
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile, highly efficient and environmentally friendly transesterification mediated by platinum dioxide and nickel oxide under essentially neutral conditions†
Binhao
Teng
,
Jiangong
Shi
and
Chunsuo
Yao
 *
*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, P. R. China. E-mail: yaocs@imm.ac.cn
First published on 26th April 2018
Abstract
A practical, facile and highly efficient transesterification reaction under essentially neutral conditions was achieved using platinum dioxide (PtO2) or PtO2/nickel oxide (NiO) as the catalyst. A number of esters and alcohols that contain various functional groups were employed. Good to excellent yields were obtained for different aromatic or aliphatic starting materials. The Pt–alcohol intermediate generated in situ facilitated the exchange of low-alcohol esters to high-alcohol esters.
Transesterification has been widely used in organic synthesis and is important in synthesising various intermediates for the synthesis of fine chemical intermediates, complex natural products, paint additives and pheromones.1 It is also a fundamental method in the modification of compounds and the introduction of functional groups.
Traditional catalysts, including acid,2 alkali3,4 and organo-metallic ones,5 present serious shortcomings in transesterification. For example, the high corrosivity of acid catalysts leads to high requirements for equipment and operation processes. Corrosive acid catalysts are gradually being replaced by mild and environmentally friendly ones because of their long reaction time, numerous side reactions and serious environmental pollution. The strong alkalinity of alkali catalysts results in poor chemoselectivity and numerous by-products in the transesterification reaction, which severely limit the application of such catalysts in actual production. Organo-metallic catalysts are difficult to use due to their difficulty of separation after the reaction, strong toxicity to animals and high residue in the environment.
Although a few novel and practical transesterification reactions have been reported in the literature,6–17 highly efficient and environmentally friendly catalysts for transesterification still need to be developed to solve the shortcomings of existing ones, such as environmental pollution, separation difficulty and poor substrate applicability.
Platinum oxide, a safe-to-handle-and-store, eco-friendly and typically solid metallic oxide, has been widely utilised as a catalyst in catalytic hydrogenation. This work is the first to reveal that platinum oxide is a convenient, highly efficient and environmentally friendly catalyst in the transesterification reaction. The transesterification reaction mediated by platinum oxide under mild and neutral conditions effectively avoids the corrosivity of acid and alkali, the high toxicity of organo-metallic catalysts and the generation of side reactions. Furthermore, the products are easy to separate.
This work also shows that a mixture of nickel oxide (NiO)/platinum dioxide (PtO2) results in the same catalytic activity to the reaction as that of PtO2. In the presence of NiO, the catalytic amount of PtO2 is sufficient to mediate the reaction effectively, and the results are similar to those of PtO2.
In an ongoing synthetic route for a natural product, we attempted to reduce the olefinic bond of the benzofuran ring in 1 by hydrogenation in the presence of PtO2 with ethanol as a solvent (Scheme 1). However, instead of the olefinic bond reduction product, the reaction produced an unprecedented transesterification product 2. This finding encouraged us to investigate and clarify the unexpected transesterification reaction.
 | ||
| Scheme 1 Catalytic hydrogenation attempt of intermediate 1. | ||
In our subsequent study, methyl 3,5-dimethoxybenzoate 3 was selected as the substrate template to examine the transesterification reaction. Methyl benzoate 3 was efficiently converted into ethyl ester 4 in 99% yield at 60 °C in ethanol with PtO2 (up to 1 equiv.) as the catalyst (Scheme 2). Filtration and evaporation of the solvent were the only work-up after the reaction. These results prompted us to explore and optimise the transesterification conditions further.
