
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An uncondensed lignin depolymerized in the solid state and isolated from lignocellulosic biomass: a mechanistic study†
Ning
Li
 a,
Yanding
Li
ab,
Chang Geun
Yoo
ac,
Xiaohui
Yang
ad,
Xuliang
Lin
ae,
John
Ralph
ab and
Xuejun
Pan
*a
a,
Yanding
Li
ab,
Chang Geun
Yoo
ac,
Xiaohui
Yang
ad,
Xuliang
Lin
ae,
John
Ralph
ab and
Xuejun
Pan
*a
aDepartment of Biological Systems Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: xpan@wisc.edu; Fax: +1-608-2621228; Tel: +1-608-2624951
bDepartment of Biochemistry, University of Wisconsin-Madison, WI 53706, USA
cBioEnergy Science Center and Bioscience Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
dInstitute of Chemical Industry of Forestry Products, Chinese Academy of Forestry, Nanjing 210042, China
eSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, 511400, China
First published on 5th June 2018
Abstract
This study demonstrated that lignin could be efficiently depolymerized in the solid state with minimal condensation and separated from biomass with high purity by dissolving and hydrolyzing cellulose and hemicelluloses, using an acidic lithium bromide trihydrate (ALBTH) system under mild conditions (with 40 mM HCl at 110 °C). The ALBTH lignins isolated from biomass sources representing the three plant classes (hardwoods, softwoods, and grasses) contained abundant uncondensed moieties (i.e., Hibbert's ketones and benzodioxanes). The benzodioxane structure was identified and confirmed for the first time in an acid-depolymerized lignin. Reactions using lignin model compounds (LMCs, guaiacylglycerol-β-guaiacyl ether and various aromatic monomers) confirmed the formation of the uncondensed moieties and revealed the synergy between LiBr and acid in inducing the crucial intermediate benzyl carbocations, which then led to cleavage of the β-O-4-aryl ether bonds to produce Hibbert's ketones, demethylation to produce benzodioxanes, and condensation reactions. Unlike in the LMC reactions, the condensation of the real lignin in biomass under ALBTH conditions was greatly diminished, possibly due to lignin remaining in the solid state, limiting its mobility and accessibility of the benzyl carbocation to the electron-rich aromatic sites for condensation. Preliminary results indicated that, because of its uncondensed nature, the ALBTH lignin was a good lignin feedstock for hydrogenolysis. This study provided a new approach to effectively isolate depolymerized lignin from lignocellulose in a less condensed form for boosting its downstream valorization.
1. Introduction
Lignin, as a major cell wall component of lignocellulosic biomass, is the most abundant aromatic biopolymer on the Earth. Lignin valorization has emerged as a consensus requirement for profitability, based on efficiently utilizing the entire lignocellulosic biomass. In view of the structural and functional nature of lignin, a broad array of potential applications is anticipated, including as dispersants, aromatic platform chemicals, fuel precursors, pharmaceutical products and ingredients for adhesives, resins, biocomposites, and carbon fibers.1–5 In efforts to separate lignin from lignocellulose and achieve value-added utilization, chemocatalytic depolymerization of lignin by cleaving the inter-unit ether linkages (mostly β-O-4-aryl ether bonds) but avoiding condensation (repolymerization) reactions is recognized to be a prerequisite.Acid is prevalently used for lignin depolymerization, cleaving β-O-4-aryl ether bonds initiated by an α- (or benzyl) carbocation intermediate. Acid-catalyzed lignin depolymerization has been extensively studied in many solvent systems, including ionic liquids6–8 and various organic solvents such as γ-valerolactone,9 dioxane,10–14 methanol,15 ethanol,16,17 formic acid,18 and acetic acid.19,20 However, acid-catalyzed lignin depolymerization is always accompanied by condensation reactions that take place either before or after the cleavage of the β-ether units. Before cleaving the β-O-4-aryl ether bond, the benzyl carbocation can attack an electron-rich aromatic ring in an electrophilic aromatic substitution reaction, leading to formation of a new C–C linkage that defines condensation.21–23 After the cleavage of the β-O-4-aryl ether bond, the resulting C2-aldehyde (a phenylacetaldehyde fragment) and Hibbert's ketone (HK) end-groups are vulnerable to aldol condensation under acidic conditions, forming other inert C–C linkages.10,24 Previous studies have explored different means of avoiding or reducing the condensation reactions by introducing trapping/capturing reagents or pretreatment by, for example, trapping the benzyl carbocation using aromatic compounds such as phenol,22,23 protecting the α-carbon from condensation by forming a 1,3-dioxane structure with the α- and γ-hydroxyls of lignin using formaldehyde12 or other aldehydes,25 or in situ capturing of the unstable post-cleavage carbonyls, e.g., the C2-aldehyde and HK, by producing acetals using diols such as ethylene glycol.10,11,15
Intrinsically, the success of lignin depolymerization to produce uncondensed lignin relies on the competition between ether cleavage and condensation. As the occurrence of condensation requires two adjacent lignin moieties or species (i.e., between the benzyl carbocation and the electron-rich aromatic ring, or between the C2-aldehydes and/or HKs), preventing the lignin moieties from direct contact can block or reduce condensation. Therefore, it is hypothesized that lignin condensation could be avoided or minimized by limiting the accessibility or mobility of the reactive lignin moieties (intermediates) by, e.g., keeping lignin in the solid state during the acid-catalyzed depolymerization (the cleavage of the β-O-4-aryl ether bond), and using mild conditions (low acid concentration and moderate temperature) such as in the LiBr system described below.
According to our previous studies, the acidic lithium bromide trihydrate (ALBTH, LiBr·3H2O) system exhibited an extraordinary ability to swell, dissolve, and hydrolyze polysaccharides (cellulose and hemicelluloses) in lignocellulose.26,27 The lignin fraction (ALBTH lignin) could be quantitatively obtained as the insoluble residue remaining after hydrolyzing the carbohydrates.28 It was found to have a low molecular weight and was highly soluble in many common organic solvents, such as THF, acetone, acetic acid/water (95/5, v/v), dioxane/water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), and DMSO (unpublished data). This feature distinguishes it from other isolated lignins such as Klason lignin and kraft lignin and indicates the uniqueness of lignin depolymerization in the ALBTH system. In addition, when the technical lignins from different pulping and biorefining processes were treated in the ALBTH system under mild conditions, the cleavage of the aryl ether bonds was also observed, leading to further reductions in molecular weight.29 It is therefore of significant interest to investigate the chemical changes of lignins in the ALBTH system, to provide new insights into the lignin depolymerization mechanisms and to determine the characteristics and potential utilization of the ALBTH lignin. In this study, the native lignin in different lignocellulosic feedstocks and lignin model compounds (LMCs) were utilized to elucidate the mechanisms and pathways of lignin depolymerization and condensation in the ALBTH system.
:1, v/v), and DMSO (unpublished data). This feature distinguishes it from other isolated lignins such as Klason lignin and kraft lignin and indicates the uniqueness of lignin depolymerization in the ALBTH system. In addition, when the technical lignins from different pulping and biorefining processes were treated in the ALBTH system under mild conditions, the cleavage of the aryl ether bonds was also observed, leading to further reductions in molecular weight.29 It is therefore of significant interest to investigate the chemical changes of lignins in the ALBTH system, to provide new insights into the lignin depolymerization mechanisms and to determine the characteristics and potential utilization of the ALBTH lignin. In this study, the native lignin in different lignocellulosic feedstocks and lignin model compounds (LMCs) were utilized to elucidate the mechanisms and pathways of lignin depolymerization and condensation in the ALBTH system.
