catalyst was X-ray amorphous after the standard calcination procedure at 350 °C for 4 h. This phase was also obtained after increasing the calcination time; however, a cubic ZrO2 phase was formed when the calcination temperature was increased to 500 °C for 4 h. The cubic ZrO2 phase was also present in the Ag/ZrO2_fsp catalyst (Fig. 1). In contrast to the other catalysts, a phase transition occurred upon the reduction of the Ag/ZrO2_fsp catalyst. After reduction at 350 °C in hydrogen (10% in N2), reflections of monoclinic ZrO2 were observed for the Ag/ZrO2_fsp catalyst and the XRD pattern became similar to that of the commercial monoclinic ZrO2 support, which was used for the preparation of the other catalysts.
 | ||
| Fig. 1 XRD patterns of (a) the Ag/ZrO2_cp and (b) the Ag/ZrO2_fsp catalysts. Solid lines correspond to calcined catalysts and dotted lines represent reduced catalysts (350 °C, 2 h, 3 L min−1, 10% H2 in N2); *cubic ZrO2, +monoclinic ZrO2. | ||
Oxidation of HMF over gold- and silver-based catalysts
First, a catalyst screening was performed in basic aqueous solution using air as the oxidant in the presence of Au- and Ag-based catalysts prepared by deposition–precipitation. It is known from the literature that HMF is unstable in basic solution, as it undergoes a polymerization to dark polymeric products (humins).14 These products were not characterized and quantified but their presence explains the difference between product yields and HMF conversion in Table 2 (i.e. a carbon balance of less than 100%). From the screening results it was observed that Au and Ag showed superior activity if they were supported on ZrO2 compared to the other support materials (Table 2, entries 1 and 2). This is reflected both by the product yields and the catalyst productivity, giving an absolute value of the product formed per noble metal content and reaction time. The trend of a higher activity of ZrO2 supported metals was previously reported by Sahu et al.22 for platinum-based catalysts. A comparison of Ag/ZrO2_dp and Au/ZrO2_dp revealed that while Au/ZrO2_dp mainly produced FDCA, the Ag/ZrO2_dp catalyst did not produce FDCA in a yield higher than in blank experiments. However, Ag/ZrO2_dp produced HFCA with a high yield of 92%. Although silver catalysts have been used in the oxidation of furfural,59 there is, to the best of our knowledge, no report yet on silver catalysts used for the oxidation of HMF.| Entry | Catalyst | HMF conversion/% | Yield/% | Productivitya/molproduct h−1 molmetal−1 | |
|---|---|---|---|---|---|
| HFCA | FDCA | ||||
| a Productivity is given as moles of product formed per mole of noble metal and time. For gold- and silver-based catalysts, the productivity is given for FDCA and HFCA production, respectively. | |||||
| 1 | Au/ZrO2_dp | 100 | 0 | 75 | 19 |
| 2 | Ag/ZrO2_dp | 100 | 92 | 5 | 37 |
| 3 | Au/TiO2_dp | 100 | 4 | 1 | 2 |
| 4 | Ag/TiO2_dp | 100 | 14 | 3 | 3 |
| 5 | Au/CeO2_dp | 100 | 0 | 46 | 11 |
| 6 | Ag/CeO2_dp | 100 | 74 | 3 | 17 |
| 7 | Au/MgO_dp | 100 | 3 | 2 | 1 |
| 8 | Ag/MgO_dp | 100 | 60 | 1 | 12 |
The different selectivity of gold- and silver-based catalysts can also be observed by comparing different support materials. Astonishingly, TiO2 supported catalysts (Table 2, entries 3 and 4) showed poor activity, although Au/TiO2 catalysts have been reported to be highly active in the literature, even with a rather low metal loading of 1 wt%.32,33 In the case of CeO2, the lower activity can be explained by the type of support used. Casanova et al.32 described a lower activity of CeO2-supported gold catalysts if ceria was not of nanoparticulate nature. The low activity for the Au catalyst supported on MgO might be explained by the presence of larger Au particles on MgO (cf. catalyst preparation).36 The relatively high activity of the Ag/MgO catalyst might be explained by the fact that larger Ag particles are known to be catalytically active, for example in ethylene epoxidation.60 A reason for the generally lower activity of the MgO-supported catalysts might be the low surface area and the high basicity of MgO. This might cause HMF degradation if this catalyst is used with additional homogeneous bases resulting in the formation of humins. A Au/MgO catalyst investigated by Gupta et al.36 was used without an additional base giving higher overall yields of oxidation products, however a higher substrate-to-metal molar ratio was used in that study.
