
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular tools for selective recovery and detection of lignin-derived molecules†
Milla
Salmela
 *a,
Hanna
Sanmark
b,
Elena
Efimova
a,
Alexander
Efimov
a,
Vesa P.
Hytönen
cd,
Urpo
Lamminmäki
b,
Suvi
Santala
a and
Ville
Santala
a
*a,
Hanna
Sanmark
b,
Elena
Efimova
a,
Alexander
Efimov
a,
Vesa P.
Hytönen
cd,
Urpo
Lamminmäki
b,
Suvi
Santala
a and
Ville
Santala
a
aLaboratory of Chemistry and Bioengineering, Tampere University of Technology, Korkeakoulunkatu 8, 33720 Tampere, Finland. E-mail: milla.salmela@tut.fi
bDepartment of Biochemistry/Biotechnology, University of Turku, Tykistökatu 6 A, 6th flr., 20520 Turku, Finland
cFaculty of Medicine and Life Sciences and BioMediTech, University of Tampere, Lääkärinkatu 1, 33520 Tampere, Finland
dFimlab Laboratories, Biokatu 4, 33520 Tampere, Finland
First published on 8th May 2018
Abstract
The pulp and paper industry together with lignocellulosic biofuel production provides plentiful streams of lignin and lignin-derived molecules (LDMs) that currently remain underutilized. The heterogeneity and complexity of lignin along with the lack of convenient tools significantly hamper its utilization. Selective separation of these LDMs from streams using specific tools would allow the recovery of aromatic compounds, as well as facilitate biological processes aiming at lignin valorization. To this end, here we report the isolation and characterization of single-chain variable fragment (scFv) antibodies against ferulate, coumarate, and caffeate, which are the molecular representatives of LDMs. Binders for the target LDMs were enriched by interrogating a synthetic scFv library with the phage display technique. As a result, scFv binders specific against each of the target molecules were obtained with affinities in the micromolar range. The selectivity of scFvs towards specific LDMs was proved by recovering caffeate from simulated LDM solution, Kraft lignin, and rice straw hydrolysate samples. Further proof of concept studies with model compounds demonstrated the applicability of antibody-based binders as a detection tool for monitoring microbial LDM conversion. Overall, this study demonstrates the potential of scFv binders as a specific toolset for lignin compound recovery and analysis.
Introduction
Currently, the chemical industry depends strongly on crude oil refinement processes designed for transportation fuels and energy production.1,2 There are, however, concerns over the economic and environmental sustainability of these industries3–5 as well as on the global sufficiency of oil deposits.6 These concerns motivate us to decrease our reliance on fossil fuels, and therefore modern refineries require new raw materials independent of the oil industry.7 For these purposes, lignin is considered a promising feedstock for a plethora of chemicals and materials. Derived from sources such as the pulp and paper industry, as well as from the emerging industry of lignocellulosic biofuel production, lignin can provide an environmentally sustainable and abundant alternative for the oil-based chemical industry.8 Furthermore, lignin valorization enables the production of both fine and bulk chemicals in a biorefinery concept.9–12 However, commercial applications and industrial scale production have been hindered due to the recalcitrant and heterogeneous nature of lignin13 and the present industrial processes discard the lignin streams mainly as a waste or combust them for heat.Lignin is an aromatic heteropolymer found embedded with cellulose and hemicellulose in lignocellulosic biomass. Plants synthesize lignin primarily from 4-hydroxyphenylpropanoids via oxidative coupling, which results in different phenylpropane structures in the polymer.14 Consequently, complex mixtures of high and low molecular weight aromatic structures, among other compounds, can be found in the lignin streams obtained from biomass pre-treatment processes. In addition, the governing aromatic structures of these technical lignins depend notably on the origin of the biomass and on the chosen treatment method.15 For instance, ferulate and coumarate are relatively dominant aromatic monomers in lignin fractions acquired from particular agricultural and grassy origins15–19 and in some cases trace amounts of caffeate have also been detected.20
These aromatic compounds are examples of industrially interesting molecules. They possess antioxidant properties, which make them valuable molecules for example for the food industry.21 In addition, in the chemical industry, the use of aromatic monomers from sustainable sources is desirable, though selective catalytic modification of these compounds is hindered due to the difficult lignin matrix these compounds reside in.22 Biological systems, however, are known to be highly specific and work in dilute solutions. Selective binding to these molecules, for example, would enable the isolation of specific aromatic compounds from a mixture of different LDMs, which might prove useful for the recovery of small molecules from complex liquors,23 dilute streams or from bioconversion processes accumulating specific aromatic compounds.24
In nature, certain bacteria have the ability to perform selective conversion of aromatic compounds from heterogeneous materials such as lignin.25,26 Bacteria, such as Acinetobacter baylyi ADP1,27Rhodococcus jostii,28 and Pseudomonas putida19,20,29 can, for example, utilize ferulate, coumarate, and caffeate as the sole carbon and energy sources. Moreover, metabolic engineering and synthetic biology approaches provide tools to convert these substrates into valuable products such as vanillin or polyhydroxy carbonates (PHA).26,27,30 An extended application of LDM selective binders could be the specific and rapid detection of LDMs from culture broths for the optimization of bioconversion processes involving lignin.
Antibodies are proteins that can bind specifically to target analytes even in complex matrices. They are commonly used in diagnostic and therapeutic applications and to some degree in environmental analyses.31–33 Single-chain variable fragment (scFv) molecules are modern antibodies engineered to contain only the smallest immunoglobulin units required for antigen recognition.34 The scFvs can be produced in simple bacterial expression systems such as Escherichia coli, and they can easily be further genetically modified, for example, to endow them with common labelling proteins such as green fluorescent protein (GFP) or alkaline phosphatase (AP). The tools of genetic engineering can be used to generate highly diverse synthetic repertoires of scFvs, from where binders against targets of interest can be isolated by techniques such as the phage display technique. Antigen-binding site architectures of the antibodies in these libraries can be designed to favor the recognition of certain types of antigens differing for instance in terms of size.35 With a suitable antibody library, specific binders can be obtained even for very small haptens, such as skatole (MW = 131.2 Da),36 which typically are very difficult targets for antibody development. Thus, scFv binders for small molecules that are relevant for the lignin industry, such as hydroxycinnamates, might also be found from synthetic antibody libraries, even though they differ only in the degree of methoxylation and the number of hydroxyl groups.
