
Fully solution-processable Cu2O–BiVO4 photoelectrochemical cells for bias-free solar water splitting†
Hyunwoo
Kim‡
 ,
Sanghyun
Bae‡
,
Dasom
Jeon
and
Jungki
Ryu
*
,
Sanghyun
Bae‡
,
Dasom
Jeon
and
Jungki
Ryu
*
Department of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: jryu@unist.ac.kr
First published on 11th June 2018
Abstract
We report a fully solution processable bias-free photoelectrochemical (PEC) cell for overall solar water splitting using a Cu2O photocathode and a BiVO4 photoanode. They were modified with catalytic multilayers (CMs) for hydrogen and oxygen evolution reactions (HER and OER), respectively, using simple and versatile layer-by-layer (LBL) assembly. The deposition of the respective CMs composed of cationic polyelectrolyte and anionic molecular polyoxometalate (POM) electrocatalysts resulted in remarkably improved performance in terms of both photocatalytic activity and stability. Detailed PEC analysis revealed that the CMs not only facilitated (1) charge transport to and (2) catalytic charge transfer at the photoelectrode/electrolyte interface, but also suppressed the deactivation of photoelectrodes, especially a labile Cu2O photocathode, by the formation of a protective coating layer. As a result, we could successfully fabricate an efficient and stable PEC cell for unassisted solar water splitting without the use of toxic/hazardous chemicals and energetic processes, which have been inevitably employed. We believe that this study provides a simple and versatile platform for the design and fabrication of various forms of electrochemical PEC devices in an environmentally friendly manner.
Introduction
Artificial photosynthesis has received a great deal of attention for decades because of its simple and elegant concept of enabling sustainable solar production of various chemical compounds.1–8 Ideally, it can be achieved using a semiconductor or molecule with a proper bandgap and band-edge positions for target-compound-specific redox reactions.2 Solar production of hydrogen requires a light-harvesting material with a bandgap larger than 1.23 eV and band-edge positions straddling the redox potentials for hydrogen and oxygen evolution reactions (HER and OER).3,4,9 However, its realization remains challenging due to the intrinsic limitations of candidate materials. For example, chalcogenide quantum dots and organometallic compounds, which have excellent visible-light harvesting properties, are mostly unstable in aqueous solutions and contain toxic or expensive elements.10–12 Most visible-light active oxide semiconductors, which are considered relatively stable, have a deep lying valence and conduction band edge position, allowing only an oxidation half reaction.13 To address these problems, a tandem photoelectrochemical (PEC) cell composed of dual photoelectrodes—a photocathode and a photoanode—has been extensively studied, such as those with Si and Fe2O3 and with Cu2O and BiVO4.14–16Functionalization of photoelectrodes is another important issue for practical application.17–38 For instance, promising OER photoanodes (e.g., Fe2O3, BiVO4, and WO3) suffer from many inherent problems: fast exciton recombination, poor electrical conductivity, and slow water oxidation kinetics.29,36–38 In the case of photocathodes, promising candidates such as Si and Cu2O are readily deactivated by the formation of an insulating oxide layer23,24 and self-redox reactions,25–28,30 respectively, and exhibit poor selectivity toward a target chemical.26–28,30 In this regard, it is necessary to functionalize both photoelectrodes with a proper charge separation/transporting material,19–22,39 protective layer,23–28,30,31 and electrocatalyst.26–28,30,37 However, conventional functionalization methods are material-specific and require harsh processing conditions, such as the use of toxic/hazardous chemicals including HF and H2SO4,22–24 vacuum/high temperature processes,23–28,30 and complex chemical synthesis.22,26 These problems necessitate the development of a new approach that satisfies the following requirements: simplicity, general applicability, and environmental friendliness. Although it was recently reported that layer-by-layer (LbL) assembly can be employed to address these problems,29,40–42 its application has been limited to the fabrication of half-cell devices requiring an external bias or sacrificial chemicals.
In this study, we report for the first time the fully solution-processable and environmentally benign fabrication of PEC cells for bias-free overall water splitting using LbL assembly. A model PEC cell was constructed using a BiVO4 photoanode and a Cu2O photocathode. They were modified with catalytic multilayers (CMs) of anionic Co- (CoPOM) and Ni-containing POM (NiPOM) electrocatalysts for OER and HER, respectively, using cationic polyethyleneimine (PEI) as an electrostatic adhesive. The deposition of CMs on the respective photoelectrode significantly improved the PEC performance due to their multiple functions: (1) providing enhanced catalytic activity and (2) facilitating the charge transport to the electrode/electrolyte interface. In particular, the OER CMs on the BiVO4 photoanode exhibited superior performance compared to well-known OER catalysts, such as cobalt phosphate (CoPi)43 and NiOOH.37 In addition, the stability of the photoelectrode was remarkably improved after the deposition of the CMs, especially for the Cu2O photocathode, because they functioned as a protective coating layer. As a result, we were able to successfully build an efficient PEC cell for unassisted overall water-splitting. We believe that this study provides an alternative green approach and a versatile platform for the flexible design and facile large-scale fabrication of unprecedented electrochemical and PEC devices.