The oxidation of the hydroxymethyl group usually proceeds via an aldehyde intermediate, 5-formyl-2-furancarboxylic acid (FFCA).37 However, FFCA has not been observed throughout the catalytic tests presented in this study. This indicates that the oxidation of the aldehyde group proceeds very rapidly over all reported catalysts active in FDCA synthesis.
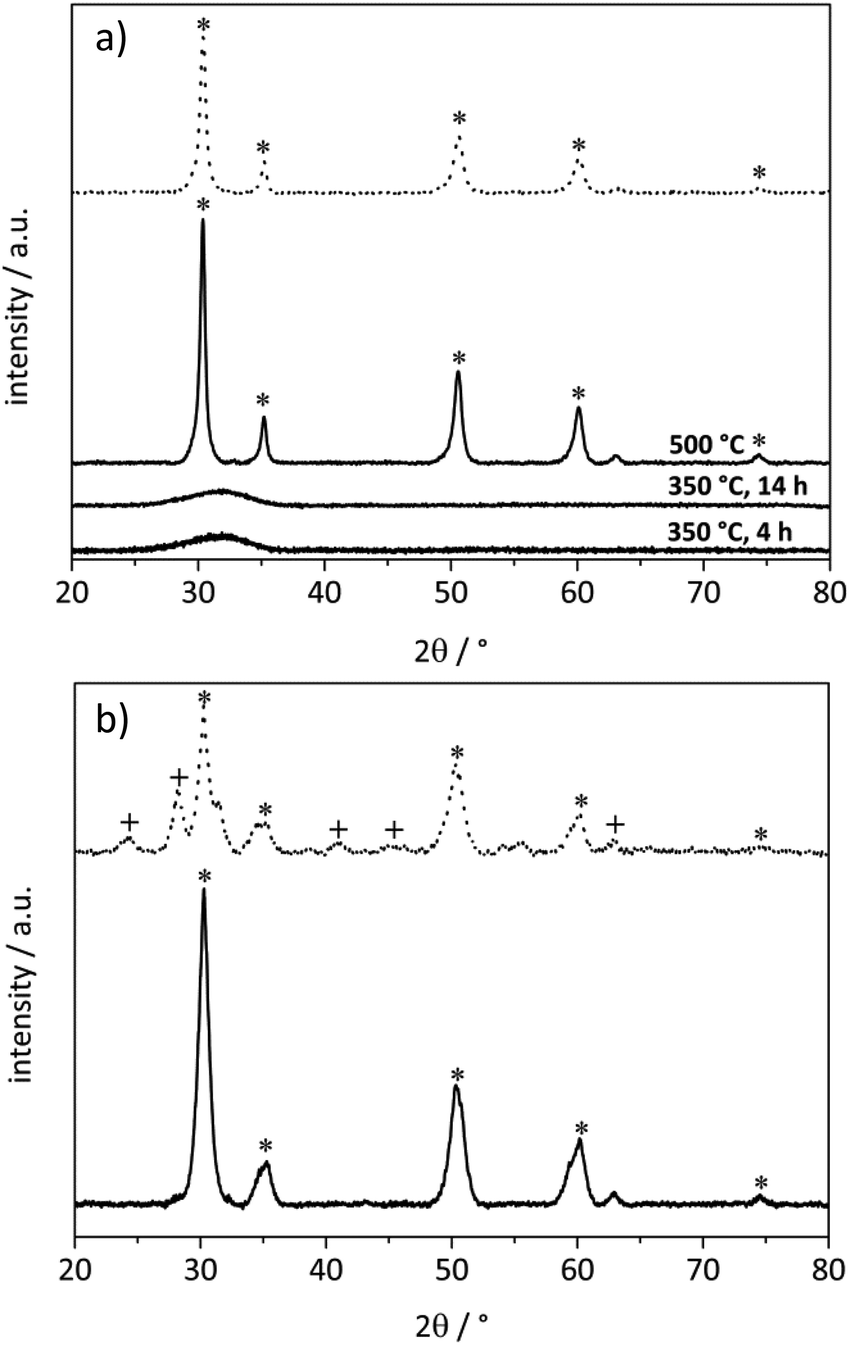
 | ||
| Fig. 2 Influence of the reaction temperature on product distributions using (a) Au/ZrO2_dp and (b) Ag/ZrO2_dp. Reaction conditions: 10 bar air pressure, 4 equivalents of NaOH, 5 h reaction time, 1 mmol HMF in 10 mL H2O, 98 mg Au/ZrO2_dp catalyst, 54 mg Ag/ZrO2_dp catalyst. | ||
The yield reached a maximum value for the different oxidation products for both catalysts and decreased due to the thermal degradation of HMF at higher temperatures. However, different selectivities of Au/ZrO2_dp and Ag/ZrO2_dp towards oxidation products are visible. Using the Au/ZrO2_dp catalyst in basic solution (Fig. 2a), the oxidation of the hydroxymethyl group became the rate-limiting step. While mainly HFCA was produced at 50 °C, HMF was further oxidized to FDCA by increasing the reaction temperature to 125 °C. In addition, the productivity reached a maximum value of 20 molFDCA h−1 molAu−1. Regarding the FDCA yield and productivity, 125 °C seemed to be optimal for the production of FDCA, while being a compromise between a short reaction time and sufficient FDCA yields. This trend is similar to the one observed by Casanova et al.32 who described 130 °C as the upper limit of the reaction. In general, increasing the temperature accelerated the reaction, however, HMF suffered from thermal degradation in basic aqueous solution,38 which was confirmed in blank experiments (Table S1†). This is also represented in the carbon balance with the degradation of HMF leading to a worse carbon balance with increasing temperature. Since HMF was quantitatively converted in almost all reactions, highly active catalysts are needed to convert HMF before its thermal polymerization in basic solution, as stated above.
Comparing these results with the Ag/ZrO2_dp catalyst at varying reaction temperatures (Fig. 2b), it was observed that the synthesis of HFCA with Ag/ZrO2_dp proceeded at significantly lower temperatures. The HFCA yield passed through a maximum at 50 °C, where HFCA was produced in an almost quantitative yield of 98% with a productivity of 39 molHFCA h−1 molAg−1. It seems that medium temperatures are beneficial for the production of HFCA, in line with other catalysts.32,36,38–40 Interestingly, at room temperature the HFCA yield was lower although HMF was fully converted. This might be due to the polymerization of HMF under basic conditions at room temperature where the catalyst was not as active as it was at slightly elevated temperatures. A further increase of the temperature lowered the HFCA yield while the FDCA yield remained at about 2%. This together with the fact that FDCA production was in the range of blank experiments in all cases showed that silver-based catalysts are not capable of oxidizing the hydroxymethyl group under the applied conditions, although silver catalysts have generally been reported to oxidize alcohols in organic solvents.61 Consequently, the selective synthesis of HFCA was possible in the presence of silver-based catalysts leaving the alcohol group untouched, which is the main difference from other catalyst systems where also the alcohol was converted to some extent.
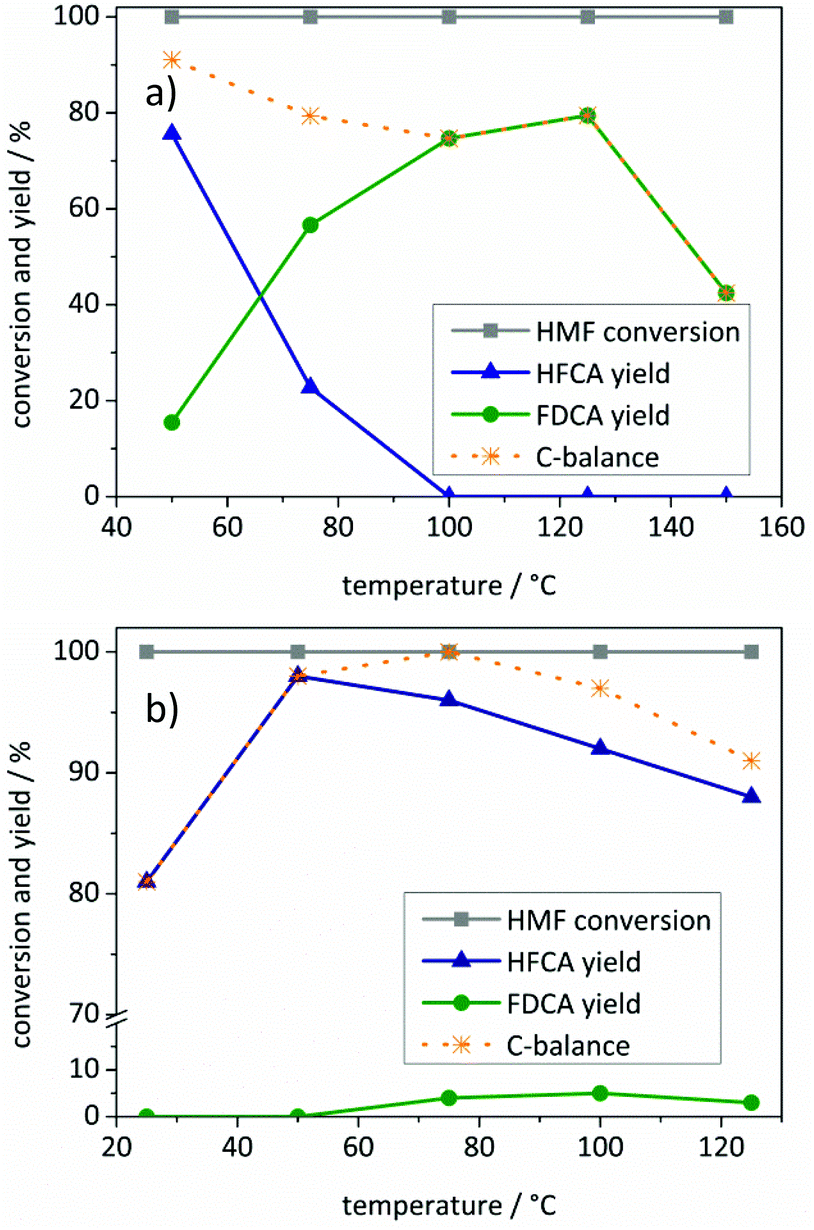
To prevent a possible degradation of HMF under highly basic conditions, the amount of NaOH added to the solution was decreased (Fig. 3). Both catalysts required the presence of a base for the oxidation of HMF. However, the influence of the added base and the observed selectivities differed when the Au/ZrO2_dp and the Ag/ZrO2_dp catalysts are compared.
