
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA one-pot biomimetic synthesis of selectively functionalized lignins from monomers: a green functionalization platform†
Nicola
Giummarella
 ab,
Claudio
Gioia
bc and
Martin
Lawoko
*ab
ab,
Claudio
Gioia
bc and
Martin
Lawoko
*ab
aDepartment of Fibre and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Teknikringen 56-58, SE-100 44, Stockholm, Sweden. E-mail: lawoko@kth.se
bWallenberg Wood Science Centre, KTH Royal Institute of Technology, Teknikringen 56-58, SE-100 44, Stockholm, Sweden
cDepartment of Civil, Chemical, Environmental and Materials Engineering, Via Terracini 28, 40131, Universita di Bologna, Italy
First published on 1st May 2018
Abstract
Lignin is the most abundant renewable source of phenolic compounds with great application potential in renewable materials, biofuels and platform chemicals. The current technology for producing cellulose-rich fibers co-produces heterogeneous lignin, which includes an untapped source of monomeric phenolics. One such monomer also happens to be the main monomer in soft wood lignin biosynthesis, namely coniferyl alcohol. Herein, we investigate the potential of coniferyl alcohol as a platform monomer for the biomimetic production of tailored functionalized oligolignols with desirable properties for material synthesis. Accordingly, a bifunctional molecule with at least one carboxyl-ended functionality is included with coniferyl alcohol in biomimetic lignin synthesis to, in one pot, produce a functionalized lignin. The functionalization mechanism is a nucleophilic addition reaction to the quinone methide intermediate of lignin polymerization. The solvent system applied was pure water or 50% aqueous acetone. Several bi-functional molecules differing in the second functionality were successfully inserted in the lignin demonstrating the platform component of this work. Detailed characterization was performed by a combination of NMR techniques which include 1H NMR, COSY-90, 31P NMR, 13C NMR, 13C APT, HSQC, HMBC and HSQC TOCSY. Excellent selectivity towards benzylic carbon and a high functionalization degree were noted. The structure of lignin was tailored through the solvent system choice, with 50% aqueous acetone producing a skeletal structure favorable for a high degree of functionalization. Finally, material concepts are demonstrated using classical thiol–ene- and Diels–Alder-chemistries to show the potential for the thermoset and thermoplastic concepts, respectively. The functionalization concept presents unprecedented opportunities for the green production of lignin-based recyclable biomaterials.
Introduction
Lignin is the most abundant renewable source of phenolic compounds at 300 billion tonnes in the biosphere.1 Due to the environmental concerns and dwindling petroleum resource, lignin is becoming increasingly interesting for fuel and material applications.2,3 Produced massively in connection with the production of cellulosic pulp, the so-called technical lignins, currently underutilized, could become highly valuable in this respect. The aromatic structure imparts rigidity in materials.4 The phenolic hydroxyls exhibit antioxidant properties5 or can be modified through several classical chemistries to introduce functionalities relevant for material synthesis. The number of recent reviews mirrors the growing interest in lignin applications and includes lignin biorefining,2 the use of lignin in polymer systems,6,7 thermo- and catalytic-valorisation of lignin8,9 and lignin-based nanoparticle systems.6,7,10 Two routes are currently being investigated for lignin applications. The first adopts the catalytic breakdown to platform monomers9 as a mean to overcome the notorious structural heterogeneity in existing technical lignins. The second adopts the use of polymeric lignin as a substrate for material synthesis subsequent to refining and/or modifications. Although progress has been made, both routes are not without challenges. The catalytic breakdown route suffers from the lack of a catalyst that can sustainably and efficiently break carbon–carbon bonds between the monolignols. In the direct use of technical lignin, on the other hand, a notorious heterogeneity with respect to the structure, molar mass and functionality is a problem to be addressed.The existing processes for chemical pulp production are a source of both monomeric and polymeric lignins, which have so far been under-exploited. The dominant process for the production of chemical pulps, the Kraft or sulphate process, produces significant amounts of mixed monomers in the process liquor e.g. coniferyl alcohol, guaiacol, vinylguaiacol, vanillin and acetovanillin.11 Dimers include stilbene type structures12 and enol ethers.13 Advances have been made in making available the polymeric fractions through technically feasible recovery to obtain a brown powder. One such technology is called Lignoboost14 and is currently applied on the industrial scale. The monomers and dimers remaining in the liquor, on the other hand, have remained less investigated and a fresh motive may be required to reverse the scenario. With the advantages of having a long tradition in pulping, existing infrastructure and existing permits, pulp mills could have an attractive position as producers of lignin-based platform monomers in addition to the already existing polymer retrieving technologies if incentives exist. One such incentive is the demonstration of the usability of key monomers. One of these monomers, coniferyl alcohol (CA), is particularly interesting from the perspective of being the monomer chosen by nature for the synthesis of softwood lignins. CA is formed under several pulping conditions which include soda-, soda-anthraquinone and kraft-anthraquinone pulping.15 Under these conditions, it is suggested that CA may react further through condensation reactions to form vinyl guaiacol. The use of flow through reactors has however more recently been shown to increase the CA concentrations.16 The production of CA during organosolv pulping with high boiling solvents (HSB) has also been shown.17
Coniferyl alcohol (CA) appears therefore to be a universal monomer produced by several existing pulping processes. If its use in materials science can be demonstrated, CA could likely become a target platform monomer.
Based on the detailed chemistry of CA polymerization to form native lignin,18 we sought to develop a green biomimetic method to functionalize lignin in one pot with its polymerization, tailoring its properties for suitability in material synthesis. Lignin polymerization from a monolignol (e.g. CA) is initiated by a redox system e.g. peroxidases/hydrogen peroxide. The formed phenoxy radicals are resonance stabilized. Radical coupling to form dimers exclusively involves β radical coupling.18 Subsequently, chain growth proceeds by either dimerization or endwise coupling of monomers. When β radicals couple, a quinone methide intermediate, an electrophile, is formed and reacts with available nucleophiles which could be intra-molecular, resulting in ring formations (e.g. ββ resinol and β5 coumaran structures) or inter-molecular as in the case of βO4. In the latter case, the competing nucleophiles are not few (Fig. 1). The addition of water is favoured as evidenced in the high content of α-hydroxylated βO4 observed by HSQC NMR studies.18,19 Minor couplings of hydroxyls from hemicelluloses and uronic acid moieties also occur leading to the formation of lignin–carbohydrate bonds of benzyl ether and benzyl ester types, the latter migrating to the Cγ (Fig. 1) as revealed by HSQC analysis.20–22 These are normally referred to as lignin carbohydrate complexes (LCC) and are responsible for the network formation in wood.23
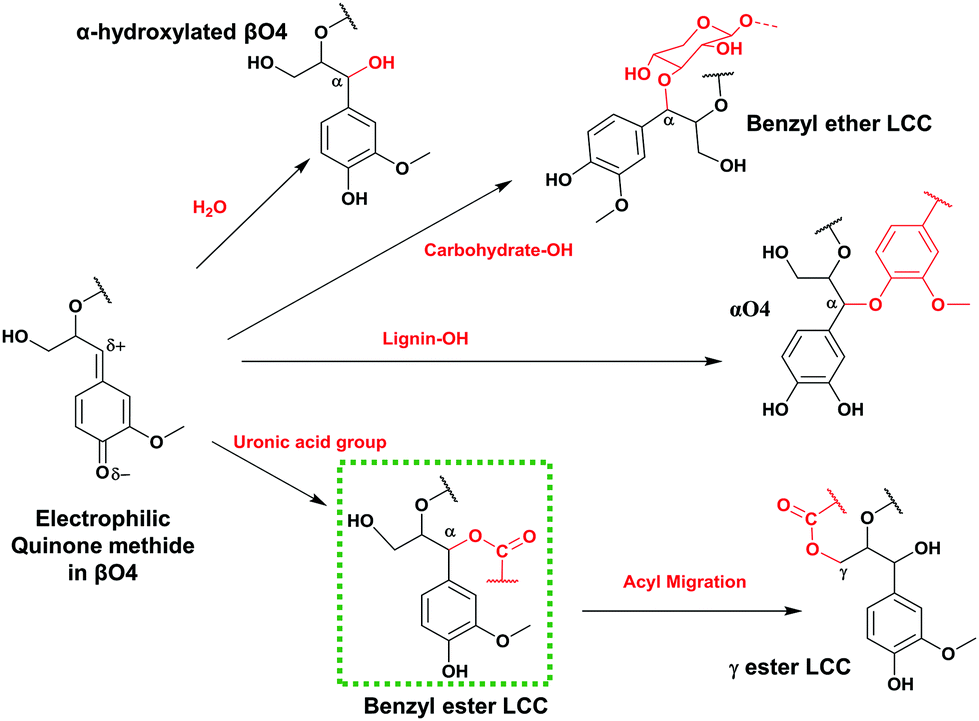 | ||
| Fig. 1 Reactions of the quinone methide electrophile in the βO4 inter-unit during lignin polymerization. | ||
Herein, we study the production of a functionalized lignin from CA inspired by the described chemistry of lignin polymerization. Synthetic lignin, commonly referred to as the dehydrogenation polymer (DHP), has been used for fundamental studies aimed at understanding the mechanisms of lignification in vivo.18,24–27 We adopt such strategies, albeit with important modifications for the purpose of green functionalization in one pot with lignin synthesis.