Results and discussion
Fully solution-processable PEC cells for visible-light-driven overall water splitting were prepared by depositing HER and OER CMs on Cu2O and BiVO4 photoelectrodes, respectively, through LbL assembly (Fig. 1). They were selected as a model photocathode15,25,27 and photoanode,15,37 respectively, and readily prepared by electrodeposition according to the method described in the literature.37,44 Despite their promising properties, they often suffer from low catalytic activity and stability under visible light illumination (Fig. 1a). For example, Cu2O is quickly deactivated to CuO and Cu by self-oxidation and reduction reactions, respectively.25–28,30 BiVO4 has poor photocatalytic activity because of the fast recombination of photogenerated excitons in the bulk and sluggish water oxidation kinetics at its interface with electrolyte.19,33,36–38,45–49 To address these problems, conventionally, they were modified with various functional materials using material-specific and harsh processes that required toxic/hazardous chemicals and consumed large amounts of energy. In particular, the stability of Cu2O is too low, so it has been utilized after the deposition of protective film, such as Al-doped ZnO and TiO2, using vacuum deposition methods.23–28,30 In this study, we utilized LbL-assembled CMs, which can be readily prepared in an environmentally friendly manner. NiPOM50 with the molecular formula of [Ni4(H2O)2(PW9O34)2]10− and CoPOM51 with that of [Co4(H2O)2(VW9O34)2]10− were used as molecular HER and OER catalysts, respectively (Fig. S1, ESI†), and integrated into the respective CMs by using cationic PEI as an electrostatic adhesive (Fig. 1b). Considering our recent report that the performance of various photoanodes can be significantly improved by modifying their surface with LbL-assembled OER CMs,29 it was anticipated that a Cu2O photocathode, a BiVO4 photoanode, and a bias-free PEC cell for overall water splitting could be readily fabricated using the LbL method in an environmentally friendly manner, without using toxic/hazardous chemicals or high-energy processes. The performance of both photoelectrodes was expected to significantly improve after the deposition of the respective CMs for the following reasons: (1) enhanced catalytic charge transfer due to the deposition of the catalysts, (2) improved charge transport to the catalysts due to the formation of interfacial dipoles, and (3) increased stability due to the formation of protective coating layer (Fig. 1c). | ||
| Fig. 1 Schematic illustrations explaining (a) the inherent problems of a PEC cell composed of a Cu2O photocathode and BiVO4 photoanode, (b) the experimental procedures for addressing them, and (c) the expected results. | ||
The fabrication of a Cu2O-based photocathode with the (PEI/NiPOM)n multilayers (i.e., HER CMs) was confirmed by scanning electron microscopy (SEM) and various spectroscopic analyses (Fig. 2a–e). Dense Cu2O thin film with cubic and octahedral crystallites on a scale of several hundred nm was uniformly deposited by electrodeposition over the entire surface of a gold-coated fluorine-doped tin oxide (FTO) substrate (Fig. 2a). The thickness of Cu2O was about 1 μm when it was deposited at a charge density of 0.98 C cm−2. The deposition of the HER CMs using the LbL method led to the formation of a uniform and conformal coating on the rough Cu2O photocathode (Fig. 2b). Analyses with UV/visible spectrophotometry, quartz crystal microbalance (QCM), and ellipsometry showed that the deposited amounts of PEI and NiPOM gradually and linearly increased with the number of bilayers BL (n) (Fig. 1a–d). The HER CMs yielded negligible absorbance (Fig. 2b inset) compared to that of the bare Cu2O photocathode (Fig. 2a inset). According to the QCM analysis, the areal molar densities of the deposited PEI and NiPOM were 5.17 × 10−9 and 2.35 × 10−9 mol cm−2, respectively, in terms of monomer concentration when 10 BL were deposited (Fig. 2c). The average thickness of each BL was 1.8 (±0.31) nm (Fig. 2d), slightly larger than the diameter of NiPOM (∼1 nm). The uniform deposition of the HER CMs was also confirmed by XPS analysis, where additional peaks corresponding to N from PEI and Ni, W, and P elements from NiPOM were observed (Fig. 2e) with a decreased Cu peak (data not shown).
 | ||
| Fig. 2 Formation of the HER CMs on the Cu2O photocathode. (a, b) Cross-sectional SEM images and (insets) UV-vis absorption spectra of (a) the Cu2O photocathodes (a) on FTO and (b) the HER CMs deposited on a slide glass. Raw and false-colored images were shown together for comparison. The deposition of the HER CMs was also confirmed by (c) QCM, (d) ellipsometry, and (e) XPS. | ||
Based on these findings, we investigated the PEC performance of the Cu2O photocathodes for visible-light-driven hydrogen evolution in the presence and absence of the HER CMs. The performance was evaluated by linear sweep voltammetry (LSV) under periodic visible-light illumination (Fig. 3a, b and Fig. S2, ESI†). It seemed that the bare Cu2O photocathode generally exhibited a higher current density than its modified counterpart. However, the bare Cu2O exhibited non-zero current density under dark conditions even at a low applied bias of 0.35 V vs. reversible hydrogen electrode (RHE), indicating the presence of unwanted side reactions. It is well-known that Cu2O can be converted and deactivated to Cu and CuO under light irradiation in the presence of water:25–28,30
| Cu2O + H2O + 2e− ⇌ 2Cu + 2OH− E° = 0.82 V vs. RHE |
| 2CuO + H2O + 2e− ⇌ 2Cu2O + 2OH− E° = 0.95 V vs. RHE |
 | ||
| Fig. 3 Improved PEC performance of the Cu2O photocathode after the deposition of the HER CMs. (a) LSV curves for the Cu2O photocathodes before and after the deposition of HER CMs under periodic light illumination. (b) The effect of the number of HER BLs on the deactivation potential (ηCu2O/Cu) and current density (JCu2O/Cu) of the Cu2O photocathode. (c) Chronoamperograms and SEM images showing the improved stability of the Cu2O photocathode after the deposition of 15 BL of the HER CMs. | ||
We also found that the LSV curve measured under light illumination exhibited a local maximum peak. Given a cathodic potential sweep for LSV analysis, this peak was attributed to the deactivation of Cu2O through its reduction to Cu. To evaluate the degree of deactivation, we defined the first cathodic peak potential as a deactivation potential (ηCu2O/Cu) and dark current density measured at an applied bias of 0.35 V vs. RHE as a deactivation current density (JCu2O/Cu). The deposition of the HER CMs led to a decrease of both light and dark current densities. As the number of BL increased, JCu2O/Cu approached zero and ηCu2O/Cu exhibited a cathodic shift (Fig. 3a and b), implying the suppression of Cu2O reduction to Cu. Interestingly, the deposition of more BL also resulted in a decrease in the intensity of a cathodic and anodic transient spike in the photocurrent (Fig. 3a and Fig. S2, ESI†). According to the literature,17,52 a reduction of these spikes indicates suppression of exciton recombination on the surface and in the bulk of a photoelectrode. These results indicate that the Cu2O photocathode with the HER CMs exhibited improved PEC performance in terms of both catalytic activity and stability.