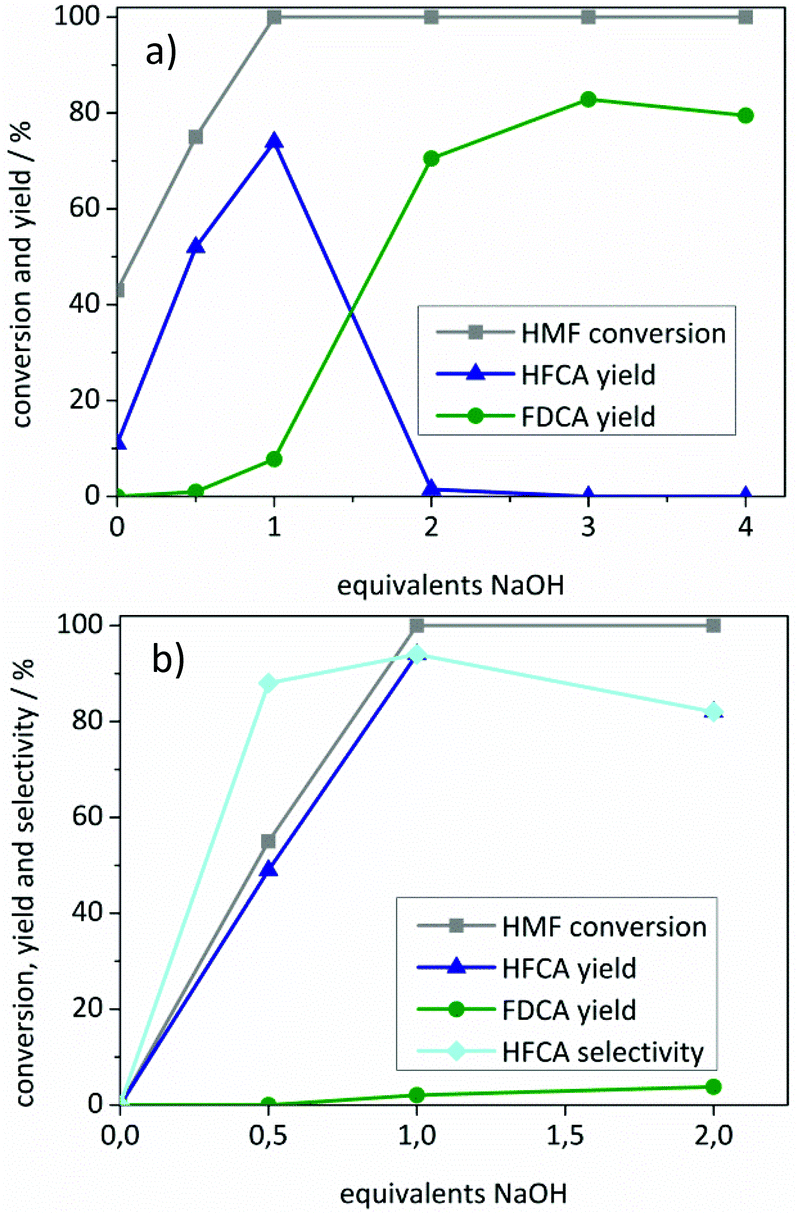 | ||
| Fig. 3 Influence of NaOH addition on the product distribution using (a) the Au/ZrO2_dp and (b) the Ag/ZrO2_dp catalyst. Reaction conditions: (a) 125 °C, 10 bar air pressure, 5 h reaction time, 1 mmol HMF in 10 mL H2O, 98 mg Au/ZrO2_dp catalyst. (b) 50 °C, 10 bar air pressure, 1 h reaction time, 1 mmol HMF in 10 mL H2O, 54 mg Ag/ZrO2_dp catalyst. | ||
HFCA was produced in 11% yield (26% selectivity) over Au/ZrO2_dp in the absence of a base (Fig. 3a). A control experiment using pure ZrO2 revealed that 39% of HMF was converted without the addition of a base at 125 °C (not shown). However, only degradation products of HMF were formed. This showed that Au/ZrO2 was active to a certain extent in HMF oxidation in the absence of a base. An increasing NaOH addition led to a more selective conversion of HMF. The HFCA yield increased to 74% at one equivalent of NaOH related to HMF. By increasing the amount of base to two equivalents, the selectivity shifted to FDCA, which indicates that at least one hydroxide ion per sidechain is necessary for the oxidation of HMF. With an excess of base, the FDCA yield increased further, probably by accelerating the reaction, and then reached a plateau. Fig. 3b shows the influence of the amount of added base on the yield of HFCA using the Ag/ZrO2_dp catalyst. HMF remained unreacted in the absence of a base contrary to the results of the Au/ZrO2_dp catalyst. However, no conversion of HMF was observed in a control experiment at 50 °C in the presence of pure ZrO2 within one hour (not shown). When no or only one equivalent of base was applied using Ag/ZrO2_dp, the HFCA yield increased linearly with HMF conversion. A pH of 7 after those reactions indicated a total consumption of the added OH− ions during the reaction. In the previous mechanistic studies on HMF oxidation over Pt and Au catalysts, oxidation of HMF via germinal diols formed by the addition of hydroxide ions to carbonyl groups has been proposed.37 Their formation seems to be crucial for the oxidation of HMF over both catalysts used in this study. In contrast to the Au/ZrO2_dp catalyst, no shift in the selectivity was observed using the Ag/ZrO2_dp catalyst, which exclusively produced HFCA.
To establish a truly sustainable process, air was used as the oxidant in all reactions, which is, due to the lower oxygen concentration, more demanding than using pure oxygen. Since the reactions were performed at the previously optimized temperature, high yields of HFCA and FDCA were already achieved at 10 bar air pressure. Therefore, the amount of catalyst was reduced compared to the standard amount of catalyst used in most reactions. As expected, using less catalyst at a pressure of 10 bar led to a lower amount of detected oxidation products for both catalysts (Fig. 4). Interestingly, the influence of air pressure on the product yields for Au/ZrO2_dp differed from the Ag/ZrO2_dp catalyst.