In the present study, antibodies capable of specific recognition of lignin compounds ferulate, coumarate, and caffeate were isolated and characterized. As a proof of concept experiment, caffeate was recovered from treated lignin containing liquors by a fast and simple column purification method utilizing a caffeate specific scFv. The additional application of the scFv binders was demonstrated with a highly specific caffeate-binding antibody by monitoring the caffeate utilization of A. baylyi ADP1 by a simple competitive enzyme-linked immunosorbent assay (ELISA).
Materials and methods
Detailed descriptions of the methods for the chemical synthesis of biotinylated target molecules, biopanning, cloning and recombinant protein expression are described in the ESI.†Phages, strains, plasmids, enzymes and biotinylated haptens
The binders for ferulate, coumarate and caffeate were isolated from a synthetic antibody phage library (ScFvM) described by Huovinen et al.35E. coli XL1-Blue (Stratagene, USA) cells were used for phage infection and production, cloning, and protein expression. The VCS-M13 helper phage (Stratagene) was used to rescue the phagemid carrying phage in biopanning. SfiI restriction sites were used to clone the scFv genes from the pEB32x phagemid to expression vectors pLK06H35 and pLK04.37 The pLK06H vector contains an ampicillin resistance marker, as well as an additional histidine tag and two SfiI sites upstream of an AP gene, whereas the pLK04H vector lacks the AP gene. All of the enzymes and buffers for cloning were purchased from Thermo Scientific, USA. Caffeate consumption studies were conducted with A. baylyi ADP1 (DSM 24194, DSMZ, Germany).The biotinylated target molecules (target haptens) were constructed by chemical synthesis. Three different conjugates were manufactured carrying ferulate, coumarate, or caffeate. Each of the molecules contained a linker with 8 PEG units and a biotin moiety. Construction of the biotinylated target molecules (biotin-PEG-[caffeate/ferulate/coumarate]) is described in detail in the ESI.†
Media components
E. coli XL-1 Blue cells were grown in Super Broth (SB) medium (tryptone 30 g L−1, yeast extract 20 g L−1, MOPS 10 g L−1, and pH 7) and supplemented with 1%w/v glucose and antibiotics (tetracycline 10 μg mL−1, ampicillin 100 μg ml−1, chloramphenicol 25 μg mL−1 or kanamycin 30 μg mL−1) when appropriate. A. baylyi ADP1 cells were grown in minimal salt medium MA/9 (Na2HPO·2H2O 5.518 g L−1, KH2OPO4 3.402 g L−1, NH4Cl 0.963 g L−1, nitrilotriacetic acid 0.008 g L−1, NaCl 1 g L−1, FeCl3 0.001 mg L−1, MgSO4 240 mg L−1, and CaCl2 11 mg L−1) supplemented with 10 mM acetate and 10 mM caffeate. For media preparation, caffeic acid (Sigma) was first dissolved in MA/9 media without supplementation (pH is adjusted to 8.5 with NaOH) and filtered through 0.2 μm filters. The pH of the stock solution decreased after the caffeic acid addition close to neutral (pH 6.5–7).Phage enrichment immunoassay
The scFv-phage library scFvM was independently enriched against the different biotinylated target molecules (biotin-PEG-[caffeate/ferulate/coumarate]) in three consecutive rounds of affinity-based biopanning using superparamagnetic beads. A more detailed description of the biopanning process is offered in the ESI.† Subsequently, a time-resolved fluorometry-based phage immunoassay was used to verify the enrichment of the target specific scFv-phage. All of the assay steps were conducted in a working volume of 200 μL of assay buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% BSA, 0.0.1% Tween20, pH 8.0) at room temperature with slow shaking on a prewashed 96-well streptavidin (SA) plate (Kaivogen, Finland). All washing steps were run four times with a Delfia plate washer (PerkinElmer, Finland) using wash buffer (5 mM Tris-HCl, 150 mM NaCl, 0.01% Tween20, pH 8.0). For target molecule immobilization, a saturating concentration of biotinylated target molecules (biotin-PEG-[caffeate/ferulate/coumarate]) was added in the streptavidin-well followed by 30 minutes of incubation and washing. Aliquots of 1 × 109 cfu mL−1 phage from all the panning reactions were added independently on the plate, which was incubated for 1 hour and then washed. Then, 25 ng of europium-labelled anti-M13 phage antibody 9E7 (Department of Biochemistry/Biotechnology, University of Turku, Finland) was added and the reactions were incubated for 30 min before washing. Thereafter, DELFIA enhancement solution (PerkinElmer) was added, incubated for 10 minutes and then the fluorescence signals were measured with time-resolved fluorescence measurement (1420 Victor Multilabel Counter (PerkinElmer) program Europium). All samples were run as duplicate. The wells without the target molecules were used as controls.Primary, secondary and competitive AP-ELISA screening assays
The enriched scFvs genes were cloned from the phagemid vector as a pool into the vector pLK06H. Depending on the target, 100–200 randomly chosen colonies were then picked from the transformation plate to express the chosen clones as scFv-AP-fusions. Later on, certain scFvs were also expressed in the single chain form using the vector pLK04H. Detailed descriptions of cloning, AP-scFv and scFv lysate preparation as well as protein expression and purification are offered in the ESI.† Assessments of the initial binding of the expressed 100–200 scFvs towards their target molecules were conducted by AP-ELISA. The assay and wash conditions were similar to the phage immunoassay. AP-scFv lysates were added on the immobilized molecules in a final dilution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, incubated for 1 hour and washed. Then, 100 μL of 1 mg ml−1 of 4-nitrophenyl phosphate disodium salt hexahydrate (pNPP) (Sigma-Aldrich) in pNNP buffer (500 mM Tris, 200 mM NaCl, 10 mM MgCl2, pH 9.0) was added into the wells and incubated for 40 min. The absorbance was measured with a Multiskan Ascent plate reader (ThermoLabsystems, Finland) at a wavelength of 405 nm. The binders enriched against coumarate and ferulate were screened against both coumarate and ferulate, whereas caffeate binders were screened against caffeate and ferulate.
:10, incubated for 1 hour and washed. Then, 100 μL of 1 mg ml−1 of 4-nitrophenyl phosphate disodium salt hexahydrate (pNPP) (Sigma-Aldrich) in pNNP buffer (500 mM Tris, 200 mM NaCl, 10 mM MgCl2, pH 9.0) was added into the wells and incubated for 40 min. The absorbance was measured with a Multiskan Ascent plate reader (ThermoLabsystems, Finland) at a wavelength of 405 nm. The binders enriched against coumarate and ferulate were screened against both coumarate and ferulate, whereas caffeate binders were screened against caffeate and ferulate.