Our strategy mimics the suggested mechanism of the formation of natural lignin carbohydrate ester bonds in plant cells (shown by the green dotted rectangle in Fig. 1).
Materials and methods
Materials and chemicals
All chemicals used were of analytical grade and purchased from Sigma-Aldrich.Size-exclusion chromatography in DMSO + 0.5% LiBr
The size exclusion chromatography (SEC) of DHPs fraction was performed on a SEC-curity 1260 system (Polymer Standards Services, Mainz, Germany) coupled to both UV and refractive index (RI) detectors. Pullulan standards of 708 kDa, 344 kDa, 194 kDa 47.1 kDa, 21.1 kDa, 9.6 kDa, 6 kDa, 1.08 kDa, and 342 Da were used for standard calibration. The preparation of the samples, separation, and the experimental setup were the same as previously reported.28NMR characterization
Quantitative 31P NMR analysis was performed as reported elsewhere29,30 phosphorylating lignin with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP). HSQC analysis was performed applying the Bruker pulse program ‘hsqcetgpsi’, on 10–15% sample solution in deuterated DMSO-d6 or acetone-d6 in the case of acetylated samples. Carbon 2 of the aromatic groups, since unsubstituted, was used as the internal standard for the semi quantification of lignin linkages.31 The peaks of DMSO (δC/δH = 39.5/2.50 ppm) or acetone (δC/δH = 29.8/2.05 ppm) signals were used as an internal reference. Quantitative analysis was carried out by the method of Zhang and Gellerstedt by applying and comparing both 13C and HSQC analyses.32 Quantitative 13C spectra were recorded with the Bruker pulse program “zgig” with a 90° pulse width using an acquisition time of 1.4 s and a relaxation delay of 1.7 s. HSQC TOCSY experiments were carried out with the Bruker pulse program “hsqcetgpml” using D9 = 90 ms. 80 scans were collected. All 2D NMR spectra were recorded and processed as described elsewhere.33 Heteronuclear multiple bond correlation experiment was carried out with the “hmbcgp” Bruker pulse program, optimizing a long-range coupling constant D6-delay 60 ms and collecting 185 scans. 13C attached proton test (APT) experiment was run with the “jmod” Bruker pulse program while COSY90 analysis with the pulse sequence “cosyqf90”, and a number of 512 s and 16 scans were collected, respectively.Coniferyl alcohol reduction from coniferyl aldehyde
Coniferyl alcohol (CA) was prepared from coniferyl aldehyde according to previous procedures.34,35 Shortly, NaBH4 (33.7 mmol) and coniferyl aldehyde (16.8 mmol) were dispersed in 180 ml of EtOAc and left to stir overnight at room temperature. Afterwards, the reaction was quenched by adding 200 ml of deionized water. The organic phase was extracted, washed 3 times with brine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume), and dried over anhydrous sodium sulfate followed by rotor evaporation. The oily bright yellow product was re-dissolved in warm dichloromethane and stored in a freezer to recrystallize overnight. Finally, the product was filtered and washed with heptane before being dried in a vacuum oven at 50 °C.
:1 volume), and dried over anhydrous sodium sulfate followed by rotor evaporation. The oily bright yellow product was re-dissolved in warm dichloromethane and stored in a freezer to recrystallize overnight. Finally, the product was filtered and washed with heptane before being dried in a vacuum oven at 50 °C.
Lignin synthesis
The synthesis of the reference dehydrogenation polymer (DHP) lignin was performed as reported elsewhere27 with horseradish peroxidase (HRP) type IV (1 mg, 250–330 units). Both hydrogen peroxide (34 mM, 20 ml in water) and coniferyl alcohol (34 mM, 20 ml in water or 50% acetone) were simultaneously injected at a constant rate (250 μl h−1) using a NE-1800 Eight Channel Programmable syringe pump for 20 hours. Modifications to the protocol above were effected for functionalized lignins as follows: in the reaction media of 20 ml of water or 50% aqueous acetone, the bi-functional monomer (0.2 M) was dissolved and the pH was adjusted to 6.5–7 before the addition of HRP. The polymerization was carried out under slow stirring (150 rpm) at room temperature. After injection, the reaction was allowed to proceed for an additional 4 h. Synthetic lignins were collected by centrifugation (20000 rpm, 30 min) after the removal of dioxane under reduced pressure, washed with water twice and lyophilized.
Acetylation of DHP-lignin
Lignin samples were acetylated according to Gellerstedt.36 An amount of roughly 100 mg lignin sample was stirred overnight at room temperature in 400 μl of acetic anhydride:pyridine solution. Afterwards, the samples were dried under reduced pressure after the sequential addition of 3–4 drops of ice cold methanol and toluene (1:1, v:v). The acetylated sample was finally kept overnight in a desiccator before being dissolved for analysis.
Diels–Alder reaction
The reaction was carried out using a 15% DMSO-d6 solution of FPA-lignin with maleimide (in equimolar ratio of functional groups). The mixture was left in closed glass vials at 50 °C for 4 days under constant stirring prior to being analysed directly by NMR and SEC.Thiol–ene reaction
Acetylated AA-lignin in the presence of 5% in weight of the AIBN [2,2′-azobis(2-methylpropionitrile)] radical initiator and an excess of M3MP [methyl 3-mercaptopropionate] (≈1:10 molar ratio of functional groups) were dissolved in a glass vial with Teflon cap with 100 μl of acetone. After the removal of the acetone under a flow of N2, the thiolene reaction proceeded overnight, in bulk, at 80 °C.
Results and discussion
Green synthesis concept
In this study, functionalized lignins were synthesized in vitro starting from monomeric compounds comprising coniferyl alcohol (CA) as the monolignol and a bi-functional monomer consisting of at least one carboxyl group. The other functionality was a variable. As a reference, the in vitro synthesized lignin, also referred to as the dehydrogenation polymer (DHPs), was produced without the added functional monomer. The solvent used is pure water or a water/acetone mixture. Under the prescribed conditions (see the Experimental section), a one-pot production of functionalized lignin oligomer is achieved. During the synthesis, the obtained polymers precipitate from the solution, probably due to the increased hydrophobicity.The method is biomimetic, facile, benign and selective. As discussed earlier, the electrophilic quinone methide formed subsequent to the βO4 coupling (Fig. 1) invokes a nucleophilic attack. Unlike the in vivo synthesis in plant cells where the mimicked reaction i.e., the lignin acylation (Fig. 2) seems to occur with low frequency,37 the functionalization strategy herein integrates a high concentration of the carboxylate with its nucleophilic character to target higher functionalization levels. Furthermore, better mobility in the system is obtained by the use of small nucleophiles. In the plant cell wall, the same reaction is suggested to involve polymeric xylan with a uronic acid group as the side chain which performs the acylation. Hence, the restriction of the mobility of polymers may also play a part in the poor acylation efficiency in vivo.
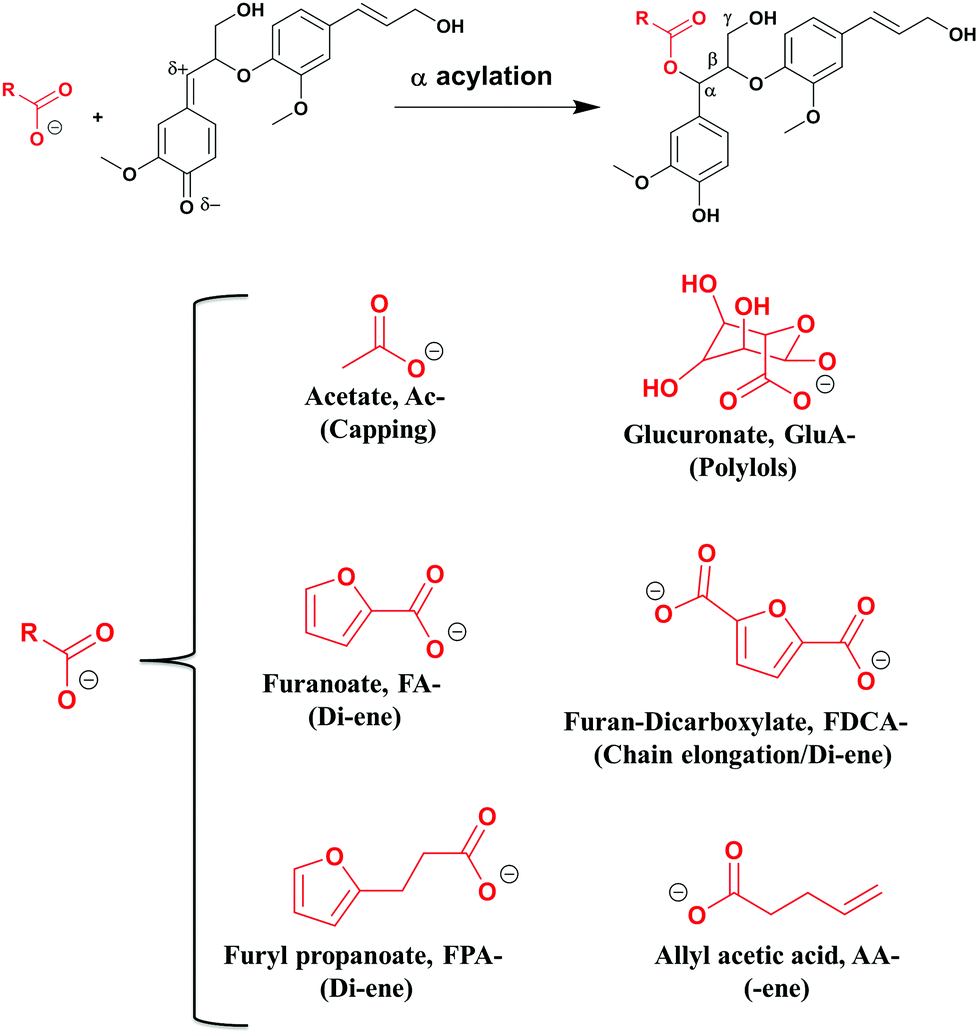 | ||
| Fig. 2 Lignin polymerization and functionalization in one pot. In red: functional molecules incorporated into the lignin skeleton; a demonstration of the platform concept. | ||
Accordingly, esterification should occur selectively at the electrophilic Cα in a newly formed βO4 moiety of lignin. The molecules tested were carefully selected to expose different functionalities (Fig. 2). O-Acetylation blocks the Cα through the capping reaction in contrast to the addition of water forming the hydroxyl (Fig. 1), hence modulating the lignin reactivity.