Encouraged by these findings, we investigated the effect of the HER CMs on the stability of the Cu2O photocathode by monitoring the changes in current-density and morphology for a prolonged time. As shown in Fig. 3c, the Cu2O photocathode with 15 BL exhibited a more stable photocurrent density of about 0.2 mA cm−2 for more than 1 hour compared to the bare counterpart at an applied bias of 0.35 V vs. RHE. Ex situ SEM analysis showed that upon the PEC test, the roughness of the bare Cu2O photocathode was rapidly increased due to the formation of nanoparticles, which can be attributed to the self-reduction of Cu2O to Cu.31 In contrast, the Cu2O photocathode with the HER CMs had a negligible morphology change even after 1 hour of the PEC test. The improved stability of the Cu2O photocathode after the deposition of the HER CMs was also confirmed by ex situ XPS analysis (Fig. S3, ESI†). XPS spectra of the bare Cu2O showed a significant shift of the peaks corresponding to Cu 2p 1/2 and Cu 2p 3/2, implying considerable change in the binding energy of Cu due to the deactivation of Cu2O by self-redox reactions. On the contrary, there was negligible change in XPS spectra of the Cu2O with the HER CMs, demonstrating the improved stability of Cu2O by the HER CMs. Although we were able to significantly improve the stability of Cu2O photocathode by the deposition of HER CMs using a simple, green solution process, further improvements are required for practical application. Nevertheless, the improved stability is quite impressive compared to relevant studies by other groups, which reported the formation of inorganic passivation layers using vacuum deposition methods.23–28,30
As a control, we also prepared a Cu2O photocathode modified with Pt nanoparticles, a well-known HER catalyst, by the electrodeposition method (Cu2O–Pt) and compared its performance with that of the Cu2O photocathode with the HER CMs (Fig. S4, ESI†). Although the Cu2O–Pt photocathode showed a much higher initial photocurrent density than the Cu2O with the HER CMs, it quickly deactivated, as shown in Fig. S4a.† SEM images showed that its deactivation was caused by both the detachment of Pt nanoparticles and conversion of Cu2O microcrystals to inactive Cu nanoparticles through self-redox reactions, which was also found in the bare Cu2O photocathode (Fig. S4b and c†). These results show that the Cu2O photocathode with the HER CMs exhibits higher HER performance and stability than conventional Pt catalysts.
Electrochemical impedance spectroscopy (EIS) and Mott-Schottky (M-S) analysis were carried out to elucidate the underlying mechanism for the observed performance improvement for the Cu2O photocathode with the HER CMs. The impedance spectra were well-fitted using a 2-RC equivalent circuit model regardless of the presence of the HER CMs (Fig. 4a, b and Table S1, ESI†). There was a negligible difference between the Rs values, which represents the series resistance related to ionic conduction through an electrolyte and electronic conduction through an external circuit. One can expect that the deposition of HER CMs facilitates catalytic charge transfer at the photocathode–electrolyte interface, resulting in a significant decrease of catalytic charge transfer resistance (R2) in the Nyquist plot (Fig. 4a) and a shift of a low frequency peak for the catalytic reaction to a higher frequency in the Bode plot (Fig. 4b). Unexpectedly, there was also a considerable decrease of the R1 value, which is related to the transport of charge carriers from a bulk photocathode to catalysts. M-S analysis was carried out to study the origin of the improved charge transport by comparing the charge carrier density (NA), flat-band potential (EFB), and Helmholtz-layer capacitance (CH) of the Cu2O photocathode before and after the deposition of the HER CMs. They can be estimated using the following M-S equation:55,56

where C and Csc are the overall and space charge capacitance, ε and ε0 are the electrode and free space permittivity, e is the elementary electric charge, E is the applied bias, k is the Boltzmann constant, and T is the absolute temperature. The negative slope of the M-S plot suggests that Cu2O exhibited p-type semiconductivity. While there was a negligible effect on NA, the deposition of the CMs led a shift of the EFB of the photocathode from 0.50 to 0.39 V (Fig. 4c). It is thought that the interfacial dipoles formed by the deposition of cationic polyelectrolytes and anionic NiPOM affect the band-structure near the photocathode–electrolyte interface29,57 and facilitate charge transport to the catalysts. In addition, we observed a significant decrease of CH, which can be attributed to the burial of more conductive Cu2O after the deposition of the less conductive58 CMs. Taking together the results of EIS and M-S analysis, we can conclude that the deposition of HER CMs improved the catalytic activity of the underlying photocathode by facilitating charge transport/transfer processes as well as its stability by rapidly scavenging photogenerated electrons and reducing its exposure to the electrolyte.