Based on the initial screening, 18 to 21 clones were chosen for a secondary screening. The assay was the same as the initial screening, except that the freshly prepared lysates were diluted to a final concentration of 1:2, run as duplicates and pNPP solution (Sigma-Aldrich, #P7998) was applied as the reactant. Using the highest signal-to-background ratio and minimal cross-reactivity as the criteria, seven scFv-APs per target molecule were chosen for sequencing.
A competitive AP-ELISA was conducted for scFv-APs with unique sequences in order to confirm the recognition of the non-biotinylated forms of the target molecules. In this assay, the lysates were incubated together with the biotinylated target molecule and increasing concentrations of the corresponding free acid (0.125–64 μM) for 1–2 h. Then, the mixture was transferred to SA-coated plates and incubated for 10 minutes. After washing the wells, pNPP solution was added and the plates were incubated for 1 h. The absorbance was measured at 405 nm. The samples were run as triplicate. Later on, similar assays were conducted with purified scFv-APs. For a caffeic acid binder a competitive assay was also performed against catechol and 4-ethylcatechol.
Evaluation of binding specificity and kinetics by biolayer interferometry
A Fortebio Octet RED384 instrument equipped with 16 parallel SA-coated optical biosensors (Fortebio, Pall Life Sciences, Menlo Park, USA) was used to further characterize the binding properties of the selected binders. The data collected were processed and analyzed using the Octet Data Analysis Software (version 7.1). For the purified binders, buffer exchange to Octet kinetic buffer (1 mg ml−1 bovine serum albumin (BSA), 0.02% Tween 20, 0.05% NaN3 in PBS) was carried out using Illustra NAP-10 columns (GE Healthcare Life Sciences, USA) according to the manufacturer's instructions. For scFv samples, kinetic buffer was supplemented with 8% glycerol.The SA biosensors were soaked prior to run for 15–60 minutes in ITC-buffer (50 mM NaPO4, 100 mM NaCl, pH 7.4). The stirring speed was set at 500 rpm and the temperature was at 27 °C. Black, tilted-bottom 384-well plates (Fortebio, Pall Life Sciences) were used with 80 μL buffer/sample volumes. The baseline for the sensors was recorded in ITC-buffer for 60 seconds. Thereafter, the synthesized ligands (∼100 μM biotin-PEG-[caffeate/ferulate/coumarate] in ITC-buffer containing 10% ethanol) were attached to the sensors in a 300 s activation step, followed by brief washing and quenching (saturation of the free streptavidins with 17 μg ml−1 biotin in ITC-buffer containing 10% ethanol) steps of 60 seconds. The baseline for the sensors in the kinetic buffer was recorded for 300 s. The association and dissociation of the scFv samples were measured for 600 s. Sensor regeneration with 1 M acetic acid (pH 2) followed by a wash step with kinetic buffer was carried out when appropriate. Sensors without immobilized ligands and functionalized sensors soaked in antibody-free kinetic buffer were used as controls.
The effect of free acids on the dissociation of the scFv-APs bound to the functionalized biosensor surface (CAF_3, CAF_5, FER_9, FER_78, COU_12, and COU_74) was studied by exposing the scFv-AP-soaked sensors to free caffeate, ferulate, and coumarate (∼500 μM in kinetic buffer containing 10% ethanol). The concentrations of scFv-APs were 1.2, 1.5, 1.6, 1.6, 1.5, and 1.6 mg mL−1 for CAF_3, CAF_5, FER_9, FER_78, COU_12, and COU_74, respectively. The dissociation of the complex in the presence of each acid or blank kinetic buffer was then recorded. The specificity towards the acids was determined by calculating the decrease of the surface depth in nanometers during the dissociation step. The decrease was proportioned to that with the blank buffer and normalized.
For determining the affinities of the scFvs CAF_3s, FER_9s, and COU_12s towards the haptens, the samples were diluted to concentrations of 0.004, 0.016, 0.06, and 0.25 mg mL−1. The biosensor analysis was carried out for each scFv sample as described above, but in the absence of the competing free ligand. Local partial fit (20 s from the beginning of the association and 60 s from the beginning of the dissociation phase) was employed to determine the binding affinities. The affinities were calculated as averages of the values determined for each concentration.
Preparation of scFv-based LDM-separation column
For selective LDM recovery, a column coupled with the caffeate binding scFv (CAF_3) was assembled. Buffer exchange for purified scFv CAF_3 was carried out with NAP buffer-exchange columns to coupling buffer (0.2 M NaHCO3, 0.5 M NaCl, pH 8.3) and glycerol was added to a final concentration of 8%. The buffer-exchanged scFvs were coupled to a sepharose matrix, in which the coupling occurs via covalent binding between the lysine residues of the antibodies and the NHS-groups of the sepharose matrix making the scFv-coupled bed reusable. The coupling was conducted according to the manufacturer's instructions using 0.4 mg of antibodies per ml bed volume, left overnight at 4 °C and blocked with 0.1 M Tris-HCl, pH 8.5. The reaction mixture was washed five times with 0.1 M Tris-HCl (pH8.5) and 0.1 M acetate buffer, 0.5 M NaCl (pH 4.5). The coupled matrix was packed into a column under gravitation and used for caffeate recovery studies. A total volume of 1 mL of sepharose matrix was used for the coupling resulting in a packed column bed of 1 ml.Caffeate recovery with scFv-based column
For the qualitative selectivity studies of the scFv CAF_3s, four different samples (caffeate, simulated LDM mixture, Kraft lignin and rice straw hydrolysate) spiked with caffeate, coumarate and ferulate were poured through the prepared column and 1 mL of the fraction samples was analyzed. Each sample was diluted in assay buffer into a total volume of 5 mL. The column was equilibrated with assay buffer (50 mM Tris-HCl, 150 mM NaCl, pH 8.0) prior to each experiment. The samples were washed with 10 ml of wash buffer (5 mM Tris-HCl, 150 mM NaCl, pH 8.0) and eluted with 3 ml of glycine buffer (pH 2.2). The collected fractions were analyzed with HPLC (Agilent 1100 series, Hewlett Packard, Germany) equipped with a fast acid H + column (Phenomex, USA), a degasser (G1322A) and an UV-detector (G1315A) using 0.005 N H2SO4 as the eluent. The pump (G1211A) flow was adjusted to 1 ml min−1, the column temperature to 80 °C, and peaks were identified at a wavelength of 310 nm by comparing the retention time and spectral profile with standards prepared for coumarate, caffeate and ferulate. For the spiked Kraft lignin and rice straw hydrolysate, the samples were analyzed throughout the spectrum of 210–900 nm.To examine the column's ability to capture caffeate, 50 μM of caffeate solution was passed through the column. The initial caffeate recovery test was rerun with the same column after re-equilibration with assay buffer. The same column was used to study caffeate recovery for a simulated mixture of LDMs containing 1–2 mM of caffeate, ferulate and coumarate (Sigma). To further test the column's capability to purify LDMs, 0.7 mg of water soluble low sulfonate containing Kraft lignin (Sigma) supplemented with caffeate, coumarate and ferulate (1–2 mM each) was used. Due to the high molecular weight of the Kraft lignin (average of 10000 g mol−1) – and the apparent lack of soluble acids that could be identified with the chosen HPLC equipment – a test with rice straw hydrolysate was also conducted. In this experiment, 50 μM of caffeate, coumarate and ferulate were mixed with 300 mg of freeze-dried rice straw hydrolysate (described by Kannisto et al., 2015),38 and passed through the column. Additionally, quantitative caffeate recovery was studied as above with a reduced washing volume (3 ml) using 2.5 μg of caffeate or 2.6 μg of caffeate dissolved with 3–5 μg of coumarate and ferulate. Caffeate recovery percentages were calculated from the elution fractions.