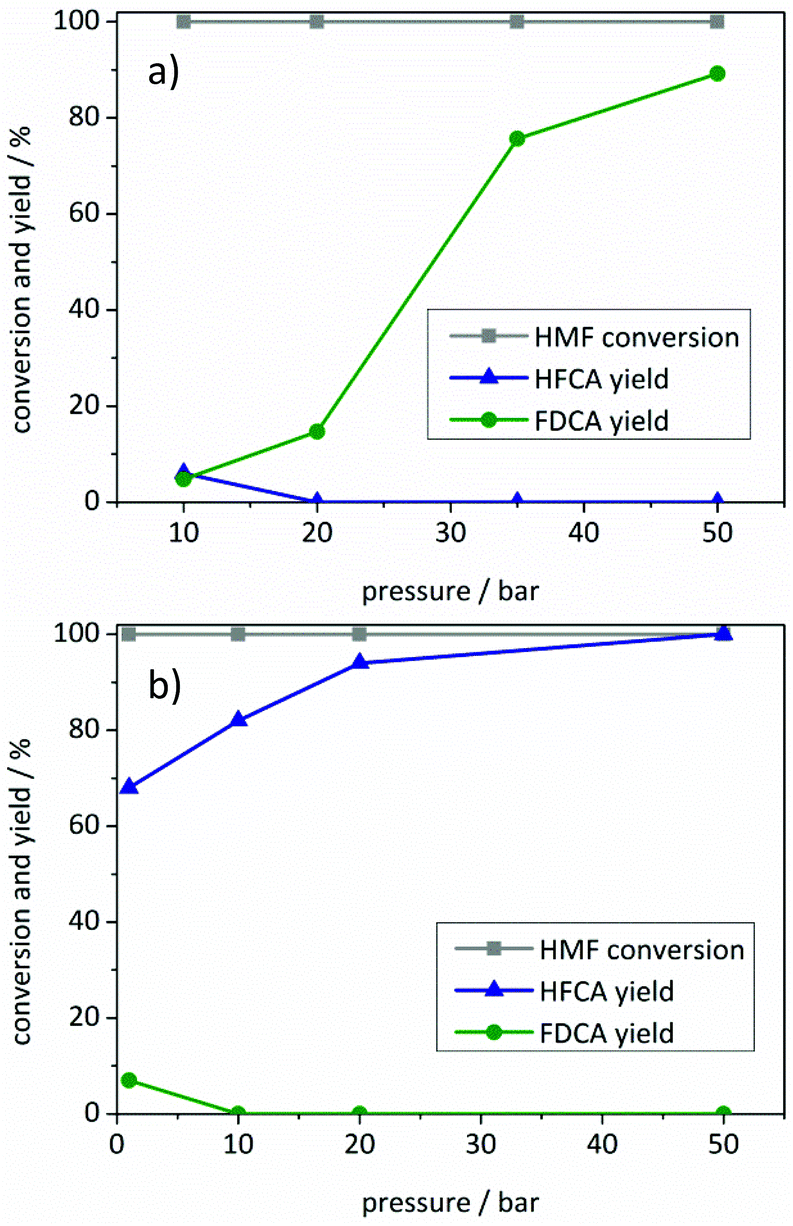 | ||
| Fig. 4 Influence of the air pressure on product distribution using (a) Au/ZrO2_dp and (b) Ag/ZrO2_dp. Reaction conditions: (a) 125 °C, 4 equivalents of NaOH, 5 h reaction time, 1 mmol HMF in 10 mL H2O, 33 mg Au/ZrO2_dp catalyst. (b) 50 °C, 4 equivalents of NaOH, 1 h reaction time, 1 mmol HMF in 10 mL H2O, 28 mg Ag/ZrO2_dp catalyst. | ||
The total yield of oxidation products remained almost constant upon increasing the air pressure to 20 bar in the presence of the Au/ZrO2_dp catalyst, where FDCA was produced with 15% yield (Fig. 4a). Further increase of the air pressure led to increased FDCA yields with a maximum yield of 89% and a maximum productivity of 67 molFDCA h−1 molAu−1 at 50 bar. This productivity is in the upper range compared to other monometallic Au catalysts described in the literature (Table S3†). In contrast to the gold catalyst and other reaction parameters mentioned above, air pressure seemed to have a significantly smaller influence on HFCA yield using the Ag/ZrO2_dp catalyst (Fig. 4b). Already at ambient air pressure, HFCA was produced with 68% yield. Increasing the air pressure from 1 bar to 50 bar led to an increase in HFCA yield. HFCA was produced quantitatively at an air pressure of 50 bar with the maximum productivity as high as 400 molHFCA h−1 molAg−1.
Recycling of both catalysts was tested by separating the catalysts after the reactions, drying them at 110 °C and using them in subsequent runs (Fig. 5). Since a slight loss of catalyst occurred during this process, the amount of HMF in the subsequent runs was adjusted to the recovered catalyst mass. Both catalysts remained active for a second cycle with a slight loss of activity in the third run. The FDCA yield decreased to 63% using the Au/ZrO2_dp catalyst in a third reaction where also 2% of HFCA were produced (Fig. 5a). Further re-use led to an increase in HFCA yield (13%) and a further decrease in FDCA yield. Since the carbon balance did not reach 100%, degradation products of HMF might block the active sites during consecutive reactions. For the Ag/ZrO2_dp catalyst, the loss in activity was less pronounced and HFCA was produced with a yield of 86% in a third reaction. The activity of both catalysts could not be restored by calcination and reduction of the catalysts, which has also been reported earlier.32 Further aspects that might be the reasons for a limited recyclability of the catalysts might be sintering of particles or leaching of the active metal. However, neither detectable amounts of gold nor of silver were found in the solutions after the reaction by means of ICP-OES performed under standard conditions (<0.5 ppm). In contrast, reflections of reduced metals appeared both for the Au/ZrO2_dp and the Ag/ZrO2_dp catalysts in XRD (Fig. S6†). This indicates a growth of the metal particles for both catalysts, which might be the reason for a decreased activity with an increasing number of runs. Especially for the Au/ZrO2_dp catalyst, this might play a significant role since it is known that particle size is a crucial parameter for Au-based catalysts.62
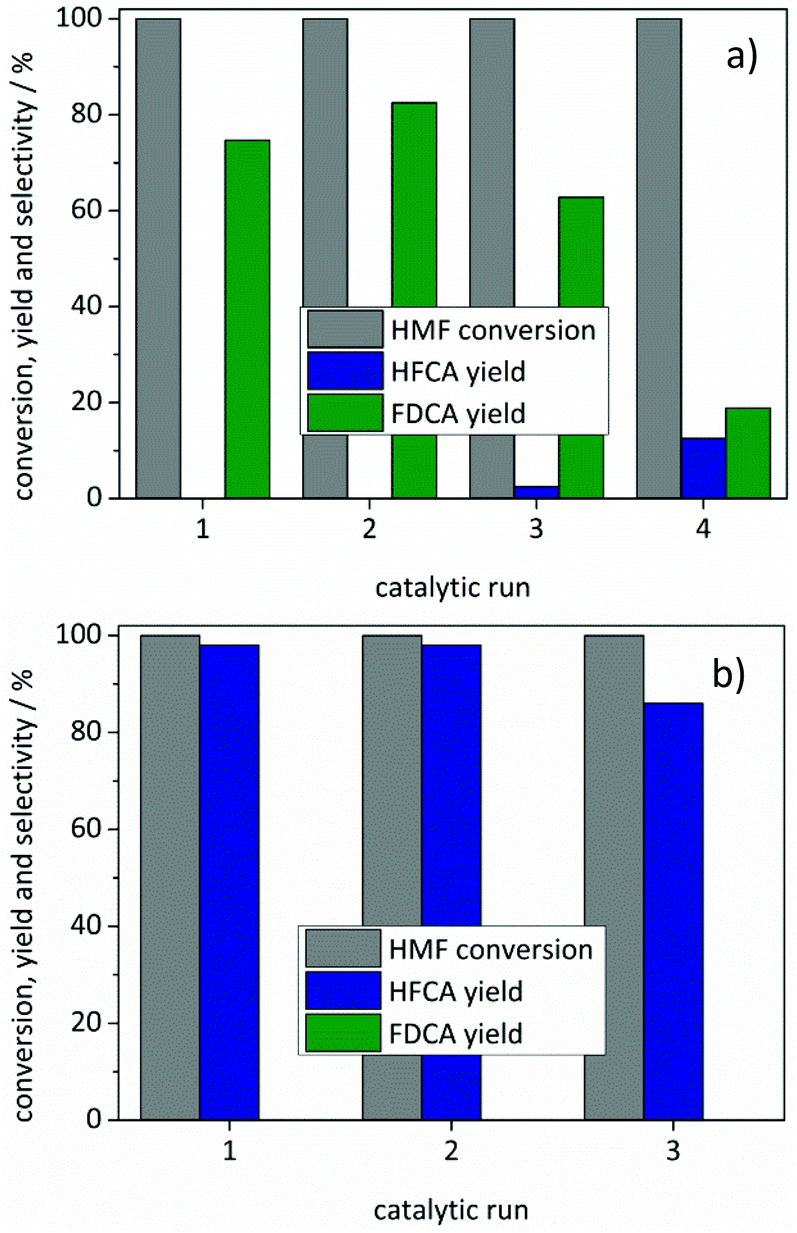 | ||
| Fig. 5 Stability tests of (a) Au/ZrO2_dp and (b) Ag/ZrO2_dp. Reaction conditions: (a) 100 °C, 10 bar air pressure, 4 equivalents of NaOH, 5 h reaction time, HMF and H2O adjusted to recovered catalyst. (b) 50 °C, 10 bar air pressure, 4 equivalents of NaOH, 5 h reaction time, HMF and H2O adjusted to recovered catalyst. | ||
In none of the studies using silver catalysts, a significant amount of FDCA was produced, which is the main difference between the commonly used noble metal catalysts and the silver catalysts described in the present study. To investigate whether silver is capable of oxidizing the alcohol group of HMF, further reactions were performed. No FDCA was produced by extending the reaction time from 5 to 17 h. Since the catalyst showed reflections of metallic silver after the reaction, a possible deactivation by sintering of silver particles was investigated. Therefore, a fresh catalyst was added to the HFCA solution produced in the first catalytic run. However, HFCA was not further converted to other products. This shows that the silver catalysts were not able to oxidize the hydroxymethyl group of HMF under the applied conditions.
Stability and active sites of silver catalysts
During this study, it was observed that the Ag/ZrO2_dp catalyst slightly deactivated upon storage for some days in air. Besides a color change of the catalyst from dark grey to a lighter brownish color, the HFCA yield decreased from 98% to 84%, which nearly corresponds to the yield of HFCA achievable with a fully calcined catalyst (82%). Although a reductive pretreatment is not necessarily required for noble metal catalysts as they are normally reduced also at high temperatures or even under reaction conditions, the used calcination temperature of 350 °C might not have been sufficient for the reduction of oxidized silver species.61 This might indicate an oxidation of the reduced catalyst in air, which is not reversible under the applied reaction conditions. As already stated above, no silver species could be observed in the XRD patterns, probably due to the low metal loading and small particle sizes. The loss