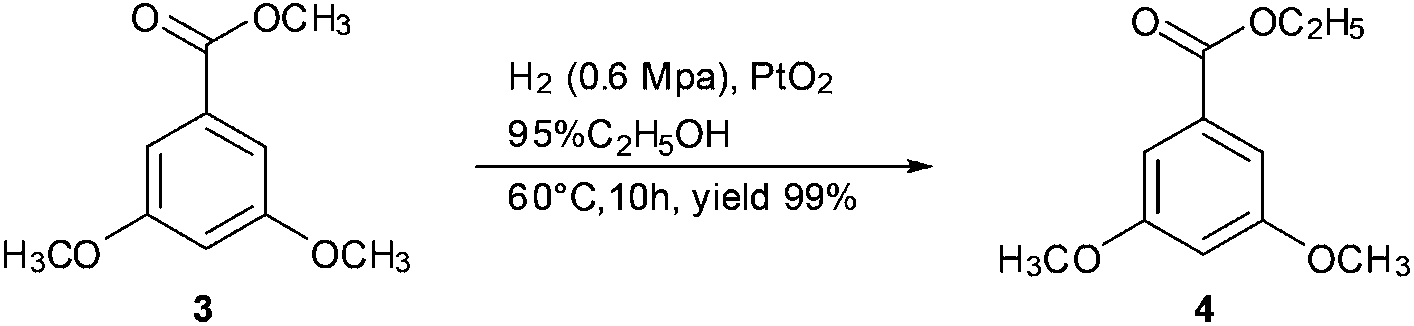 | ||
| Scheme 2 The template reaction of transesterification. | ||
Thereafter, a series of reaction conditions were screened, and the results are shown in Table 1. In the transesterification of 3, the reaction proceeded smoothly with 1.0 equiv. of PtO2 as the catalyst, whereas poor results were obtained when PtO2 was used at less than 1.0 equiv. (entry 1). An excellent yield was also obtained when 95% ethanol was replaced with anhydrous ethanol (entry 3). This result indicates that water was not required for the reaction process. The reaction proceeded smoothly, and an excellent yield was obtained when hydrogen pressure was reduced to 0.1–0.2 MPa from 0.6 MPa (entry 4). However, in the absence of hydrogen gas (entry 6), the transesterification reaction did not proceed, and the unchanged starting material was recovered quantitatively. These pieces of evidence suggest that hydrogen gas was indispensable for the reaction, and the reaction would proceed smoothly if the hydrogen pressure is not less than 0.1 MPa. Further experimentation revealed that temperature was important to the reaction rate (entry 5), and room temperature was sufficient for the reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (equiv.) | H2 pressure (MPa) | Additive | Time | Yieldb |
| a Reaction conditions: Methyl 3,5-dimethoxybenzoate (30 mg), ethanol (10 ml), temperature 60 °C. b Isolated yields. c Temperature 25 °C. d No reaction. e PtO2 reacts with H2 in EtOH for 2 h, and then H2 was replaced with N2 and methyl 3,5-dimethoxybenzoate was added. f 50 wt%. | |||||
| 1 | PtO2(0.43) | 0.6 | 5% H2O | 6 h | 46% |
| 2 | PtO2(1.00) | 0.6 | 5% H2O | 6 h | 99% |
| 3 | PtO2(1.00) | 0.6 | — | 6 h | 99% |
| 4 | PtO2(1.00) | 0.1–0.2 | — | 6 h | 99% |
| 5c | PtO2(1.00) | 0.6 | — | 72 h | 88% |
| 6 | PtO2(1.00) | — | — | 36 h | N.R.d |
| 7 | — | 0.1–0.2 | — | 36 h | N.R. |
| 8 | PtO2(1.00) | 0.1–0.2e | — | 6 h | 99% |
| 9 | Pt/C(1.00) | 0.1–0.2 | — | 36 h | N.R. |
| 10 | Pd/C(1.00)f | 0.1–0.2 | — | 36 h | N.R. |
| 11 | Pt(1.00) | 0.1–0.2 | — | 36 h | N.R. |
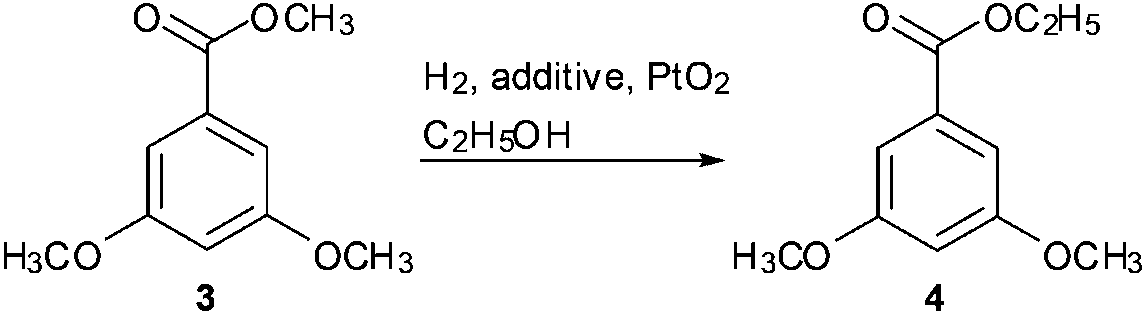
In the absence of catalyst PtO2, the production of 4 was not detectable (entry 7). To ascertain the role of hydrogen and PtO2, another experiment was designed and conducted. Initially, PtO2 reacted with hydrogen in ethanol for 2 h, and then hydrogen was replaced with nitrogen and compound 3 was added (entry 8). The reaction proceeded completely, uncovering that the role of hydrogen was only to reduce PtO2, and not to affect the process of transesterification. Pt(0) and Pt/C were tested as catalysts (entries 9 and 11) in the reaction to determine if Pt(0) is actually the effective catalyst. However, the transesterification product was not detected, indicating that intermediate valence Pt could be an effective catalyst in this reaction. In addition, another transition-metal catalyst (Pd) was also examined, but it failed to provide detectable product 4 (entry 10).