Caffeate consumption studies
The applicability of the scFv-AP-based immunoassay as a monitoring tool for substrate conversion studies was tested with the caffeate binder designated as CAF_3. For these purposes, A. baylyi ADP1 was pre-grown overnight on MA/9 minimal salt media supplemented with 10 mM Na-acetate and 10 mM caffeate. From the pre-culture, 50 ml cultivation (same as pre-culture) was inoculated to the initial OD600 of 0.02, incubated at 30 °C and shaken at 300 rpm. The samples were collected every 2 h for a duration of 12 hours. The growth was monitored with optical density measurements and 1 ml of culture supernatant was stored at −20 °C for substrate analysis.Caffeate depletion was analyzed with competitive AP-ELISA. 0.1 μM of biotinylated caffeate and 14 μg mL−1 of the caffeate binder CAF_3 were mixed together with a 1:625 dilution of the collected samples in a final volume of 150 μL. Free caffeic acid concentrations ranging from 1 μM to 32 μM were used for the calibration curve.
To convert the ELISA absorbance values to mM, a 4 PL curve fit39 was applied on the calibration curve. Eqn (1) was used to solve the sample concentration by replacing λ with the absorbance value and solving x from the equation. The values c (inflection point) and b (coefficient for slope steepness) were approximated with the Matlab 2016 software data fitting tool. For data fitting, the least squares method was used.
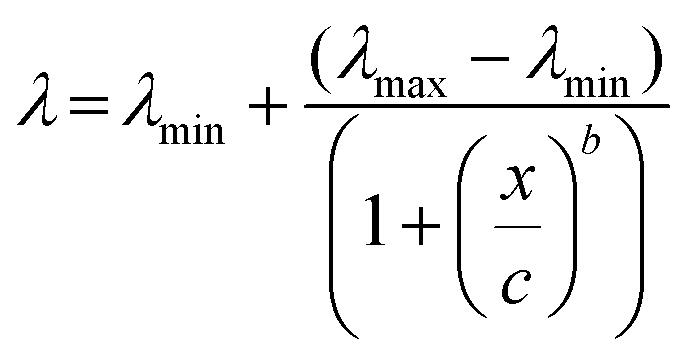 | (1) |
Additional caffeate concentration measurements were conducted with UV-Vis at 280 nm (NanoDrop 2000 Spectrophotometer, Thermo Fischer Scientific). The acetate concentrations were measured with a HPLC (Shimadzu, USA) equipped with an RID-10A detector, an SIL-20AC HT autosampler and a DGU-20A3 degasser. The temperature of the Resex™ RHM-Monosaccharide H + column (Phenomex, USA) was adjusted to 25 °C and the flow rate to 0.6 mL min−1. 0.01 N H2SO4 was used as the mobile phase.
Results and discussion
In principle, phage displayed scFv-libraries provide a potential source of binders towards any molecule of interest. LDMs such as hydroxycinnamates are the molecules of interest due to their potential use as building blocks for the chemical industry or as substrates in biotechnological processes. Their selective conversion to valuable end products is possible by certain microorganisms, even though to many organisms used in current biorefinery processes even low concentrations of these molecules can be toxic owing to the antioxidant nature of the molecules.23 In addition to their potential as substrates for bioconversion processes, aromatic LDMs are also valued as flavors, fragrances and antioxidants.21,40 As these compounds are found in heterogeneous mixtures in treated lignin fractions, a system targeting these highly similar molecules specifically and directly from hydrolysates could prove useful for the recovery of particular LDMs. Such recovery might be useful also in cases, where the heterogeneous LDMs are converted into specified aromatics through microbial processes.In this study, we used a synthetic scFvM phage library, specially designed to favor the recognition of low molecular weight compounds to select binders against three structurally related biotinylated targets (LDMs) with a very low molecular weight. The biotinylated target-molecule conjugates for coumarate, ferulate and caffeate were chemically synthesized for our LDM-binder development. Successful antibody development requires conjugates with unambiguously defined structures, together with an appropriately spaced linker between the biotin moiety and the target molecule. Without these conjugates, it would be necessary to use immobilized carrier proteins, such as BSA, during antibody selection. Typically, the target haptens attach variably around the carrier protein resulting in different epitope structures. Undesirably, this phenomenon produces a more miscellaneous group of binders, whereas biotinylated conjugates produce binders of more uniform quality.