in activity of stored samples could be partially regained by calcination and reduction of the catalyst for a second time (88% HFCA yield). This finding raised the question of the true nature of the active species in the oxidation reaction in terms of oxidation state and nature of active sites.As has been shown in the literature, homogeneous silver catalysts are able to oxidize furanic compounds like furfural.63 However, no silver dissolution was detected by means of ICP-OES (<0.5 ppm) in the reaction solutions. Nevertheless, control experiments were performed: first, AgNO3 was used as a model system for a soluble silver catalyst at 50 °C and 10 bar air pressure. AgNO3 was added at 1 mol%, which was the same amount as for a supported 2 wt% catalyst. Under these conditions, HFCA was produced with a yield of 59%. This shows that either soluble silver ions or in situ formed nanoparticles might be a catalytically active species in the oxidation of HMF. Directly after the addition of AgNO3 to the basic HMF solution, a black precipitate was formed, which indicates the reduction of the silver ions, most probably by the aldehyde functionality of HMF.64 In addition, a mechanism for HMF oxidation catalyzed by gold and platinum nanoparticles proposed by Davis et al.37 claims that the electrons of HMF oxidation are taken up by the metal particles, further keeping the metal in the reduced state. Next, AgNO3 was added to a basic suspension containing HMF and ZrO2. The reaction was performed analogously to the previous experiment at 10 bar air pressure and 50 °C. In this reaction, 61% of HFCA was produced and, as before, no silver was detected by ICP-OES in solution after the reaction. However, XRD and XRF confirmed the presence of metallic silver particles on the separated solid (see Fig. S8†). This material was then used as a catalyst in a subsequent reaction, where HFCA was produced in a high yield of 92%, indicating that indeed reduced silver particles might be responsible for HMF oxidation. The generally lower HFCA yield using AgNO3 might be explained by a rather slow particle growth compared to the fast HMF polymerization. A comparable pathway of metallic nanoparticles forming upon reduction of a homogeneous catalyst by the substrate as the active species has been reported for the Au-catalyzed oxidation of dibenzylamine.65
Despite the obtained evidence for the formation of metallic nanoparticles, dissolved silver species might still be catalytically active in the oxidation of HMF, e.g. via leaching and subsequent redeposition onto the support. To gain information on the degree of silver leaching, the Ag/ZrO2_dp catalyst was stirred under reaction conditions without the addition of HMF as a reducing agent at different pH values and the silver content was analyzed via ICP-OES (Fig. 6a). The results showed that significant leaching took place below pH 12, while a slight leaching was recognized above pH 12. Only at pH 12, no silver was found in solution by means of ICP-OES. Oxidative dissolution of silver from silver nanoparticles is a subject of current research.66–68 In general, different mechanisms for silver dissolution have been proposed; however, a common feature is a strong effect of pH on silver dissolution. For example, Sotiriou et al.66 studied the oxidative dissolution for Ag/SiO2 prepared by impregnation and flame-spray pyrolysis. It was found that the degree of silver dissolution was the same for both preparation methods and that small silver particles were more prone to dissolution. Comparing the amount of silver with the particle size suggested silver dissolution from one to two monolayers of Ag2O on silver particles.66 Reduction of both samples in hydrogen led to a decrease in dissolution by minimizing the fraction of oxidized silver. In support of the assumptions made by Sotiriou et al.,66 a small fraction of oxidized silver after preparation was observed by X-ray absorption spectroscopy of flame-made Ag/SiO2 catalysts.69 The formation of a stabilizing layer of Ag2O which is poorly soluble and therefore stable at high pH might be an explanation for a decreasing silver content at high pH (Fig. 6a).68 After these initial leaching experiments, the reaction solutions of the experiments using the Ag/ZrO2_dp catalyst with the initial pH values of 8, 12 and 14 were tested in the oxidation of HMF (Table S2†). No conversion of HMF was observed using the solutions with the initial pH values of 8 and 12, which showed that no reaction took place if either the base (pH 8) or soluble silver species (pH 12) were missing. In contrast, HFCA was produced using the solution with an initial pH value of 14, where obviously enough silver and hydroxide ions were present to convert HMF into HFCA. However, only 62% of HMF was converted and HFCA was produced with a low selectivity of 24% (15% yield). These results show that silver leached from the catalyst with no reducing agent (HMF) present in solution. However, leaching occurred to a large extent at pH values, where HMF could not be converted in further experiments. At the high pH values (pH 14) that were also applied for the supported catalysts, soluble Ag species showed inferior activities and selectivities compared to the reactions where the heterogeneous catalysts were used.