Under the optimum reaction conditions, the versatility of the transesterification of model methyl benzoate 3 with a series of alcohols (ethanol, 1-butanol, 1-octanol, benzyl alcohol, isopropanol, t-butanol and phenol) was explored further. The results shown in Table 2 are generally good to excellent. When chain or benzyl alcohols were used (entries 1–4) in the reaction, the corresponding esters were obtained in excellent yields. However, when hindered alcohols or phenols (secondary, tertiary alcohol or phenol, entries 5–7) were employed, poor results or no products were observed. These experiments suggest that sterically hindered alcohols lead to low esterification rates for the reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Alcohol | Time | Product (R) | Yieldb |
| a Reaction conditions: Methyl 3,5-dimethoxybenzoate (30 mg), catalyst PtO2 (1.0 equiv.), alcohol (10 ml), H2 pressure (0.1–0.2 MPa), temperature 60 °C. b Isolated yields. c No reaction. | ||||
| 1 | C2H5OH | 6 h | C2H5 | 99% |
| 2 | n-C4H9OH | 15 h | C4H9 | 97% |
| 3 | n-C8H17OH | 15 h | C8H17 | 97% |
| 4 | C6H5CH2OH | 15 h | CH2C6H5 | 99% |
| 5 | (CH3)2CHOH | 36 h | CH(CH3)2 | 56% |
| 6 | (CH3)3COH | 36 h | — | N.R.c |
| 7 | C6H5OH | 40 h | — | N.R. |

These results enabled us to further investigate this obviously different transesterification caused by the alcohol with a different steric hindrance. Methyl ester 3 was treated with a mixture of ethanol and 1-butanol and a mixture of ethanol and isopropanol under the present conditions. The results are shown in Scheme 3. Transesterification products were obtained in a higher yield with lower alcohols and alcohols with smaller hindrance than the corresponding alcohol. This suggested that reactivity was related to the steric hindrance of alcohols, and the alcohols with small steric hindrance had strong reactivity. However, the results in Table 2 show that low alcohols (the alcohol with less carbons) can be transesterificated by high alcohols, which indicates that high alcohols (the alcohol with more carbons) have stronger reactivity than low alcohols. According to this conclusion, high-alcohol esters should be the major products in the experiment of Scheme 3. However, the obtained results were incompatible.