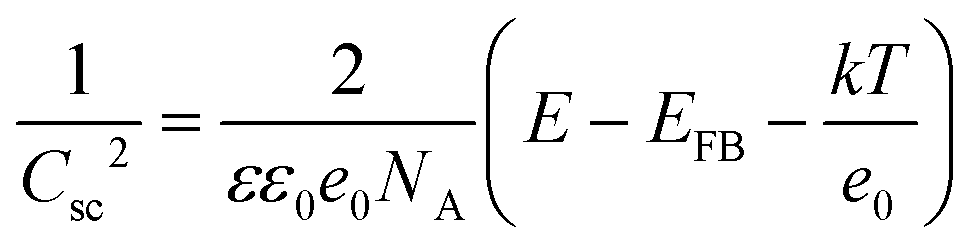
 | ||
| Fig. 4 Analysis of the underlying mechanism for the improved PEC performance of the Cu2O photocathode after the deposition of the 15 BL of the HER CMs. (a, b) EIS analysis was carried out under visible light illumination and represented in the form of (a) Nyquist and (b) Bode plots. (c) The charge carrier densities (NA), flat-band potentials (EFB), and Helmholtz-layer capacitance (CH) of the Cu2O photocathodes were estimated before and after the deposition of the HER CMs using M-S analysis. | ||
The improved charge separation kinetics after the deposition of the HER CMs was indirectly demonstrated by calculating the charge separation efficiency (∅sep) through comparison between the photocurrent densities in the presence and absence of Na2S2O8, an efficient electron scavenger.53,54 ∅sep was calculated using the following equation:

To build a bias-free PEC cell, we also prepared a photoanode for solar water oxidation by modifying the surface of nanoporous BiVO4 with (PEI/CoPOM)n multilayers (i.e., OER CMs). SEM and transmission electron microscopy (TEM) images showed that the OER CMs were uniformly and conformally coated even on a highly porous BiVO4 photoanode (Fig. 5a and b). The OER CMs were readily distinguished from the underlying BiVO4 photoanode by contrast difference in the TEM images. The morphological change of BiVO4 photoanodes after the modification was also investigated using high-resolution TEM (HRTEM), scanning TEM (STEM), and elemental mapping analyses. As shown in Fig. S6 (ESI†), the OER CMs were conformally and uniformly coated even on the surface of nanoporous BiVO4 photoanodes. Considering that it might be more difficult to uniformly coat nanoporous BiVO4 photoanodes than relatively flat Cu2O photocathodes with the corresponding CMs, it is thought that the HER CMs could also be readily deposited on the surface of Cu2O photocathodes. The gradual and linear growth of the OER CMs was confirmed again with UV/visible absorbance spectroscopy, QCM, ellipsometry, and XPS analysis (Fig. S7, ESI†). According to ellipsometry, the average thickness of each BL on the BiVO4 photoanode was 4.4 (±0.7) nm, much thicker than that on the Cu2O photocathode. Given that the structure and size of CoPOM are very similar to those of NiPOM, the observed difference in the average BL thickness is attributed to the morphological difference of BiVO4 and Cu2O photoelectrodes: 4.4 nm for nanoporous BiVO4vs. 1.8 nm for dense Cu2O.
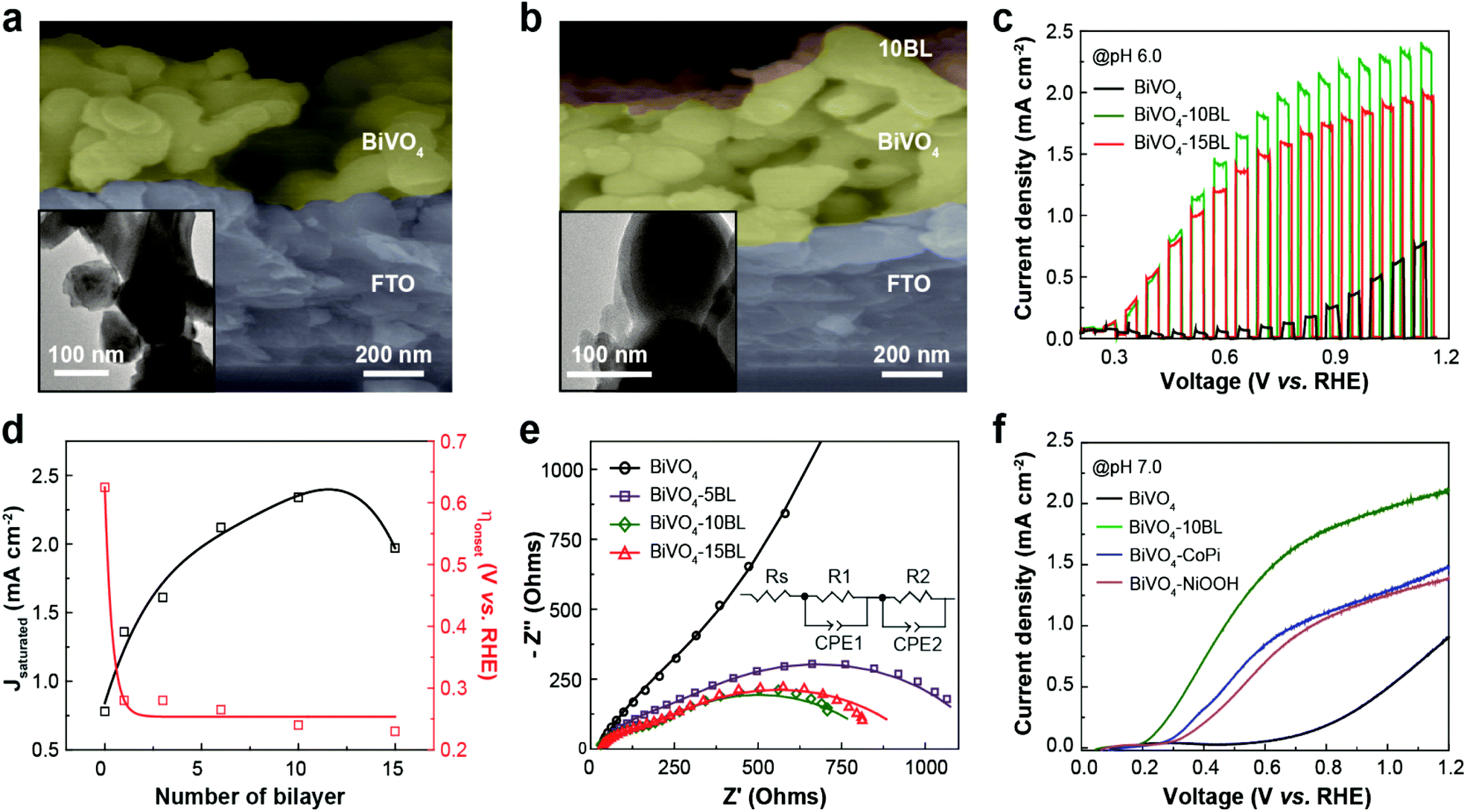 | ||
| Fig. 5 Formation of the OER CMs and their influence on the PEC performance of the BiVO4 photoanodes. (a, b) Cross-sectional SEM and (insets) TEM images of the BiVO4 photoanode (a) before and (b) after the deposition of 10 BL of the OER CMs. (c) LSV curves and (d) summarized results showing the effect of the OER CMs on the PEC performance of the BiVO4 photoanode. (e) EIS analysis was carried out to elucidate the mechanism underlying the performance improvement. (f) LSV curves showing the superior performance of the OER CMs on the BiVO4 photoanode compared to well-known OER catalysts, such as CoPi and NiOOH. | ||