Glucuronylation introduces polyols that could further be functionalized. Acylation with furan derivatives introduces the di-ene functionality which is useful for classical Diels–Alder reactions and could find use in e.g. recyclable polymers/materials. Finally, acylation with allyl acetate introduces the ene-functionality of interest in further reactions to form e.g. thermosets. These material concepts are elaborated in a later section.
Synthesis in the pure water system
For this system, the performed syntheses were O-acetylation (AC-), O-glucuronation (GluA-), acylation with a furanoate (FA-) and with furan dicarboxylate (FDCA) as shown in Fig. 2. These choices were dictated by both the solubility of the monomers in water and their sustainability. These monomers can be produced from biomass-based sugars38,39 and are hence attractive. The yields of the formed polymer are calculated based on the starting weight of CA (Table 1). In our investigation, a carboxylate/CA ratio of 6:1 (mol/mol) gave higher yields of functionalized lignins compared with other trials done at ratios of 15:1 and 1.5:1.
| M n (Da) | M w (Da) | Đ | DP | Yield of precipitation (%) | |
|---|---|---|---|---|---|
| Abbreviations of functionalized lignins can be found in Fig. 1.a The mass of the precipitated FDCA-lignin is higher than the starting amount of CA due to the co-precipitation of the FDCA monomers with the lignin. Đ: Dispersity. DP: Degree of Polymerization. | |||||
| Reference (water) | 950 | 1500 | 1.6 | 5 | 59 |
| Ac-lignin (water) | 1050 | 2000 | 1.4 | 6 | 63 |
| FA-lignin (water) | 1100 | 1500 | 1.3 | 6 | 19 |
| FDCA-lignin (water) | 1750 | 3500 | 2 | 10 | 106a |
| GluA-lignin (water) | 1200 | 1700 | 1.4 | 6 | 34 |
| Reference (50% acetone) | 1700 | 3250 | 1.9 | 10 | 90 |
| FPA-lignin (50% acetone) | 1500 | 3450 | 2.3 | 8 | 45 |
| AA-lignin (50% acetone) | 1000 | 1550 | 1.6 | 6 | 90 |
In terms of the yields of precipitation, FDCA-lignin was the highest followed by the O-acetylation-lignin. FA- and GluA-lignins gave lower yields (Table 1). These could probably be optimized and more focused optimizations are a topic of future investigations. It should be noted that only the precipitated polymers were included for yield determination.
Studies of the molecular weight performed by size exclusion chromatography (SEC) (Table 1, ESI: Fig. S1†) gave for the reference sample a number average molecular weight, Mn, of about 900 Dalton, giving a degree of polymerization (DP) between 5 and 6.
The polymerization was probably terminated by precipitation due to increased hydrophobicity in the aqueous solvent. With the exception of FDCA-lignin, the degrees of polymerization of the synthesized oligomers were all in the same order of 5–6, suggesting that the onset of precipitation of lignin was independent of the functionality inserted. The reaction with furan dicarboxylic acid (FDCA) resulted in a polymer with a DP ∼10. This is consistent with that both the carboxylates in the bi-functional molecule may have coupled to two lignin molecules resulting in chain elongation. This precipitation did not occur for the FDCA-lignin polymer at DP 5–6 which was likely because the charge was maintained until the second acylation forming a charge-neutral molecule was complete.
Hence, tailoring of the molar mass of the functionalized lignins by the multi-functionality in the carboxylate function of monomers is an interesting option. The polydispersity of all formed polymers in this system was less than or equal to 2. Both the molar mass and its dispersity are important considerations for material synthesis as they have ramifications for both the processing and the properties of the material synthesized. Broad polydispersities for instance yield inhomogeneous materials. High molar mass, on the other hand, leads to entanglements affecting homogeneous processing. The obtained molar mass and dispersities in this work are appropriate for material synthesis as we recently demonstrated for polymer systems based on fractionated technical lignin.4
Synthesis in the water–acetone system
We hypothesized for the pure water syntheses that the increased hydrophobicity of lignin during polymer growth can lead to precipitation in water when the degree of polymerization (DP) is between 5 and 6 for the reference and the mono-carboxylate functionalized monomers. This hypothesis was tested on the reference lignin synthesis (only CA as the monomer) by the inclusion of acetone in the system (water/acetone 1:1, v:v). Accordingly, a higher molecular weight is expected. Acetone was chosen as the co-solvent for several reasons; it is a good lignin solvent, it is compatible with the redox system40 and it is a green and sustainable solvent.41 Size exclusion chromatography (SEC) studies confirmed the hypothesis as the DP of the reference was about 10 (Table 1, ESI: Fig. S1†). Thus, the molar mass of the polymers can be tailored by the polarity difference of the solvent systems applied. Changing the solvent composition also allows for the expansion in the portfolio of functional monomers to extend beyond pure water soluble compounds. The functionalizations using this solvent system were performed with 3-(2-furyl)propanoic acid (FPA) and allyl acetic acid (AA), which were only soluble when ionized. SEC studies of the functionalized polymers however showed no significant difference when compared with the water-synthesized polymers (Table 1) indicating that the effect of the solvent polarity difference on the DP observed for the reference lignins was offset in these cases by the stronger hydrophobicity of the functionalized lignins. Another interesting feature of the synthesized lignins was the physical appearance. The products had a whitish yellow colour in contrast to the dark brown colour of Kraft lignin (Fig. 3). The colour of Kraft lignin has been attributed to conjugated double bonds and quinoid structures, and may set limits for its application.42
 | ||
| Fig. 3 Physical appearance of the synthetic lignin reference (left) and functionalized lignins (right) compared with kraft lignin (middle). | ||
NMR studies