 | ||
| Scheme 3 Chemoselective transesterification of methyl benzoate 3 with a mixture of different alcohols. | ||
On the basis of these discussions, a plausible mechanistic pathway for the transesterification process is proposed and depicted in Scheme 4. Supported by the previous literature on Pd18–21 and other transition-metal catalysts,22–25 we divided the course of the transesterification reaction into two stages, namely, intermediate generation and intermediate reaction. In the first stage, PtO2 was initially reduced to Pt(II) in the presence of hydrogen. Simultaneously, Pt(II) was inserted into the H–OR bond of the alcohol through an annular transition state to produce Pt–alcohol intermediate a. In the second stage, intermediate a reacted with ester b (ester of low alcohols) to produce an unstable intermediate c through another annular transition state, which eliminated an HPt–OR’ molecule to generate transesterification product d following the generation of Pt(0) through the reductive elimination of Pt–alcohol e.
 | ||
| Scheme 4 Plausible mechanism of transesterification catalyzed by PtO2. | ||
On the basis of the possible mechanism mentioned above and the experimental results shown in Scheme 3, we speculated that the Pt–alcohol intermediate formed from low alcohols was more stable than those formed from high alcohols. Therefore, in the intermediate generation stage (first stage), low alcohols showed stronger reactivity than high alcohols, resulting in the production of a low-alcohol intermediate. In the intermediate reaction stage (second stage), the reaction rate decreased significantly when an alcohol was replaced with a sterically hindered one. Moreover, the high-alcohol intermediate could be replaced with a low one because the low alcohol–Pt intermediate was more stable than the high alcohol one, and the low alcohol is easy to leave to facilitate the reaction process. To substantiate this hypothesis, three high-alcohol esters were treated with PtO2 and ethanol under similar conditions, and the predicted results were obtained. Table 3 shows that the transesterification reaction of the high-alcohol esters to the low-alcohol esters was not observed.

Several experiments were conducted to verify the assumption that low-alcohol esters can be exchanged to high-alcohol esters. When the ethyl esters were treated with n-butanol as a solvent under the present conditions, the desired products were obtained in excellent yields (Table 4). These results show that besides methyl ester, other low-alcohol esters can also be transformed into high-alcohol esters with high yields.

These results prompted us to conceive that the formation of different Pt–alcohol intermediates is a reversible reaction, and this conception was validated by the subsequent control experiments (Scheme 5). Firstly, PtO2 was treated with excess C4H9OH under the transesterification conditions for 3 h. Second, the same amount of ethanol was added and stirred for another 4 h. Lastly, methyl ester 3 was added. If the generation of the Pt–alcohol intermediate is an irreversible reaction, then the major transesterification product would be butyl ester in the reaction. However, the result showed that the reaction product was a mixture of ethyl and butyl esters. Similarly, before the addition of C4H9OH, treatment of PtO2 with excess ethanol produced a similar result (Scheme 5). These results suggest that the formation of different Pt–alcohol intermediates was a reversible reaction.
 | ||
| Scheme 5 Reversible reactions of different Pt–alcohol intermediates. | ||
The esterification reaction of carboxylic acid with ethanol under similar conditions was conducted to understand the transesterification procedure, and the results are summarised in Table 5. Several different acids were examined in our experiments, but no desired esterification products were detected. These results suggest that the mechanism of this transesterification reaction was irrelevant to acids other than that of the well-known esterification catalysed by acid.