The PEC performance of the BiVO4 photoanode for visible-light-driven water oxidation was measured under visible-light illumination in the presence and absence of OER CMs (Fig. 5c, d and Fig. S8, ESI†). Regardless of the presence of the CMs, back-side illumination (substrate–electrode side) resulted in a much higher photocurrent density than front-side illumination (electrolyte–electrode side) (Fig. S8, ESI†), due to the suppression of exciton recombination.59,60 Similar to the case of the Cu2O photocathode, the BiVO4 photoanode exhibited an excellent performance when the OER CMs were deposited. The PEC performance of the BiVO4 photoanode was highly dependent on the number of BL. The best performance was observed when 10BL of the OER CMs were deposited. After the deposition, the onset potential for solar water oxidation was shifted from 0.63 to 0.24 V vs. RHE, and the photocurrent density at 1.23 V vs. RHE was increased from 0.78 to 2.34 mA cm−2. It is noteworthy here that the observed cathodic shift in the onset potential of about 400 mV is one of the largest cathodic shifts for the BiVO4 photoanode. While the onset potential remained almost constant for the BiVO4 photoanode with the OER CMs, the photocurrent density rapidly increased up to 10 BL and decreased thereafter.
According to EIS analysis (Fig. 5e and Table S2†), the considerably improved performance of the BiVO4 photoanode after the deposition of the OER CMs resulted from (1) the improved charge transport to the interface and (2) enhanced catalytic activity, similar to the case of the Cu2O photocathode with the HER CMs. ∅sep for the BiVO4 photoanodes in the presence and absence of the OER CMs was calculated using a method similar to the case of the Cu2O photocathodes. Note that Na2SO3 was used as a hole scavenger for BiVO4. As shown in Fig. S9 (ESI†), ∅sep of the BiVO4 photoanode was significantly improved at both a low and high applied bias after the deposition of the OER CMs. The underlying mechanism for the improved ∅sep is unclear at this stage, requiring further studies using spectroscopic analysis such as transient absorption and photoluminescence spectroscopies. As a result, our OER CMs exhibited superior performance for solar water oxidation compared to well-known OER catalysts (Fig. 5f), such as cobalt phosphate (CoPi)43 and nickel oxyhydroxide (NiOOH).37 In this study we will focus on the fabrication of fully solution-processable PEC devices with the CMs and additional studies on the principles underlying the outstanding performance we observed will be reported soon.
Based on individual testing results of the Cu2O photocathode and BiVO4 photoanode with the respective CMs, we fabricated a bias-free PEC cell for overall solar water splitting (Fig. 6a). Before the modification with the respective CMs, there was no overlap between the independently measured LSV curves for both photoelectrodes (Fig. 6b). Note that the maximum operating current of a PEC cell can be estimated from this overlap.15 Thus, a PEC cell with bare Cu2O and BiVO4 would not work without any external bias even under light conditions. In contrast, there was a significant overlap of the LSV curves after the deposition of the respective CMs due to the significant shift of the onset potentials and huge increase of the photocurrent densities (Fig. 6b), suggesting the possibility of unassisted solar water splitting. Indeed, the PEC cell composed of the Cu2O photocathode and BiVO4 photoanode operated without any applied bias under continuous visible-light illumination when they were modified with the respective CMs (Fig. 6c). Although the measured current density was lower than the estimated value, it remained stable for at least 2 hours without noticeable degradation due to the improved stability of the Cu2O photocathode. The incident photon-to-current conversion efficiency (IPCE) of the respective photoelectrodes was measured and exhibited a wavelength-dependent response similar to their absorbance spectra (Fig. 6d). When the Cu2O photocathode was illuminated through the BiVO4 photoanode, the PEC cell exhibited a maximum IPCE of 1.46% in the near-UV and blue-light spectral region.