A combination of both one-dimensional (13C NMR, 13C APT, 1H NMR, 31P NMR) and two-dimensional NMR techniques (HSQC, HMBC, HSQC TOCSY NMR, COSY-90) was applied for detailed structural analysis. For the reference sample (Fig. 4), two quantification methods were applied and the results are reported in Table 2. In the first 13C NMR-HSQC analysis, the method described by Zhang and Gellerstedt32 was used. In this method, the cluster quantification of signals with similar T2-relaxation is performed using the aromatic carbon signals as the internal reference. The quantification is then used in the HSQC where the peaks are better resolved to allow more specific inter-unit quantification. The second method utilizes only the HSQC.31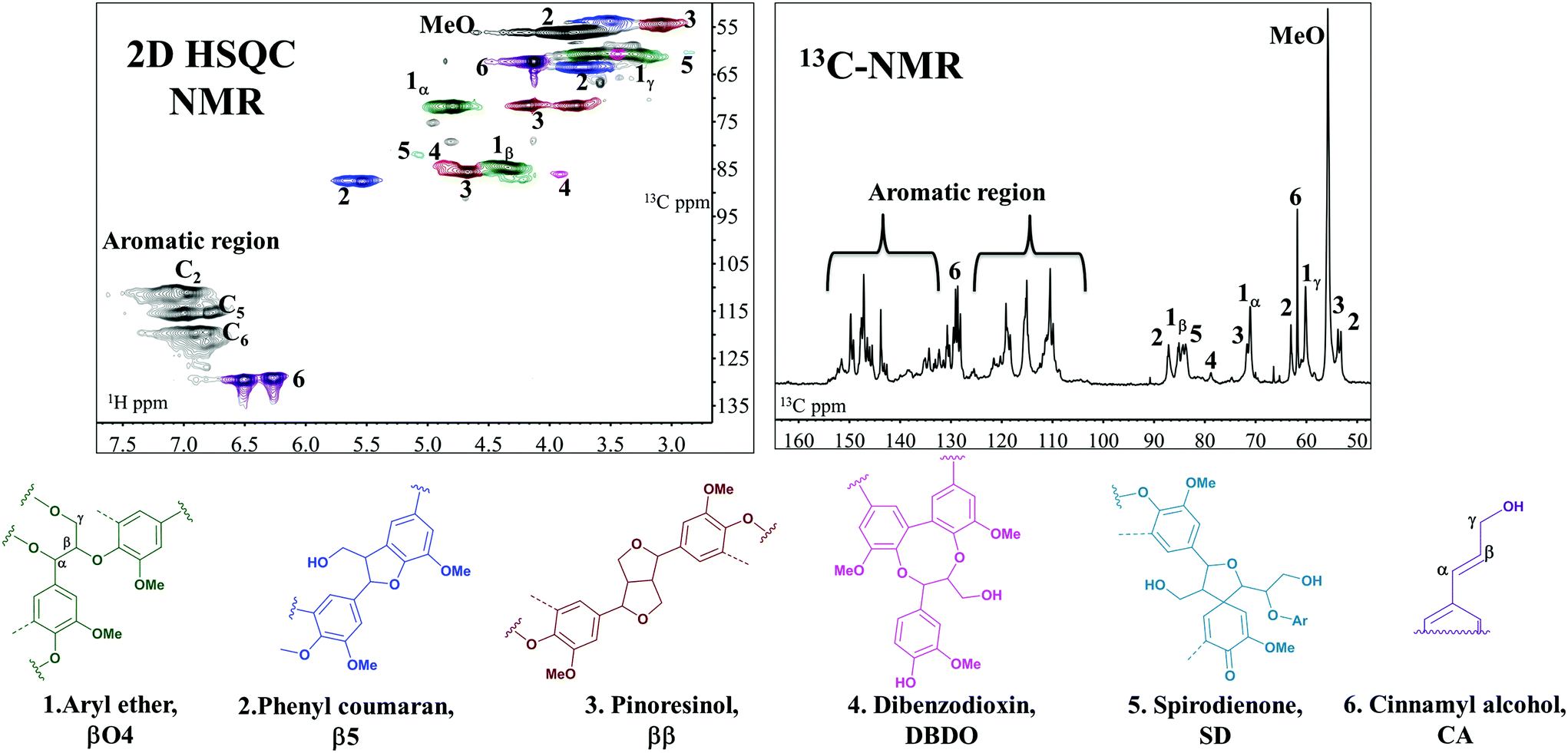 | ||
| Fig. 4 2D HSQC (left) and 13C NMR (right) expansion spectra of the DHP reference precipitated in acetone 50%. Structures (1–6) are representative of the main inter-monolignol linkages and the end group. | ||
| Structure | 13C-HSQC | HSQC-C2 aromatic |
|---|---|---|
| DBDO = dibenzodioxicin, SD = spirodienone, CA = cinnamyl alcohol (as expressed in Fig. 4). | ||
| βO4 | 30 | 32 |
| β5 | 20 | 24 |
| ββ | 16.4 | 18 |
| DBDO | 2.6 | 2 |
| SD | 2 | 2 |
| CA | 26 | 20 |
Here, the C2 aromatic signal, which is never substituted in native lignin, is used as the internal reference. Both methods are semi-quantitative. In our studies, the difference in the values obtained between the two methods was not that significant (Table 2). For this reason, the second method was used for the other samples since the NMR running time is much less. In general, the HSQC NMR data (Table 3, ESI: 13C/1H assignment in Table S1–S3,† spectra presented in Fig. S3–S10†) showed a predominance of βO4, ββ, β5 couplings as expected.18
| HSQC semi-quantification | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Error (%) | % | ||||||||
| Referencea (water) | Ac-L | GluA-L | FA-L | FDCA-L | Referencea (acetone 50%) | FPA-L | AA-L | ||
| L = Lignin. The numbering of lignin structures is consistent with Fig. 4 whereas the nomenclature of functionalized lignin can be found in Fig. 2. 1α-ester = Cα/Hα of benzyl ester; — = Not detecteda 31P NMR spectra available in ESI, Fig. S11 and S12. | |||||||||
| 1 | ±2.0 | 21 | 9 | 19 | 12 | 8 | 32 | 7 | 5 |
| 1α-ester | ±1.5 | — | 16 | 2 | 7 | 5 | — | 22 | 34 |
| 2 | ±2.5 | 27 | 25 | 30 | 28 | 31 | 24 | 24 | 16 |
| 3 | ±2.0 | 24 | 25 | 23 | 23 | 20 | 19 | 28 | 14 |
| 4 | ±0.5 | 2 | 2 | 2 | 1 | — | 3 | 8 | 4 |
| 5 | ±0.5 | 1 | 1 | 1 | 1 | — | 2 | 1 | 2 |
| 6 | ±2.5 | 26 | 25 | 29 | 30 | 32 | 25 | 30 | 22 |
| 31P NMR | |||||||||
| Error (mmol g−1) | mmol g−1 | ||||||||
| Aliphatic-OH | ±0.12 | 3.17 | 3.29 | 5.04 | 4.89 | 10.8 | 3.68 | 2.55 | 3.34 |
| C5 condensed para-OH | ±0.07 | 0.87 | 0.92 | 0.83 | 1.61 | 3.41 | 0.56 | 0.40 | 0.61 |
| Guaiacyl para-OH | ±0.04 | 0.71 | 0.77 | 0.54 | 1.18 | 1.82 | 0.68 | 0.57 | 0.89 |
The 55 and 4O5 structures are formed from cross couplings of preformed oligolignols, with the former being predominantly in the dibenzodioxin structure (DBDO).18 Accordingly, the presence of the dibenzodioxin structure (structure 4, Fig. 4) in a lignin molecule implies that the DP of the molecule is at least 5.
A clear difference is observed in the amounts of βO4 linkages of the references for the two solvent systems with the acetone/water system having a 30% higher content (Table 3). Hence, the change in solvent polarity affected the inter-unit composition of lignin and demonstrated the possibilities to tailor the backbone structure of lignin. It is also observed that each functionalization results in a lignin skeleton with a unique inter-unit composition (Table 3). For instance, the functionalization with GluA and FDCA in the pure water system leads to elevated levels of phenylcoumaran (β5) structures. FPA functionalization in the acetone/water system showed the highest levels of pinoresinol (ββ) and dibenzodioxins.
These differences highlight the role of different synthesis environments in the outcome of the lignin structure, also elaborated in the literature.18,26 Specific to our study, such effects could result, for instance, from the solvent polarity, small pH differences or effects from the nucleophile strength of functional monomers. Here, deeper studies are required to investigate the possibilities to tune the lignin structure during its synthesis through parametric design. Such structural tailoring is interesting from the view point of tailoring material properties. In this connection, our recent studies of epoxy-resins fabricated from well-defined Kraft lignin fractions alluded to the role of the molecular structure in material properties.43
Selectivity of functionalization
HSQC studies confirmed that the functionalization for all compounds occurred with excellent benzylic carbon (Cα) selectivity over Cγ of lignin, which is consistent with the proposed mechanism of in vivo benzyl ester formation (Fig. 1). The Cα/Hα correlations in these benzyl esters appear at around 13C/1H: 74.8/5.92 ppm (Fig. 5, ESI: C/H assignment in Tables S1 and S3†). The absence of signals from βO4 in Cγ esterified structures attested to the selectivity. These signals, if present, would appear between 13C/1H: 63 and 64/3.8–4.3 ppm.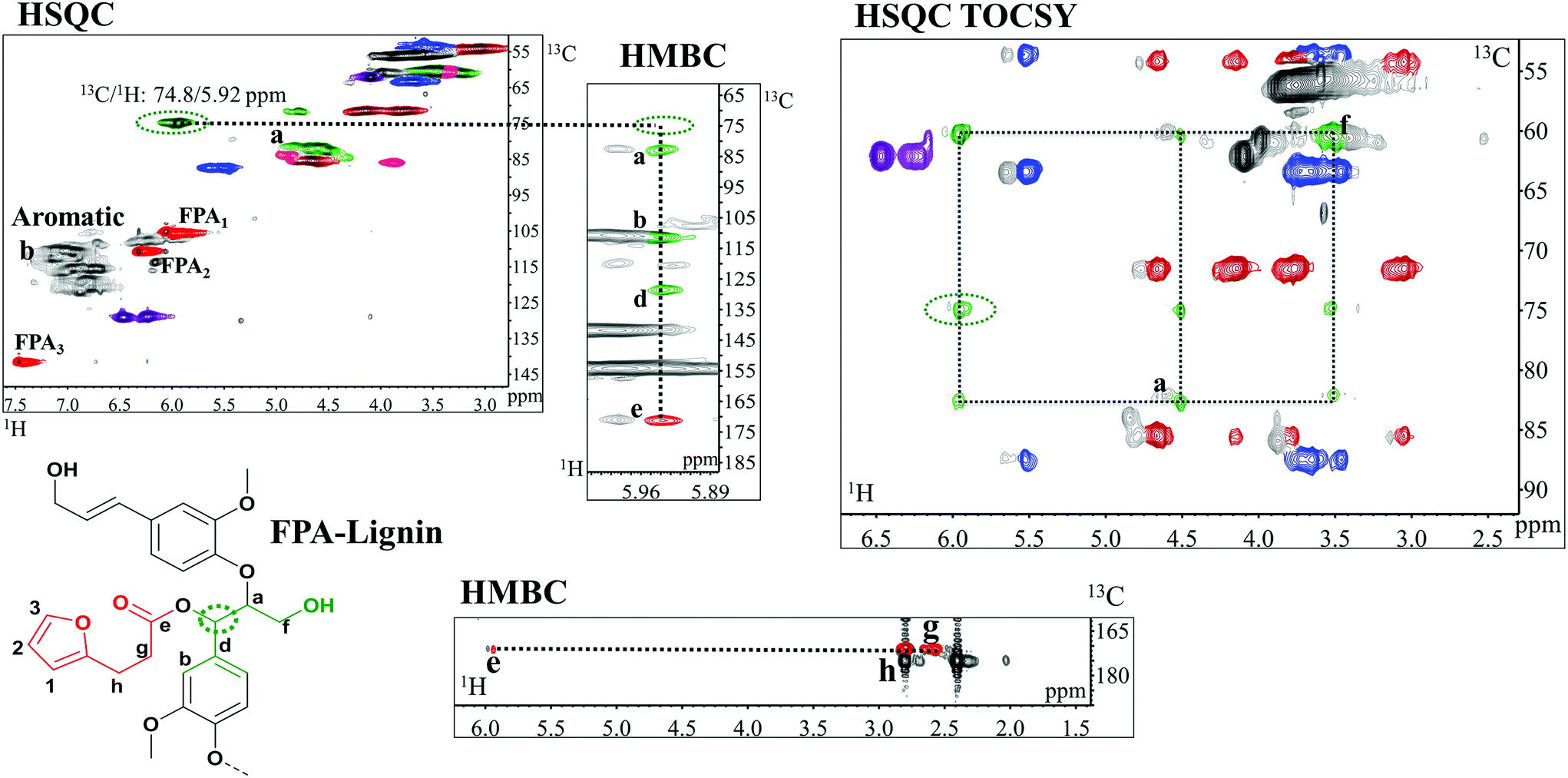 | ||
| Fig. 5 Expanded spectra of HSQC, HMBC and HSQC TOCSY of FPA-lignin functionalized in the water/acetone system. HMBC shows 3 bonds of the correlation of proton in the α-position (circled in green dots) with carbons in a(Cβ), b(C2), c(C6), d(C1) and carbonyl (e) which in turn correlates with the aliphatic signals in g and h of the functional group. HSQC TOCSY shows the correlation of the α proton with those of the β (a) and hydroxylated γ (f) in βO4 structures confirming the α selectivity of functionalization. Colours of lignin substructures are expressed in Fig. 4. Numbers and letters are clarified in the figure. Extended HSQC TOCSY and HMBC spectra of FPA-lignin are available in ESI: Fig. S13,† respectively. | ||
Diagnostic analyses of the assigned structures were obtained through the HMBC and HSQC TOCSY studies of acetone/water-synthesized functional polymers (Fig. 5).