Under the optimised reaction conditions, the effect of a series of diversely substituent methyl esters on the transesterification reaction was explored. Table 6 shows that all the methyl esters that bear various substituted groups, including electron-donating groups (Me, MeO, N(Me)2) and electron-withdrawing groups (F, CF3) on the benzene ring, showed high reactivity in the transesterification reaction under optimal reaction conditions. Aside from benzoate esters, other aromatic acid esters, such as 2-furoate ester and 2-naphthoate ester, also showed high reactivity in the reaction. The effects of different substituent sites at the phenyl ring of the benzoate esters were also explored (Table 6). Notably, neither electron-donating nor electron-withdrawing substituents at different sites of the phenyl ring exerted a significant effect on the transesterification reaction. All of the transesterification products were obtained in excellent yields.
|
|
|||
|---|---|---|---|
| Entry | R | Time | Yieldb |
| a Reaction conditions: Methyl ester (1.0 equiv.), catalyst PtO2 (35 mg, 1.0 equiv.), ethanol (10 ml), H2 pressure (0.1–0.2 MPa), temperature 60 °C. b Isolated yield. c The yields were determined by 1H NMR spectroscopy. d In this entry, extra PtO2 (2 equiv.) was added in 13 h and 27 h, respectively. e Few fluorine was removed (33%). f The olefinic bond was reduced. | |||
| 1 | o-CH3OC6H4 | 16 h | 97% |
| 2 | m-CH3OC6H4 | 16 h | 98% |
| 3 | p-CH3OC6H4 | 16 h | 98% |
| 4 | 3,5-(OCH3)2C6H3 | 6 h | 99% |
| 5 | C6H5 | 6 h | 99%c |
| 6 | 3,4-(OCH3)2C6H3 | 14 h | 99% |
| 7 | p-(CH3)2NC6H4 | 40 h | 88%d |
| 8 | p-CH3C6H4 | 14 h | 98% |
| 9 | 3,5-(CH3)2C6H3 | 14 h | 97% |
| 10 | p-CH3OC6H4CH2 | 14 h | 99% |
| 11 | p-FC6H4 | 6 h | 99%c |
| 12 | m-FC6H4 | 6 h | 96%e |
| 13 | p-CF3C6H4 | 16 h | 99%c |
| 14 | 2-Furyl | 6 h | 98%c |
| 15 | 2-Naphthyl | 13 h | 99% |
| 16 |
p-CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC6H4 CHC6H4 |
13 h | 97%f |
Our study also revealed that the reaction substrates were not restricted to aromatic esters. Table 6 shows that aliphatic esters also produced the corresponding products in high yields (Table 6, entry 10). In the case of transesterification of the electron-rich amino-substituted acid (Table 6, entry 7), the reaction rate was comparatively sluggish in the presence of 1.0 equiv. of PtO2 (46.6% yield). This phenomenon could be due to the competing reaction of the amino group and Pt–alcohol intermediate at the reaction site of an ester. In light of this hypothesis, extra PtO2 (2.0 equiv.) was added to the reaction mixture to provide an additional Pt–alcohol intermediate to compete with the amino group at the reaction site. As a result, an ester exchange product was obtained in high yield (88%), as expected. The results shown in Table 6 reveal that the reaction can be widely used for substrates with different structures.
In the course of the ester exchange reaction, the only role of hydrogen was to reduce PtO2, and it did not participate in the transesterification process, as shown in Table 1, entry 8. This result suggests that the reaction condition was suitable for ester containing reductive groups, such as an olefinic bond, nitro group and aldehyde group. For these esters, transesterification could be performed successfully whilst the reductive group is unchanged.
As shown in Table 6, entry 16, PtO2 initially reacted with hydrogen in ethanol for 2 h. Then, hydrogen was replaced with nitrogen, and the substrate methyl ester was added (Scheme 6). The desired product 7 with a double bond was obtained in excellent yield. By treating the corresponding substrates in the same manner, the transesterification products of nitro compound 8 and aldehyde compound 9 could be synthesized easily (Scheme 6).