2. Experimental section
2.1 Chemicals
Chips of hardwood (aspen, poplar and eucalyptus) and softwood (Douglas fir), and coarsely chopped grasses (switchgrass and corn stover) were ground with a Wiley-mill, and the fraction between 20 and 100 mesh was collected as lignocellulosic materials for this study. Chemicals including 3,4,5-trimethoxylbenzyl alcohol (TMBA), guaiacol (GA), creosol (CS), anhydrous lithium bromide (LiBr), hydrochloric acid (HCl), dimethylsulfoxide (DMSO-d6), pyridine-d5, pyridine, tetrahydrofuran (THF), acetonitrile, and N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) were purchased from Fisher Scientific (Pittsburgh, PA), VWR (Radnor, PA), and Sigma Aldrich (St Louis, MO). Guaiacylglycerol-β-guaiacyl ether (GG) was synthesized and provided by Dr Gaojin Lyu with ∼99% purity, as determined by NMR.302.2 Isolation of the ALBTH lignin fraction from biomass
The ground biomass was mixed with the ALBTH solution (60 wt% LiBr containing 40 mM HCl) at room temperature in a 40 mL glass vial fitted with a Teflon-lined screw cap; a magnetic stir-bar was added. The mixture in the vial was heated in an oil bath at 110 °C with magnetic stirring (400 rpm) to hydrolyze cellulose and hemicelluloses to soluble fractions. After the preset reaction time, the reaction was quenched by placing the vial into an ice-water bath. The insoluble ALBTH lignin fraction was collected by vacuum filtration on a filtering crucible and thoroughly washed with deionized (DI) water. The solid sample was dried at 50 °C under vacuum for subsequent analyses.2.3 Reactions of lignin model compounds (LMCs) in ALBTH
Various aromatic monomers and dimers (including GA, CS, TMBA, and GG) were used as LMCs to investigate the reaction mechanism in the lithium bromide trihydrate (60 wt%) medium with or without an acid catalyst. A dilute acid solution (10 mM HCl) in the absence of LiBr was used as the control. Due to the limited solubility of LMCs in 60% LiBr at room temperature, LMCs (individually or in pairs) were first dissolved in dioxane prior to mixing with ALBTH (the final ratio of dioxane to ALBTH was ∼5/95, v/v). The mixture was then sonicated for 5 min to obtain a homogeneous suspension of LMCs in ALBTH. During subsequent heating, the suspension became a transparent solution. The reaction was conducted following the same procedure as for the biomass above but with reduced reaction severity (temperature: 100 °C and acid concentration: 10 mM HCl). During the reaction, aliquots were taken for analysis at preset intervals. After the reaction, the mixture was extracted triply with CH2Cl2 to recover products. The CH2Cl2 phase was washed with water to remove the salts. The aqueous phase was filtered to collect the insoluble products (especially from the GG reactions). The insoluble products were washed with DI-water and dissolved in acetone. The products in CH2Cl2 and acetone were combined, dried over anhydrous Na2SO4, and the solvents were removed by rotary evaporation at reduced pressure. Samples were stored at −4 °C for subsequent analyses.2.4 Acetylation of ALBTH lignin
Lignin (0.1 g) was dispersed/dissolved in 1 mL pyridine in a 20 mL glass vial, and then acetic anhydride (1 mL) was added. The mixture was sealed and allowed to react at room temperature for 72 h. Lignin acetate was precipitated in acidic water (pH < 2) with constant stirring, collected by filtration, washed with DI-water, and vacuum-dried.2.5 NMR spectroscopic analysis
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCE III 500 MHz spectrometer equipped with a DCH (13C-optimized) cryoprobe (Billerica, MA). The lignin and GG reaction samples (unacetylated) were dissolved in 0.5 mL of dimethylsulfoxide-d6 (DMSO-d6) or DMSO-d6/pyridine-d5 (4:1, v/v). Heteronuclear single-quantum coherence (HSQC) spectra were recorded at 25 °C using Bruker's standard hsqcetgpsisp2.2 pulse program (acquisition times 200 ms and 8 ms in 1H and 13C dimensions, respectively; inter-scan relaxation delay 1 s). HSQC-total correlation spectroscopy (TOCSY) spectra were recorded at 25 °C using Bruker's standard hsqcdietgpsispp2 pulse program (inter-scan relaxation delay 2 s; mixing time 80 ms). Data processing was performed using Bruker's Topspin 3.5 software to a final matrix size of 2k × 1k data points.
The relative abundance of each interunit linkage Ri, appearing in the aliphatic region of the HSQC spectra, from lignin samples was calculated using the following equation:
| Ri% = Ii/[IA + IB + IC + IHK + IBD] × 100% |
2.6 Gel-permeation chromatographic (GPC) analysis
The number-average and weight-average molecular weights (Mn and Mw, respectively) of the ALBTH lignins (after acetylation) and the products of LMCs were estimated on a Dionex ICS-3000 system equipped with three tandem 300 mm × 7.8 mm (L. × I.D.) Phenogel 5U columns (10000, 500, and 50 Å, respectively) and a 50 mm × 7.8 mm (L. × I.D.) Phenogel 5U guard column (Phenomenex, Torrance, CA). The eluent was an isocratic 100% THF (HPLC grade without a stabilizer) at a flow rate of 1.0 mL min−1 and the column temperature was maintained at 30 °C. Each sample (∼0.5%) in THF was injected after passing through a 0.45 μm filter and the fractionated lignin in the column eluent was detected using a variable wavelength detector (VWD) at 280 nm. The apparent molecular weight was calibrated using polystyrene standards and GG at 254 nm.
2.7 Gas chromatography-mass spectrometric (GC-MS) analysis
Qualitative analysis of the LMC reaction products was conducted on a GC-MS system (GCMS-QP 2010S, Shimadzu Co., Addison, IL) with an SHRXI-5MS column (30 m × 0.25 mm, L. × I.D., with 0.25 μm film thickness). The injection port was at 280 °C, and the carrier gas was helium at a flow rate of 8 mL min−1 in a split mode (split ratio, 5:1). The programmed GC oven temperature profile started with a 30 °C isotherm for 5 min, followed by a 5 °C min−1 ramping to 220 °C and subsequent 10 °C min−1 ramping to 300 °C, then held at the temperature for 5 min. The products from the GG reaction were silylated to improve the sample volatility. Specifically, an approximately 50 μL sample was mixed and reacted with a 500 μL N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA)/pyridine mixture (1:1, v/v) at room temperature for 2 h.
2.8 High-performance liquid chromatographic (HPLC) analysis
The LMC reaction products were quantitated on a Dionex ICS-3000 system equipped with a YMC-Triart C18 column (250 mm × 4.6 mm, L. × I.D.) at 30 °C and a VWD set at 210 nm. Both samples and standards were diluted with 20% acetonitrile in water to ensure full dissolution and filtered through a 0.45 μm syringe filter prior to injection. The mobile phase was an isocratic acetonitrile/water solution (20/80, v/v) with addition of 0.1% phosphoric acid at 0.8 mL min−1. The column was reconditioned using an isocratic acetonitrile/water solution (60/40, v/v) for 10 min after each run.3. Results and discussion
3.1 Formation of HK and BD moieties from lignin depolymerization in the ALBTH system
Feedstocks (aspen, poplar, eucalyptus, Douglas fir, switchgrass, and corn stover) representing the three major biomass classes (hardwoods, softwoods, and grasses) were treated in the ALBTH system under mild reaction conditions (110 °C, LiBr 60 wt% and 40 mM HCl) to investigate lignin depolymerization in the process of ALBTH treatment. By completely dissolving and hydrolyzing cellulose and hemicelluloses, the ALBTH treatment allowed quantitative isolation of lignin in the solid state from biomass,28 as shown in Table S1.† The GPC-estimated molecular weights of the lignins are listed in Table 1 and compared with those of the corresponding isolated lignins, such as milled wood lignin (MWL) and cellulolytic enzyme lignin (CEL), estimated under similar chromatographic conditions.31–34 MWL and CEL fractions are generally considered to incur only minor modifications to the native lignin and therefore acceptable to represent the native lignin in the biomass. However, the molecular weights of MWL and CEL are slightly lower than that assumed for native lignin, because they only represent the solvent-extractable (low molecular weight) portions of the native lignin.| Biomass | Treatment | M n | M w | PDI |
|---|---|---|---|---|
| The molecular weights of all the lignin samples (both in this study and in the literature) were estimated after acetylation (with either acetic anhydride or acetyl bromide) using gel-permeation chromatography (GPC) under similar conditions (a UV detector, THF as the eluent, and polystyrene as the standards for calibration). | ||||
| Poplar | MWL31 | 4100 | 10000 |
2.4 |
| ALBTH-30 min | 1200 | 2900 | 2.5 | |
| ALBTH-120 min | 1100 | 3200 | 2.9 | |
| Aspen | MWL32 | 4500 | 23300 |
5.2 |
| ALBTH-30 min | 1000 | 2200 | 2.2 | |
| Eucalyptus | CEL33 | 5500 | 17200 |
3.1 |
| ALBTH-120 min | 1100 | 2500 | 2.2 | |
| Douglas fir | CEL33 | 5500 | 21800 |
4.0 |
| ALBTH-30 min | 1100 | 3700 | 3.3 | |
| ALBTH-120 min | 1100 | 3600 | 3.3 | |
| Corn stover | CEL34 | 1400 | 10100 |
7.4 |
| ALBTH-30 min | 1200 | 4100 | 3.3 | |
| Switchgrass | MWL33 | 2100 | 5100 | 2.5 |
| ALBTH-120 min | 1000 | 3100 | 3.1 | |
The weight-average molecular weight (Mw) dropped to below 3000 Da for the hardwood (aspen, poplar, and eucalyptus) ALBTH lignins and 3700 Da for the softwood (Douglas fir), after treatment in ALBTH for 30 min. These Mw values are markedly lower than those of isolated lignins (CEL and MWL, 10K–25K Da). The results suggest that the native lignin in woody biomass was extensively depolymerized in the ALBTH system. Extending the ALBTH treatment to 120 min, the Mw of the lignins did not decrease further, suggesting that the cleavage of the β-O-4-aryl ether bonds was less prevalent and/or that lignin condensation was occurring, as discussed below. Similarly, the native lignin in grassy biomass such as corn stover and switchgrass was depolymerized to low molecular weight polymers (Mw 3100–4100 Da) in the ALBTH system.