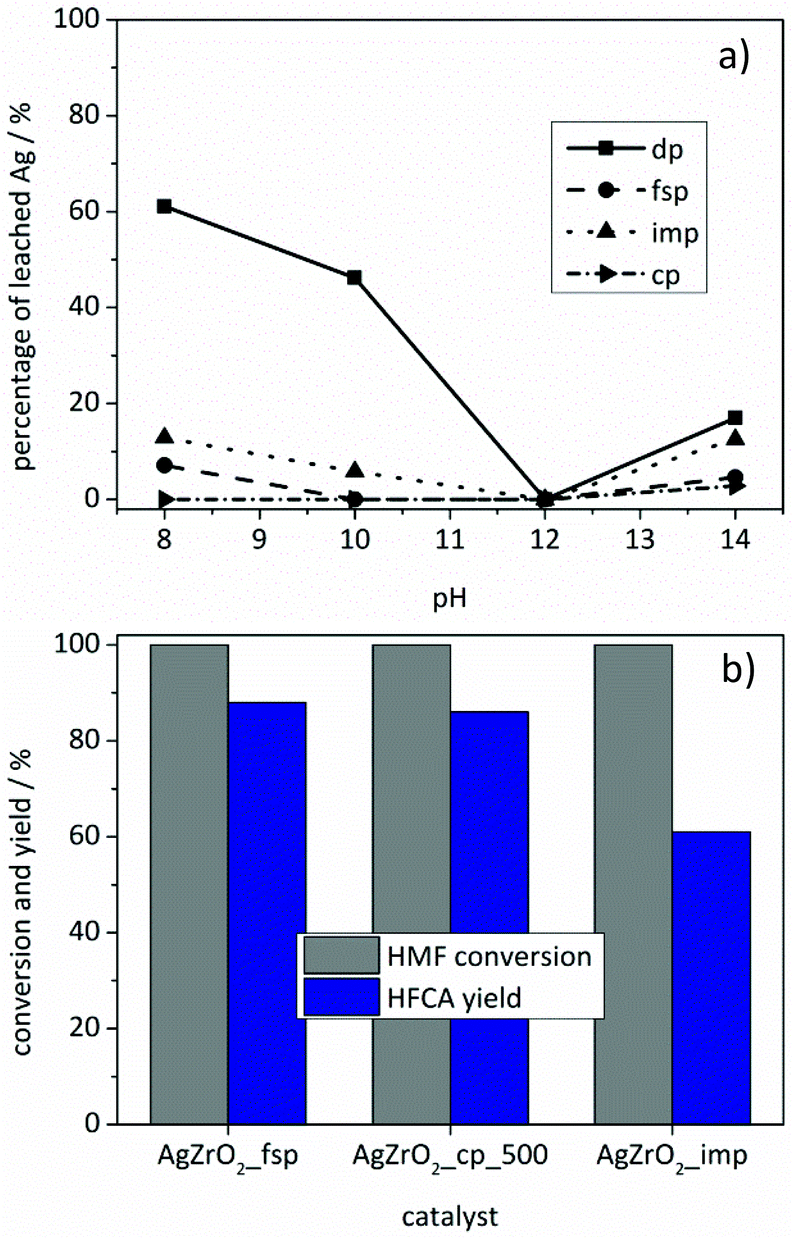 | ||
| Fig. 6 Comparison of (a) the leaching behavior and (b) catalytic activities (b) of Ag/ZrO2 catalysts prepared by different methods. Reaction conditions: (a) 50 °C, 10 bar air pressure, adjustment of pH with NaOH, 1 h, 54 mg catalyst. (b) 50 °C, 10 bar air pressure, 4 equivalents of NaOH, 1 h reaction time, 1 mmol HMF in 10 mL H2O, 54 mg catalyst. | ||
These experiments provided a first information on the active species of silver catalysts in the oxidation of HMF. Our results thus indicate that the reaction takes place on reduced silver particles and further studies hinted at a predominantly heterogeneous mechanism. However, concerning the mechanism i.e. the role of silver particles in the catalytic reaction, further studies have to be performed. Silver catalysts are often employed in selective oxidation reactions and therefore, the formation of different silver oxygen species and their appearance in gas phase oxidation mechanisms have been studied extensively.70–74 In general, their formation strongly depends on the pretreatment conditions especially at high temperatures. Three main silver oxygen species are generated upon exposure of silver to oxygen. At lower temperatures (below 330 °C), surface adsorbed oxygen (Oα) is formed.70,72 Increasing temperatures first lead to bulk dissolved oxygen (Oβ) before strongly bound surface oxygen species (Oγ, 630 °C) are generated. These species are crucial in some reactions. For example, a silver catalyst calcined at high temperature was more active in the selective oxidation of CO in hydrogen than a reduced catalyst, proving silver oxygen species to be active in this case.73 Given that a reduction in hydrogen atmosphere leads to more active catalysts in HMF oxidation (see above) and a possible formation of a thin layer of Ag2O on silver particles at high pH,68 it can be speculated that a surface layer oxide is involved in the catalytic cycle. The formation of this surface silver oxide at high pH might also explain the HFCA yield only being moderately influenced by increasing air pressures (Fig. 4). In a recent publication, Durndell et al.75 studied the oxidation of cinnamaldehyde over platinum catalysts and found PtO2 to be an active species. Because of the lack of information on reactions in the liquid phase, further studies of silver catalysts under HMF oxidation conditions are very important. For this purpose, operando X-ray absorption spectroscopy is expected to be a powerful tool.76–78
Variation of the preparation method
In order to minimize the silver leaching, different preparation methods were applied. Preferably catalysts with a stronger interaction of the silver particles with the support should be used. Catalysts were prepared by flame-spray pyrolysis, co-precipitation and impregnation. The catalysts were tested under the optimized reaction conditions for silver-based catalysts. As Fig. 6b shows, the catalysts differed in activity with the Ag/ZrO2_fsp catalyst giving the best results. Comparison of the Ag/ZrO2_cp catalysts calcined at different temperatures revealed that the Ag/ZrO2_cp_500 catalyst gave 86% yield of HFCA whereas the Ag/ZrO2_cp_350 catalyst produced HFCA in 42% yield (not shown). Most Ag/ZrO2 catalysts prepared in this study produced HFCA in comparable yields, apart from Ag/ZrO2_imp which was the only catalyst that produced HFCA with a lower yield of 61% (Fig. 6b). Since the catalysts differed in silver loadings (Table 1), the productivity toward HFCA is another reasonable parameter for catalyst comparison. Ag/ZrO2_imp gave the lowest productivity of 61 molHFCA h−1 molAg−1. The productivity was 103 molHFCA h−1 molAg−1 for the Ag/ZrO2_fsp and 91 molHFCA h−1 molAg−1 for the Ag/ZrO2_cp_500 catalyst, which corresponds to their performance described in Fig. 6b. Since Ag/ZrO2_dp was the most active catalyst (productivity of 164 molHFCA h−1 molAg−1 at identical reaction conditions), but featured the lowest silver content, the preparation method strongly influenced the catalytic activity. Deposition–precipitation seemed to be optimal for achieving a high activity per silver content.In addition, the catalysts prepared by different methods differed in their leaching behavior (Fig. 6a). The general trend in leaching behavior as a function of pH remained the same. However, all catalysts were significantly less prone to leaching compared to the Ag/ZrO2_dp catalyst. For Ag/ZrO2_fsp (≤7%) and Ag/ZrO2_imp (≤13%), less Ag leaching was observed. These preparation methods might have led to a stronger binding of silver particles or even an encapsulation of silver in ZrO2, which might prevent the oxidation of silver and, therefore, promote a lower degree of leaching at low pH. As discussed above, an oxide layer on silver particles may be the reason for silver leaching at low pH.66 Since almost no leaching occurred using the Ag/ZrO2_cp_500 catalyst (3% at pH 14), it was tested in several consecutive runs. However, a loss of activity was recognized after two runs leading to a yield of 58% of HFCA in the third reaction (Fig. S9†).