 | ||
| Fig. 6 A bias-free PEC cell composed of a Cu2O photocathode with HER CMs and a BiVO4 photoanode with OER CMs. (a) A photograph of the bias-free PEC cell tested in this study. (b) LSV curves of the Cu2O photocathodes and BiVO4 photoanodes measured with and without the respective CMs. (c) Operation of the PEC cells with and without the CMs in the absence of any external bias. (d) IPCE spectra showing the wavelength-dependent performance of the half- and full-cell devices. | ||
In the present study, we successfully fabricated a bias-free PEC cell for overall water splitting using only simple and environmentally friendly solution processes. Conventional bias-free PEC cells are fabricated using complex modification methods (e.g., vacuum deposition, complex synthetic processes, etc.) and expensive co-catalysts (Pt, IrOx, RuO2, etc.).22–28,30 We found that a simple LbL-based approach enabled us to readily fabricate an efficient bias-free PEC cell without the use of any toxic/hazardous chemicals or expensive elements. Although the stability of the PEC cell needs to be further improved for practical application, the present study provides insight and flexibility for the design and fabrication of PEC cells. First, we found that CMs composed of cationic polyelectrolytes and anionic POM-based electrocatalysts play multifunctional roles: (1) facilitating charge separation/transport in the semiconductor electrode by the dipole effect, (2) improving catalytic charge transfer efficiency at the electrode–electrolyte interface, and (3) enhancing the stability of the underlying electrode by rapidly scavenging photogenerated charge carriers and forming a protective coating layer. Second, in principle, the present LbL-based method can be applied to integrate various functional materials, such as light-harvesting components40–42 and charge transporting/separating materials.61 Lastly, the present method can be readily scaled-up for practical application, as all the processes are solution-compatible and do not require complex and expensive processes/conditions. For the reasons delineated above, we believe that the present study can also provide insight into the design and fabrication of novel energy storage and conversion devices.
Conclusions
In summary, we successfully fabricated an efficient and stable bias-free PEC cell for overall water splitting using the LbL method for the deposition of the respective CMs on a Cu2O photocathode and a BiVO4 photoanode in an environmentally friendly manner. The detailed PEC analysis revealed that the CMs have multifunctional roles in addition to simple catalyst loading effects. As a result, a Cu2O photocathode and a BiVO4 photoanode exhibited significantly improved PEC performance after modification with the respective CMs. In particular, the stability of the labile Cu2O photocathode was considerably enhanced. Based on these results, an efficient and stable bias-free PEC cell was readily prepared using only simple and environmentally friendly solution processes. We anticipate that the present study can provide not only a green alternative approach but also a novel platform for the design and fabrication of various electrochemical and PEC energy conversion devices that show excellent performance.Experimental
Materials
Unless stated otherwise, all chemicals were purchased from Sigma Aldrich (St Louis, MO, USA). L-(+)-Lactic acid was bought from Alfa Aesar (MA, USA). KI was purchased from Acros Organics (NJ, USA). The gold-coated quartz crystal disk was obtained from Stanford Research Systems (CA, USA).Deposition of photoelectrodes on a FTO
Cu2O photocathodes39 and BiVO4 photoanodes21 were fabricated by electrodeposition on a clean FTO substrate. A precursor solution for Cu2O was prepared by dissolving 0.4 M CuSO4·5H2O and 3 M lactic acid in deionized (DI) water. After the pH of the solution was adjusted to 13, electrodeposition was carried out at 30 °C and −0.4 V vs. Ag/AgCl for 20 minutes. A FTO coated with 100 nm-thick Pt was used as a counter electrode. A precursor solution for the deposition of BiOI was prepared by mixing 0.04 M Bi(NO3)3·5H2O solution in 0.4 M KI solution (50 ml) and 0.23 M p-benzoquinone solution (20 ml) dissolved in absolute ethanol. After adjusting the pH of the precursor solution to 1.7, electrodeposition of BiOI was conducted at −0.1 V vs. Ag/AgCl for 5 minutes. A 0.2 M vanadyl acetylacetonate solution in dimethyl sulfoxide was uniformly placed on the BiOI electrodes and then annealed in air at 450 °C for 2 hours at a ramping rate of 2 K min−1. The annealed electrodes were cooled to room temperature and soaked in a 1 M NaOH solution for 30 min and then rinsed with DI water to obtain BiVO4 by removing excess V2O5.Synthesis of POM water oxidation/reduction catalysts
CoPOM sodium salt was synthesized according to the literature.41 Next, 1.2 g of Co(NO3)2·6H2O and 0.6 g of Na2WO4·2H2O were dissolved in 0.5 M sodium acetate buffer (120 mL, pH 4.8). After vigorous mixing, 0.27 g of NaVO3 was added to the solution. After refluxing the solution for 2 hours at 80 °C, hot brown mixture was vacuum-filtered to remove precipitates. After storing the filtered solution in a refrigerator for 3 to 4 days, dark brown solid crystals were formed and extracted by filtration and vacuum-drying. NiPOM potassium salt was synthesized using a similar method.40 Then, 0.1 M Na2WO4·2H2O and 0.011 M Na2HPO4 were dissolved in 100 mL of DI water. After the pH of the solution was adjusted to 7.0 using a concentrated acetic acid, it was slowly mixed with an aqueous solution containing 0.022 M Ni(OOCCH3)4·H2O (50 mL). After 2.5 hours of refluxing, any precipitate was removed by vacuum filtration. Then 4 g of K(OOCCH3) was added to the filtered solution for precipitation, and NiPOM was obtained by vacuum filtration and overnight vacuum drying.Deposition of the CMs on a photoelectrode by LbL assembly