Enrichment of LDM specific scFv pools
Highly specific scFvs towards three common LDMs were isolated from a total number of 6 × 109 independent clones stored in a scFvM phage-library, and used in a novel approach to recover, detect, and quantify LDMs. These scFvs act as binders towards phenolic acids, coumarate, ferulate and caffeate. From an immunoassay perspective, the phenolic acid concentrations of lignin hydrolysates19,20 are rather high. Consequently, scFvs with a moderate affinity can be successfully used as binders, however high specificity is necessary due to the variability of compounds present in technical lignins.The phages with the desired properties were enriched with target-molecule coated magnetic beads. To minimize the unspecific binding to the paramagnetic bead material, the streptavidine-coated beads were changed to neutravidin (NA) in the second round and back to SA in the third round. According to the obtained data from the phage immunoassay, ferulate and coumarate binders were enriched in the 2nd and 3rd panning rounds without significant unspecific binding to SA (Fig. 1). For the caffeate binding phage also unspecific binding to SA occurred. Within all of the samples, no further enrichment was observed between the 2nd and 3rd rounds, though the overall yield of the phage increased towards the 3rd round.
 | ||
| Fig. 1 The results of phage immunoassay. The bars represent the relative fluorescence signals (RFU) obtained from the assay. White bars = phages incubated with the target molecule immobilized on an SA-coated microtiter well; grey bars = background signal from the SA-well without the target molecule. | ||
LDM positive clones
After phage enrichment, as the phage pool contains scFvs with varying binding characteristics, the binders with the desired characteristics were screened. An immunoassay (AP-ELISA) was used to screen 96–192 randomly chosen scFv-AP clones per target molecule for recognition and differentiation between similar molecules. The initial screening revealed 86 positive clones out of 96 clones for ferulate and 84 positive clones out of 96 clones for coumarate (ESI Fig. 7–9†). Interestingly, active binders against caffeate were also found with a relatively high frequency (56 positive clones out of 192 clones), despite the apparent enrichment for the streptavidin in this panning. A clone was determined positive when the absorbance signal for the well containing the target molecule was higher than that of the biotin saturated background control wells (= S/B > 1.5). From these positive clones, for each target molecule, approximately 20 clones were chosen for further validation based on the S/B and low cross-binding signal (ESI Fig. 7–9†).According to the secondary screening, for ferulate targeted binders some degree of cross-binding occurred with the majority of the clones (16/20 clones), though ferulate specific clones were also obtained (4/20 clones). The specific recognition ability of most of the binders targeted against caffeate and coumarate structures was verified by the AP-ELISA. Nearly all of the clones selected against coumarate showed a low background as well as low cross-binding (19/21 clones), though 1/21 clones with cross-binding to ferulate and 1/21 clones with no signal at all were observed. Most of the clones selected against caffeate showed selective binding without cross-binding (12/18 clones), though 3/18 clones indicated a high background and cross-binding, whereas 3/18 clones showed no signal at all (ESI Fig. 10–12†).
Characterization of the LDM specific ScFv-APs and scFvs
Further scFv characterization included sequencing for unique clones as well as binding specificity and affinity studies. Seven to eight clones per target molecule with the highest specific signal – compared to the background and cross-signals according to the secondary screening – were chosen to be sequenced. The sequencing data (ESI Table 1†) revealed 2/8 unique sequences among binders targeted against caffeate and 3/7 for binders targeted against ferulate, whereas for binders targeted against coumarate 6/7 sequences were unique.| Binder | Target antigen | Relative dissociation | |||
|---|---|---|---|---|---|
| No competitor | Free caffeate | Free ferulate | Free coumarate | ||
| CAF_3 | Caffeate | 1.0 | 2.0 | 1.0 | 1.0 |
| CAF_5 | 1.0 | 1.2 | 0.8 | 0.9 | |
| FER_9 | Ferulate | 1.0 | 1.2 | 1.5 | 1.1 |
| FER_78 | 1.0 | 1.3 | 1.3 | 1.1 | |
| COU_12 | Coumarate | 1.0 | 1.6 | 1.4 | 1.7 |
| COU_74 | 1.0 | 1.6 | 1.5 | 1.7 | |
All the binders showed preference towards their cognate target haptens, although clear differences in specificities were observed. The caffeate binders CAF_3 and CAF_5 showed the highest specificity, whereas for the ferulate and coumarate binders also slight cross-binding occurred. In the AP-ELISA, the coumarate binders showed very little indication of cross-binding to the biotinylated ferulate, whereas the binding profiles of caffeate and ferulate binders indicated similar behavior against the biotinylated and free form of the targeted molecule. In studies where single model compounds are used, the cross-reactivity with other similar compounds is not an issue and this type of detection is applicable even for quantitative conversion studies. However, the slight cross-binding might prove problematic when more complex lignin structures are used as a substrate or if multiple hydroxycinnamates are monitored at the same time. To overcome this obstacle, the tools of genetic engineering can be used to further optimize the binding characteristics of the obtained antibodies. In addition, a generic binder, which could identify all of the hydroxycinnamates equally and simultaneously, might also be useful. Such generic binders have previously been developed for molecules such as sulphonamides and fluoroquinolones.41,42 Nonetheless, the specific behavior of the caffeate binder CAF_3 was clearly proved in these experiments indicating the applicability of scFvs as specific tools in lignin research.
Based on the specificity profile, one AP-scFv binder against each target LDM (CAF_3, FER_9, and COU_12) was selected for additional competitive AP ELISA experiments. All of the selected binders recognized also the non-biotinylated (free acid) form of the molecule (Fig. 2), similar to the biolayer interferometry experiments, even though biotinylated target molecules were used in the panning and screening steps. This implies that the paratope–epitope pair occurs at the aromatic ring structure of the molecule. This claim is further supported by the fact that the different hydroxycinnamates – differing only in the degree of methoxylation or in the number of hydroxyl groups found in the aromatic ring structures – were recognized to a lesser degree when compared to the target molecule in the cross-binding experiments. Additionally, the CAF_3 binder is specific for the caffeic acid structure, including the alkyl chain of the molecule. Catechol and 4-ethylcatecol have similar aromatic substitution to caffeic acid differing only in the alkyl substituents. In the presence of catechol or 4-ethylcatechol, the scFv CAF_3 showed no competitive binding to these compounds, in contrast to the free caffeic acid (ESI 14†). According to the data collected from the competitive immunoassay, the IC50 values were approximately 3 μM for the caffeate binder, 7 μM for the coumarate binder and 17 μM for the ferulate binder. The reliable recognition for these scFv-AP binders under the chosen conditions spans in the region of 1–20 μM.