Considering the limited number of studies focusing on the targeted synthesis of HFCA, the silver-based catalysts presented in this study show a superior performance compared to other routes, i.e. the Cannizzaro reaction49 or the synthesis of HFCA in toluene using pure oxygen.52 Moreover, the silver-based catalytic oxidation of HMF to HFCA reported herein opens up an attractive alternative to the more frequently applied noble metal-catalyzed production of FDCA. The silver-based catalysts did not only show an excellent performance in the production of HFCA, they also required a smaller amount of base for almost quantitative HFCA yields (1 eq., see Fig. 3). Ongoing work in our laboratories aims – among others – at obtaining a deeper mechanistic understanding and at using HFCA as a monomer for bio-based polymers.
Conclusions
In summary, highly active gold and silver catalysts have been successfully applied in the oxidation of HMF to FDCA and HFCA, respectively. Reaction conditions were optimized for the production of FDCA and HFCA, both of which could be produced in high yields. In particular, for the production of HFCA the route reported here appears much easier than in other studies where biocatalytic oxidation or Cannizzaro reactions have been utilized. In addition, water was used as the solvent, air as the oxidant and supported metal nanoparticles as heterogeneous catalysts. This, together with the achieved high selectivities towards the desired products, drives the reaction more towards a green process.In general, ZrO2 supported catalysts gave higher yields compared to other support materials. While, e.g., the Au/ZrO2_dp catalyst enabled the selective synthesis of FDCA, the Ag/ZrO2_dp catalyst produced HFCA in almost quantitative yields under very mild conditions and with little amounts of added base. Further experiments with the Ag/ZrO2_dp catalyst showed that silver leached off the support in the absence of HMF and that soluble silver species can be catalytically active for the synthesis of HFCA to a certain extent. However, control experiments evidenced that the reaction was mostly heterogeneously catalyzed. By variation of the preparation method for Ag/ZrO2 catalysts, leaching could be significantly reduced demonstrating the potential for a future continuous process. With the investigated silver catalysts, the synthesis of HFCA in high yields with water as the solvent was possible, opening up an attractive route to produce a promising polymer precursor based on renewable resources.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded and supported by KIT. The authors thank Wu Wang, Dr. Sabrina Müller, and the Karlsruhe Nano Micro Facility (KNMF) at KIT for providing STEM-EDX measurements. They also thank Angela Beilmann (BET), Hermann Köhler (XRF and ICP-OES) and Matthias Stehle (support during the synthesis of the flame-made catalyst). The KIT synchrotron provided beamtime at the CAT-ACT beamline and Dr. Anna Zimina and Dr. Tim Prüßmann are gratefully acknowledged for the XAS measurements.Notes and references
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef PubMed.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 4734–4739 RSC.
- M. Chatterjee, T. Ishizaka, A. Chatterjee and H. Kawanami, Green Chem., 2017, 19, 1315–1326 RSC.
- B. Saha, C. M. Bohn and M. M. Abu-Omar, ChemSusChem, 2014, 7, 3095–3101 CrossRef PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- H. Hirai, J. Macromol. Sci., Part A: Pure Appl.Chem., 1984, 21, 1165–1179 CrossRef.
- M. Ventura, A. Dibenedetto and M. Aresta, Inorg. Chim. Acta, 2018, 470, 11–21 CrossRef.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- T. Miura, H. Kakinuma, T. Kawano and H. Matsuhisa, US Patent0232815, 2007 Search PubMed.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 826–830 CrossRef PubMed.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515–6518 CrossRef PubMed.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef PubMed.
- W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102–111 CrossRef.
- C. M. d. Diego, W. P. Schammel, M. A. Dam and G. J. M. Gruter, US Patent8519167, 2013 Search PubMed.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef.
- H. Ait Rass, N. Essayem and M. Besson, Green Chem., 2013, 15, 2240–2251 RSC.
- Z. Miao, T. Wu, J. Li, T. Yi, Y. Zhang and X. Yang, RSC Adv., 2015, 5, 19823–19829 RSC.
- W. Niu, D. Wang, G. Yang, J. Sun, M. Wu, Y. Yoneyama and N. Tsubaki, Bull. Chem. Soc. Jpn., 2014, 87, 1124–1129 CrossRef.
- R. Sahu and P. L. Dhepe, React. Kinet., Mech. Catal., 2014, 112, 173–187 CrossRef.
- X. Han, L. Geng, Y. Guo, R. Jia, X. Liu, Y. Zhang and Y. Wang, Green Chem., 2016, 18, 1597–1604 RSC.
- C. Zhou, W. Deng, X. Wan, Q. Zhang, Y. Yang and Y. Wang, ChemCatChem, 2015, 7, 2853–2863 CrossRef.
- B. Siyo, M. Schneider, J. Radnik, M.-M. Pohl, P. Langer and N. Steinfeldt, Appl. Catal., A, 2014, 478, 107–116 CrossRef.
- Z. Zhang, J. Zhen, B. Liu, K. Lv and K. Deng, Green Chem., 2015, 17, 1308–1317 RSC.
- Y. Wang, K. Yu, D. Lei, W. Si, Y. Feng, L.-L. Lou and S. Liu, ACS Sustainable Chem. Eng., 2016, 4, 4752–4761 CrossRef.
- Y. Y. Gorbanev, S. Kegnæs and A. Riisager, Top. Catal., 2011, 54, 1318–1324 CrossRef.