 | ||
| Scheme 6 Transesterification of esters with reductive groups. | ||
Five methyl benzoates of different substituents (Table 6; entries 3, 5, 8, 10 and 11) were treated with ethanol under the same conditions to understand the effect of substituents on the reaction rate (Fig. 1). These results revealed that aromatic esters with electron-withdrawing groups, aromatic esters without substituents on the benzene ring and aliphatic esters proceeded completely in less than 1 h (TOF > 1 h−1) in the ester exchange reaction. In the case of transesterification of methyl esters with electron-donating groups, the reaction rate was comparatively sluggish (TOF: 0.689 h−1). This result is due to the reduction of the positive electricity of carbonyl carbon caused by the electron-donating groups, which leads to the reduced reactivity of the Pt–alcohol intermediate during transesterification. Moreover, when the substituent group was replaced with electron-rich amino group (TOF: 0.272 h−1), the transesterification reaction proceeded incompletely because of the competing reaction of the amino group with the Pt–alcohol intermediate in the ester exchange reaction. For transesterification products, a good yield can also be obtained after the addition of extra PtO2 in the reaction.
 | ||
| Fig. 1 Rates of transesterification reaction. | ||
Fig. 1 shows that the reaction rate was obviously slow when the substituent group exhibited an electron-donating effect during the transesterification of methyl benzoate of different substituents on the benzene ring with ethanol. These pieces of evidence indicate that the stage of intermediate generation (first stage) is rapid, and the stage of intermediate reaction (second stage) is the rate-determining step.
Transesterification catalysed by PtO2 and hydrogen is a convenient, highly effective and eco-friendly reaction. However, the exorbitant price and unrecyclability of PtO2 are the obvious drawbacks of this reaction. Thus, exploring a cheap succedaneum of PtO2 is important. As a congener of noble platinum, nickel is a naturally abundant and cheap element with a low CO2 footprint, and it possesses a unique capability to produce novel or challenging transformations.26 Thus, the mixture of NiO/PtO2 and a hydrogen catalytic system was attempted in the transesterification reaction for atomic economy. To our satisfaction, this system showed excellent catalytic activity to the reaction (Table 7, entry 3). NiO was an effective additive, but it could not catalyse the reaction independently in the absence of PtO2, as shown in Table 7. Moreover, in the presence of NiO, the catalytic amount of PtO2 (largest TON: 31) was sufficient to mediate the reaction effectively, and all the results are similar to those of PtO2. Moreover, screening of the reaction conditions showed that the amount of PtO2 controlled only the reaction rate but not the product yield (Table 7; entries 3, 4 and 6). The temperature controlled the reaction rate and the product yield. For this reaction, 0.1 equiv. of PtO2 and a temperature of 93 °C–97 °C were suitable conditions.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (equiv.) | H2 pressure (MPa) | Additive (equiv.) | Temp. | Time | Yieldb |
| a Reaction conditions: Methyl 3,5-dimethoxybenzoate (30 mg: 0.000153 mol), ethanol (10 ml). b Isolated yields. c No reaction. | ||||||
| 1 | — | 0.1–0.2 | PtO2(0.10) | 60 °C | 18 h | N.R.c |
| 2 | NiO(1.00) | 0.1–0.2 | — | 60 °C | 72 h | N.R. |
| 3 | NiO(1.00) | 0.1–0.2 | PtO2(0.10) | 60 °C | 15 h | 39% |
| 4 | NiO(1.00) | — | PtO2(0.10) | 60 °C | 18 h | N.R. |
| 5 | NiO(1.00) | 0.1–0.2 | PtO2(0.20) | 60 °C | 20 h | 40% |
| 6 | NiO(1.00) | 0.1–0.2 | Pt (0.20) | 60 °C | 72 h | N.R. |
| 7 | NiO(1.00) | 0.6 | PtO2(0.10) | 75 °C | 72 h | 83% |
| 8 | NiO(1.50) | 0.6 | PtO2(0.05) | 75 °C | 15 h | 80% |
| 9 | NiO(1.20) | 0.7–0.8 | PtO2(0.10) | 95 °C | 15 h | 99% |
| 10 | NiO(1.20) | 0.7–0.8 | PtO2(0.03) | 95 °C | 59 h | 92% |

Furthermore, a possible mechanism of the catalytic system (PtO2/NiO) was proposed as follows. As shown in Scheme 7, it can be speculated that the alkoxys of the different metal–alcohol intermediates may be exchanged with each other in the presence of hydrogen. Therefore, the alkoxy of Ni–alcohol intermediate f exchanged with that of Pt–alcohol intermediate e to regenerate new Pt–alcohol intermediate a, which enables the transesterification reaction to proceed in succession. In this reaction, the role of NiO is the regeneration of the Pt–alcohol intermediate, which was initially reduced to Ni(II) in the presence of hydrogen, and then inserted into the H–OR bond of the alcohol to produce Ni–alcohol intermediate f.