HMBC not only provides the proof of connectivity between atoms in a spin system but also the proof of connectivity between different spin systems. Specifically, with reference to acylation herein (Fig. 5), the connected C atoms align on the proton axis of the Cα signal (circled in green dots in the figure). Signals b, c and d are proton correlations with aromatic C2, C6 and C1, respectively. More interesting is the alignment of the correlation of the α protons with the carbonyl carbon (signal e) from a different spin system, implying that the carboxylate is indeed coupled to the Cα. Furthermore, the long range correlations of the carbonyl carbon, e, with the protons at positions g and h of the molecule assert that the carbonyl originates from the FPA (Fig. 5). In the HSQC TOCSY, the correlation of the α protons with those of the β (a) and hydroxylated γ (f) in βO4 structures are confirmed (Fig. 5) and attest to the α selectivity of functionalization. The alignments of signals from the lignin skeleton are also shown and are consistent with previous studies.44
The functionalization efficiency
Since functionalization occurs in the βO4 sub-units, the functionalization efficiency (FE%) can be calculated as a function of the total βO4 according to eqn (1).Calculation of the efficiency of functionalization (FE%):
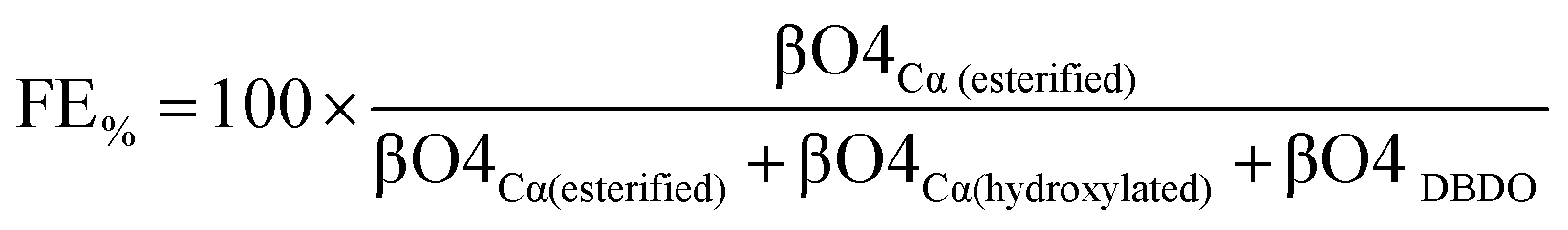 | (1) |
Accordingly, using the data in Table 3, the highest values calculated were obtained for the synthesis performed in the water/acetone system, with the AA acylation showing the highest efficiency of 79%, O-acetylation 59% and FPA 59%. The polymers from the water/acetone system were more efficiently functionalized. This is a reasonable expectation given that this system produced more βO4 inter-units than when pure water was used as the solvent (Table 3), and functionalization occurs in such units. The major competing reaction which is the addition of water leads to the formation of a Cα hydroxylated βO4 moiety with the 13C/1H correlation at 71.4/4.71 ppm in the HSQC spectrum. Halving the amount of water as in the acetone/water system should somewhat lower the probability for this reaction. However, the number of moles of water present in both solvent systems is still a few orders of magnitude higher than that of the carboxylates. The carboxylate:water molar ratio in the pure water system is 1:800 and in the acetone/water system it is 1:400. That the addition of carboxylate could competitively occur at such superior stoichiometry in favour of water is likely due to the carboxylate being a stronger nucleophile than water. When compared to the other carboxylates, the favourable acylation of lignin with AA and FPA is probably due to their stronger nucleophilic character. In addition, the longer aliphatic chains carrying the reactive end group in FPA improves the accessibility when compared to e.g. FA and FDCA.
Unravelling molecular structures
The degree of functionalization is here defined as the number of inserted functionalities per 100 monolignols. The AA-lignin has a degree of functionalization of 34% and a DP of 6 (Table 2). In addition, the cinnamyl alcohol (structure 6, Fig. 4) content is about 22%. Put together, the average molecule is tri-functional in the -ene functionality (2-enes from the allyl acetate inserted group and one from the cinnamyl group). A plausible structure is shown in Fig. 6 (left). Similarly, FPA-lignin fractions have a DP of about 6–8 and a functionalization degree of about 20%, meaning that they on average had one functional group inserted per molecule. Furthermore, they also have one cinnamyl alcohol end group on average per molecule (≥20% cinnamyl structure Table 3). The FPA-lignin polymer is thus bi-functional in the ene-functionality but mono-functional in the di-ene functionality as shown in Fig. 6 (right).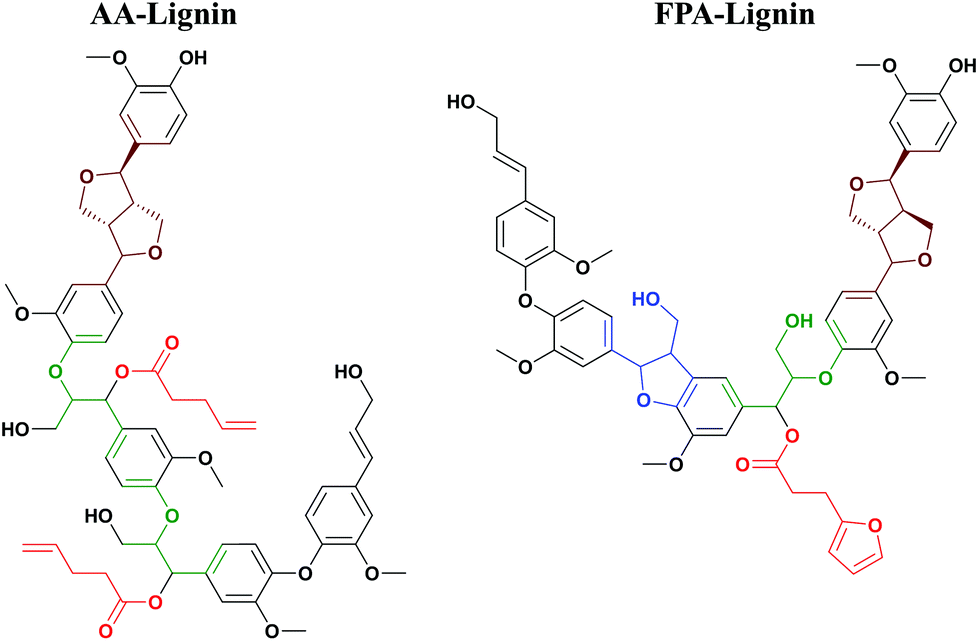 | ||
| Fig. 6 Typical structures of AA-lignin and FPA-lignin. | ||
Testing material concepts
The AA-and FPA-lignin possess interesting functionalities for material applications. In the following section, we demonstrate a few material concepts but will not embark on material synthesis as this would require detailed studies and are a subject of future work.Thiol–ene reaction test
Thiol–ene chemistry consists of the addition reaction of thiols on alkenes. Often promoted by radical initiators, it is performed under mild conditions usually providing high yields and harmless by-products.45 In materials science, it has been shown to be a versatile tool with applications in surface modification, grafting, polymerization of thermoplastic materials and curing of thermosetting resins. Although such a class of reactions were studied for more than a century,46 renewed interest is attributed to the possibilities it offers for the production of biopolymers47 and bio-based materials.48Specifically in relation to lignin, we recently fabricated lignin-based thiol–ene thermosets from a pre-allylated ethanol-soluble Kraft lignin fraction.4
Herein, we investigate the feasibility of the thiol–ene reaction in the green synthesis of allylated lignin (AA-lignin, Fig. 6, left). It should be noted here that the choice of a monothiol in our work was made intentionally to avoid crosslinking.
In classical thiol–ene based thermosets, bi- tri- or multi-functional thiols are used. The reason for our choice was to enable solution state NMR studies, since this would not only allow for the confirmation that the thiol addition had occurred, but also enable detailed qualitative and quantitative analyses of structural changes that may occur in the lignin skeleton during the reaction. In addition, the selectivity of the reaction can be studied. The reaction was performed on AA-lignin which was mixed with an excess of methyl 3-mercaptopropionate (M3MP) and left to react in bulk for 24 hours at 120 °C.4 This first attempt was on the AA-lignin without further modification, while being aware of the possibilities of phenoxy radical generation from lignin. The HSQC study did not show any changes in the lignin structure that would have arisen from radical coupling reactions of resonances to the phenoxy radical (ESI: Fig. S14†). Instead, new signals assigned to Cγ esters on the lignin appearing at 13C/1H: 63.5/4.17–4.27 ppm were observed. More specifically, the reaction involved the primary alcohol on the cinnamyl structure (structure 6, Fig. 4), as observed from the expected shift of the Cβ in this structure from 13C/1H: 128.6/6.23 ppm to 124.4/6.1 ppm (ESI: Fig. S14†). A trans-esterification reaction between Cγ hydroxyls in structure 6 and the ester-ended thiol had occurred. No signals typical of the expected thiol reaction with double bonds were identified. Thus, for improved selectivity, the pre-acetylation of the AA-lignin was adopted to cap reactive hydroxyls. The thiol–ene reaction was then performed on the acetylated AA-lignin and in the presence of a radical initiator, AIBN (5%).
The reaction was carried out at a lower temperature, 80 °C, since AIBN has 10 hours half-life at 65 °C and kd(s−1) = 3.2 × 10−5 at 70 °C;49 a small amount of DMSO-d6 (4% wt/vol%) was added to help the dissolution of the reaction mixture and facilitate NMR studies. Interestingly, after 12 hours, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the cinnamyl structure was fully thiolated while the double bond in the esterified AA was in principle unreacted, even in the excess of thiol. This selectivity could be the result of the higher electron density in the CC bond of the cinnamyl structure imposed by the presence of the electron-donating aromatic ring. The terminal allyl group in the inserted AA only reacted when the thiol–ene reaction was carried out solvent-free and for a longer running time (18 hours). The disappearance of unsaturated carbon peaks (Fig. 7a, signals 6α–6β of cinnamyl alcohol and signals a–b of the allyl end group) suggests the formation of new aliphatic bonds due to the thiolene reaction. The HSQC analysis of AA-lignin after the thiolene reaction shows a diagnostic signal at 13C/1H: 45.9/3.17 ppm. HMBC confirmed the addition of thiol in the β position of cinnamyl alcohol manifested in the alignment of Cα(e), Cγ(g) and C1(d) with the β proton in lignin (Fig. 7a). The confirmation of thiol addition to AA, on the other hand, was not possible by HMBC due to the crowded overlapping of C/H signals in the aliphatic region. However, HSQC TOCSY (Fig. 7b) combined with 13C APT (ESI: Fig. S15†) was the key analysis to confirm the thiol addition to the allyl functionality in AA. The result supported an anti Markovnikov reaction50 which would create diagnostic methylene signals in the same spin system. More specifically, HSQC TOCSY revealed cross peak correlation between the newly formed aliphatic signals at 13C/1H: 31.1/2.47 ppm (h), 28.7/1.53 ppm (i), 23.8/1.62 ppm (l) as shown in Fig. 7b, while 13C APT confirmed that all were methylene signals. Overall, these reactions demonstrated that thiols could be inserted into the structures selectively following an acetylation capping reaction of reactive hydroxyls. Bi- or multi-functional thiols could be used instead and serve as cross-linkers to produce lignin-based thiol–ene thermosets.4
C double bond in the cinnamyl structure was fully thiolated while the double bond in the esterified AA was in principle unreacted, even in the excess of thiol. This selectivity could be the result of the higher electron density in the CC bond of the cinnamyl structure imposed by the presence of the electron-donating aromatic ring. The terminal allyl group in the inserted AA only reacted when the thiol–ene reaction was carried out solvent-free and for a longer running time (18 hours). The disappearance of unsaturated carbon peaks (Fig. 7a, signals 6α–6β of cinnamyl alcohol and signals a–b of the allyl end group) suggests the formation of new aliphatic bonds due to the thiolene reaction. The HSQC analysis of AA-lignin after the thiolene reaction shows a diagnostic signal at 13C/1H: 45.9/3.17 ppm. HMBC confirmed the addition of thiol in the β position of cinnamyl alcohol manifested in the alignment of Cα(e), Cγ(g) and C1(d) with the β proton in lignin (Fig. 7a). The confirmation of thiol addition to AA, on the other hand, was not possible by HMBC due to the crowded overlapping of C/H signals in the aliphatic region. However, HSQC TOCSY (Fig. 7b) combined with 13C APT (ESI: Fig. S15†) was the key analysis to confirm the thiol addition to the allyl functionality in AA. The result supported an anti Markovnikov reaction50 which would create diagnostic methylene signals in the same spin system. More specifically, HSQC TOCSY revealed cross peak correlation between the newly formed aliphatic signals at 13C/1H: 31.1/2.47 ppm (h), 28.7/1.53 ppm (i), 23.8/1.62 ppm (l) as shown in Fig. 7b, while 13C APT confirmed that all were methylene signals. Overall, these reactions demonstrated that thiols could be inserted into the structures selectively following an acetylation capping reaction of reactive hydroxyls. Bi- or multi-functional thiols could be used instead and serve as cross-linkers to produce lignin-based thiol–ene thermosets.4
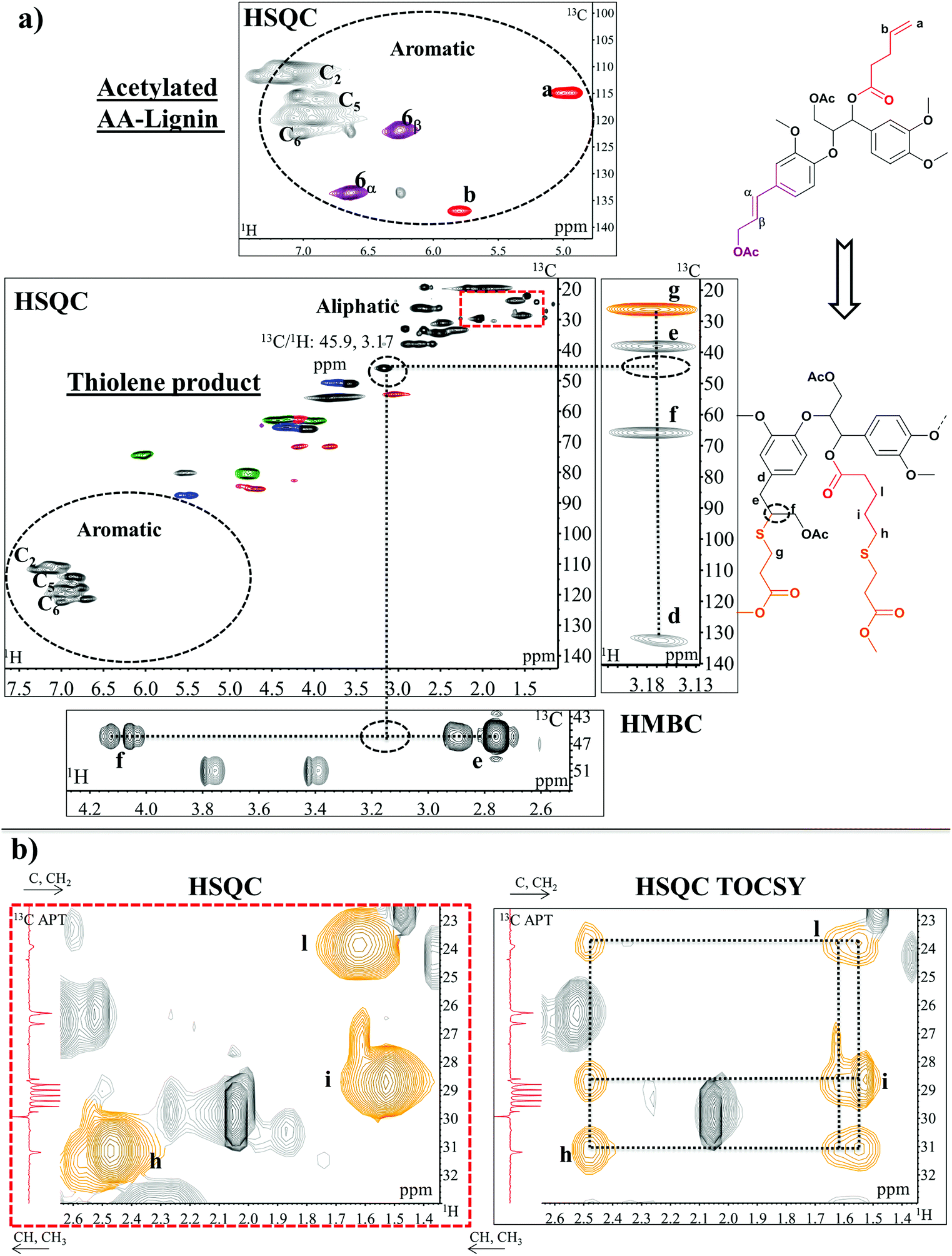 | ||
| Fig. 7 Expanded HSQC, HMBC (a) and HSQC TOCSY (b) spectra of acetylated AA-lignin before and after the thiolene reaction. (a) In the aromatic region, the disappearance of unsaturated carbon peaks (6α-6β of cinnamyl alcohol and a-b of the allyl end group) after the thiolene reaction is evident. HMBC shows the alignment of Cα(e), Cγ(g) and C1(d) with β proton of the reacted cinnamyl alcohol whereas (b) HSQC TOCSY reveals the cross peak correlation between newly formed methylene signals h, I, l. Extended HSQC TOCSY and HMBC spectra of AA-lignin are available in the ESI: Fig. S16.† Colours of lignin substructures are expressed in Fig. 4. | ||
Diels–Alder [4 + 2] cycloaddition reaction test
The Diels–Alder reaction (DA) is a powerful approach for the fabrication of complex molecular architectures. Although it proved to be an essential instrument for the synthesis of bioactive natural products, in materials science it represents one of the most exploited pathways for the construction of reversible materials. In particular, furan-based DA systems have been deeply exploited for the fabrication of linear macromolecules presenting reversible polycondensation processes or in reversible cross-linking resins.51The Diels–Alder reaction was recently demonstrated on Kraft lignin functionalization and FPA.52 The stepwise synthesis however included non-green chemicals. Nevertheless, the reaction was shown to form a gel at 70 °C in DMSO after several days. The retro-reaction reforming the solution occurred at 120 °C.
Herein, we intended to achieve chain elongation and subjected the benignly synthesized di-ene (FPA-lignin) to a Diels–Alder reaction with maleimide monomer (M) as di-enophile.
The optimized conditions were 50 °C, 4 days reaction for a 15% wt/volume solution of DMSO-d6. Under these conditions, a conversion of about 60% was obtained as typified by the signals associated with the DA product (Fig. 8: HSQC). The 13C/1H correlation appears at 135.2–137.8/6.3–6.4 ppm for the newly formed CC bond (cross peaks in d and e), two signals at 78.0/5.12 and 79.8/5.0 ppm for the carbons bridged by oxygen (cross peak a) and a couple of signals at 49.6/2.74, 51.3/2.94 and 50.5/3.15, 49.3/3.55 ppm assigned to the newly formed saturated carbons (cross peaks b and c). The conversion was estimated by dividing the sum of the volume integrals of the signals at 49.3/3.55 and 51.3/2.94 ppm corresponding to the 13C/1H correlation of the atom marked b for endo- and exo-products, with a characteristic signal of unreacted FPA-lignin (FPA 3, 13C/1H: 141.5/7.47 ppm).
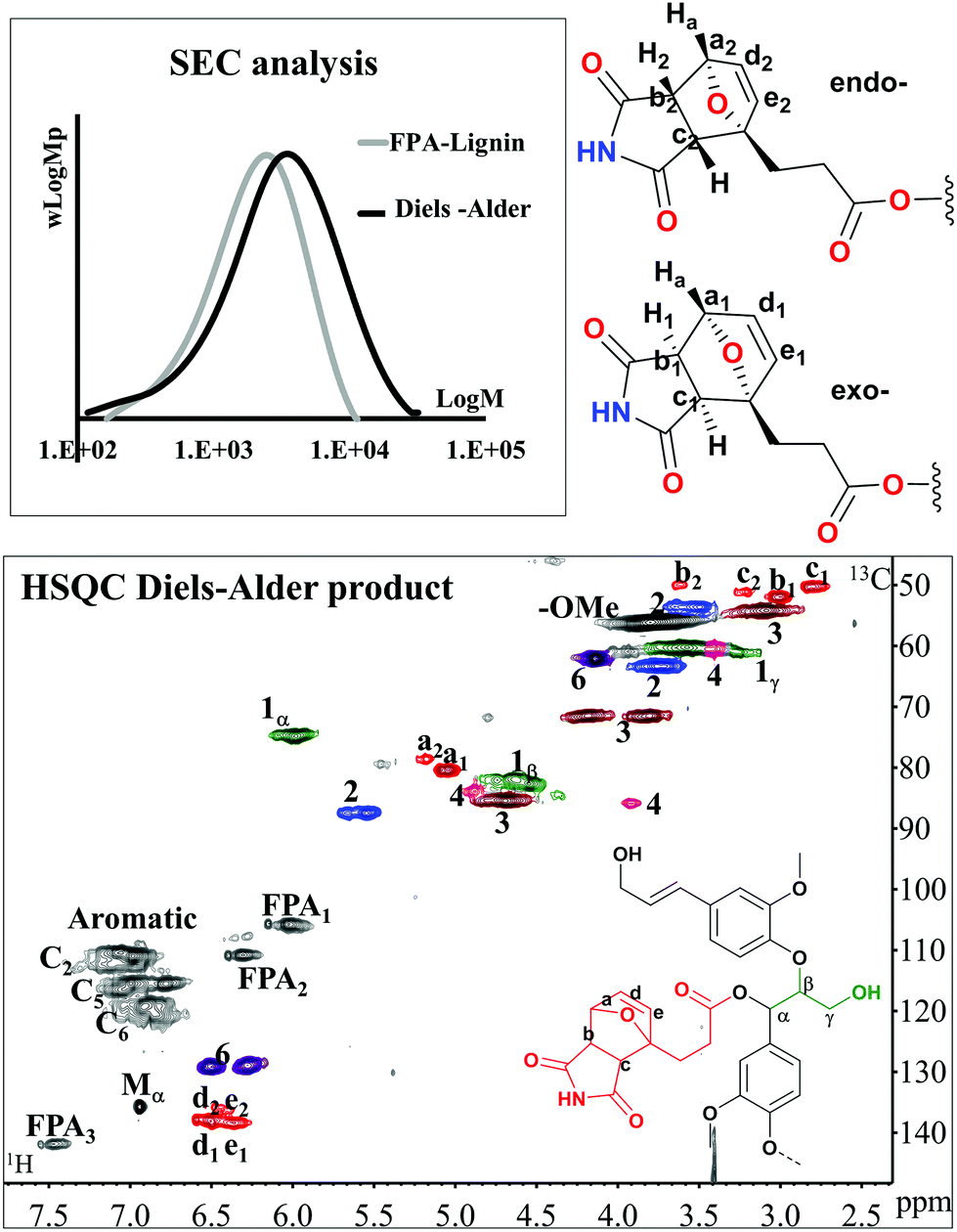 | ||
| Fig. 8 SEC of FPA-lignin before and after the Diels–Alder reaction and the HSQC spectrum of the Diels–Alder product. The subscripts 1 and 2 stand for exo- and endo- diastereoisomers, respectively, whose orientation between the proton in a (Ha) and b (H1–H2) is shown. | ||
A thorough analysis on model compounds by 1H and COSY-90 NMR allowed the discrimination of such signals relative to the exo- and endo-forms of the adduct (ESI: Fig. S17 and S18†).53 Specifically, the exo-adduct accounts for 82% (Fig. 8, signals a1–e1) while the endo adduct (Fig. 8, signals a2–e2) accounts only for 18%. The retro DA reaction occurred at 130 °C but was also accompanied by the cleavage of the ester linkage between lignin and FPA. SEC traces suggested an increase in molar mass resulting from the DA reaction (Fig. 8: SEC).
Thus, here, we demonstrated that the DA reaction on a green functionalized lignin had occurred without affecting the lignin skeletal structure. This specific DA product could find use in chain extension chemistries if reacted with bi-functional amines. Such a reaction constitutes the ring opening of the M ring and could find applications in thermoplastics. The disassembly aspect of the retro-DA reaction observed here presents the added advantage of the recyclability of such materials.