- Y. Y. Gorbanev, S. Kegnæs and A. Riisager, Catal. Lett., 2011, 141, 1752–1760 CrossRef.
- J. Artz and R. Palkovits, ChemSusChem, 2015, 8, 3832–3838 CrossRef PubMed.
- L. Zheng, J. Zhao, Z. Du, B. Zong and H. Liu, Sci. China: Chem., 2017, 60, 950–957 CrossRef.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef PubMed.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675 CrossRef PubMed.
- J. Cai, H. Ma, J. Zhang, Q. Song, Z. Du, Y. Huang and J. Xu, Chem. – Eur. J., 2013, 19, 14215–14223 CrossRef PubMed.
- Z. Miao, Y. Zhang, X. Pan, T. Wu, B. Zhang, J. Li, T. Yi, Z. Zhang and X. Yang, Catal. Sci. Technol., 2015, 5, 1314–1322 RSC.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- T. Pasini, M. Piccinini, M. Blosi, R. Bonelli, S. Albonetti, N. Dimitratos, J. A. Lopez-Sanchez, M. Sankar, Q. He, C. J. Kiely, G. J. Hutchings and F. Cavani, Green Chem., 2011, 13, 2091–2099 RSC.
- S. Albonetti, T. Pasini, A. Lolli, M. Blosi, M. Piccinini, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, G. J. Hutchings and F. Cavani, Catal. Today, 2012, 195, 120–126 CrossRef.
- A. Villa, M. Schiavoni, S. Campisi, G. M. Veith and L. Prati, ChemSusChem, 2013, 6, 609–612 CrossRef PubMed.
- Z. Gui, W. Cao, S. Saravanamurugan, A. Riisager, L. Chen and Z. Qi, ChemCatChem, 2016, 8, 3636–3643 CrossRef.
- X. Han, C. Li, Y. Guo, X. Liu, Y. Zhang and Y. Wang, Appl. Catal., A, 2016, 526, 1–8 CrossRef.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef.
- H. Choudhary and K. Ebitani, Chem. Lett., 2016, 45, 613–615 CrossRef.
- A. C. Braisted, J. D. Oslob, W. L. Delano, J. Hyde, R. S. McDowell, N. Waal, C. Yu, M. R. Arkin and B. C. Raimundo, J. Am. Chem. Soc., 2003, 125, 3714–3715 CrossRef PubMed.
- M. Munekata and G. Tamura, Agric. Biol. Chem., 1981, 45, 2149–2150 Search PubMed.
- K. Mitsukura, Y. Sato, T. Yoshida and T. Nagasawa, Biotechnol. Lett., 2004, 26, 1643–1648 CrossRef PubMed.
- Y.-Z. Qin, Y.-M. Li, M.-H. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3722 RSC.
- E.-S. Kang, D. W. Chae, B. Kim and Y. G. Kim, J. Ind. Eng. Chem., 2012, 18, 174–177 CrossRef.
- T. Reichstein, Helv. Chim. Acta, 1926, 9, 1066–1068 CrossRef.
- B. W. Lew, U.S. Patent3326944, 1967 Search PubMed.
- Z. Zhang, B. Liu, K. Lv, J. Sun and K. Deng, Green Chem., 2014, 16, 2762–2770 RSC.
- R. Grabowski, J. Słoczyński, M. Śliwa, D. Mucha, R. P. Socha, M. Lachowska and J. Skrzypek, ACS Catal., 2011, 1, 266–278 CrossRef.
- M. Høj, K. Linde, T. K. Hansen, M. Brorson, A. D. Jensen and J.-D. Grunwaldt, Appl. Catal., A, 2011, 397, 201–208 CrossRef.
- A. Wolf and F. Schüth, Appl. Catal., A, 2002, 226, 1–13 CrossRef.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231, 105–114 CrossRef.
- J. Li, J. Chen, W. Song, J. Liu and W. Shen, Appl. Catal., A, 2008, 334, 321–329 CrossRef.
- N. Sasirekha, P. Sangeetha and Y.-W. Chen, J. Phys. Chem. C, 2014, 118, 15226–15233 CrossRef.
- Q. Tian, D. Shi and Y. Sha, Molecules, 2008, 13, 948 CrossRef PubMed.
- O. Deutschmann, H. Knözinger, K. Kochloefl and T. Turek, Ullmann's Encyclopedia of Industrial Chemistry, 2009 Search PubMed.
- M. J. Beier, T. W. Hansen and J.-D. Grunwaldt, J. Catal., 2009, 266, 320–330 CrossRef.
- J.-D. Grunwaldt, C. Kiener, C. Wögerbauer and A. Baiker, J. Catal., 1999, 181, 223–232 CrossRef.
- M. Liu, H. Wang, H. Zeng and C.-J. Li, Sci. Adv., 2015, 1, e150002 Search PubMed.
- K.-S. Chou and C.-Y. Ren, Mater. Chem. Phys., 2000, 64, 241–246 CrossRef.
- L. Aschwanden, T. Mallat, J.-D. Grunwaldt, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 300, 111–115 CrossRef.
- G. A. Sotiriou, A. Meyer, J. T. Knijnenburg, S. Panke and S. E. Pratsinis, Langmuir, 2012, 28, 15929–15936 CrossRef PubMed.
- B. Molleman and T. Hiemstra, Langmuir, 2015, 31, 13361–13372 CrossRef PubMed.
- B. Molleman and T. Hiemstra, Environ. Sci.: Nano, 2017, 4, 1314–1327 RSC.
- S. Hannemann, J.-D. Grunwaldt, F. Krumeich, P. Kappen and A. Baiker, Appl. Surf. Sci., 2006, 252, 7862–7873 CrossRef.
- X. Bao, M. Muhler, B. Pettinger, R. Schlögl and G. Ertl, Catal. Lett., 1993, 22, 215–225 CrossRef.
- H. Schubert, U. Tegtmeyer, D. Herein, X. Bao, M. Muhler and R. Schlögl, Catal. Lett., 1995, 33, 305–319 CrossRef.
- V. I. Bukhtiyarov, M. Hävecker, V. V. Kaichev, A. Knop-Gericke, R. W. Mayer and R. Schlögl, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 235422 CrossRef.
- Z. Qu, M. Cheng, W. Huang and X. Bao, J. Catal., 2005, 229, 446–458 CrossRef.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer and P. C. Redfern, Science, 2010, 328, 224–228 CrossRef PubMed.
- L. J. Durndell, C. Cucuzzella, C. M. A. Parlett, M. A. Isaacs, K. Wilson and A. F. Lee, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.02.052.
- J.-D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 25586–25589 CrossRef PubMed.
- C. Keresszegi, J.-D. Grunwaldt, T. Mallat and A. Baiker, Chem. Commun., 2003, 2304–2305 RSC.
- P. J. Ellis, I. J. Fairlamb, S. F. Hackett, K. Wilson and A. F. Lee, Angew. Chem., 2010, 122, 1864–1868 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization results and further experiments. See DOI: 10.1039/c8gc01340c |
| ‡ Due to the high basicity of MgO, the pH of the suspension was already at a value of over 10. Therefore, the deposition–precipitation method 2 was modified by addition of the noble metal precursor solution without additional NaOH. |
| This journal is © The Royal Society of Chemistry 2018 |