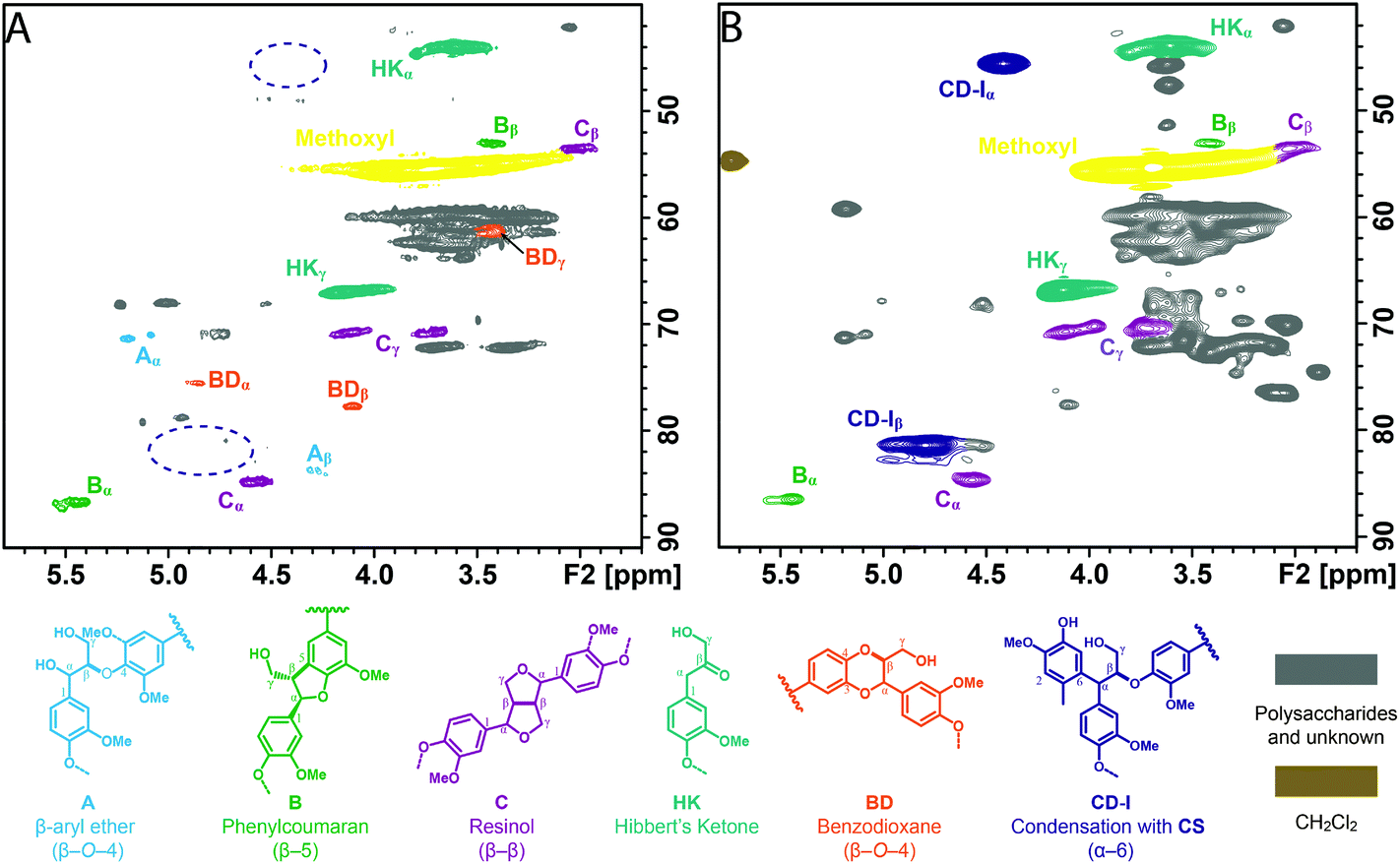
To reveal the structural changes in lignin during the depolymerization, the ALBTH lignins from aspen, eucalyptus, Douglas fir, and corn stover were characterized using HSQC NMR. Their spectra were compared with those from the whole cell wall (WCW) of the corresponding raw biomass, as shown in Fig. 1 (the aliphatic region) and Fig. S1† (the aromatic region). Many of the major contours in the δC/δH 65–85/2.5–4.5 ppm region of the WCW spectra (left column) assigned to the non-anomeric carbohydrates are mostly absent in the ALBTH lignin spectra (right column), verifying the excellent ability of ALBTH to dissolve and hydrolyze the carbohydrates from the biomass to leave a relatively pure lignin fraction. As shown in Fig. 1A, C, E, and G (left column), the native biomass lignins had abundant β-ether units A (with their characteristic β-O-4-aryl ether linkages) along with small amounts of phenylcoumarans B (β-5) and resinols C (β-β), which are in agreement with other studies.35,36 After a mild ALBTH treatment, the signals from the β-ether units were attenuated and some new chemical structures including the HK and BD moieties were revealed. The assignment of the Hibbert's ketone moiety HK was made by the cross peaks at δC/δH 44.5/3.67 ppm (α-position) and 67.1/4.19 ppm (γ-position) according to the study on LMCs,13 and the assignment of the trans-benzodioxane moiety BD was made by the cross peaks at δC/δH 75.6/4.86 ppm (α-position) and 77.5/4.07 ppm (β-position) according to the study on C-lignin and C-DHP polymers derived from caffeyl alcohol polymerization.37 Notably the resonance peaks from both moieties were distinctive from other lignin signals and were consistently observed in the ALBTH lignins from all woody biomass species used in this study. The ALBTH lignin from grasses (corn stover, Fig. 1H) only had HK, but not BD moieties resolved, possibly due to its low β-O-4-aryl ether content and the presence of p-coumarates and ferulates. The identification of HK and BD moieties suggests that the condensation to form the inert C–C linkages was less significant (or reduced) during the cleavage of the β-O-4-aryl ether bonds in ALBTH. More evidence and discussion will be provided below from the LMC study. A striking feature of the reaction was the generation of BD units, obviously from β-ether units and involving demethylation. As far as we are aware, this is the first time that BD units have been detected from an acid-catalyzed lignin depolymerization. BD units were originally discovered in vanilla seed coats that make their lignins entirely from caffeyl alcohol,37 with its “C-lignin” being recently positioned as an ideal lignin structure that can survive acidic treatments but can be readily depolymerized to valuable 4-propanolcatechol and its derivatives.38,39 These observations suggest that the ALBTH system has the uniqueness to yield uncondensed and valuable lignin moieties/structures, compared with other acidic solvent systems for lignin depolymerization. The presence of G, S, and H units in lignin before/after ALBTH treatment is most readily discerned from the aromatic regions of the HSQC spectra and can be verified with reference to Fig. S1.† Notably, the formation of HK and BD moieties significantly changed the chemical shifts of G and S units in the aromatic regions.
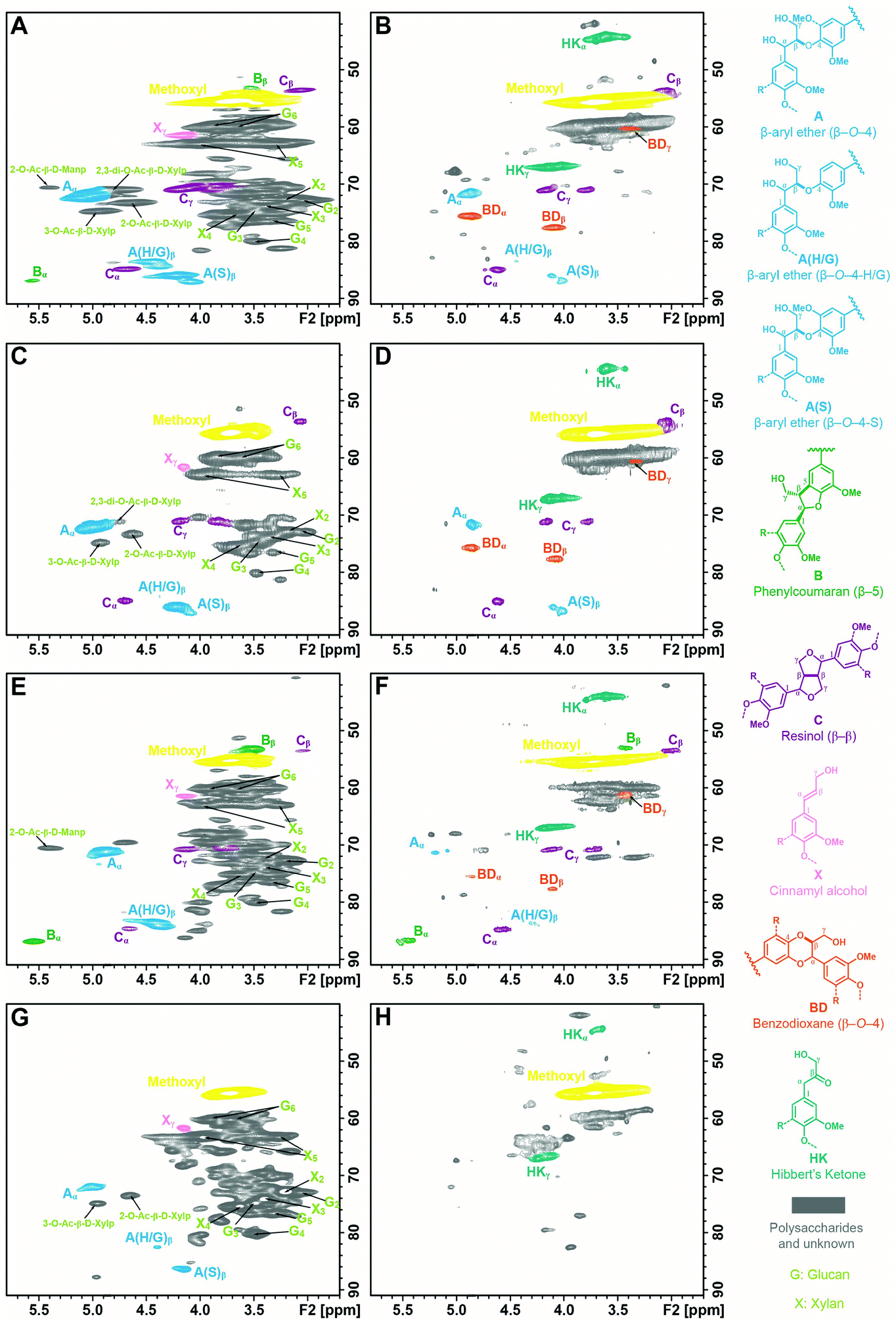 | ||
| Fig. 1 The aliphatic regions of 2D 1H–13C correlation (HSQC) spectra of the ball-milled plant cell walls dispersed in DMSO-d6/pyridine-d5 (A: aspen, C: eucalyptus, E: Douglas fir, and G: corn stover) and the lignins isolated by acidic lithium bromide trihydrate treatment in DMSO-d6 (D: eucalyptus and F: Douglas fir) and in DMSO-d6/pyridine-d5 (B: aspen and H: corn stover). Correlation signals are categorized and color-coded by the type of aromatic units. (A, β-O-4-aryl ether; B, phenylcoumaran (β-5); C, resinol (β-β); X, cinnamyl alcohol end-group; HK, Hibbert's ketone; and BD, benzodioxane). | ||
Using semi-quantitative analysis, the relative abundance of various units in the lignins was estimated, as shown in Table 2. The phenylpropanoid units in hardwood species such as aspen were mainly the β-O-4-aryl ether A and resinol C moieties with a trace amount of the phenylcoumaran B moiety. After ALBTH treatment, the relative abundance of the resinol C moiety remained unchanged, while the β-ether A decreased from 91% to 25% and phenylcoumaran B was no longer detectable. Notably, HK and BD (as novel uncondensed lignin depolymerization products) formed in 46% and 23% levels, respectively. For softwood species (e.g., Douglas fir), the relative level of resinol C rose from 4% to 8%, whereas the β-ether A and phenylcoumaran B moieties decreased from 79% and 18% to 13% and 18%, respectively. These results suggested that the ease of cleaving the lignin linkages during the ALBTH treatment followed the order of β-ether > phenylcoumaran > resinol (i.e., A > B > C). In other words, the ALBTH system was able to cleave the β-ether A in lignin fairly selectively, leading to uncondensed HK and BD moieties. Notably, it was observed that the hardwood lignin had a much higher level of the BD moiety (over 23%) than the softwood lignin (5%) after the ALBTH treatment. Apparently, the S type lignin (abundant in hardwood) has more chances to demethylate one of the two methoxyl groups to form a BD moiety than the G type lignin in softwood that has only one methoxyl group per unit.
| Lignin unit | C | B | A | HK | BD | |
|---|---|---|---|---|---|---|
| Aspen | WCW | 6.3 | 2.6 | 91.1 | ||
| ALBTH | 5.2 | 25.3 | 46.3 | 23.1 | ||
| Eucalyptus | WCW | 5.0 | 95.0 | |||
| ALBTH | 9.8 | 31.0 | 33.6 | 25.6 | ||
| D. fir | WCW | 3.6 | 17.5 | 78.9 | ||
| ALBTH | 7.8 | 17.9 | 13.2 | 56.1 | 5.0 | |
As mentioned above, the acid-catalyzed lignin depolymerization has been extensively studied in the systems such as acidic ionic liquid,6 aqueous dilute acid,40 steam explosion,41 and in acidic organic solvents such as methanol, GVL, and dioxane.9,11,13 However, the HK from cleavage of the β-ether units was not observed or barely visible in the HSQC NMR spectra of the lignins from these processes. There are presumably two reasons. First, the competing C6–C2 type cleavage might be more favorable than the C6–C3 type cleavage in the traditional acid systems (especially in H2SO4-catalyzed systems), resulting in abundant C2-aldehyde end-groups that promptly succumb to aldol condensations.10,42–44 This was supported by the finding that the cleavage pathways were dependent on the types of acid and solvent systems used.45 Second, the HK (C3 type ketone) end-groups might be preferentially stabilized or prevented from condensation in the ALBTH system because, in the solid state, the lignin moiety was less accessible and less mobile, limiting the opportunity for condensation reactions, as further discussed below. In summary, the observations above suggest that the ALBTH system is able to selectively cleave the β-O-4-aryl ether bonds under mild reaction conditions and generate depolymerized and less condensed lignins containing novel HK and BD moieties. Compared with other acidic systems, the ALBTH system showed considerable potential to isolate “good” lignin from lignocellulosic biomass for valorization.
3.2 Reactions of LMCs in ALBTH
Guaiacylglycerol-β-guaiacyl ether (GG), a popular β-O-4-aryl ether model compound, was used to investigate lignin depolymerization in ALBTH. The decomposition of GG and the yield of guaiacol (GA) are presented as a function of the reaction time in Fig. 2. GG and GA were quantitated by HPLC (Fig. S2†). In order to investigate the possible synergy of LiBr and acid in ALBTH, the GG reaction was conducted at 100 °C in three disparate solvent systems: acidic lithium bromide trihydrate (ALBTH) with 10 mM HCl, neutral lithium bromide trihydrate (NLBTH) without acid, and an aqueous dilute acid (DA) solution with 10 mM HCl but without LiBr. In ALBTH (Fig. 2A), GG was completely consumed within 10 min, yielding 34% GA; extending the reaction, no additional GA was released. Without an acid catalyst, the conversion of GG was slow in NLBTH (Fig. 2B). The GG content (both the erythro and threo diastereomers) gradually decreased with time, and over 80% conversion was reached at 180 min. In this case, the erythro-isomer was depleted more rapidly, as has been previously observed under alkaline degradation conditions.46 However, the yield of GA was negligible (less than 2%). This observation indicated that most GG was slowly transformed in NLBTH, but no GA was released, possibly because either the β-O-4-aryl ether bonds were not cleaved, or the released GA promptly condensed with GG decomposition products. The analysis results of GG condensation products in NLBTH mentioned later apparently support the fact that the former was the primary reason. The results shown in Fig. 2B also verified that the presence of acid was essential to the effective cleavage of the β-ether. However, acid alone (10 mM HCl in water) without LiBr did not break down GG at 100 °C (Fig. 2C). The total GG content in the system remained without apparent change over 3 h and without formation of GA, although threo-GG partially isomerized to erythro-GG, resulting in the apparent equilibrium favoring the erythro-isomer. Notably, higher reaction temperatures and higher acid concentrations (e.g., 150 °C with 200 mM H2SO4) were usually necessary to catalyze the cleavage of GG in the aqueous dilute acid.40 The observation of the effective cleavage of GG under milder conditions (very low acid concentration and moderate temperature, 10 mM HCl and 100 °C) in lithium bromide trihydrate was presumably ascribed to (1) the synergy between the acid and LiBr in cleaving the β-O-4-aryl ether bond, and (2) the enhanced acidity of the Brønsted acid in LiBr trihydrate.47 The aforementioned results demonstrated that both acid and LiBr trihydrate synergistically depolymerized lignin in the ALBTH system under mild conditions. A similar synergistic effect of the acid and LiBr trihydrate in ALBTH was reported in the hydrolysis and dehydration of polysaccharides to furfurals.47,48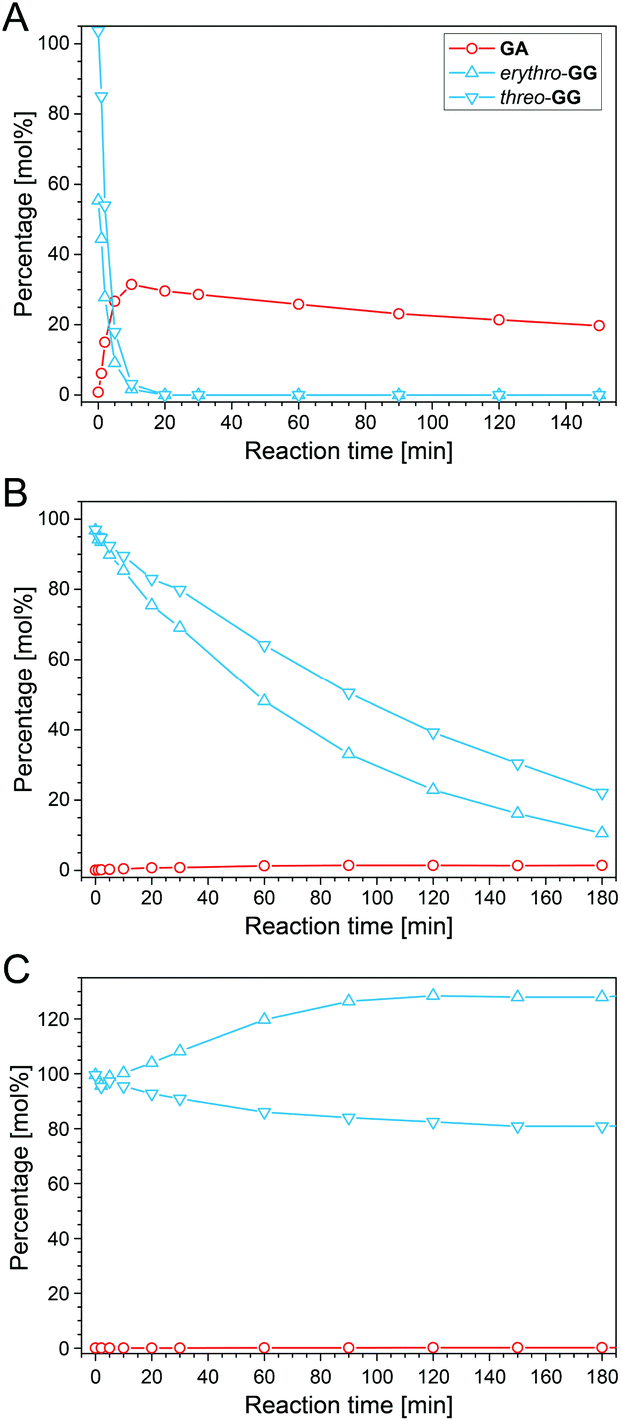 | ||
| Fig. 2 Reactions of guaiacylglycerol-β-guaiacyl ether (GG) in acidic lithium bromide trihydrate (A), neutral lithium bromide trihydrate without acid (B), and acidic water solution (C). Reaction conditions: 100 °C, 1 g L−1GG dimer concentration, and 10 mM HCl for A and C. | ||
The structures of the GG products in ALBTH and NLBTH were further characterized by HSQC NMR analysis. HK and BD moieties were detected from the ALBTH-treated GG (Fig. 3A), whereas only a negligible amount of HK but no BD was identified from the NLBTH-treated GG (Fig. 3B). HK was also detected in the HPLC analysis of GG treated in both ALBTH and NLBTH (Fig. S2†). The formation of HK and BD was further confirmed by the GC-MS analysis (Fig. S3B†) of the ALBTH-treated GG, in which a HK monomer and 4-(3-(hydroxymethyl)-2,3-dihydrobenzo-[b][1,4]dioxin-2-yl)-2-methoxyphenol (a BD dimer) were detected. These verified our previous observation that both HK and BD moieties are formed during lignin depolymerization in ALBTH. No BD was identified in NLBTH-treated GG, suggesting that acid was crucial to generate the uncondensed BD structures in the LiBr trihydrate system. The relatively low abundance of the BD moieties from GG was consistent with our biomass HSQC NMR results above, in which G units (softwood lignin) were less prone to form the BD structures than S units (hardwood lignin).
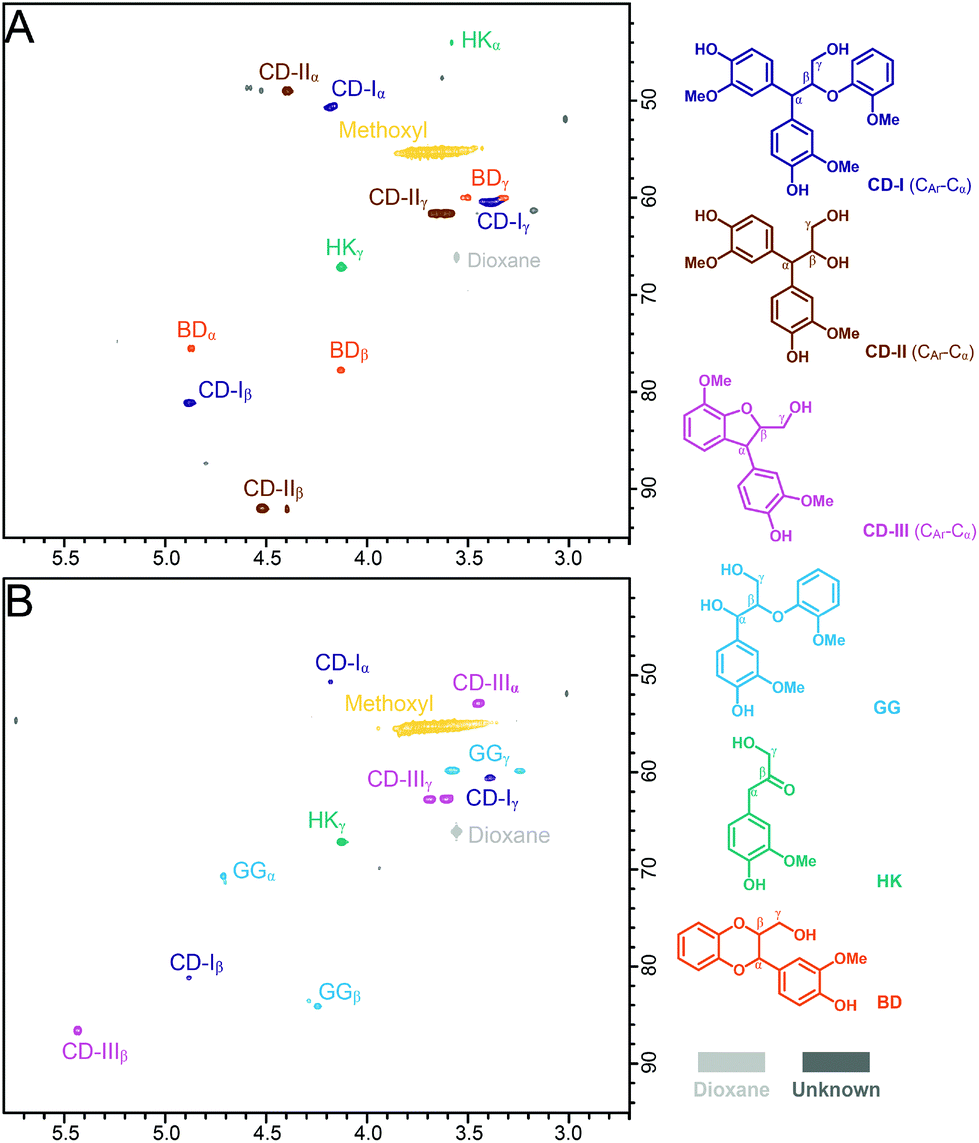 | ||
| Fig. 3 HSQC NMR spectra of GG reaction products in the LiBr trihydrate reaction at 100 °C with 10 mM HCl for 10 min (A) and without acid (B) for 240 min. | ||
Condensation products from GG were also detected in the HPLC analysis (Fig. S2†) and further confirmed by GPC analysis (Fig. S4†). Both ALBTH and NLBTH systems yielded higher molecular weight fractions from GG. As shown in Fig. S4,† the sample from the ALBTH system had a broad peak A (Mw: 750 Da, Mn: 630 Da) and a sharp peak B (Mw: 290 Da, Mn: 290 Da) with the A to B intensity ratio (A/B) = 2.1. For the sample from the NLBTH system, the peaks A and B represented the Mw/Mn of 780/660 and 290/280, respectively, with A/B = 4.3. These results indicate that the products from the NLBTH-treated GG had more condensed structures and slightly higher molecular weights than that from the ALBTH-treated GG. This is in agreement with the results shown in Fig. 2B where GG was consumed but did not release GA, implying that condensation was predominant over cleavage of the β-O-4-aryl ether bond in NLBTH.
The structures of the condensation products from GG were identified by HSQC NMR (Fig. 3) in which the assignments were made based on the GC-MS results shown in Fig. S3† and HSQC-TOCSY NMR results shown in Fig. S5.† In the ALBTH-treated GG sample, the diphenylmethane-type condensation products were unveiled, including the 4-(3-hydroxy-1-(3-hydroxy-4-methoxyphenyl)-2-(2-methoxyphenoxy) propyl)-2-methoxyphenol trimer (CD-I type condensation) and the 3-(4-hydroxy-3-methoxyphenyl)-3-(3-hydroxy-4-methoxyphenyl)-propane-1,2-diol dimer (CD-II type condensation) (Fig. 3A, S3B, and S5A†). In the NLBTH-treated GG sample, the phenyl-dihydrobenzofuran type condensation products (e.g., 4-(2-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-3-yl)-2-methoxyphenol, a dimer from intramolecular condensation, CD-III type condensation) were the most abundant together with a small amount of CD-I type condensation products (Fig. 3B, S3A, and S5B†). This observation further established that the cleavage of the β-O-4-aryl ether bond was not the dominant reaction of GG in NLBTH. It was apparent that condensation mostly occurred via the α-benzyl carbocation by its electrophilic aromatic substitution on the electron-rich aromatic rings, forming a new C–C linkage, which resulted in characteristic correlation signals from the α-position of CD-I, CD-II, and CD-III type condensation products at δC/δH 50.8/4.18, 48.9/4.40, and 53.0/3.44 ppm, respectively. The identification of condensation products mentioned earlier brings new insight into the competition between the lignin depolymerization and condensation via the α-benzyl carbocation intermediate as catalyzed by both LiBr trihydrate and acid.
3.3 Proposed mechanisms of lignin reactions in the LiBr trihydrate system
Based on the results and discussions mentioned above, pathways of lignin depolymerization to form BD and HK moieties and condensation reactions in the LiBr trihydrate system are proposed, as illustrated in Fig. 4 and S6.† In this system, the benzyl carbocation is logically the essential intermediate toward various products. There are two feasible routes to form the benzyl carbocation. Route I, which has been extensively elucidated in acidic systems, is initiated by acid-induced protonation of the benzyl alcohol and followed by dehydration to form the α-carbocation. Alternatively, in Route II, LiBr is postulated to catalyze the slow formation of the benzyl carbocation via reversible substitution of the α-hydroxyl by the bromide in the system. Route II is proposed based on the observation that condensation reactions occurred when GG was treated in NLBTH (LiBr alone without acid). In addition, Route II was supported by the experiment in which 3,4,5-trimethoxylbenzyl alcohol (TMBA) was treated in the NLBTH system. As shown in Fig. S7A,† both the α-brominated intermediate of TMBA and its subsequent condensation product were detected, suggesting that LiBr alone did lead to the formation of the α-carbocation which then attacked the electron-rich 2/6-position on the aromatic ring of TMBA, resulting in the condensation products. The benzyl carbocation can then be subsequently transferred from the α- to the β-position either by the small equilibrium in Route III via an enol ether intermediate or in Route IV via direct hydride shift.42 Following trapping of the β-carbocation by H2O, cleavage of the β-O-4-aryl ether bond occurs, releasing the HK and GA moieties. As GA was barely detected in the absence of acids, both Routes III and IV were presumed to be exclusively acid-catalyzed processes.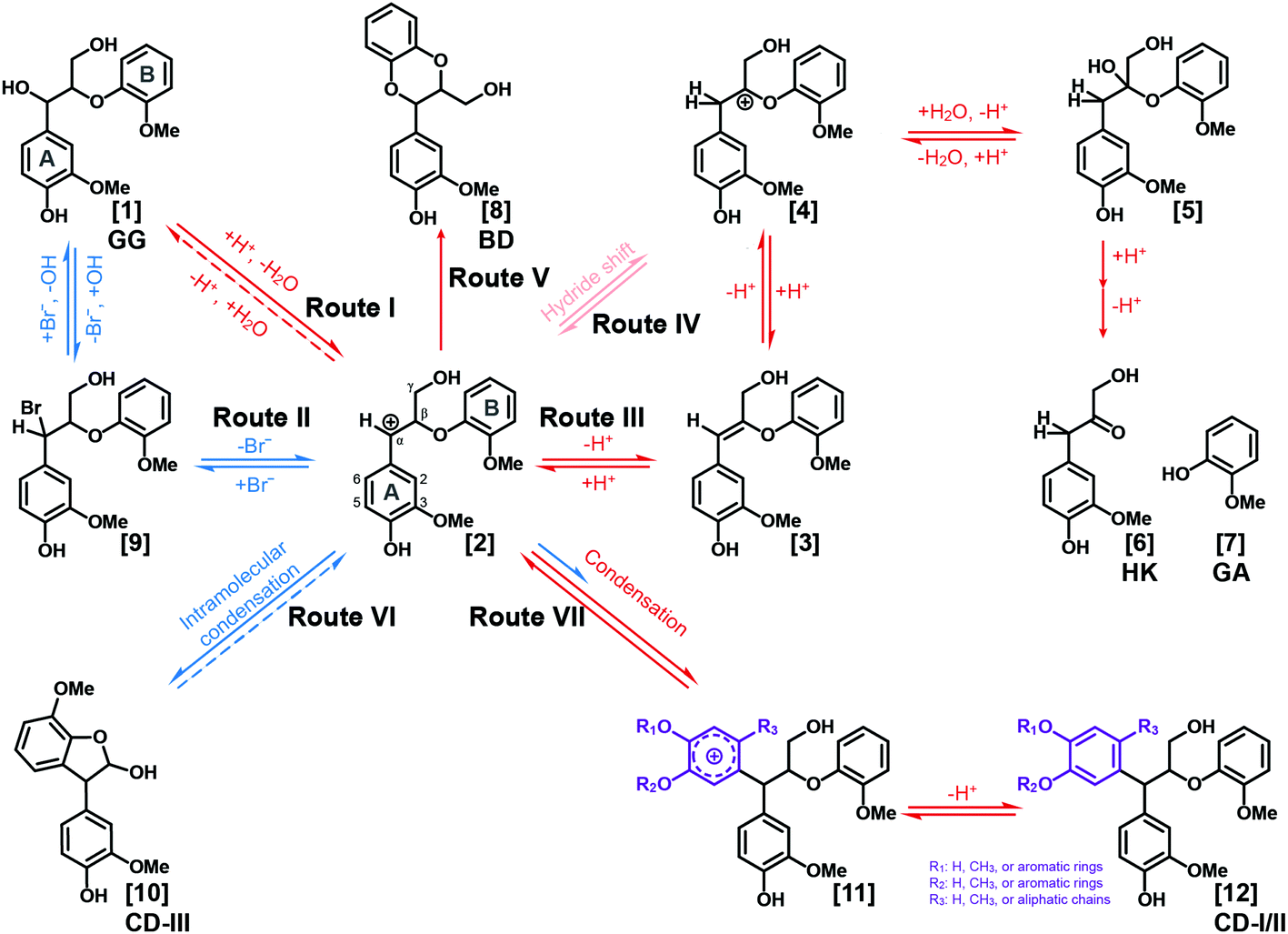 | ||
| Fig. 4 Proposed lignin depolymerization and condensation mechanisms. | ||
The reactive benzyl carbocation intermediate can also lead to the formation of BD moieties via Route V, which is supported by the results mentioned above that the BD structures were identified in the products from both native lignins and GG in ALBTH. The nucleophilic methoxyl oxygen on the GA ring of GG can intramolecularly trap the benzyl carbocation, resulting in a BD moiety from β-O-4-aryl ethers after demethylation. It seems that trapping the benzyl carbocation by C3/5-methoxyl is the first step in BD formation followed by demethylation. Otherwise, other demethylation structures besides BD should have been identified from the GC-MS and NMR analyses. In addition, no BD moiety is detected in the NLBTH system, suggesting the essentiality of an acid to the BD formation. It is worth mentioning that the detection of the BD moiety has never been reported in the products from the acidolysis of lignin or LMCs. The conversion of β-O-4-aryl ethers to BD (an acid-tolerant structure) suggests the possibility of a new strategy for lignocellulosic biomass valorization in the ALBTH system, i.e., hydrolyzing polysaccharides efficiently to their component sugars26,28 and converting all lignin β-ethers to benzodioxanes in an isolated lignin that is then converted to catechol-type monomers by hydrogenolysis.38
Based on the identified condensation products from the GG studies above, two condensation routes are proposed. In Route VI (Fig. 4), intramolecular condensation occurs via electrophilic aromatic substitution by the benzyl carbocation on the electron-rich C5-carbon on the B-ring. The resultant phenyl-dihydrobenzofuran structure (CD-III) was detected only in the NLBTH system, the analog of which was also reported in the previous model compound study using 1-phenyl-2-phenoxy-1,3-propanediol (PD) and 1-(4-hydroxyphenyl)-2-phenoxy-1,3-propanediol (HH) in dilute H2SO4.40,44,49 However, the CD-III structures were not detected from GG dimers under acidic conditions in several LMC studies including our own. The observation in this study suggested that the presence of acid might greatly inhibit the intramolecular condensation of GG from occurring in the LiBr trihydrate system, possibly due to the overwhelming prevalence of intermolecular condensation. As proposed in Route VII, the benzyl carbocation readily attacks the electron rich carbon on the aromatic rings. The reactive sites on the ring may vary depending on the ring substituents. For example, the favorable sites are the C1 position (para to the hydroxyl substituent) for GA (R1: H, R2: CH3, and R3: H) and the C6 position (para to the methoxyl substituent) for lignin units (R1: H, R2: CH3, and R3: aliphatic side chain) shown in Fig. 4. Three factors may affect the regioselectivity of the electrophilic substitution (condensation) of the aromatic rings: (1) the electron donating substituents activate the aromatic rings mostly at their para and ortho positions and their activation power follows the order OH > OMe > methyl ≈ side chain of lignin; (2) the substituents that form extra resonance structures with the aromatic rings favor ortho/para substitution; and (3) larger substituents (e.g., aliphatic side chains) sterically inhibit the substitution at their ortho positions.
In analogy with traditional acid-catalyzed lignin depolymerization,50,51 the trapping of a benzyl carbocation by the aromatic rings is considered to be the major condensation reaction in the ALBTH system. Each of the aromatic rings of GG, even after condensation, has active electron-rich sites, which are able to condense with a new benzyl carbocation to form C–C linkages. As a result, the intermolecular condensation reactions could generate high molecular weight products, such as those detected in the GPC analysis (Fig. S4†).
In order to further probe the condensation reactions occurring in ALBTH, TMBA (a benzyl carbocation generator) was reacted with GA and creosol (CS), and the products were analyzed with GC-MS, as shown in Fig. S7B and S7C.† The results confirmed the proposed Route VII (Fig. 4) in which the C–C condensation mostly occurred between the benzyl carbocation and electron-rich aromatic rings. Creosol (CS) is a monomeric lignin-mimic without the carbocation-forming ability that, alone, was stable in ALBTH and NLBTH (∼98% recoverable at 100 °C). When CS was introduced into the GG system in LiBr trihydrate, CS was significantly consumed, indicating that CS reacted with GG (Fig. 5). In the ALBTH system (Fig. 5A), the maximum yield of GA decreased significantly from 31.4 to 10.7% when CS was added, indicating that the addition of CS inhibited the cleavage of the β-O-4-aryl ether bond in GG that would generate GA. This is because, under the acidic condition, the protonation of the benzyl alcohol of GG readily occurs and yields the benzyl carbocation, and its trapping by the excess creosol in the system forms the Cα–Caryl linkage, which apparently blocks the pathway for cleavage of the β-O-4-aryl ether linkage via the analog of Route III or IV. In NLBTH (Fig. 5B), the GG consumption was much slower than that in ALBTH, as only LiBr contributed to the formation of the benzyl carbocation. In this case, the presence of CS produced no significant changes to GG consumption, indicating that the rate-limiting step of the condensation reactions in NLBTH is formation of the benzyl carbocation.
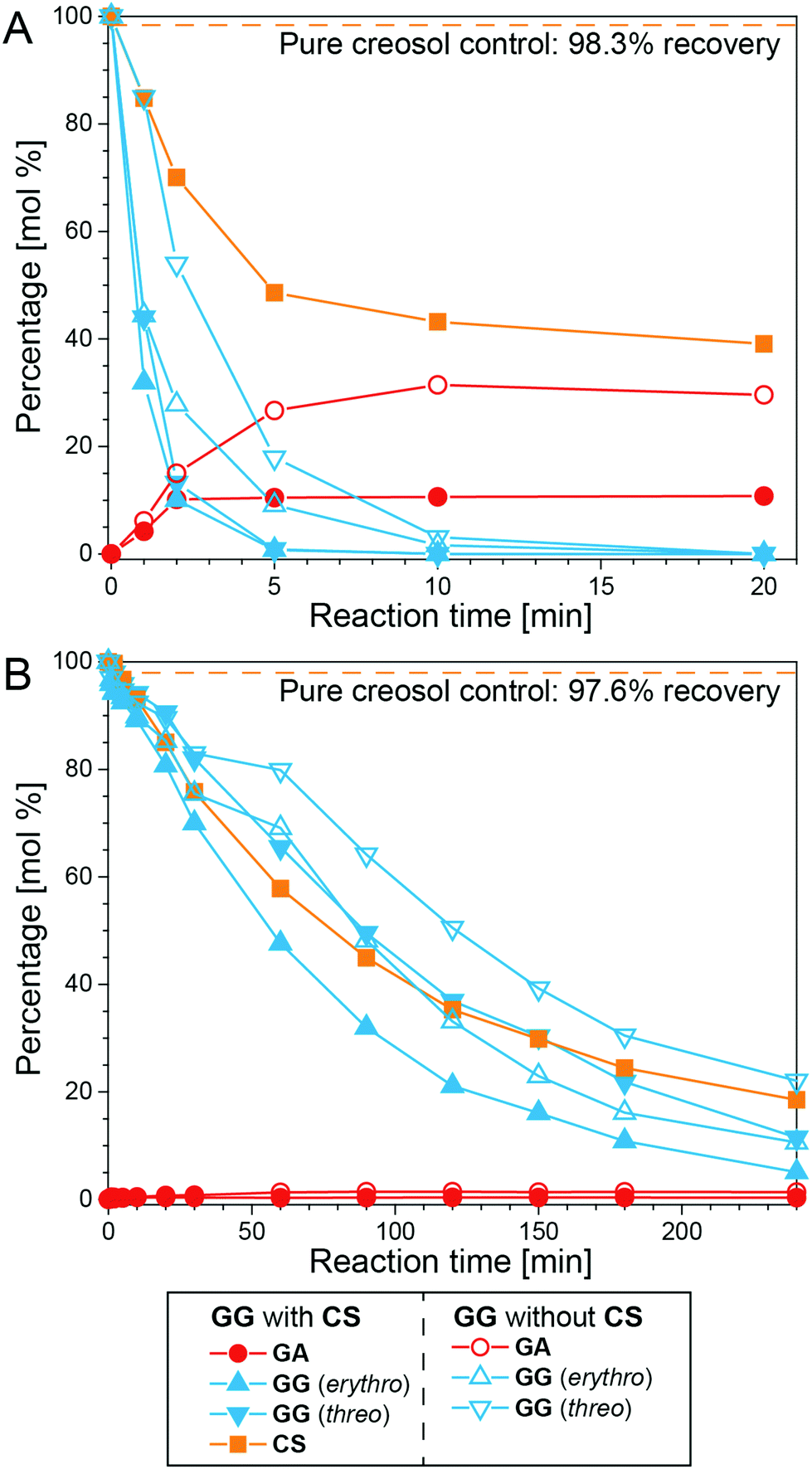 | ||
| Fig. 5 The reaction of GG with CS in ALBTH with 10 mM HCl (A) and NLBTH (B). Reaction conditions: 100 °C, GG to CS ratio: 1/1 (w/w); no addition of CS as the control. | ||
To summarize the findings of the mechanistic studies using LMCs, both LiBr and acid can induce the formation of the benzyl carbocation intermediate, which is crucial to lignin depolymerization and BD formation. Although LiBr exerts synergistic effects on the formation of the benzyl carbocation, the acid dominates the catalysis of both the formation of the benzyl carbocation and the subsequent cleavage of the β-O-4-aryl ether bonds to yield HK and demethylation to yield BD moieties. The undesirable competing pathway is the formation of C–C bonds between the benzyl carbocation and the electron-rich aromatic rings, which is catalyzed by both LiBr and acid.
3.4 Suppressed lignin condensation in the ALBTH system
In any process of lignin isolation and depolymerization for downstream valorization, condensation is intrinsically undesirable. Although the LMC studies above demonstrated the possible condensation reactions occurring in ALBTH and NLBTH systems, the condensation products (containing Cα–Caryl bonds), interestingly, remained at a minimal level in the real ALBTH lignins. In the HSQC NMR spectra (Fig. 1B, D, F, and H), the 1H/13C correlation signals at δC/δH (45–52)/(4.0–5.0) ppm from the assignment of the condensation structures at the Cα position were negligible. This observation indicated that the C–C condensation between the α-carbocation and an aromatic carbon was greatly suppressed during the ALBTH treatment of lignocellulosic biomass. The condensation disparity between the real lignin and LMCs might result from their solubility, status, and mobility in ALBTH (i.e., the accessibility of electron-rich aromatics to the benzyl carbocation). In the case of the LMC reactions, the small aromatic molecules are either well dispersed or are dissolved in the system. Once the benzyl carbocation is formed, the nearby electron-rich (nucleophilic) aromatic carbons can readily succumb to attack by the carbocation, resulting in the Cα–Caryl condensation products. On the other hand, lignin and lignin moieties are insoluble and tend to coil and aggregate as large particles in the ALBTH medium. The benzyl carbocation and the electron-rich aromatic rings from larger lignin polymers are therefore less mobile and have fewer chances to react with each other because of the steric and physical hindrance. Instead, the intramolecular cleavage of β-O-4-aryl ether bonds and the formation of BD from the benzyl carbocation are promoted. As a result, the ALBTH lignin has abundant uncondensed moieties (HK and BD) with limited condensation structures, which is consistent with our findings that the ALBTH lignin has low molecular weight and good solubility in many organic solvents.In order to verify the hypothesis that condensation is unfavorable in real lignin due to limited mobility and accessibility, the lignocellulosic biomass (e.g., Douglas fir) was treated in ALBTH in the presence of a monomeric lignin model nucleophile (CS). As expected, the resultant lignin featured significant correlation signals at δC/δH 45.4/4.41 ppm (α-position) and 81.4/4.80 ppm (β-position) due to condensation with CS (Fig. 6B), whereas the lignin without CS had negligible signals corresponding to condensation structures (Fig. 6A, the same as Fig. 1E). The results indicated that CS, as a small and mobile nucleophile, condensed readily with the lignin benzyl carbocation and formed Cα–Caryl bonds; in contrast the condensation between two real lignin polymers was limited. These observations confirmed that the aromatic carbons and the benzyl carbocation in the solid-state lignin are less accessible to each other, contributing to the limited condensation reactions in ALBTH.
 | ||
| Fig. 6 Evaluation of condensation in ALBTH lignin from Douglas fir without creosol (A) and with addition of creosol (B, 5.5/1, w/w) at 110 °C. | ||
To demonstrate whether the uncondensed nature of the ALBTH lignin benefits its downstream valorization, preliminary hydrogenolysis of the ALBTH lignin from poplar was conducted using a Pd/C catalyst in methanol under 40 bar H2 at 220 °C for 6 h. For comparison, Klason lignin (representing a condensed lignin) from poplar and native lignin from poplar were also hydrogenolyzed under the same conditions. As shown in Table S2,† over 96.0% of the ALBTH lignin was converted to a methanol-soluble lignin oil. Under the same conditions, only 29.6% of the Klason lignin was converted into the oil, whereas 63.7% was retained as an insoluble residue. On the other hand, 93.1% of the native lignin in poplar was hydrogenolyzed to an oil. These observations suggested that the ALBTH lignin had a comparable response to hydrogenolysis as with the native lignin, and was much better than the Klason lignin. These preliminary results verified that the ALBTH lignin may be a good lignin feedstock for downstream valorization, via hydrogenolysis, for example.