 | ||
| Scheme 7 Plausible mechanism for the mixture catalytic system PtO2/NiO/hydrogen. | ||
The present reaction conditions were utilised to evaluate the transesterification on preparative scales. Experiments were performed with 3 (1.00 g, 8.0 mmol), NiO (1.00 equiv.) and PtO2 (0.10 equiv.) at 95 °C under 0.7–0.8 MPa of hydrogen pressure (Scheme 5). At the conclusion of the reactions, the reaction mixture was filtered, and the filtrate was evaporated in vacuum to produce product 4 with 96% yield. This result indicates that the reaction has a wide applicability.
With the optimum reaction conditions established, the application of various substituents on the benzene ring and alcohol was evaluated for this reaction (Table 8). The mixed catalytic system had a similar catalytic activity to that of PtO2, but the amount of precious metal oxide PtO2 was reduced to a catalytic amount, which is highly suitable for atom economy. Compared with the traditional method, this approach is more eco-friendly and has a higher yield. It is suitable for the production of various types of fine chemical intermediates, complex natural products, paint additives and pheromones. The results of this work are not only theoretically important but also possess a potential application value in industrial chemistry.
|
|
||||
|---|---|---|---|---|
| Entry | R | Alcohol | Time | Yieldb |
| a Reaction conditions: Methyl ester (1.0 equiv.), catalyst PtO2 (3.5 mg, 0.1 equiv.), NiO (1.2 equiv.), alcohol (10 ml), H2 pressure (0.7–0.8 MPa), temperature 95 °C. b Isolated yield. | ||||
| 1 | 3,5-(OCH3)2C6H3 | Ethanol | 15 h | 99% |
| 2 | 3,5-(CH3)2C6H3 | Ethanol | 23 h | 99% |
| 3 | p-CH3OC6H4 | Ethanol | 31 h | 97% |
| 4 | p-CF3C6H4 | Ethanol | 15 h | 93% |
| 5 | p-CH3OC6H4CH2 | Ethanol | 15 h | 98% |
| 6 | 3,5-(OCH3)2C6H3 | Butanol | 18 h | 99% |
| 7 | 3,5-(OCH3)2C6H3 | Phenylcarbinol | 40 h | 39% |

Conclusions
In summary, we developed a practical, facile and highly efficient method for transesterification using PtO2 or NiO/PtO2 as the catalyst under neutral conditions in the presence of hydrogen. The conversion product was obtained in small- to gram-scale synthesis without the need for inconvenient work-up or complicated purification procedures. A number of esters and alcohols that bear various functional groups were employed, and good to excellent yields were obtained for different aromatic or aliphatic starting materials (up to 99% in most cases). The success of this transesterification relied on the in situ generation of Pt–alcohol intermediates from Pt(II) and alcohols, which facilitated the exchange of low-alcohol esters to high-alcohol esters. The catalyst PtO2 exhibited significant activity, and only the catalytic amount of PtO2 was required in the presence of NiO for the reaction. Overall, this method is convenient, chemoselective and environmentally friendly, and it can be employed in the synthesis of active natural products and industrially relevant processes, such as synthesis of drugs and production of chemical products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Key Project of National Natural Science Funds of China (NNSFC 81630094) and the CAMS Innovation Fund for Medical Sciences (CIFMS 2016-I2M-2-002). The authors are grateful to the department of Instrumental Analysis, Institute of Materia Medica, Chinese Academy of Medical Sciences, and Peking Union Medical College for measuring the NMR and HRMS spectra.Notes and references
- A. Solhy, J. H. Clark, R. Tahir, S. Sebti and M. Larzek, Green Chem., 2006, 8, 871 RSC.