Aqueous solutions of anionic POM catalysts (1 mM) and cationic PEI (3 mM) in a phosphate buffered saline were prepared to deposit the CMs on the respective photoelectrode using the LBL method. For one cycle, the respective photoelectrode was dipped in the PEI and POM solutions for 5 minutes each. Washing with DI water was conducted 3 times for 30 seconds each between dipping processes. As a control, Pt nanoparticles were electrodeposited on the surface of Cu2O photocathode under the following condition: a precursor solution of 1 mM K2PtCl4 in deionized water, applied bias of −0.1 V vs. Ag/AgCl, deposition time of 15 min.Characterization
Morphology and elemental composition of photoelectrodes were characterized by an S-4800 SEM (Hitachi High-Technologies, Japan), a JEM-2100 TEM (JEOL, Japan), and K-alpha X-ray photoelectron spectroscopy (XPS) (Thermo Fisher Scientific, Massachusetts, USA). The deposition of the respective CMs was confirmed with analytical tools such as a V-730 UV-Visible spectrophotometer (JASCO, Japan), an EC-400 and M-2000 V ellipsometry (J.A. Woollam Co. Inc., USA), and a QCM200 quartz crystal microbalance (QCM) (Stanford Research Systems, Sunnyvale, CA, USA).PEC characterization
PEC performance was evaluated by linear sweep voltammetry (LSV) in the absence and presence of visible light irradiation with a WMPG1000 multichannel potentiostat/galvanostat (WonA Tech Co. Ltd, Korea) in a three-electrode configuration: a working electrode, photoelectrodes with and without LbL modification; a reference electrode, Ag/AgCl; a counter electrode, FTO coated with 100 nm-thick Pt; scan rate, 10 mV s−1. The PEC test was carried out in a 0.1 M phosphate buffer with 0.5 M Na2SO4 (pH 6.0) under visible light illumination with a 300 W Xe lamp equipped with a 400 nm cut-on filter (100 mW cm−2). Cu2O photocathode and BiVO4 photoanode were illuminated from the front and back sides, respectively. For a bias-free PEC cell, the Cu2O photocathode with the HER CMs was connected to and illuminated through the BiVO4 photoanode with the OER CMs without any external bias. Electrochemical impedance spectroscopy was carried out using a SP-150 (Bio-Logic Science Instruments, France). Cu2O photocathodes (or BiVO4 photoanodes) were analyzed under the following condition: applied bias of 0.35 (0.35) V vs. RHE, amplitude of 25 (20) mV, and frequency range from 0.1 (0.1) Hz to 100 (100) kHz. A M-S analysis of Cu2O photocathodes was performed at 1 kHz in the dark. All measurements were done at least three times for statistical analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program (2015R1C1A1A02037698 and 2018R1D1A1A02046918), the Nano-Material Technology Development Program (2017M3A7B4052802), and the Technology Development Program to Solve Climate Changes (2017M1A2A2087630) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT of Korea. This work was also supported by the 2018 Research Fund (1.180014.01) of UNIST (Ulsan National Institute of Science and Technology).Notes and references
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef.
- R. I. Pinhassi, D. Kallmann, G. Saper, H. Dotan, A. Linkov, A. Kay, V. Liveanu, G. Schuster, N. Adir and A. Rothschild, Nat. Commun., 2016, 7, 12552 CrossRef PubMed.
- P. Dias, T. Lopes, L. Meda, L. Andrade and A. Mendes, Phys. Chem. Chem. Phys., 2016, 18, 5232–5243 RSC.
- J. Ryu, S. H. Lee, D. H. Nam and C. B. Park, Adv. Mater., 2011, 23, 1883–1888 CrossRef PubMed.
- J. Ryu, D. H. Nam, S. H. Lee and C. B. Park, Chem. – Eur. J., 2014, 20, 12020–12025 CrossRef PubMed.
- R. Li, Chin. J. Catal., 2017, 38, 5–12 CrossRef.
- X. Li, J. Yu, S. Wageh, A. A. Al-Ghamdi and J. Xie, Small, 2016, 12, 6640–6696 CrossRef PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- R. Matheu, I. A. Moreno-Hernandez, X. Sala, H. B. Gray, B. S. Brunschwig, A. Llobet and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 11345–11348 CrossRef PubMed.
- M. Seol, J. W. Jang, S. Cho, J. S. Lee and K. Yong, Chem. Mater., 2013, 25, 184–189 CrossRef.
- R. Bin Wei, P. Y. Kuang, H. Cheng, Y. B. Chen, J. Y. Long, M. Y. Zhang and Z. Q. Liu, ACS Sustainable Chem. Eng., 2017, 5, 4249–4257 CrossRef.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 RSC.
- H. Dotan, N. Mathews, T. Hisatomi, M. Grätzel and A. Rothschild, J. Phys. Chem. Lett., 2014, 5, 3330–3334 CrossRef PubMed.
- P. Bornoz, F. F. Abdi, S. D. Tilley, B. Dam, R. Van De Krol, M. Graetzel and K. Sivula, J. Phys. Chem. C, 2014, 118, 16959–16966 CrossRef.
- J.-W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu, A. Javey, J. Guo and D. Wang, Nat. Commun., 2015, 6, 7447 CrossRef PubMed.
- F. F. Abdi and R. Van De Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CrossRef.
- W. He, R. Wang, L. Zhang, J. Zhu, X. Xiang and F. Li, J. Mater. Chem. A, 2015, 3, 17977–17982 RSC.