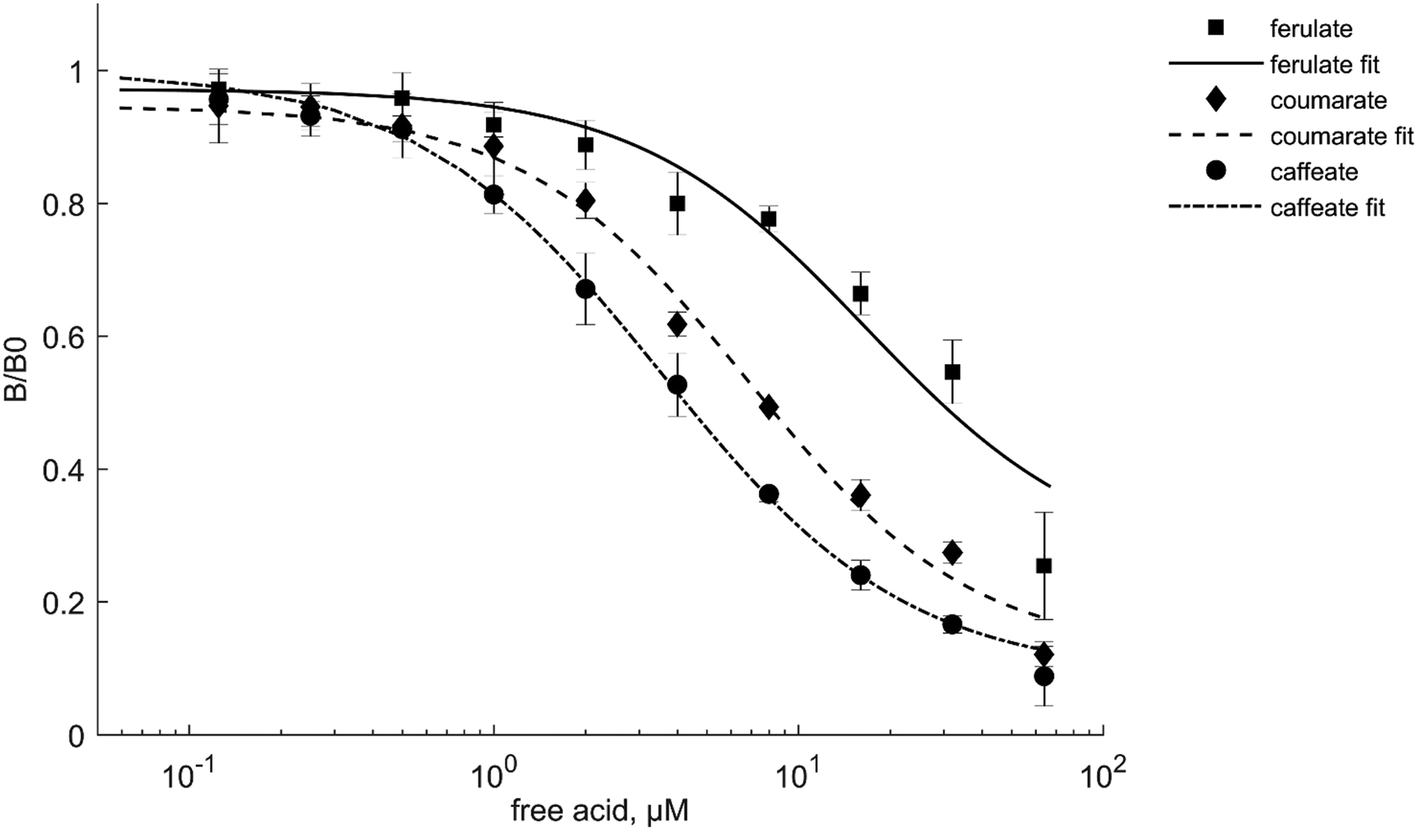 | ||
| Fig. 2 The binding of scFv-APs FER_9, COU_12 and CAF_3 towards target molecules. In this competitive assay, the selected binders were mixed together with the biotinylated target molecule and increasing concentrations of the corresponding free acid (0.125–64 μM). The binders bound to the corresponding free acid were washed away and the absorbance was measured. The absorbance signals of the competitive binding results are scaled to relative absorbance B/B0 (B = measured signal, B0 = max. signal). White = ferulate binder FER_9, blue = coumarate binder COU_12, and red = caffeate binder CAF_3. The standard deviation of triplicates is also shown in the figure as error bars. | ||
Notably, with multiple screening and selection methods, binders with specific binding profiles towards the target molecules were successfully obtained. Regardless of the high background in the phage immunoassay, it was also possible to isolate a highly specific antibody for caffeate, which could distinguish between the hydroxyl and methoxy groups of ferulate and coumarate.
 | ||
| Fig. 3 Determination of the binding affinity and dissociation kinetics for the caffeate (CAF_3s), ferulate (FER_9s), and coumarate (COU_12s) binders. Local partial fits were employed to estimate the affinities. The KD and Kdiss values are presented as an average of the affinities estimated for four different binder concentrations (three for FER9s). The determination was based on the association and dissociation of the binders to/from the biotinylated LDMs (the selected hydroxycinnamates) attached to streptavidin-coated optical biosensors. | ||
The micromolar affinity range is convenient for their use in both target compound monitoring and recovery, considering the relevant concentrations of hydroxycinnamates in the lignin-containing liquors as well as to allow efficient elution of the target compound in hydroxycinnamate recovery.
LDM separation from heterogeneous mixtures
One potential approach for the valorization of lignin-based streams is the selective and high recovery of monomeric compounds such as phenolic acids. The commonly used methods for phenolic compound recovery include polyvinyl resins or activated carbon matrices.43–45 The phenolic compounds bind to the matrix either via hydrophobic interactions or by hydrogen bonding from the aromatic or carboxyl group of the phenolic compound allowing non-selective adsorption onto the matrix. For example, da Costa Lopes et al. (2016) developed a method for purifying phenols extracted from biomass with ionic liquids using polyvinyl resins.46 The recovered fraction was further purified with supercritical CO2. However, there were no attempts to selectively recover a given target molecule from the fraction resulting in a mixture of phenolic acids.On the other hand, the adsorption and desorption conditions in a matrix can be optimized to favor the recovery of certain molecules. For example, the selective recovery of a ferulic acid from enzymatically treated sugar-beet pulp was studied by Couteau & Mathaly (1998) by optimizing a fixed-bed system utilizing activated carbon.47 In their study, 50% ferulic acid purity was achieved. As another example, ferulic acid recovery studies conducted by Tilay et al. (2008) revealed 50.89% purity and 57.97% recovery efficiency for polymeric adsorbents.48 However, by using more complex methods, such as subsequent preparative HPTLC, the purity of the recovered ferulic acid fraction was increased to 95.35%.48 Additionally, the molecular imprinting technique can be used to increase the selectiveness of the resin. As an example, Michailof et al. (2008) used molecularly imprinted polymers to recover caffeic acid, 4-HBA and other phenolic compounds with recovery efficiencies between 9–51%. For the caffeic acid imprinted polymer strong competition for binding sites occurred also with p-coumaric acid.49
To examine the potential of the antibody use in specific LDM recovery in the context of lignin valorization, the selectivity of the chosen scFvs was examined in a proof of principle experiment by separating caffeate from a heterogeneous mixture of soluble compounds. Caffeate recovery from different mixtures spiked with caffeate was demonstrated by using an affinity column prepared with immobilized CAF_3 scFv. Elution of the caffeate molecules was achieved with pH change, followed by immediate equilibration of the column to refold the covalently bound antibodies, thus making the column matrix reusable.
After verifying that the prepared column captures caffeate (ESI 14†), the column was used to recover caffeate from simulated LDM solution, kraft lignin and rice straw hydrolysate spiked with caffeate, coumarate and ferulate. Caffeate was recovered with high purity from all of the samples without traces of ferulate, coumarate or other detectable compounds proposing the system to be highly selective towards caffeate (ESI Fig. 15–17†).
In order to quantitatively determine the caffeate recovery rate of the column, the maximum theoretical binding capacity of the scFv-coupled bed was determined. Assuming that all of the scFvs were coupled functionally to the matrix and each scFv binds the targeted molecule in a molar ratio of 1:1, the binding capacity of the column was estimated to be 2.6 μg of caffeate per 1 ml of sorbent in the column. Thus, samples containing 2.5 μg per 1 ml column of caffeate were used for quantitative recovery experiments. Very high recovery rates were obtained for the simulated LDM mixture (94%) (Table 2) and also for the caffeate solution (77%). Coumarate and ferulate were found from the flow and wash fractions of the LDM mixture verifying that they are not retained in the column.
| LDM model compound solution | |||
|---|---|---|---|
| Caffeate (μg) | Coumarate (μg) | Ferulate (μg) | |
| Sample | 2.45 | 3.84 | 5.62 |
| Flow through | 0.00 | 2.24 | 3.16 |
| Wash through | 0.00 | 1.54 | 2.14 |
| Elution 1 | 0.24 | 0.00 | 0.00 |
| Elution 2 | 1.57 | 0.00 | 0.00 |
| Elution 3 | 0.50 | 0.00 | 0.00 |
| Recovery % from column | 94 | 0.00 | 0.00 |
| Recovery % total | 94 | 98 | 94 |
Caffeate binder as a monitoring tool
Hydroxycinnamates, such as p-coumarate, ferulate, and caffeate, are found in plant cells and technical lignin structures. These molecules are potential substrates for bacterial lignin upgrading schemes and simple tracking methods of these substrates are of great interest. For example, the specific detection of these molecules directly from the culture medium and other complex matrices is an appealing approach in bacterial conversion studies. To this end, experiments with the highly specific caffeate binder CAF_3 were conducted to verify its applicability as a monitoring tool for substrate conversion.A. baylyi ADP1 was cultivated with acetate and caffeate as the carbon sources. According to the culture sample analysis performed by the immunoassay, the samples collected from A. baylyi ADP1 cultures showed the subsequent utilization of acetate and caffeate (Fig. 4). This is well in correlation with the known substrate preferences of A. baylyi; its aromatic catabolism is repressed in the presence of acetate which is consequently utilized before the aromatics.50 The developed immunoassay quantified the concentration of caffeate in the cultivation samples up to the time point of 10 hours. Thereafter, the concentrations were below the detection limit. The calibration curve of the immunoassay gave a linear response between 4–10 μM for the caffeate binder CAF_3 and the caffeate concentrations from the samples were further confirmed with UV-Vis 208 nm, supporting the immunoassay results (Fig. 4).
 | ||
| Fig. 4 Upper figure: Substrate consumption and growth of A. baylyi ADP1 batch culture over a time course of 12 hours. Closed diamonds represent acetate concentration in mM and blue circles caffeate concentration in mM (immunoassay). The black asterisks represent the bacterial growth measured as the optical density at 600 nm. The open circles indicate the caffeate concentration as mM when measured with UV-Vis at 280 nm. Bottom figure: the calibration curve used for calculating the caffeate concentration. Open circles represent the absorbance measured at 405 nm in different caffeate concentrations and the dotted line the coefficient of variation. The black dashed line represents the data fitting of the obtained absorbance values over varying free acid concentrations. | ||
Conclusions
The structural complexity and heterogeneity of lignin make its further utilization and analysis very challenging. Due to the high binding specificity, antibody-based tools could provide a unique means for the recovery, detection and monitoring of a variety of lignin compounds even from complex matrices. In this study, binders against three lignin-derived hydroxycinnamates ferulate, coumarate and caffeate were successfully isolated from a synthetic antibody phage library. These scFvs can be used to isolate and collect individual chemically pure substances from a complex mixture of lignin-derived compounds as was demonstrated with the specific caffeate-binding antibody. Furthermore, the binder against caffeate was successfully employed to track caffeate consumption in a bacterial bioprocess. This proof of concept study shows the potential of antibodies for the recovery of small molecules from dilute and challenging streams.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Academy of Finland (grant no. 311986, 310188, 310135) and Fortum Foundation (grant no. 201600065).References
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310 RSC.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef.
- J. W. Lee, D. Na, J. M. Park, J. Lee, S. Choi and S. Y. Lee, Nat. Chem. Biol., 2012, 8, 536–546 CrossRef PubMed.
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056–5058 CrossRef PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef PubMed.
- N. Abas, A. Kalair and N. Khan, Futures, 2015, 69, 31–49 CrossRef.
- R. Liguori and V. Faraco, Bioresour. Technol., 2016, 215, 13–20 CrossRef PubMed.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef PubMed.
- M. K. Bhat and S. Bhat, Biotechnol. Adv., 1997, 15, 583–620 CrossRef PubMed.
- L. R. Lynd, P. J. Weimer, W. H. van Zyl and I. S. Pretorius, Microbiol. Mol. Biol. Rev., 2002, 66, 739–739 CrossRef.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef.
- B. Hahn-Hägerdal, M. Galbe, M. F. Gorwa-Grauslund, G. Lidén and G. Zacchi, Trends Biotechnol., 2006, 24, 549–556 CrossRef PubMed.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef PubMed.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- F. Abdelkafi, H. Ammar, B. Rousseau, M. Tessier, R. El Gharbi and A. Fradet, Biomacromolecules, 2011, 12, 3895–3902 CrossRef PubMed.
- S. Sun, J. Wen, S. Sun and R.-C. Sun, Biotechnol. Biofuels, 2015, 8, 37 CrossRef PubMed.
- M. Li, C. Foster, S. Kelkar, Y. Pu, D. Holmes, A. Ragauskas, C. M. Saffron and D. B. Hodge, Biotechnol. Biofuels, 2012, 5, 38 CrossRef PubMed.
- E. M. Karp, C. T. Nimlos, S. Deutch, D. Salvachúa, R. M. Cywar and G. T. Beckham, Green Chem., 2016, 18, 4750–4760 RSC.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 Search PubMed.
- M. P. Kähkönen, A. I. Hopia, H. J. Vuorela, J.-P. Rauha, K. Pihlaja, T. S. Kujala and M. Heinonen, J. Agric. Food Chem., 1999, 47, 3954–3962 CrossRef.
- X. Ren, P. Wang, X. Han, G. Zhang, J. Gu, C. Ding, X. Zheng and F. Cao, ACS Sustainable Chem. Eng., 2017, 5, 6548–6556 CrossRef.
- G. Garrote, J. M. Cruz, A. Moure, H. Domínguez and J. C. Parajó, Trends Food Sci. Technol., 2004, 15, 191–200 CrossRef.
- C. Civolani, P. Barghini, A. R. Roncetti, M. Ruzzi and A. Schiesser, Appl. Environ. Microbiol., 2000, 66, 2311–2317 CrossRef PubMed.
- O. Y. Abdelaziz, D. P. Brink, J. Prothmann, K. Ravi, M. Sun, J. García-Hidalgo, M. Sandahl, C. P. Hulteberg, C. Turner, G. Lidén and M. F. Gorwa-Grauslund, Biotechnol. Adv., 2016, 34, 1318–1346 CrossRef PubMed.
- D. Salvachúa, E. M. Karp, C. T. Nimlos, D. R. Vardon and G. T. Beckham, Green Chem., 2015, 17, 4951–4967 RSC.
- D. Parke and N. Ornston, Appl. Environ. Microbiol., 2003, 69, 5398–5409 CrossRef PubMed.
- Z. Mycroft, M. Gomis, P. Mines, P. Law and T. D. H. Bugg, Green Chem., 2015, 17, 4974–4979 RSC.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CrossRef PubMed.
- P. D. Sainsbury, E. M. Hardiman, M. Ahmad, H. Otani, N. Seghezzi, L. D. Eltis and T. D. H. Bugg, ACS Chem. Biol., 2013, 8, 2151–2156 CrossRef PubMed.
- M. Siddiqui, Indian J. Pharm. Sci., 2010, 72, 12 CrossRef PubMed.
- D. A. Blake, X. Zhu and B. Ban, in The New Uranium Mining Boom: Challenge and Lessons Learned, 2011, pp. 467–476 Search PubMed.
- K. Nishi, M. Ishiuchi, K. Morimune and H. Ohkawa, J. Agric. Food Chem., 2005, 53, 5096–5104 CrossRef PubMed.
- Z. A. Ahmad, S. K. Yeap, A. M. Ali, W. Y. Ho, N. B. M. Alitheen and M. Hamid, Clin. Dev. Immunol., 2012, 2012, 1–15 CrossRef PubMed.
- T. Huovinen, M. Syrjänpää, H. Sanmark, E. C. Brockmann, A. Azhayev, Q. Wang, M. Vehniäinen and U. Lamminmäki, Protein Eng., Des. Sel., 2013, 26, 683–693 CrossRef PubMed.
- J. Leivo, J. Mäkelä, J. Rosenberg and U. Lamminmäki, Anal. Biochem., 2015, 492, 27–29 CrossRef PubMed.
- T. Huovinen, M. Julin, H. Sanmark and U. Lamminmäki, Plasmid, 2011, 66, 47–51 CrossRef PubMed.
- M. S. Kannisto, R. K. Mangayil, A. Shrivastava-Bhattacharya, B. I. Pletschke, M. T. Karp and V. P. Santala, Biotechnol. Biofuels, 2015, 8, 198 CrossRef PubMed.
- J. W. A. Findlay and R. F. Dillard, AAPS J., 2007, 9, E260–E267 CrossRef PubMed.
- J. Kyselka, D. Rabiej, M. Dragoun, F. Kreps, Z. Burčová, I. Němečková, J. Smolová, M. Bjelková, A. Szydłowska-Czerniak, Š. Schmidt, L. Šarman and V. Filip, Eur. Food Res. Technol., 2017, 243, 1633–1644 CrossRef.
- T. Korpimäki, E. C. Brockmann, O. Kuronen, M. Saraste, U. Lamminmäki and M. Tuomola, J. Agric. Food Chem., 2004, 52, 40–47 CrossRef PubMed.
- J. Leivo, U. Lamminmäki, T. Lövgren and M. Vehniäinen, J. Agric. Food Chem., 2013, 61, 11981–11985 CrossRef PubMed.
- E. Conde, P. Gullón, A. Moure, H. Domínguez and J. C. Parajó, Food Bioprod. Process., 2009, 87, 208–214 CrossRef.
- E. Conde, A. Moure, H. Domínguez, M. H. Gordon and J. C. Parajó, J. Agric. Food Chem., 2011, 59, 9158–9165 CrossRef PubMed.
- S. Dupoiron, M.-L. Lameloise, M. Pommet, O. Bennaceur, R. Lewandowski, F. Allais, A. R. S. Teixeira, C. Rémond and H. Rakotoarivonina, Ind. Crops Prod., 2017, 105, 148–155 CrossRef.
- A. M. da Costa Lopes, K. G. João, A. R. C. Morais, E. Bogel-Łukasik and R. Bogel-Łukasik, Sustainable Chem. Processes, 2013, 1, 3 CrossRef.
- D. Couteau and P. Mathaly, Bioresour. Technol., 1998, 64, 17–25 CrossRef.
- A. Tilay, M. Bule, J. Kishenkumar and U. Annapure, J. Agric. Food Chem., 2008, 56, 7644–7648 CrossRef PubMed.
- C. Michailof, P. Manesiotis and C. Panayiotou, J. Chromatogr., A, 2008, 1182, 25–33 CrossRef PubMed.
- T. Zimmermann, T. Sorg, S. Y. Siehler and U. Gerischer, J. Bacteriol., 2009, 191, 2834–2842 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc00490k |
| This journal is © The Royal Society of Chemistry 2018 |