- J. Otera, Chem. Rev., 1993, 93, 1449 CrossRef CAS.
- M. Blumel, J. M. Noy, D. Enders, M. H. Stenzel and T. V. Nguyen, Org. Lett., 2016, 18, 2208 CrossRef CAS PubMed.
- O. R. Suárez-Castillo, L. A. Montiel-Ortega, M. J. Fragoso-Vázquez, M. Meléndez-Rodríguez and M. Sánchez-Zavala, Tetrahedron Lett., 2008, 49, 996 CrossRef.
- J. Xiang, A. Orita and J. Otera, Adv. Synth. Catal., 2002, 344, 84 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664 CrossRef CAS PubMed.
- G. A. Grasa, T. Güveli, R. Singh and S. P. Nolan, J. Org. Chem., 2003, 68, 2812 CrossRef CAS PubMed.
- M. Hatano, Y. Furuya, T. Shimmura, K. Moriyama, S. Kamiya, T. Maki and K. Ishihara, Org. Lett., 2010, 13, 426 CrossRef PubMed.
- M. Hatano and K. Ishihara, Chem. Commun., 2013, 49, 1983 RSC.
- K. Ishihara, M. Niwa and Y. Kosugi, Org. Lett., 2008, 10, 2187 CrossRef CAS PubMed.
- Y. Maegawa, T. Ohshima, Y. Hayashi, K. Agura, T. Iwasaki and K. Mashima, ACS Catal., 2011, 1, 1178 CrossRef CAS.
- G. W. Nyce, J. A. Lamboy, E. F. Connor, R. M. Waymouth and J. L. Hedrick, Org. Lett., 2002, 4, 3587 CrossRef CAS PubMed.
- Y. Ogura, T. Terashima and M. Sawamoto, Polym. Chem., 2017, 8, 2299 RSC.
- M. Selva, S. Guidi and M. Noè, Green Chem., 2015, 17, 1008 RSC.
- K. P. Dhake, P. J. Tambade, Z. S. Qureshi, R. S. Singhal and B. M. Bhanage, ACS Catal., 2011, 1, 316 CrossRef CAS.
- Z. Song, X. Jin, Y. Hu, B. Subramaniam and R. V. Chaudhari, ACS Sustainable Chem. Eng., 2017, 5, 4718 CrossRef CAS.
- L. Xu, Y. Wang, X. Yang, X. Yu, Y. Guo and J. H. Clark, Green Chem., 2008, 10, 746 RSC.
- T. Kitanosono, M. Miyo and K. Shu, ACS Sustainable Chem. Eng., 2016, 4, 6101 CrossRef CAS.
- A. Kumar, G. K. Rao and A. K. Singh, RSC Adv., 2012, 2, 12552 RSC.
- M. Mao, L. Zhang, Y. Z. Chen, J. Zhu and L. Wu, ACS Catal., 2016, 7, 181 CrossRef.
- A. D. Zotto and D. Zuccaccia, Catal. Sci. Technol., 2017, 7, 3934 Search PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC.
- J. Schranck and A. Tlili, ACS Catal., 2018, 8, 405 CrossRef CAS.
- J. R. Webb, S. A. Burgess, T. R. Cundari and T. B. Gunnoe, Dalton Trans., 2014, 45, 16646 Search PubMed.
- N. A. Weires, D. D. Caspi and N. K. Garg, ACS Catal., 2017, 7, 4381 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data (PDF). See DOI: 10.1039/c8gc00500a |
| This journal is © The Royal Society of Chemistry 2018 |