DOI:
10.1039/C8GC01092G
(Paper)
Green Chem., 2018,
20, 3279-3286
The CO2 capturing ability of cellulose dissolved in NaOH(aq) at low temperature†
Received
6th April 2018
, Accepted 13th June 2018
First published on 14th June 2018
Abstract
Herein, we explore the intrinsic ability of cellulose dissolved in NaOH(aq) to reversibly capture CO2. The stability of cellulose solutions differed significantly when adding CO2 prior to or after the dissolution of cellulose. ATR-IR spectroscopy on cellulose regenerated from the solutions, using ethanol, revealed the formation of a new carbonate species likely to be cellulose carbonate. To elucidate the interaction of cellulose with CO2 at the molecular level, a 13C NMR spectrum was recorded on methyl α-D-glucopyranoside (MeO-Glcp), a model compound, dissolved in NaOH(aq), which showed a difference in chemical shift when CO2 was added prior to or after the dissolution of MeO-Glcp, without a change in pH. The uptake of CO2 was found to be more than twice as high when CO2 was added to a solution after the dissolution of MeO-Glcp. Altogether, a mechanism for the observed CO2 capture is proposed, involving the formation of an intermediate cellulose carbonate upon the reaction of a cellulose alkoxide with CO2. The intermediate was observed as a captured carbonate structure only in regenerated samples, while its corresponding NMR peak in solution was absent. The reason for this is plausibly a rather fast hydrolysis of the carbonate intermediate by water, leading to the formation of CO32−, and thus increased capture of CO2. The potential of using carbohydrates as CO2 capturing agents in NaOH(aq) is shown to be simple and resource-effective in terms of the capture and regeneration of CO2.
Introduction
Designing renewable materials for the capture of CO2 is timely and of great importance for environmental challenges. Today, there are several materials capable of efficiently capturing CO2 such as different types of amines and MOFs (metal–organic frameworks).1 However, many of these systems suffer from being expensive or from being difficult to synthesise, and are to some extent not sustainable since a lot of energy is needed to regenerate CO2 and to recycle the capturing agent. In this aspect, another interesting system for capturing CO2 is NaOH(aq). Aqueous hydroxides are in general very efficient CO2 capturing agents. In theory, to capture 1 ton of CO2, 0.9 tons of NaOH are needed in comparison with the 1.39 tons of the commonly used amine, monoethanolamine (MEA). The capture of CO2 in NaOH(aq) occurs through absorption and conversion of CO2 by the reaction with hydroxide ions, resulting in different species depending on the pH of the solution. At a pH above 9, all CO2 is converted to carbonate ions, CO32−, forming sodium carbonate, Na2CO3, a reaction consuming two moles of NaOH for each mole of Na2CO3 formed. However, in a recent study it was shown that the CO2 capturing ability of NaOH(aq) was greatly increased by the addition of glycerol, suggesting the occurrence of two simultaneous reactions in the NaOH(aq)/glycerol-system.2 Almost simultaneously, a similar study was published where it was found that the mass transfer coefficient for CO2 in NaOH(aq) could be doubled if glycerol was added and thereby increase the chemical absorption of CO2, even though the viscosity increased with the addition of glycerol.3 Furthermore, Song et al. implied, based on a series of studies performed in 1927 by Faurholt,4 that glycerol is deprotonated in NaOH, and as glyceroxide, it is capable of competing with the hydroxide ions in the reaction with CO2. The final products would then be Na2CO3 and glycerolcarbonate. Interestingly, it was found that the reaction kinetics between glyceroxide and CO2 was 6–7 times faster than the reaction between the hydroxide ions and CO2, and hence a larger amount of CO2 could be captured in a system containing both NaOH and glycerol.2 On the other hand, this phenomenon has also been attributed to the ability of the individual C–O bonds in CO2 being able to interact with the polar groups of the glycerol through hydrogen bonding.5,6 Furthermore, glycerol and carbohydrates, with a larger number of hydroxyl groups, have been shown to have a higher affinity for CO2 than, e.g. poly(ethylene glycol)(PEG),5 which is claimed to be a CO2 capturing polymer through a Lewis base–Lewis acid mechanism between the carbon in CO2 and the ether oxygen in PEG. Clearly, there are different explanations for the CO2 capturing ability of alcohols in general and of alcohols in NaOH(aq) in particular. From this point of view, it is interesting to note similarities to our recent work which reports on a previously overlooked feature of the NaOH(aq) system used for the dissolution and reshaping of cellulose into regenerated (precipitated) cellulose materials. Our study showed that CO2, readily dissolved in NaOH(aq), to a certain extent sorbs onto the dissolved cellulose.7 The sorption of CO2 onto dissolved cellulose was proven using ATR-IR (Attenuated Total Reflectance – Infrared) spectroscopy on samples regenerated in ethanol. Ethanol, as such, has been shown to preserve chemisorbed CO2 in synthesised organic carbonates, whilst precipitation in water promotes the desorption of CO2,8 which was also observed in our recent study. Both the preservation of sorbed CO2 as well as the desorption, depending on the type of regenerating agent used, is promising for the application of using cellulose as a sustainable CO2 capturing agent. As mentioned before, there is to our knowledge still no mechanistic understanding of the CO2 capture in these systems, and that is why many questions remain. The focus of this work has therefore been set on investigating the molecular interactions that occur between CO2 and cellulose in its dissolved state in NaOH(aq). Essential in this investigation is the molecular state of the CO2 when interacting with cellulose dissolved in NaOH(aq) (i.e. CO2(aq) or CO32−). Particular emphasis has, thus, been placed on comparative studies exploring the consequences of adding CO2 pre or post-dissolution of cellulose in a NaOH(aq) system. In the case of CO2 added prior to the dissolution of cellulose, the high alkalinity of the system can be assumed to completely convert CO2 into CO32−, before the cellulose is added to the NaOH(aq). On the other hand, when CO2 is added to a system already containing dissolved cellulose (post-dissolution addition), CO2 in its aqueous state could interact with the dissolved and, most likely, deprotonated cellulose (cellulose alkoxides) and possibly form cellulose carbonate. Microcrystalline cellulose (MCC) and a model compound, MeO-Glcp, were therefore employed in ATR-IR spectroscopy and NMR studies in order to elucidate the interactions occurring between CO2 and cellulose dissolved in NaOH(aq). The model compound MeO-Glcp allows for an increased sensitivity and accuracy in the NMR measurements compared to cellulose. A recent study by Bialik et al.9 reports on partial deprotonation of the hydroxyl groups in cellobiose in NaOH(aq), especially on the C2 carbon, which could provide a reaction site for CO2(g) forming a cellulose carbonate. The deprotonation or dissociation constants of different sugars were actually studied already in the 1950s10 and are in line with the observed results in the more recent study by Bialik et al. The effects of deprotonation were therefore studied in terms of comparing the addition of CO2(g) to MeO-Glcp dissolved in a NaCl(aq) or NaOH(aq) solution. Furthermore, CO2(g) was also added as a function of time (30, 60 or 120 seconds) before or after the addition of MeO-Glcp, to monitor the effects of various CO2-species as well as the uptake of CO2.
Results and discussion
Reversible sorption of CO2 on MCC
Initially, ocular observations were made on the influence of CO2 addition on the dissolved state of MCC in NaOH(aq), owing to the fact that NaOH(aq) itself is able to absorb CO2.11 Roughly one litre of CO2 was percolated into 10 ml of NaOH(aq) solution before or after the dissolution of 4 wt% MCC, denoted as NaOH + CO2 + MCC and NaOH + MCC + CO2, respectively. The optimal dissolution conditions for cellulose in NaOH(aq) have been found to be −5 °C and 8 wt% NaOH,12 and were thus employed here. The transparent solutions were then stored for two weeks at room temperature. After two weeks, the reference sample without added CO2 still remained as a liquid solution while the NaOH + CO2 + MCC had turned opaque but surprisingly the NaOH + MCC + CO2 had formed a gelled structure (Fig. 1). Hence, percolating CO2 disrupts the stability of the MCC solutions, and interestingly, the effect is even more significant when CO2 is added after the dissolution of MCC in NaOH(aq), thus enabling a direct interaction between the dissolved MCC and the freshly added CO2 as CO2(aq), before the conversion to CO32− takes place. On the other hand, pre-dissolution addition of CO2, leading to the conversion of CO2 to CO32− prior to the interaction with MCC, seems to be of minor importance.
 |
| | Fig. 1 MCC solutions after two weeks at room temperature. Reference sample NaOH(aq) + MCC (left), NaOH(aq) + CO2 + MCC (middle), and NaOH(aq) + MCC + CO2 (right). | |
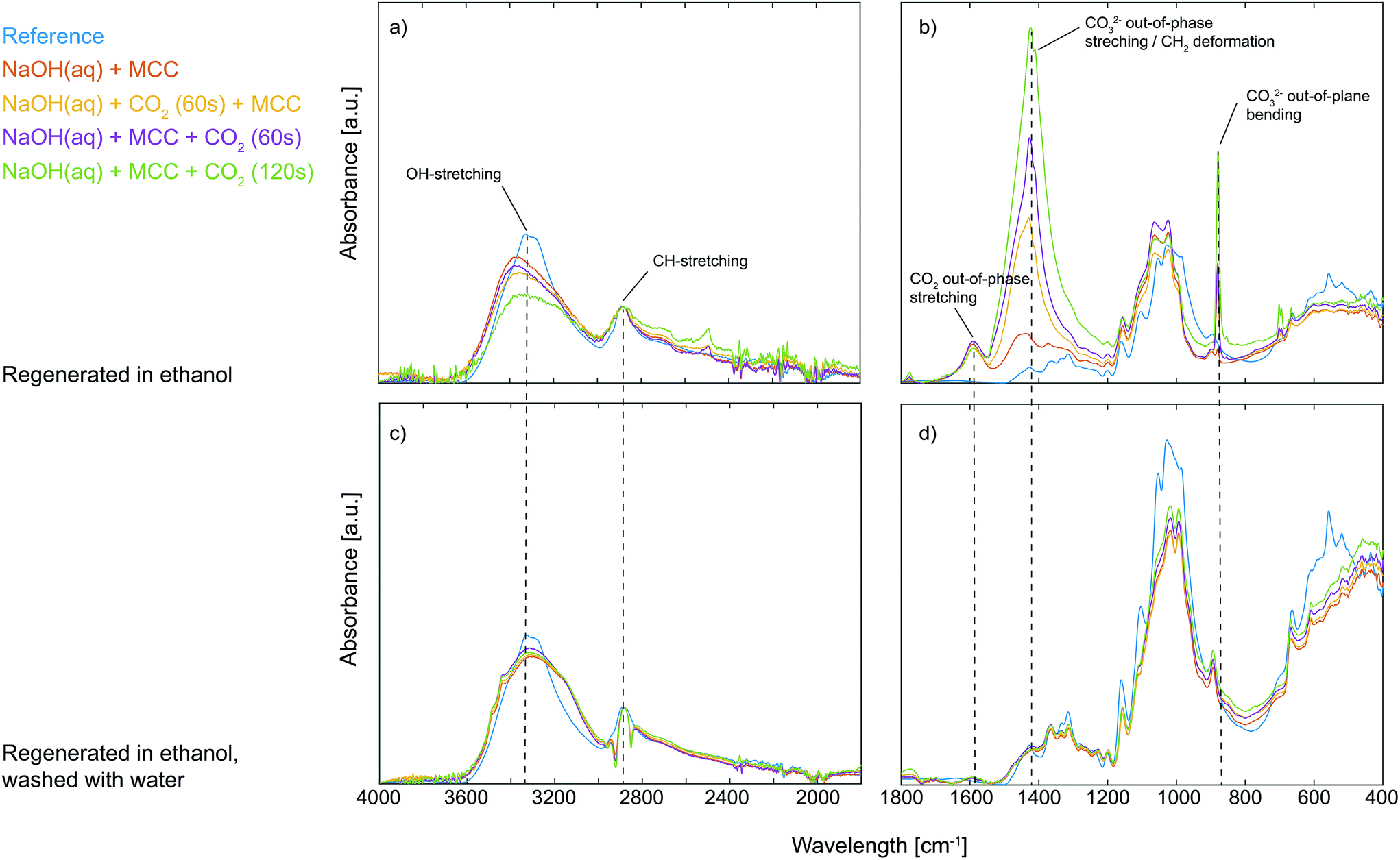
The visible difference in the dissolved state of MCC was further characterised by regenerating the solutions with ethanol followed by solid state analysis using ATR-IR spectroscopy. The gelled state of MCC observed upon post-dissolution addition of CO2 suggests specific interactions between deprotonated MCC and CO2, possibly through the formation of an organic carbonate which in turn can give rise to carbonate bridges.13 Moreover, it is well known that synthesised organic carbonates are preserved during work-up in ethanol but decompose if instead water is used.8 If an organic carbonate is formed through the reaction of deprotonated MCC and CO2, it is expected to be observed as a new waveband around 1590 cm−1 in the ATR-IR spectrum, similar to the structure found for carboxymethylated cellulose, corresponding to the out-of-phase stretching in the ionic state.14 In addition to this, it is important to remember that all the studied solutions, including those where no CO2 was added purposely, contained a certain amount of post-dissolution introduced CO2 due to the diffusion of CO2 present in the surrounding air. From the ATR-IR spectroscopy analysis it could indeed be concluded that a new structure was introduced on MCC through the addition of CO2 as a new waveband at 1590 cm−1 appeared, both for the samples only in contact with air CO2 as well as those subjected to CO2 percolation (Fig. 2). Moreover, compared to the reference MCC, the addition of CO2 to the MCC solutions (both pre and post-dissolution) resulted in a decrease in the OH-stretching around 3300 cm−1 as well as a change in the waveband around 1000 cm−1 in the ATR-IR spectra, corresponding to the primary alcohol absorption band of the MCC.15 These changes are attributed to a difference in interactions between the hydroxyl groups in MCC and indicate a change in the crystalline structure, probably due to the interruption of the hydrogen bonding pattern.16 Additionally, the spectra reveal that solutions subjected to pre or post-dissolution addition of CO2 to a different extent show the formation of Na2CO3, giving rise to new wavebands at 1427 cm−1 and 880 cm−1 (see the ESI† for comparison with the ATR-IR spectrum for Na2CO3). Nonetheless, the new waveband at 1590 cm−1 observed for the ethanol regenerated cellulose samples is not present in the spectrum for Na2CO3, indeed proving the formation of a new species of CO2 (CO2 out-of-phase stretching) associated with the presence of MCC when dissolved in NaOH(aq). Interestingly, the formation of this new species seems to be competing with the formation of Na2CO3 as the increased percolation time of CO2 does not lead to an increase in the intensity of the CO2 out-of-phase stretching but rather the opposite: the highest intensity of this signal is observed for the MCC subjected to the post-dissolution addition of CO2 for 60 s or the one only subjected to CO2 from the surrounding air. The reason for this could possibly be an effect of deterioration of the dissolved state of the MCC, which is the result of consuming OH− ions due to the extensive CO2 addition and the formation of Na2CO3. As already mentioned, a change in the structure of the primary alcohol on MCC could be observed in the spectra, this is why the corresponding change for the CH2-group on MCC carrying the primary alcohol should be observable as well. Unfortunately, the waveband for CH2 deformation on MCC is overlapping with the CO32− out-of-phase stretching (originating from the Na2CO3) and with that said, it is not possible to declare whether this band carries information on the interactions between MCC and CO2 added pre- or post-dissolution. However, sorption of CO2 on MCC is evidently taking place when dissolved in NaOH(aq),7 which explains why the stability of the sorbed CO2 was evaluated. The regenerated MCC was hence washed with water, filtered and dried before analysis once again using ATR-IR spectroscopy (Fig. 2). From this it was confirmed that the sorption of CO2 on MCC indeed is reversible as almost no traces of the waveband for the CO2 out-of-phase stretching could be detected after washing with water. This indicates that the sorption of CO2 on MCC in NaOH(aq) is both simple and sustainable in terms of capturing CO2 as well as the subsequent regeneration by the use of a water-based system.
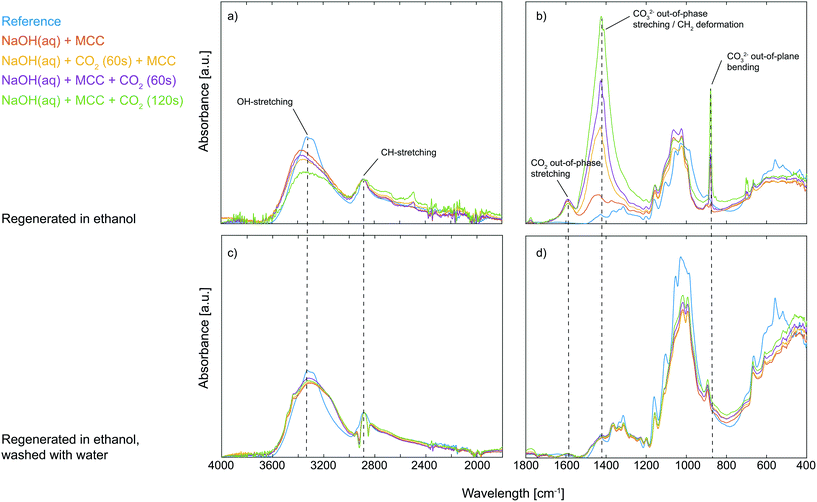 |
| | Fig. 2 ATR-IR spectra from 4000–1800 cm−1 and 1800–400 cm−1 of untreated reference MCC (blue), MCC dissolved in NaOH(aq) (red), MCC dissolved in NaOH(aq) with 60 s pre-dissolution addition of CO2 (yellow), MCC dissolved in NaOH(aq) with 60 s post-dissolution addition of CO2 (purple) and MCC dissolved in NaOH(aq) with 120 s post-dissolution addition of CO2 (green). All solutions were regenerated in ethanol and dried (a and b) and subsequently washed with water and dried (c and d). | |
Impact of CO2 on a model system
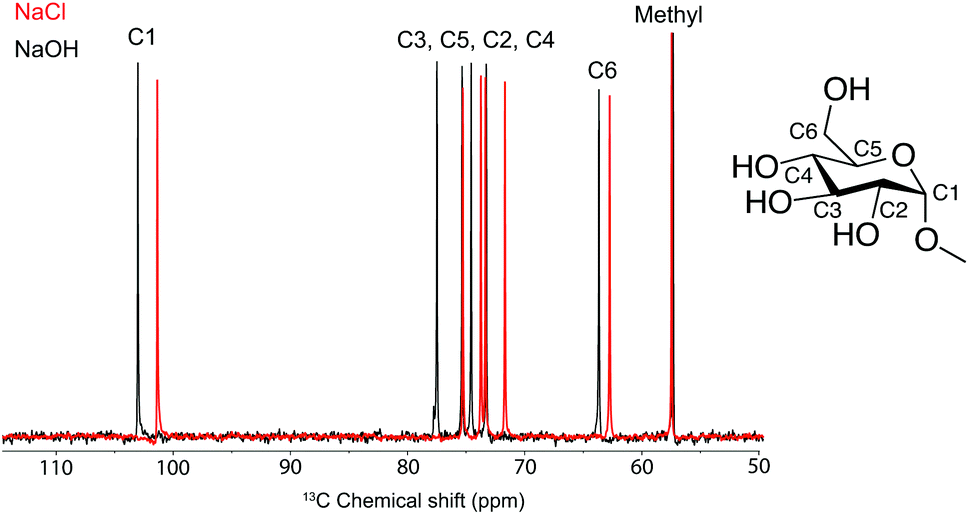
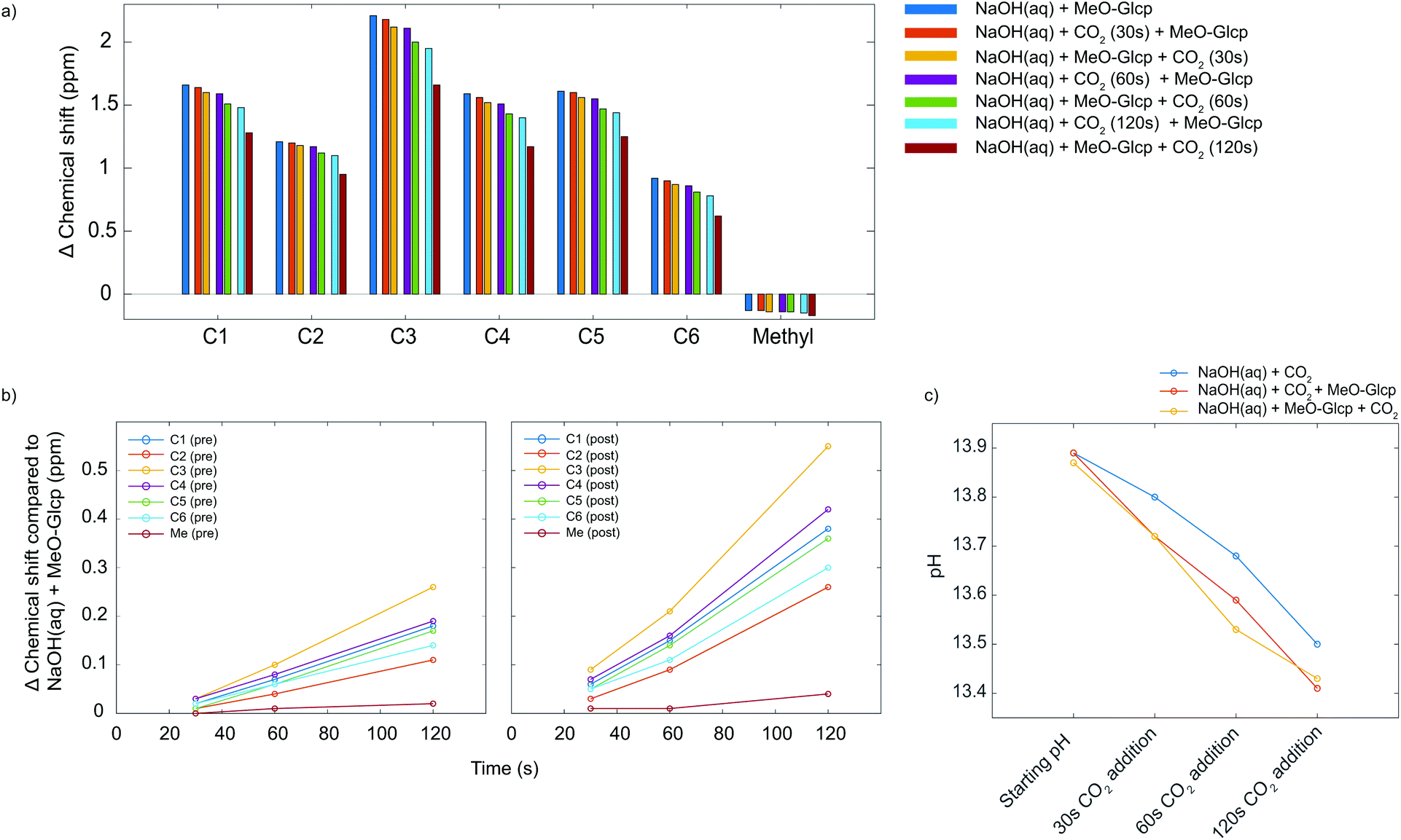
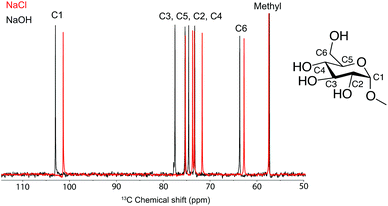
Although ATR-IR spectroscopy provides structural information, many questions are still unresolved such as in which state CO2 interacts with MCC in a NaOH(aq) solution when the sorption occurs. Characterisation using solution NMR (nuclear magnetic resonance) has been shown to be a promising method to study similar systems,17 but for polymers such as MCC, the observed peaks are unfortunately broad and featureless. The model compound methyl α-D-glucopyranoside (MeO-Gclp) was therefore instead used, which unlike unprotected glucose or cellobiose does not ring open. The MeO-Glcp was dissolved in either NaCl or NaOH to elucidate the role of the solvent in the sorption of CO2 in the MCC/NaOH-system. The 13C NMR spectra of MeO-Glcp dissolved in NaCl(aq) and NaOH(aq) (Fig. 3) showed that all carbon peaks in the MeO-Glcp were shifted to higher chemical shift values upon dissolution in NaOH(aq) compared to NaCl(aq), except for the methyl group bonded to C1, which was shifted from 57.8 to 57.6 ppm. The observed change in the chemical shifts agrees well with the chemical shift changes reported previously by Bialik et al.9 for cellobiose, which was explained as a consequence of the deprotonation of one or several of the hydroxyl groups on the glucose ring. This phenomenon has also been discussed earlier by Isogai, who ascribed it to a dissociation of the hydroxyl groups in NaOH(aq) resulting in an electron-deshielding effect on the carbon atoms.18 The largest change in the chemical shift was observed for the carbons at the C1 and C3 positions, indicating that the hydroxyl group in position C2 is deprotonated to the highest extent. In contrast to this, the position C6 in MeO-Glcp was observed as being the least affected when dissolved in NaOH(aq) compared to NaCl(aq). Having established the deprotonation of the MeO-Glcp in NaOH(aq), the effect of adding CO2 pre or post-dissolution of MeO-Glcp was evaluated by percolating CO2 for 30, 60 and 120 s into the solutions. Noteably, no precipitation of Na2CO3 was observed. In general, the addition of CO2(g) to NaOH(aq) causes a change in pH due to the consumption of OH− ions. Thus, as expected, all the carbon peaks in MeO-Glcp were shifted to lower chemical shifts upon the addition of CO2(g), disregarding pre or post-dissolution addition (Fig. 4a).
 |
| | Fig. 3
13C NMR spectra of MeO-Glcp dissolved in NaCl(aq) and NaOH(aq). All measurements were recorded at +5 °C in D2O. Inset shows the chemical structure of MeO-Glcp. | |
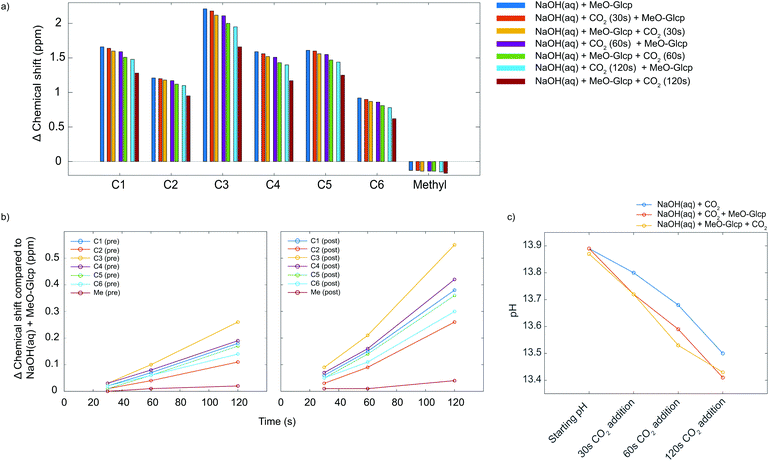 |
| | Fig. 4 (a) The difference in chemical shift for MeO-Glcp in NaOH(aq) with pre or post-dissolution addition of CO2 in comparison with MeO-Glcp in NaCl(aq). All measurements were recorded at +5 °C in D2O. (b) An extension of the difference in chemical shift for MeO-Glcp in NaOH(aq) with pre or post-dissolution addition of CO2 in comparison with MeO-Glcp in NaOH(aq). (c) The change in pH for 0.5 M NaOH(aq) with MeO-Glcp and pre or post-dissolution addition of CO2. | |
Interestingly though, it was observed that the pre or post-dissolution addition of CO2(g) did not result in an identical change of the chemical shifts of the carbon peaks (Fig. 4b). Namely, the post-dissolution addition of CO2 resulted in a larger chemical shift change of the carbon peaks. An explanation for this could partly be due to a difference in change of pH or due to an interaction between the deprotonated MeO-Glcp and the added CO2(g), as well as a combination of both. As the interaction between the available OH− ions in the solvent and the added CO2 is assumed to be identical both in the case of pre and post-dissolution addition it should thus give rise to the same change in chemical shift as a result of the pH change. The same effect could additionally be observed in the corresponding 1H spectra, with the difference that the protons were shifted in the opposite direction as they become shielded, instead, during deprotonation and by the addition of CO2. Furthermore, the pH of the solutions was measured in order to investigate if a variation in pH was the cause for the differences in chemical shifts for the MeO-Glcp with pre or post-dissolution addition of CO2. When measuring pH, a limiting factor is the accuracy of the electrode, especially at high pH. In addition to this, temperature has a large influence on the accuracy of the measurement and becomes even more important at low temperature. The pH was therefore measured for a 0.5 M NaOH(aq) solution at 10 °C (Fig. 4c). The initial pH of the 0.5 M NaOH(aq) was found to be 13.87 and decreased to 13.79 when MeO-Glcp was added, which supports the results discussed above pointing out the deprotonation of MeO-Glcp in NaOH(aq). From the pH decrement, a degree of deprotonation could be estimated to be around 15% which further suggests the pKa for the majority of the MeO-Glcp hydroxyls to be higher than 13.8. On the other hand, the pKa is probably quite close to 13.8 since the pH scale is logarithmic. Moreover, when CO2 was added to the solutions the pH decreased as expected but interestingly enough the decrement was equal for the solutions with pre or post-dissolution addition of CO2, which means that an equal amount of OH− ions is consumed in both cases. Therefore, the observed increase in chemical shift change for the solution with post-dissolution addition of CO2 could not be due to a larger change in pH but rather a difference in interaction with the added CO2.
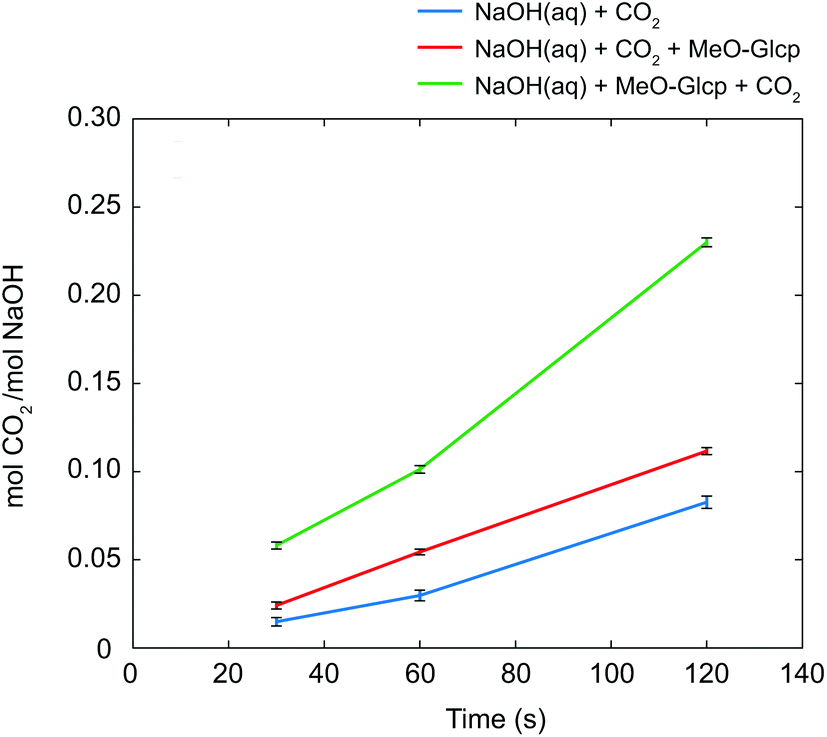
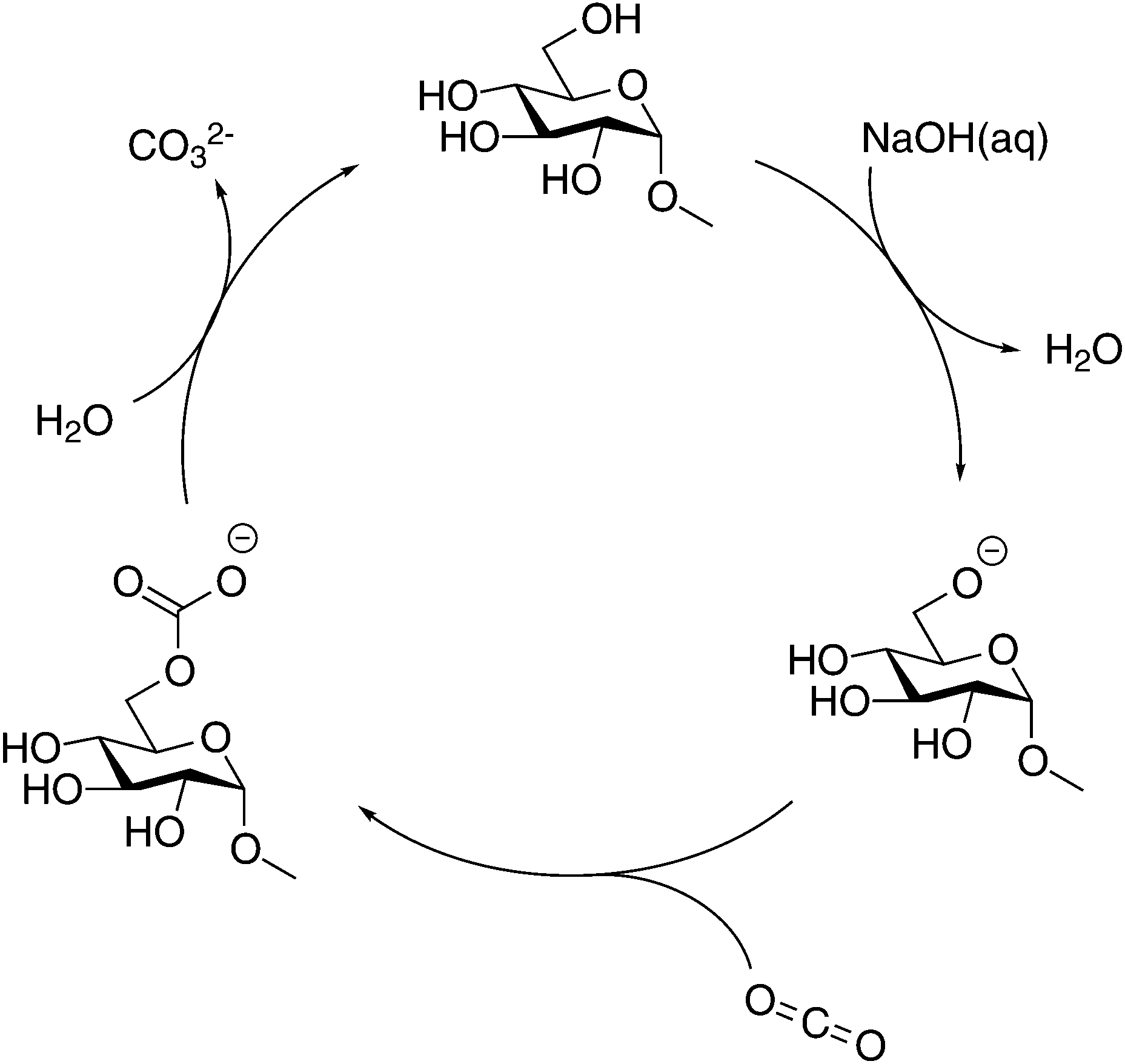
As discussed in the Introduction section, several studies on the increased capture of CO2 in NaOH(aq) have been based on the hypothesis that a deprotonated alcohol reacts with CO2 in the formation of an organic carbonate19,20 which in 13C NMR should result in a new peak around 150 ppm. No such peak could be observed in the present study, this is why complementary NMR measurements were made on both glycerol and PEG in NaOH(aq) in order to elucidate the origin of the CO2 sorption mechanism of alcohols and ethers. As previously reported, both the presence of glycerol and PEG increases the CO2 capturing ability of NaOH(aq) and several explanations have been presented on this phenomenon, e.g. the formation of glycerol carbonate2 or as an interaction between CO2 and terminal hydroxyl groups5,6 or the ether brigde.21 Surprisingly, no such interactions could be observed in the complementary NMR measurements (see the ESI†), thereby suggesting another mechanism for the increased CO2 capture in NaOH(aq). On the other hand, the occurrence of a CO32− peak at 171.0 ppm in NaOH(aq) could clearly be observed in 13C NMR, showing that CO2(g) dissolves and converts, as expected, into CO32− in the NaOH(aq) at this high pH (which was also true for the complementary measurements on glycerol and PEG in NaOH(aq)). Moreover, the addition of CO2 to NaOH(aq) of different durations (30, 60 or 120 s) correlated well with the integral of the CO32− peak. From the peak integral of CO32−, the amount of absorbed CO2 could be calculated which is shown as mol CO2/mol NaOH in Fig. 5. Comparing the amount of absorbed CO2 in the system MeO-Glcp/NaOH(aq) with the pre-dissolution addition of CO2 to pure NaOH(aq) with the same amount of CO2 added, the amount was observed to be larger, indicating either a stabilising effect of MeO-Glcp on CO2 (hypothetical coordination through hydrogen bonding) dissolved in NaOH or/and an additional post-dissolution addition of CO2 from the surrounding air facilitated by the presence of MeO-Glcp in the solution. Interestingly, in the case of MeO-Glcp with the post-dissolution addition of CO2, the amount of absorbed CO2 increased even more profoundly and was shown to be more than twice as large as the amount of absorbed CO2 in pure NaOH(aq). If this would be the result of CO2 (aq) and OH− ions converting to CO32− in the presence of MeO-Glcp, it would have been visible as a larger decrease in pH which was not the case according to the pH measurements. Instead, the increased formation of CO32− in the presence of deprotonated MeO-Glcp at rather constant pH indicates a decisive role of deprotonated glucose (working as a catalyst) in this conversion. Lastly, a concluding reference measurement was made on the MeO-Glcp dissolved in NaCl(aq) with the addition of CO2, both pre and post-dissolution. The result showed no effect on either the 1H nor 13C chemical shifts, nor on the integral intensities. A tiny 13C peak of bicarbonate was however observed, indicating that a small amount of CO2 dissolves in NaCl(aq) but without influencing the chemical shifts of MeO-Glcp. From the results obtained with NMR spectroscopy in combination with the pH measurement, it is evident that the dissolution, and most probably the deprotonation, of MeO-Glcp in NaOH(aq) give rise to specific interactions with CO2 or CO32− dissolved in NaOH(aq) and, thus increases the amount of CO2 that can be sorbed in NaOH(aq). Even though the anticipated MeO-Glcp-carbonate peak was lacking in the NMR spectroscopy measurements, the ATR-IR analysis of precipitated samples clearly showed the introduction of an organic carbonate on MCC when dissolved in NaOH(aq) and subjected to CO2(g). An explanation for this is most likely the capture, during precipitation in ethanol, of the intermediate state of the reaction between a deprotonated hydroxyl on MeO-Glcp and CO2(aq), yielding a MeO-Glcp-carbonate intermediate. The MeO-Glcp-carbonate intermediate, which probably is in equilibrium with the free CO32− ions in solution, is further readily hydrolysed by water and thereby converted to CO32− (Fig. 6). The observed increased tendency of gelation of MCC solutions upon post-dissolution addition of CO2 observed in the ocular investigation could thus be the result of an interaction between the cellulose chains via the intermediate (carbonate bridges) or a re-protonation of the cellulose hydroxyl groups when the intermediate is hydrolysed. Overall, this process is thus a more lean conversion of CO2 to CO32− going via the MeO-Glcp-carbonate intermediate with water as the hydrolysing agent (instead of OH−) leaving the pH of the system largely unaffected. Consequentially, more CO2 can be captured in the presence of MeO-Glcp or similar alcohols due to their catalytic role in the conversion of CO2 to CO32−. This hypothesis is in line with the results previously reported and concluded by both Faurholt in 1927 and Song in 2017,2,4 where alcohols such as methanol and glycerol were observed to increase the amount of CO2 captured in an aqueous hydroxide to a larger extent compared to a system without alcohol. Altogether, these results prove the potential of using cellulose as a mediator in water-based systems for CO2 capture.
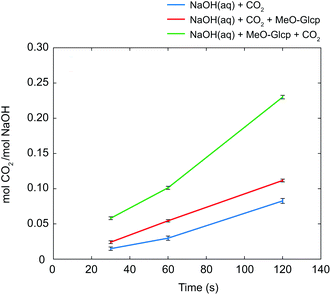 |
| | Fig. 5 Amount of CO2 captured calculated from the integral of CO32− at 171 ppm and reported as mol CO2/mol NaOH with errorbars for NaOH(aq) with the addition of CO2 (blue), NaOH(aq) with pre-dissolution addition of CO2 (red) and NaOH(aq) with post-dissolution addition of CO2 (green). All NMR measurements were recorded at +5 °C in D2O. | |
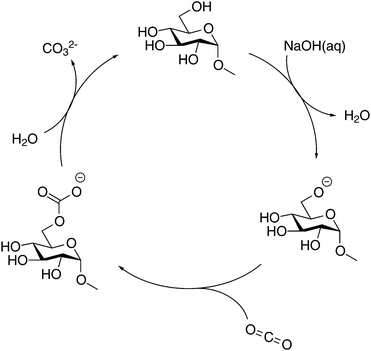 |
| | Fig. 6 Possible mechanism for conversion of CO2 to CO32− mediated by MeO-Glcp as the catalyst. | |
Conclusions
In summary, this work elucidates the sorption of CO2 in NaOH(aq) in the presence of the model compound MeO-Glcp or MCC. The CO2 capturing capacity of NaOH(aq) was evaluated in terms of pre or post-dissolution addition of CO2 and was shown, using NMR spectroscopy, to increase drastically when MeO-Glcp was dissolved in NaOH(aq) prior to the addition of CO2. As a consequence, a significant capture of CO2 in the system could be observed as CO32− in NMR and ATR-IR measurements, while retaining rather constant pH. The potential of using MCC as a CO2 capturing agent was further evaluated in terms of regeneration of the captured CO2 and proved to be efficient through just washing the regenerated MCC samples with water. The increased CO2 capturing ability is explained according to the present findings on the specific interaction of a deprotonated carbohydrate and freshly added CO2, likely leading to the formation of an organic carbonate intermediate, which was observable on regenerated MCC as CO2 out-of-phase stretching (1590 cm−1) using ATR-IR spectroscopy. The corresponding carbonate peak in the NMR measurements was absent, which indicates a rather fast hydrolysis of the intermediate by water, leading to a lean conversion of CO2 to CO32− in terms of the amount of OH− consumed. Simultaneously, a deprotonated carbohydrate is regenerated upon the release of CO32−, and is able to continue into the conversion of CO2 to CO32− and thus works as a catalyst. In contrast to this, the possibility of a re-protonation of the carbohydrate also exists, which instead would quench the catalytic cycle. From this point of view, it is plausible to suggest that the formation of an intermediate state could be the reason for the observed tendency of MCC to form a gel upon the post-dissolution addition of CO2. On one hand, an intermediate could cause an interaction with a neighbouring cellulose chain in solution through a so-called carbonate bridge hence causing the system to gel. While on the other hand, a re-protonation of the MCC would disrupt the dissolved state, leading to the precipitation of the MCC and phase separation into a gel. These findings could play an important role not only in the development of sustainable and efficient CO2 capturing agents but also in the innovation of functional bio-based materials.
Experimental
Materials and methods
Microcrystalline cellulose (MCC) Avicel PH-101, with a degree of polymerisation of 260, was purchased from FMC BioPolymer and used without further treatment. Methyl α-D-glucopyranoside (<99%), NaOH (<98%), NaCl and D2O (99.9%) were purchased from Sigma Aldrich and used as received. Dissolution of MCC was carried out by dispersing MCC in deionised water and pre-cooling it to +5 °C. The dispersed MCC was then added to a NaOH aqueous solution with a temperature of −5 °C, and stirred for 1 h. The solutions had a transparent appearance at the end of this procedure; the final concentrations were 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:88 wt% MCC:NaOH:H2O. The MCC solutions were regenerated using ethanol. The regenerated materials were washed with ethanol until neutral and dried in vacuo, and were later washed with water, filtered and dried in an oven at 105 °C. Solutions were prepared by dissolving MeO-Glcp in D2O at +5 °C. The MeO-Glcp solution was then added to either NaOH or NaCl in D2O, maintaining a temperature of −5 °C. The final concentrations of the MeO-Glcp and the NaOH or NaCl in D2O were 0.4 M and 3.0 M, respectively. CO2 was added for 30, 60 or 120 s to the solutions pre or post-dissolution of MCC or MeO-Glcp by immersing a syringe into the solutions. The syringe was connected to a tube containing CO2, mounted with a flow regulator set to approximately 1 l min−1. pH measurements were carried out by pre-cooling NaOH(aq) with a concentration of 0.5 M at +10 °C. The pH was then measured pre or post-dissolution of MeO-Glcp (4 wt%) in combination with the addition of CO2 for 30, 60 and 120 s pre or post-dissolution of the MeO-Glcp according to method above.
:8:88 wt% MCC:NaOH:H2O. The MCC solutions were regenerated using ethanol. The regenerated materials were washed with ethanol until neutral and dried in vacuo, and were later washed with water, filtered and dried in an oven at 105 °C. Solutions were prepared by dissolving MeO-Glcp in D2O at +5 °C. The MeO-Glcp solution was then added to either NaOH or NaCl in D2O, maintaining a temperature of −5 °C. The final concentrations of the MeO-Glcp and the NaOH or NaCl in D2O were 0.4 M and 3.0 M, respectively. CO2 was added for 30, 60 or 120 s to the solutions pre or post-dissolution of MCC or MeO-Glcp by immersing a syringe into the solutions. The syringe was connected to a tube containing CO2, mounted with a flow regulator set to approximately 1 l min−1. pH measurements were carried out by pre-cooling NaOH(aq) with a concentration of 0.5 M at +10 °C. The pH was then measured pre or post-dissolution of MeO-Glcp (4 wt%) in combination with the addition of CO2 for 30, 60 and 120 s pre or post-dissolution of the MeO-Glcp according to method above.
Characterisation
Fourier Transform Infrared (FT-IR) spectra were obtained on a PerkinElmer Frontier equipped with an Attenuated Total Reflectance (ATR) sampling accessory (PIKE Technologies GladiATR). Samples were placed on top of the ATR crystal and secured using a metal clamp to ensure consistent pressure; they were measured with a resolution of 4 cm−1 and 32 scans. All spectra were corrected against air, baseline corrected and normalised against the CH-band around 2890 cm−1 which was shown not to be affected by the different treatments in the experiments. All spectra are shown without an absorbance scale. All NMR experiments were run on an 800 MHz magnet equipped with a Bruker Avance HDIII console and a TXO cryoprobe. 13C NMR spectra were recorded with a low angle radio frequency pulse to minimize relaxation-weighting using a single pulse experiment with 1H decoupling during acquisition. Hence, the repetition delay and number of scans were set to 33.0 s and 128, respectively, for monitoring the amount of dissolved CO2 while a repetition delay of 5 s was used for the observation of chemical shift differences. A capillary containing D2O with 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) was placed inside the tube as the internal reference. The amount of CO2 captured was converted from the CO32− peak integral using the peak integrals of MeO-Glcp or DSS. The pH was measured using a HACH HQ430D Multimeter with an Intellical PHC705A1 pH probe.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is part of the framework of Avancell – Center for Fiber Engineering, which is a research collaboration between Södra Innovation and Chalmers University of Technology. The authors are grateful to the Södra Skogsägarna Foundation for Research, Development and Education for their financial support.
References
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- D. Song and G. T. Rochelle, Chem. Eng. Sci., 2017, 161, 151–158 CrossRef.
- C.-Y. Chiang, D.-W. Lee and H.-S. Liu, J. Taiwan Inst. Chem. Eng., 2017, 72, 29–36 CrossRef.
- C. Faurholt, Z. Phys. Chem., 1927, 126, 85–104 Search PubMed.
- O. Aschenbrenner and P. Styring, Energy Environ. Sci., 2010, 3, 1106–1113 RSC.
- P. Raveendran, Y. Ikushima and S. L. Wallen, Acc. Chem. Res., 2005, 38, 478–485 CrossRef PubMed.
- M. Gunnarsson, H. Theliander and M. Hasani, Cellulose, 2017, 24, 2427–2436 CrossRef.
-
A. P. N. Franchimont, KNAW Proceedings, 1910, vol. 12, pp. 303–304.
- E. Bialik, B. Stenqvist, Y. Fang, Å. Östlund, I. Furó, B. Lindman, M. Lund and D. Bernin, J. Phys. Chem. Lett., 2016, 7, 5044–5048 CrossRef PubMed.
- J. Thamsen, Acta Chem. Scand., 1952, 6, 270–284 CrossRef.
- F. Lucile, P. Cézac, F. Contamine, J.-P. Serin, D. Houssin and P. Arpentinier, J. Chem. Eng. Data, 2012, 57, 784–789 CrossRef.
- T. Budtova and P. Navard, Cellulose, 2016, 23, 5–55 CrossRef.
- T. Elschner, K. Ganske and T. Heinze, Cellulose, 2013, 20, 339–353 CrossRef.
-
P. Larkin, Infrared and Raman Spectroscopy, Elsevier, Oxford, 2011, pp. 73–115 Search PubMed.
- Y. Maréchal and H. Chanzy, J. Mol. Struct., 2000, 523, 183–196 CrossRef.
- T. Kondo and C. Sawatari, Polymer, 1996, 37, 393–399 CrossRef.
- D. Bernin and N. Hedin, Curr. Opin. Colloid Interface Sci., 2018, 33, 53–62 CrossRef.
- A. Isogai, Cellulose, 1997, 4, 99–107 CrossRef.
- J. Thamsen, Acta Chem. Scand., 1956, 10, 1165–1171 CrossRef.
- B. Smidt and J. Thamsen, Acta Chem. Scand., 1956, 10, 1172–1176 CrossRef.
- S. P. Nalawade, F. Picchioni, J. H. Marsman and L. Janssen, J. Supercrit. Fluids, 2006, 36, 236–244 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc01092g |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Diana
Bernin
a,
Diana
Bernin