4. Conclusions
In this study, we illustrated how the treatment of lignocellulose in the ALBTH system achieved depolymerization and separation of lignin in its solid state without dissolution after nearly quantitative hydrolysis of polysaccharides to sugars. A pure lignin could be isolated from various woody and grass biomass sources, also essentially quantitatively. The lignin was extensively depolymerized with a minimal level of condensation. The most vulnerable β-O-4-aryl ether bonds were selectively cleaved/transformed to uncondensed HK and BD moieties. Notably, BD was identified and confirmed for the first time in an acid-depolymerized lignin; its appearance suggests the possibility of using simpler routes toward catechol-type monomers that may be attractive for lignin valorization.Model compound studies using GG revealed that the formation of the benzyl carbocation is the prerequisite for both depolymerization and condensation. LiBr and HCl in the ALBTH system synergistically catalyze the formation of the benzyl carbocation, which subsequently leads to either an HK moiety via the cleavage of the β-O-4-aryl ether bond or a BD moiety via the demethylation of methoxyls on the aromatic rings. The benzyl carbocation may also attack the electron-rich aromatic rings of lignin, resulting in undesirable condensation products. However, we found that the level of condensation (formation of Cα–Caryl bonds) in real ALBTH lignin was surprisingly low. It was postulated that solid-state lignin reactions in ALBTH suppressed the condensation due to the reduced mobility and accessibility of the rigid lignin moieties (benzyl carbocations and electron-rich aromatic carbons) to each other. These findings together with preliminary hydrogenolysis results suggested that the ALBTH method may provide a new strategy to separate a useful, less condensed lignin from biomass for downstream valorization.
Author contributions
XP initiated the study. NL and XP designed the experiments and completed most of the data analyses and writing. NL conducted most of the lab work. YL, JR, CY, XY, and XL contributed to the data analyses and discussions. CY also made contributions to the whole-cell-wall NMR experiments. YL performed the GC-MS analysis of the GG reaction products, and YL and JR contributed to the interpretation and assignment of some NMR data, in particular the confirmation of the BD structure. All authors were involved in writing or proofing the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support from the National Science Foundation (NSF) (CBET 1159561), the U.S. Department of Agricultural (USDA) National Institute of Food and Agriculture, and the McIntire Stennis project (1006576) are acknowledged. Dr Gaojin Lyu kindly provided the β-ether lignin model compound used in this study. The NMR analysis was conducted in the Magnetic Resonance Facility in the Chemistry Department at the University of Wisconsin–Madison. Ning Li would like to thank the China Scholarship Council (CSC) for partially supporting his PhD study at the University of Wisconsin–Madison. Yanding Li and John Ralph were funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409).References
- B. M. Upton and A. M. Kasko, Chem. Rev., 2015, 116, 2275–2306 CrossRef PubMed.
- N. Mahmood, Z. Yuan, J. Schmidt and C. C. Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- Z. Sun, B. L. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef PubMed.
- G. F. De Gregorio, C. C. Weber, J. Gräsvik, T. Welton, A. Brandt and J. P. Hallett, Green Chem., 2016, 18, 5456–5465 RSC.
- B. J. Cox and J. G. Ekerdt, Bioresour. Technol., 2012, 118, 584–588 CrossRef PubMed.
- B. J. Cox, S. Jia, Z. C. Zhang and J. G. Ekerdt, Polym. Degrad. Stab., 2011, 96, 426–431 CrossRef.
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Energy Environ. Sci., 2015, 8, 2657–2663 RSC.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef PubMed.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Slawin, J. G. de Vries, P. C. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef PubMed.
- D. M. Miles-Barrett, A. R. Neal, C. Hand, J. R. Montgomery, I. Panovic, O. S. Ojo, C. S. Lancefield, D. B. Cordes, A. M. Slawin, T. Lebl and N. J. Westwood, Org. Biomol. Chem., 2016, 14, 10023–10030 RSC.
- T. Yokoyama, J. Wood Chem. Technol., 2014, 35, 27–42 CrossRef.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Green Chem., 2017, 19, 2774–2782 RSC.
- S. Aziz and K. Sarkanen, Tappi J., 1989, 72, 169–175 Search PubMed.
- X. Pan, C. Arato, N. Gilkes, D. Gregg, W. Mabee, K. Pye, Z. Xiao, X. Zhang and J. Saddler, Biotechnol. Bioeng., 2005, 90, 473–481 CrossRef PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef PubMed.
- H. Nimz and R. Casten, Eur. J. Wood Wood Prod., 1986, 44, 207–212 CrossRef.
- X.-J. Pan and Y. Sano, Holzforschung, 1999, 53, 590–596 Search PubMed.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef PubMed.
- M. Funaoka, T. Kako and I. Abe, Wood Sci. Technol., 1990, 24, 277–288 CrossRef.
- K. Shimada, S. Hosoya and T. Ikeda, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef.
- T. Imai, T. Yokoyama and Y. Matsumoto, J. Wood Sci., 2011, 57, 219–225 CrossRef.
- W. Lan, M. Talebi Amiri, C. M. Hunston and J. Luterbacher, Angew. Chem., Int. Ed., 2017, 130, 1–6 Search PubMed.
- X. Pan and L. Shuai, US9187790B2, 2015 Search PubMed.
- L. Shuai, Ph.D. Thesis, University of Wisconsin-Madison, 2012.
- N. Li, X. Pan and J. Alexander, Green Chem., 2016, 18, 5367–5376 RSC.
- X. Yang, N. Li, X. Lin, X. Pan and Y. Zhou, J. Agric. Food Chem., 2016, 64, 8379–8387 CrossRef PubMed.
- Y. Liu, G. Lyu, X. Ji, G. Yang, J. Chen and L. A. Lucia, BioResources, 2016, 11, 5816–5828 Search PubMed.
- J.-Y. Kim, E.-J. Shin, I.-Y. Eom, K. Won, Y. H. Kim, D. Choi, I.-G. Choi and J. W. Choi, Bioresour. Technol., 2011, 102, 9020–9025 CrossRef PubMed.
- M. S. Jahan and S. P. Mun, Bangladesh J. Sci. Ind. Res., 2010, 44, 271–280 CrossRef.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef.
- L. Zoia, A. W. King and D. S. Argyropoulos, J. Agric. Food Chem., 2011, 59, 829–838 CrossRef PubMed.
- C. Heitner, D. Dimmel and J. Schmidt, Lignin and lignans: Advances in chemistry, CRC press, 2016 Search PubMed.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef PubMed.
- F. Chen, Y. Tobimatsu, D. Havkin-Frenkel, R. A. Dixon and J. Ralph, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1772–1777 CrossRef PubMed.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv., 2018 Search PubMed , submitted.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- M. R. Sturgeon, S. Kim, K. Lawrence, R. S. Paton, S. C. Chmely, M. Nimlos, T. D. Foust and G. T. Beckham, ACS Sustainable Chem. Eng., 2013, 2, 472–485 CrossRef.
- G. Wang and H. Chen, Sep. Purif. Technol., 2016, 157, 93–101 CrossRef.
- H. Ito, T. Imai, K. Lundquist, T. Yokoyama and Y. Matsumoto, J. Wood Chem. Technol., 2011, 31, 172–182 CrossRef.
- L. H. Hoo, K. V. Sarkanen and C. D. Anderson, J. Wood Chem. Technol., 1983, 3, 223–243 CrossRef.
- S. Yasuda and K. Ota, Holzforschung, 1987, 41, 59–65 CrossRef.
- T. Imai, T. Yokoyama and Y. Matsumoto, J. Wood Chem. Technol., 2012, 32, 165–174 CrossRef.
- J. R. Obst, Holzforschung, 1983, 37, 23–28 CrossRef.
- S. Sadula, O. Oesterling, A. Nardone, B. Dinkelacker and B. Saha, Green Chem., 2017, 19, 3888–3898 RSC.
- C. G. Yoo, S. Zhang and X. Pan, RSC Adv., 2017, 7, 300–308 RSC.
- C. Zhang, H. Li, J. Lu, X. Zhang, K. E. MacArthur, M. Heggen and F. Wang, ACS Catal., 2017, 7, 3419–3429 CrossRef.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef PubMed.
- L. Shuai and B. Saha, Greem Chem., 2017, 19, 3752–3758 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc00953h |
| This journal is © The Royal Society of Chemistry 2018 |