Potential of and criteria for the functionalization concept
Our efforts were focused on functionalization through esterification. However, based on the functionalization mechanism, nucleophiles other than carboxylates could be used. Such nucleophiles need to meet certain criteria which would include solubility and compatibility with the solvent- and redox systems, respectively. Strategies for material concepts are not limited to those demonstrated in our work but could extend to include grafting from-, grafting to- and other types of chemistries. Caution must however be taken to tame the lignin reactivity towards selective reactions. For instance, selective capping reactions of hydroxyls subsequent to functionalization may be required. The potential to tailor the molar mass of the lignin could also be exploited depending on the application. The choice of monomers is also another important variable and is not limited to coniferyl alcohol. Obviously, other monomers such as sinapyl alcohol from hardwood or coumaryl alcohol can be used. These can be applied to tailor the backbone structure of lignin. The use of sinapyl would for instance elevate the content of aryl ethers and pinoresinol structures. The concept may also be extended to dimers or oligomers that meet the criteria of the Cα–Cβ conjugation on a unit with free phenolic hydroxyl groups. Limitations could arise from factors such as sterical hindrance. Interestingly, structures such as coniferyl aldehydes which have the Cα–Cβ conjugation do not polymerize under these conditions. This is probably due to the extended conjugation to Cγ carbonyls in these structures. However, the option to reduce such structures to the corresponding coniferyl alcohols also offers possibilities to increase the yields of coniferyl alcohols. Such oxidized monomers are also present in technical lignins and could also be produced through other catalytic oxidative processes currently under investigation. Thus, in a lignin-first biorefinery context, the potential for high yield production of coniferyl alcohol is not far-fetched. With respect to environment benignity, the green functionalization strategy is further supported by the natural recyclability potential manifested in the backbone structure of the synthesized lignins possessing inter-unit similarities to native lignin. Furthermore, in analogy to native lignin networks, benzylic carbon is significant for the network formation in the synthesis and an important disassembly point in the proposed recycling philosophy.Conclusions
In the present work, we investigated the possibilities for the adoption of coniferyl alcohol as a platform monomer in the synthesis of lignin-based materials. Coniferyl alcohol was selected based not only on the production potential in existing pulping technologies, but also on its uniqueness from the viewpoint of being nature's chosen monomer for softwood lignin biosynthesis. Accordingly, we report a concept for the production of selectively functionalized lignins in one pot with lignin synthesis using a biomimetic approach.A bi-functional molecule, with at least one carboxylate functionality, was included in the biomimetic synthesis of lignin from coniferyl alcohol, in one pot, to obtain a functionalized lignin oligomer, with degrees of polymerization (5–10), which is well suited for material synthesis. Detailed characterization using several NMR techniques, such as 1H NMR, 31P NMR, 13C APT, COSY-90, HSQC, HMBC and HSQC TOCSY, confirmed that lignin functionalization occurred through the esterification of the carboxylate functionality on the studied molecules to the benzylic carbons in lignin, and with excellent selectivity. The other functionality on the bi-functional molecule was variable and included ene- di-ene- carboxylate- and polyol-functionalities demonstrating the platform nature of the adopted strategy. High degrees of functionalization were obtained for certain compounds and were attributed to their higher nucleophile strengths.
On selected functionalized lignins, material concepts were tested. More specifically, thiol–ene- and Diels–Alder cycloaddition-chemistries were chosen to demonstrate the possibilities for thermoset synthesis and chain extension chemistries, respectively.
For the thiolene test, the selected molecule was the lignin esterified with allylacetic acid (AA-lignin), which was tri-functional in the ene-functionality (two originating from the allylacetic acid and one from the lignin cinnamyl end group). It was shown that a pre-acetylation of AA-lignin was required to tame the lignin reactivity in order to obtain the desired reaction. Thus, thiol addition to the double bonds was successfully done, demonstrating the thermoset resin concept. For the Diels–Alder demonstration, a lignin esterified with furylpropanoic acid (FPA-lignin), which was mono-functional in the di-ene functionality, was used without further modification, together with maleimide monomer as the di-enophile. The cyclo-addition reaction was successfully performed. Here, we envision further reaction of the Diels–Alder product with other compounds e.g. diamines in chain extension chemistries for e.g. thermoplastic applications.
The proposed functionalization concept presents unprecedented possibilities for greener production of sustainable lignin-based materials with ramifications for environmental benignity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Knut and Alice Wallenberg Foundation and the authors are grateful for the financial support by the Wallenberg Wood Science Center.Notes and references
- M. Funaoka, M. Keigo and A. Mitsuru in Encyclopedia of Polymeric Nanomaterials, Springer-Verlag, Berlin Heidelberg, 2015, pp. 1080–1098 Search PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 6185 CrossRef PubMed.
- A. Gandini, Macromolecules, 2008, 41(24), 9491–9504 CrossRef.
- M. Jawerth, M. Johansson, S. Lundmark, C. Gioia and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5(11), 10918–10925 CrossRef.
- C. Pouteau, P. Dole, B. Cathala, L. Averous and N. Boquillon, Polym. Degrad. Stab., 2003, 81(1), 9–18 CrossRef.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- W.-J. Liu, H. Jiang and H.-Q. Yua, Green Chem., 2015, 17(11), 4888–4907 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. V. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55(29), 8164–8215 CrossRef PubMed.
- Y. Zhao, H. Tan, Y. Xu and L. Zou, Bioresour. Technol., 2016, 214, 496–503 CrossRef PubMed.
- L. Lowendahl, G. Petersson and O. Samuelson, Sven. Papperstidn., 1978, 12(81), 392–396 Search PubMed.
- D. Schmitt, N. Beiser, C. Regenbrecht, M. Zirbes and S. R. Waldvogel, Sep. Purif. Technol., 2017, 181, 8–17 CrossRef.
- G. Gellerstedt and E. V. Lindfors, Nord. Pulp Pap. Res. J., 1987, 2(2), 71–75 Search PubMed.
- P. Tomani, Cellul. Chem. Technol., 2010, 44(1–3), 53–58 Search PubMed.
- R. D. Mortimer, J. Wood Chem. Technol., 1982, 2, 383–415 CrossRef.
- A. D. Venica, C.-L. Chen and J. S. Gratzl, Holzforschung, 2008, 62, 627–636 Search PubMed.
- T. Kishimoto and Y. Sano, Holzforschung, 2001, 55, 611–616 CrossRef.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph and J. H. Christensen, Phytochem. Rev., 2004, 3, 29–60 CrossRef.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2004, 52(7), 1850–1860 CrossRef PubMed.
- M. Y. Balakshin, E. A. Capanema and H.-m. Chang, Holzforschung, 2007, 61, 1–7 CrossRef.
- E. Capanema, M. Balakshin, R. Katahira, H.-m. Chang and H. Jameel, J. Wood Chem. Technol., 2014, 35(1), 17–26 CrossRef.
- D. V. Evtuguin, B. J. Goodfellow, P. Neto and N. Terashima, Proceeding 13th ISWPC, 2005, vol. 2, pp. 439–444.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Biomacromolecules, 2005, 6(6), 3467–3473 CrossRef PubMed.
- L. L. Landucci, J. Wood Chem. Technol., 2000, 20, 243–264 CrossRef.
- B. Cathala, B. Chabbert, C. Joly, P. Dole and B. Monties, Phytochemistry, 2001, 56, 195–202 CrossRef PubMed.
- N. Terashima, R. H. Atalla, S. A. Ralph, L. L. Landucci, C. Litpierre and B. Monties, Holzforschung, 1996, 50, 9–14 CrossRef.
- T. Warinowski, S. Koutaniemi, A. Kärkönen, I. Sundberg, M. Toikka, L. K. Simola, I. Kilpeläinen and T. H. Teeri, Front. Plant Sci., 2016, 7, 1523 Search PubMed.
- A. Duval, F. Vilaplana, C. Crestini and M. Lawoko, Holzforschung, 2015, 70(1), 11–20 Search PubMed.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef.
- D. S. Argyropoulos, Res. Chem. Intermed., 1995, 21(3), 373–395 CrossRef.
- M. Sette, R. Wechselberger and C. Crestini, Chem. – Eur. J., 2011, 17, 9529–9535 CrossRef PubMed.
- L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37–45 CrossRef PubMed.
- N. Giummarella, L. Zhang, G. Henriksson and M. Lawoko, RSC Adv., 2016, 6, 42120–42131 RSC.
- F. Lu and J. Ralph, J. Agric. Food Chem., 1998, 46, 1794–1796 CrossRef.
- H. Kim and J. Ralph, J. Agric. Food Chem., 2005, 53, 3693–3695 CrossRef PubMed.
- G. Gellerstedt, Gel permeation chromatography, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Heidelberg, Germany, 1992 Search PubMed.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2016, 4, 5319–5326 CrossRef.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef PubMed.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its Many by-products, Elsevier, The Netherlands, 2000 Search PubMed.
- A. M. Klibanov, Trends Biotechnol., 1997, 15, 97–101 CrossRef PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehadad and P. J. Dunne, Green Chem., 2016, 18, 288 RSC.
- H. Zhang, Y. Bai, B. Yu, X. Liu and F. Chen, Green Chem., 2017, 19, 5152–5162 RSC.
- C. Gioia, G. Lo Re, M. Lawoko and L. A. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef PubMed.
- J. Ralph, Y. Zhang and R. M. Ede, J. Chem. Soc., Perkin Trans. 1, 1998,(16), 2609–2614 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef PubMed.
- T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef.
- T. O. Machado, C. Sayer and P. H. H. Araujo, Eur. Polym. J., 2017, 86, 200–215 CrossRef.
- O. Turunc and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2013, 115, 41–54 CrossRef.
- Thermal Initiators: Decomposition Rate and Half-Life, https://www.sigmaaldrich.com/content/dam/sigma aldrich/docs/Aldrich/General_Information/thermal_initiators.pdf?utm_source=redirect&utm_medium=promotional&utm_campaign=insite_thermal_initiators.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135(28), 10334–10337 CrossRef PubMed.
- A. Gandini, Polim.: Cienc. Tecnol., 2005, 15, 95 CrossRef.
- A. Duval, H. Lange, M. Lawoko and C. Crestini, Green Chem., 2015, 17, 4991 RSC.
- J. H. Cooley and R. V. Williams, J. Chem. Educ., 1997, 74(5), 582 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEC chromatograms, C/H HSQC assignment; 31P NMR, 13C APT, 2D HSQC, HMBC and HSQC TOCSY spectra. See DOI: 10.1039/c8gc01145a |
| This journal is © The Royal Society of Chemistry 2018 |