- B. Cheng, J. Yang, H. Cho and J. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20032–20039 CrossRef PubMed.
- M.-K. Son, L. Steier, M. Schreier, M. T. Mayer, J. Luo and M. Grätzel, Energy Environ. Sci., 2017, 10, 912–918 RSC.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef PubMed.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef PubMed.
- S. Li, P. Zhang, X. Song and L. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 18560–18565 CrossRef PubMed.
- J. Azevedo, S. D. Tilley, M. Schreier, M. Stefik, C. Sousa, J. P. Araújo, A. Mendes, M. Grätzel and M. T. Mayer, Nano Energy, 2016, 24, 10–16 CrossRef.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef PubMed.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Graetzel, Adv. Funct. Mater., 2014, 24, 303–311 CrossRef.
- C. G. Morales-Guio, S. D. Tilley, H. Vrubel, M. Graẗzel and X. Hu, Nat. Commun., 2014, 5, 3059 CrossRef PubMed.
- D. Jeon, H. Kim, C. Lee, Y. Han, M. Gu, B.-S. Kim and J. Ryu, ACS Appl. Mater. Interfaces, 2017, 9, 40151–40161 CrossRef PubMed.
- C. G. Morales-Guio, L. Liardet, M. T. Mayer, S. D. Tilley, M. Grätzel and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 664–667 Search PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef PubMed.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nano Lett., 2011, 11, 1928–1933 CrossRef PubMed.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, J. Mater. Chem. A, 2017, 5, 6825–6831 RSC.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, E. A. Santori, D. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, J. Am. Chem. Soc., 2011, 133, 1216–1219 CrossRef PubMed.
- I. Oh, J. Kye and S. Hwang, Nano Lett., 2012, 12, 298–302 CrossRef PubMed.
- B. D. Chernomordik, H. B. Russell, U. Cvelbar, J. B. Jasinski, V. Kumar, T. Deutsch and M. K. Sunkara, Nanotechnology, 2012, 23, 194009 CrossRef PubMed.
- T. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef PubMed.
- R. P. Antony, P. S. Bassi, F. F. Abdi, S. Y. Chiam, Y. Ren, J. Barber, J. S. C. Loo and L. H. Wong, Electrochim. Acta, 2016, 211, 173–182 CrossRef.
- J. Zhang, Z. Liu and Z. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 9684–9691 CrossRef PubMed.
- D. M. Guldi, F. Pellarini, M. Prato, C. Granito and L. Troisi, Nano Lett., 2002, 2, 965–968 CrossRef.
- D.-C. Lee, G. M. Morales, Y. Lee and L. Yu, Chem. Commun., 2006, 100–102 RSC.
- F. Nishiyama, T. Yokoyama, T. Kamikado, S. Yokoyama and S. Mashiko, Appl. Phys. Lett., 2006, 88, 253113 CrossRef.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef PubMed.
- C. M. McShane, W. P. Siripala and K. S. Choi, J. Phys. Chem. Lett., 2010, 1, 2666–2670 CrossRef.
- Y. Luo, G. Tan, G. Dong, H. Ren and A. Xia, Appl. Surf. Sci., 2016, 364, 156–165 CrossRef.
- S. Singh, R. Sharma and B. R. Mehta, Appl. Surf. Sci., 2017, 411, 321–330 CrossRef.
- S. G. Hosseini and S. Safshekan, Chin. J. Catal., 2017, 38, 710–716 CrossRef.
- C. Wu, Y. Fang, A. H. Tirusew, M. Xiang, Y. Huang and C. Chen, Chin. J. Catal., 2017, 38, 192–198 CrossRef.
- C. Regmi, Y. K. Kshetri, T.-H. Kim, R. P. Pandey, S. K. Ray and S. W. Lee, Appl. Surf. Sci., 2017, 413, 253–265 CrossRef.
- H. Lv, W. Guo, K. Wu, Z. Chen, J. Bacsa, D. G. Musaev, Y. V. Geletii, S. M. Lauinger, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015–14018 CrossRef PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu, C. L. Hill and G. Djamaladdin, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef PubMed.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CrossRef.
- Z. Xu, H. Wang, Y. Wen, W. Li, C. Sun, Y. He, Z. Shi, L. Pei, Y. Chen, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2018, 10, 3624–3633 CrossRef PubMed.
- R.-L. Lee, P. D. Tran, S. S. Pramana, S. Y. Chiam, Y. Ren, S. Meng, L. H. Wong and J. Barber, Catal. Sci. Technol., 2013, 3, 1694 RSC.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef PubMed.
- Y. Yang, D. Xu, Q. Wu and P. Diao, Sci. Rep., 2016, 6, 35158 CrossRef PubMed.
- H. Li, R. Liu, W. Kong, J. Liu, Y. Liu, L. Zhou, X. Zhang, S.-T. Lee and Z. Kang, Nanoscale, 2014, 6, 867–873 RSC.
- K. Lee, S. Lee, H. Cho, S. Jeong, W. D. Kim, S. Lee and D. C. Lee, J. Energy Chem., 2018, 27, 264–270 CrossRef.
- Y. Liang, T. Tsubota, L. P. A. Mooij and R. Van De Krol, J. Phys. Chem. C, 2011, 115, 17594–17598 CrossRef.
- L. Zhang, L. O. Herrmann and J. J. Baumberg, Sci. Rep., 2015, 5, 16660 CrossRef PubMed.
- C. L. Hsu, C. Te Lin, J. H. Huang, C. W. Chu, K. H. Wei and L. J. Li, ACS Nano, 2012, 6, 5031–5039 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc00681d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |