
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphitization of Miscanthus grass biocarbon enhanced by in situ generated FeCo nanoparticles†
Ian
Major
ab,
Jean-Mathieu
Pin
a,
Ehsan
Behazin
 ab,
Arturo
Rodriguez-Uribe
a,
Manjusri
Misra
ab and
Amar
Mohanty
*ab
ab,
Arturo
Rodriguez-Uribe
a,
Manjusri
Misra
ab and
Amar
Mohanty
*ab
aBioproducts Discovery and Development Centre, Department of Plant Agriculture, Crop Science Building, University of Guelph, Guelph, N1G 2W1, Ontario, Canada. E-mail: mohanty@uoguelph.ca
bSchool of Engineering, Thornbrough Building, University of Guelph, Guelph, N1G 2W1, Ontario, Canada
First published on 23rd April 2018
Abstract
Catalytic graphitization of biocarbon prepared from Miscanthus grass was possible through chemical treatment with iron(III) nitrate, Fe(NO3)3, and cobalt(II) nitrate, Co(NO3)2. The pyrolysis monitoring, until the temperature of 900 °C, permitted for the primary analysis of the effects of the generated catalysis species on biomass degradation. Then both Raman spectroscopy and X-ray diffraction (XRD) allowed for the observation of microstructural changes between the samples treated with metal catalysts and the untreated samples. The samples treated with both iron and cobalt nitrates are twice as efficient at forming ordered graphite than the treated sample with the catalysts used separately. This may be due to FeCo nanoparticles generated in situ during pyrolysis. These results show that the morphology and allotropy of renewable biocarbon can be tailored in such a way that it can be applied to design smart materials.
Introduction
Various allotropes of ordered carbon including graphite and graphene structures have been heavily investigated in recent years for a variety of applications. Applications of these carbons range from steel-making,1 to pencils,2 to anodes in supercapacitors and lithium ion batteries.3 As the need for these materials increases, the effects on the environment should be taken into consideration.4 The major drawback with common uses of these types of carbon is that they are frequently obtained from petroleum based sources, such as petroleum coke and coal, and are therefore unrenewable and unsustainable for the environment.5 Another concern with graphite is related to its very energy intensive process of fabrication. This is commonly done through expensive methods such as mining or through synthetic pathways such as the Acheson process, which use silica and high temperatures.6As a start to moving away from petroleum based carbon, biomass has been used as an alternative carbon source to modify the properties of polymer composites such as the impact strength and Young's modulus in many recent studies.7,8 Biomass offers an easily renewable, inexpensive resource that can be converted to biocarbon at relatively low temperatures.6 Processes using biomass are considered to be “carbon-neutral” because the emissions let out during pyrolysis were originally absorbed from the atmosphere.9 Biocarbon is a porous material with a high surface area,10 which can be increased further by both physical or chemical processes using steam activation, ball milling,11 or potassium hydroxide (KOH), respectively, to produce surface areas up to 1500 m2 g−1.12 As highlighted above, biocarbon is versatile since its structure can be tailored.13 It has the potential to be a substitute in many of the same applications that petroleum based carbons are currently used for including composite fillers,14 electrical sensors, and capacitor anodes.3 To make it an efficient, renewable source of graphite, many studies have investigated methods involving metal catalysts to create a graphitic structure throughout the biocarbon. Biocarbon can be prepared from any kind of organic matter such as grasses,14 wood,13 fruit peels,4 and even human hair.15 For example, Thompson et al.13 used wood sawdust as a raw material to create their biocarbon, and iron nitrate as a catalyst to produce carbon nanotubes.
In this investigation, Miscanthus powder is used as a biomass source to prepare biocarbon through pyrolysis. Miscanthus grass is a perennial grass that does not require high soil quality or a fertilizer. It is a hardy, resilient plant that is used as a biofuel feedstock or polymer filler due to its high amounts of crop yield.16,17 New uses for Miscanthus have been discovered after pyrolysis, where Miscanthus biocarbon can be used as a renewable source of carbon.14 The low cost of production and seasonal growth patterns could potentially make Miscanthus a promising crop for transitioning to a sustainable future.
Due to the dominant amorphous structure of carbon in biocarbon there is a need to use a catalyst to promote the formation of a graphitic structure at temperatures less than 1000 °C.18–20 In the literature, many different metals such as nickel,21 magnesium,22 titanium,22 copper,22 manganese,23 and chromium21 have been used as attempted catalysts to create ordered carbon structures, but often iron and cobalt are among those shown to produce the best results.21–23 Rodriguez et al.24 described the mechanism with which iron catalysts promote the growth of graphene layers. The graphene layers are able to grow on specific faces of the iron particle, and begin stacking afterwards. The presence of the metal nanoparticle provides a starting point for the carbon to grow out from, drastically increasing the order found throughout the material. These metals are viable catalysts because they are relatively abundant and therefore are of lower cost compared to metals that are less common. This investigation compares the catalytic efficiency of iron(III) nitrate and cobalt(II) nitrate used separately and together for the conversion of Miscanthus grass into graphitic biocarbon. The use of a hybrid catalyst system is motivated by several reasons. Firstly, as a general feature, it is known that the combination of different metals can generate multi-metal complexes or clusters that exhibit a new catalytic activity. These kinds of clusters are generally used in heterogeneous catalysis, especially to catalyse hydrogenation reactions using water as a substrate. Typical examples are the supported zeolites.25 Secondly, the rationality behind using both iron and cobalt nitrate is related to the fact that the nitrogen doped FeCo alloy is a very efficient catalyst for oxygen reduction reactions, which could enable the reduction of temperature of carbonization during the pyrolysis.26
After biomass impregnation and drying with the respective metal solutions, the effect of the catalysts on the Miscanthus pyrolysis was monitored and modeled in a thermogravimetric analyzer. Then the obtained samples were deeply analyzed, highlighting the chemical change operating through the pyrolysis process, as well as the morphology and composition of the resulting biocarbons.
Materials and methods
Materials
Miscanthus grass powder (<2 mm fibres) from Competitive Green Technologies in Leamington, Ontario was used as a biomass in this study. Iron nitrate, Fe(NO3)3, (99.95%, Sigma-Aldrich) and cobalt nitrate, Co(NO3)2, (≥98%, Sigma-Aldrich) were used as received.Miscanthus grass treatment
7.18 × 10−3 moles of either iron or cobalt were used (2 wt% and 2.1 wt%, respectively) to treat the biomass. The metal nitrate solution was prepared by making a 10 wt% concentration aqueous solution. 20 g of Miscanthus was mixed with 80 mL of distilled water, and then the metal solution was poured in. The mixture was left to stir for 3 hours, on a hot plate at 80 °C. It was then removed, and placed in an oven overnight to dry at 80 °C. The mixed treatment catalyst was prepared using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iron(III) nitrate moles to cobalt(II) nitrate moles, for a total of 7.18 × 10−3 moles of metal, to be compared to the other treatments. The mixed metal solution was prepared by making a 10 wt% concentration aqueous solution of Fe(NO3)3 + Co(NO3)2. The overall preparation with biomass was the same as the previous process.
:1 mixture of iron(III) nitrate moles to cobalt(II) nitrate moles, for a total of 7.18 × 10−3 moles of metal, to be compared to the other treatments. The mixed metal solution was prepared by making a 10 wt% concentration aqueous solution of Fe(NO3)3 + Co(NO3)2. The overall preparation with biomass was the same as the previous process.
Characterization techniques
Thermogravimetric analysis (TGA) was carried out using a TA Instruments Q500. The samples were prepared at 10 °C min−1 to temperatures of 500 °C, 700 °C and 900 °C, to be used for characterization. All TGA were carried out under a nitrogen atmosphere (N2) at a flow rate of 40 mL min−1. The generation of magnetic materials (i.e. FeCo nanoparticles and graphite) has been followed by using a TGA 5500 from TA Instruments. A scan at the rate of 10 °C min−1 until 900 °C has been achieved with an active electromagnet, under 25 mL min−1 of N2. The impregnation efficiency has been checked by using adsorption results from the N2 isotherm (77 K) with an Autosorb iQ (Quantachrome, FL, USA) machine. The pore volume distribution was determined using the NLDFT (slit pore) analysis model for carbon. Fourier-transform infrared (FTIR) spectroscopy was used to observe the structural differences between biocarbon samples prepared from different metals. The apparatus used was a Thermo Scientific Nicolet™ 6700 FTIR, in attenuated total reflectance (ATR) mode. 64 scans at a resolution of 4 cm−1 were recorded for all spectra. The blank was done in air. Scanning electron microscopy/energy-dispersive X-ray spectroscopy (SEM/EDS) was conducted to obtain pictures of the treated Miscanthus and resulting biocarbon samples, as well as analyze the elemental composition of each. SEM/EDS was conducted using a Phenom ProX scanning electron microscope. Biocarbon samples were placed on carbon tape sample holders and inserted into the machine. Spectra were recorded at 10 kV, for 120 seconds each, and at magnifications between 290× and 5900×. The elemental composition was taken as the average of 4 range analyses for each sample. Raman spectroscopy was used to analyze the relative amounts of graphite and disorder in each sample. Raman spectra were recorded using a DXR™ 2 Raman microscope from Thermo Fisher Scientific, at 10× magnification. The aperture was set to a 50 μm pinhole. The laser power was set to 5 mW, at a wavelength of 532 nm. X-ray diffraction (XRD) patterns have been recorded with a Panalytical X'Pert Pro using CuKα radiation (λ = 1.54060 Å). The High-Resolution Transmission Electron Microscope (HRTEM) used is a FEI Titan LB 80–300, operated at 300 kV. This microscope is equipped with a CEOS image corrector and has a point resolution of 0.8 Å.Results and discussion
The first step of this investigation is related to the impregnation efficiency. As a first attempt, in order to determine if there is any agglomeration of crystallized metal on the Miscanthus surface, we performed SEM analysis on the treated fibers (for the case of Co + Fe nitrate catalyst). As observed in Fig. S1 (ESI†), even if the iron and cobalt elements are detected by EDS (Table S1 (ESI†)), we cannot distinguish macro metal agglomerates from the impregnated fiber being similar to the unimpregnated fiber. This is a first sign of a homogeneous repartition of the metal species on the fiber at the molecular level. Secondly, we were interested to check if the metal catalyst succeeded to reach the smallest pores present in the system, in the fiber core. It could assure a relative homogeneity in the catalysed pyrolysis (i.e. uniformity in terms of catalytic efficiency both inside the fiber and on its surface). Good insight was provided by the adsorption results from the N2 isotherm. Fig. S2† presented in the ESI† exhibits the calculated pore volume distribution of both raw Miscanthus and Co + Fe impregnated Miscanthus. There is a noticeable disappearance/decrease of the pore volume population related to a half pore width in between 40 and 190 Å, and appearance of a new population in between 20 and 30 Å. These differences could be associated with an efficient catalyst filling in the pores, with the new population being attributed to the pores that are only partially filled by the catalyst. As a result, the catalyst seemed to reach the macro and mesopores present in Miscanthus, a sign of a good impregnation methodology. After this first statement about the impregnation efficiency, we will focus on the pyrolysis step. The weight variation as well as the rate of change in weight percent throughout the pyrolysis of each sample can be seen in Fig. 1. As observed in Fig. 1 the majority of the weight change occurs between roughly 200 °C and 400 °C. To get more information about the precise conversion of the samples, the derivative curves were analyzed, seen in Fig. 2. It can be seen that there are some common trends across treatments. The initial weight loss up to about 130 °C can be attributed to the residual water evaporation. The rapid weight loss seen around 290 °C can be associated with the degradation of hemicellulose in the Miscanthus.27,28 The next rapid weight loss at around 340 °C is due to the degradation of the cellulose,27,28 while the shoulder on the curve between 400 °C and 500 °C is a result of the degradation of the lignin.27 We notice a sharp peak that occurs exclusively in the iron treated sample just above 600 °C. This peak is not related to any artefacts since it appears in every replicate we made. Thus it is a possible result of the iron reactions which release gas and occur above 600 °C, according to the mechanism and reaction shown below.29 | ||
| Fig. 1 Pyrolysis thermograms of treated Miscanthus with different metals. | ||
 | ||
| Fig. 2 The associated weight derivatives of treated Miscanthus with different metals. | ||
Hydrolysis:
| Fe(NO3)3 + 3H2O → Fe(OH)3 + 3HNO3 |
Dehydration:
| 2Fe(OH)3 → Fe2O3 + 3H2O |
Reduction:
| Fe2O3 + H2 → 2Fe + 3H2O |
| Fe2O3 + 3CO → 2Fe + 3CO2 |
| 2Fe2O3 + 3C → 4Fe + 3CO2 |
The thermograms produced by the cobalt treated sample look slightly different from the iron and untreated homologs, with a small peak at 167 °C, a shoulder at 256 °C and a peak at 311 °C. These differences suggest that a new set of reactions may be occurring between the Co(NO3)2 and the biomass at low temperatures. The changes occur mainly between 150 °C and 350 °C, indicating that it may be related to the degradation of hemicellulose and cellulose.30,31 On another hand, the sample treated with both iron and cobalt shows characteristics resembling the curves of those treated with iron and cobalt individually. It shows a similar hemicellulose peak to the iron treated sample, and shifted peaks at lower temperatures similar to those of the cobalt treated sample. Herein, the peak at 159 °C may correspond to that at 167 °C in the cobalt sample, and the peak at 222 °C may be related to the shoulder at 256 °C. The hemicellulose peak is lower in the samples treated with iron or iron and cobalt because when hemicellulose degrades, CO is produced,32 which the iron oxides react with.29
In order to have more information about the different catalysts’ effect on the ligno-cellulosic degradation, FTIR analysis was performed on all samples at the temperature of 500 °C as seen in Fig. 3. It is known that as heat treatment temperature (HTT), or pyrolysis conversion, increases the amount of functional groups decreases, and therefore peak absorbance intensity decreases in the spectra. Spectra disparity between the samples is already a proof highlighting the catalyst efficiency. The broad peak near 1030 cm−1 in each sample can be associated with C–O stretching found in ethers.33 This could be from either aryl or alkyl ethers that may be present after the depolymerisation of cellulose and hemicellulose, and the dehydration of levoglucosan.27 The small, sharp peak observed at 1435 cm−1 in the untreated sample is caused by C–O–H bending,34 which could possibly be a result of products such as oligosaccharides formed during the depolymerisation of cellulose and hemicellulose.27,28 The peak at 1580 cm−1 can be seen in all of the samples, and represents C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic bonds.35 These could be from aromatic lignin since it is possible that not all lignin has been fully degraded at this temperature.27 Evidence of this can be seen in Fig. 2, showing the lignin degradation occurring from about 400 °C to 600 °C. These bonds could also be present because of the newly formed groups of C6 rings that may later contribute to either the graphitic or amorphous content throughout the structure.36 This will be discussed further with respect to Raman spectroscopy in the following section. The sharp peak observed at 1740 cm−1 in the untreated sample corresponds to CO stretching,34,35 which may be present after the depolymerisation of cellulose and subsequently levoglucosan,28 or after the depolymerisation of hemicellulose which eventually degrades into furfural.27 The broad band seen in all samples from 2842–3000 cm−1 can be attributed to the C–H aliphatic bonds that may be present due to methyl groups seen throughout lignin degradation.27 All of these bands seem to disappear or become indistinguishable at temperatures above 500 °C, due to the loss of functional groups at higher temperatures.
C aromatic bonds.35 These could be from aromatic lignin since it is possible that not all lignin has been fully degraded at this temperature.27 Evidence of this can be seen in Fig. 2, showing the lignin degradation occurring from about 400 °C to 600 °C. These bonds could also be present because of the newly formed groups of C6 rings that may later contribute to either the graphitic or amorphous content throughout the structure.36 This will be discussed further with respect to Raman spectroscopy in the following section. The sharp peak observed at 1740 cm−1 in the untreated sample corresponds to CO stretching,34,35 which may be present after the depolymerisation of cellulose and subsequently levoglucosan,28 or after the depolymerisation of hemicellulose which eventually degrades into furfural.27 The broad band seen in all samples from 2842–3000 cm−1 can be attributed to the C–H aliphatic bonds that may be present due to methyl groups seen throughout lignin degradation.27 All of these bands seem to disappear or become indistinguishable at temperatures above 500 °C, due to the loss of functional groups at higher temperatures.
 | ||
| Fig. 3 Comparison of FTIR spectra of treated and untreated Miscanthus grass pyrolyzed at 500 °C. | ||
After this first insight revealing the catalytic efficiency of these metal salts on biomass decomposition, we will focus on the characterization and mechanism involved in the graphitic biocarbon creation. Firstly, SEM and EDS data, available in Table 2, were used to gather information on both biocarbon morphology and elemental composition of the various samples. As seen in the micrographs, there is no significant difference in the appearance between samples of different temperatures or treatments. The biocarbon shows a long, fibrous structure, which is often broken into smaller pieces. The surfaces of the particles have a tree bark-like appearance around the outside, covering a porous structure inside that resembles a honeycomb structure, in good agreement with the literature.37 The largest particles observed had a length of roughly 300 μm. From EDS analysis it was determined that the metals were homogeneously dispersed throughout the biocarbon particles, rather than grouped together as separate masses. Silicon was found consistently in the samples, as a result of the ash present. The ash percentage, for the untreated biocarbon, was calculated according to ASTM standard E-1131, and was found to be 18.2%, which is reasonable when compared to the published values of Miscanthus biocarbon.37 In general it can be seen that the oxygen content decreases with pyrolysis temperature, which is expected. Because of the release of volatile organic matter, the percentage of metal in the sample increases as temperature increases. This matches the trend from the TGA results, seen in Table 1, which shows the metal percentage increasing, based on the calculated yield of biocarbon, and weight of the metals. It appears that there is more iron present than cobalt, though this could be due to the iron carbide species being closer or part of the surface of the particles, while the cobalt is more embedded, to provide a site for graphite sheet growth.
| Temperature | Co + Fe BC | Co BC | Fe BC | Untreated BC | |
|---|---|---|---|---|---|
| % Yield | 500 °C | 30.7 | 31.1 | 32.6 | 28.8 |
| 700 °C | 24.8 | 25.7 | 27.9 | 25.7 | |
| 900 °C | 22.2 | 24.4 | 25.2 | 24.1 | |
| % Metal | 500 °C | 6.7 | 6.8 | 6.1 | 0 |
| After | 700 °C | 8.3 | 8.2 | 7.2 | 0 |
| Pyrolysis | 900 °C | 9.3 | 8.7 | 7.9 | 0 |
| Fe BC | Co BC | Fe + Co BC | Untreated BC | |
|---|---|---|---|---|
 Carbon; Carbon;  oxygen; oxygen;  iron; iron;  cobalt; cobalt;  silicon. silicon. |
||||
| 500 °C |

|

|

|

|
| 700 °C |

|

|

|

|
| 900 °C |

|

|

|

|
Raman spectroscopy is a powerful tool to highlight the allotropic carbon structures forming throughout the graphitization process. The spectra, in Fig. 4(A), were analyzed between 800 cm−1 and 1800 cm−1 to give insight into the relative amounts of graphite. In total, 10 bands were used to fit the spectra, based on the work of Keown et al.38 as seen in Fig. 4(B). Traditionally, 2–5 bands are reported,14,19,39 however in this case a 10 peak model provides a composite curve that is a much closer fit to the given spectrum. Each of these 10 peaks are defined and explained in the literature, and correspond to specific chemical structures within the biomass, as shown in Table 3. In this investigation, analysis was performed based on the G and D peaks. The G and D bands were used to calculate the intensity ratio, ID/IG, defined as the area of the D peak over the area of the G peak. This intensity ratio is frequently used as a measure of graphitization,20,39 with lower ratios indicating lower disorder and higher amounts of graphite. The G peak was defined as the band positioned between 1520 cm−1 and 1600 cm−1, and is known to represent sp2 bonding in graphitic carbon.40 The D peak was defined as the band between 1320 cm−1 and 1365 cm−1, and represents disorder in the carbon structure.40 The results of the intensity ratio analysis are shown in Fig. 4(C). Looking at the trends across temperatures, it is clear that the intensity ratio is the highest at 500 °C for all treated samples, and tends to be the lowest at 900 °C. Each of the treated samples show similar values at 700 °C and 900 °C. This trend is the opposite of that observed in the untreated sample with increasing HTT. The Raman spectra of the untreated samples resemble the typical spectra of pyrolyzed biocarbon.38,41 The intensity ratio of the untreated sample increases drastically from 500 °C to 900 °C, indicating that with increasing temperature comes increasing disorder, in the absence of a catalyst. Jones et al.20 explained this increasing disorder as a result of gas species including CH4, CO2, CO and H2O forming throughout pyrolysis up to this point. These gasses evolve throughout the structure, promoting disorder throughout the structure until after 1000 °C. Then, when thermal degradation is complete, a reordering of the carbonaceous structure begins to take place.20 In the presence of a catalyst however, reactions with these gasses occur,29 which may promote the reordering of the carbon structure at lower temperature. These results for both treated and untreated samples agree well with the trends observed by Maldonado-Hódar et al.21 When comparing the treatments with each other, it can be seen that at any temperature, iron treated samples have lower and therefore better intensity ratios than cobalt treated samples. The results also show that at the higher HTTs of 700 °C and 900 °C, all treated samples show lower intensity ratios than the untreated sample. Note that at 500 °C, the untreated sample appears to have higher graphitization than the samples treated with cobalt, or both cobalt and iron. This may be due to the zerovalent cobalt and intermediate cobalt carbide species formed before the precipitation of graphite.42 The metals may be initially promoting disorder in the carbon structures, since the catalytic effects on graphitization are theorized to occur only above 500 °C.29,42 Finally, it is observed that at higher HTTs, samples treated with a mixture of both iron and cobalt together perform better in terms of graphitization than samples using either one of the metals individually. The results of this treatment, particularly at 900 °C, are comparable to the results found by Maldonado-Hódar et al.21 at 1800 °C with the use of a chromium catalyst on carbon aerogels prepared from carbonizing formaldehyde and resorcinol. This result also shows similar graphitization to that of Jones et al.20 at temperatures up to about 1600 °C, using naphthalene as a carbon source, and without a catalyst. These results suggest that the catalysts are able to substantially lower the HTT to achieve similar graphitization to those at much higher temperatures, thereby reducing the energy required during production. This also suggests that using both catalysts together results in a more efficient catalytic species. Further evidence of the graphitic structure can be found in the broad band around 2700 cm−1 (Fig. 4(D)). The band at 2700 cm−1 is a result of second order resonance from the D band, and can be called the G′, 2D or D* band.40,43,44 This band is characteristic of stacked graphene layers in the sample.45,46 The results show the presence of this band in all the treated samples, and the absence of it in the untreated sample, indicating the occurrence of some graphene stacking. Increased graphitization produces higher and more well defined peaks in this region, as shown by Pawlyta et al.47 When comparing these bands across treatments, it is clear that these bands are the most well defined in the treatment using iron and cobalt together. This agrees with the results shown by the intensity ratios, indicating the highest amounts of graphitization. The bands of this sample are not nearly as sharp as those of pure graphene sheets or graphite,40,45,46 however they do resemble the shape of the bands in spectra from carbon black and soot samples.47,48
 | ||
| Fig. 4 Data from Raman spectroscopy, showing full spectra for each of the 900 °C heat treated samples (A), a sample deconvolution using the 10 peak curve fit (B), a comparison of the intensity ratios (C), and the 2D band of each of the 900 °C heat treated samples (D). | ||
| Band name | Band position (cm−1) | Description |
|---|---|---|
| Gl | 1700 | Carbonyl group CO |
| G | 1590 | Graphite E2g, aromatic ring breathing, CC |
| Gr | 1540 | Aromatics with 3–5 rings, amorphous carbon structures |
| Vl | 1465 | Methylene or methyl, semi-circle breathing of aromatic rings, amorphous carbon structures |
| Vr | 1380 | Methyl group, semi-circle breathing of aromatic rings, amorphous carbon structures |
| D | 1300 | D band on highly ordered carbonaceous materials, C–C between aromatic rings and aromatics with 6 or more rings |
| Sl | 1230 | Aryl-alkyl ether, para-aromatics |
| S | 1185 | Caromatics–Calkyl, aromatic (aliphatic) ethers, C–C on hydroaromatic rings, hexagonal diamond carbon sp3, C–H on aromatic rings |
| Sr | 1060 | C–H on aromatic rings, benzene (ortho-di-substituted) ring |
| R | 960–800 | C–C on alkanes and cyclic alkanes, C–H on aromatic rings |
| 2D | 2700 | π bonds in graphene sheets along the c axis |
Additional information on biocarbon graphitization could be achieved through the corroboration of Raman results and XRD analysis. Furthermore, this technique can also provide important insights about the metal species that were formed during the reaction and which play a fundamental role in the graphitization enhancement. As a first observation, the trends of the XRD results reflect those of Raman spectroscopy, in terms of graphitic content (Fig. 5(A)). Graphite is shown most distinctly by the (002) peak near 2θ = 26.0°. This peak can be seen clearly in all treated samples. In the untreated sample, a broad band exists, with no distinct peak at 2θ = 26.0°, indicating high amounts of disordered carbon. Both the broad band and the sharp band exist in all treated samples. Thus we proposed a comparison of their relative area ratios, as a measure of the graphitic content, after their very good fitting into two Gaussian peaks. The results of this analysis are shown in Table 4, and compared well with the intensity ratios from Raman spectroscopy at 900 °C. In both these tests, the samples treated with cobalt and iron together showed the highest amount of graphitization. Also seen in Table 4 is the crystallite size of the graphite in nanometers for each sample (Lc), calculated using the Scherrer equation, based on the (002) peak. From this, the interlayer spacing (d002) was calculated, and used to find the graphitization degree parameter (g), used by Maldonado-Hódar et al.21 The g values found indicate the presence of graphite in all samples, and match the results obtained by Liu et al.29 In this case, the ratio of the peaks may give a more precise indication of graphitization in each sample, as it takes into account the relative amount of graphitic content as well as disorder, rather than relying solely on the size of the graphite sheets. XRD analysis was also helpful in determining the iron and cobalt species present. Zerovalent iron was likely present, shown by the (110) peak at 2θ = 44.7°.29 It can be seen in the samples that were treated with iron that Fe3C was formed, based on the peaks around the (110) peak, at 2θ = 42.9°, 43.7°, and 45.9°, as expected based on the findings of Thompson et al.13 The formation of this may be a result of iron reacting with gasses being formed during pyrolysis including CH449 or potentially through an intermediate Fe3N phase.50 The XRD pattern of the samples treated with iron and cobalt gives evidence of a Co/Fe alloy forming with face centered cubic (fcc)51 or body centered cubic (bcc),52 according to the (110), (200), and (211) peaks at 2θ = 45.0°, 65.5° and 83.0° respectively.53 Though the (110) peak can also be related to zerovalent iron, in this case the existence of the (200) and (211) peaks suggests that in this sample it may be due to a Co/Fe alloy as well.54 Based on the (110), (200), and (211) peaks, the Scherrer equation was used to calculate the nano-crystallite size of the Fe/Co alloy particles. The average size was found to be 27.9 nm, or 27.4 nm using the modified Scherrer equation proposed by Monshi et al.55 Both of these are in the normal range for typical Fe/Co alloys.53,55,56
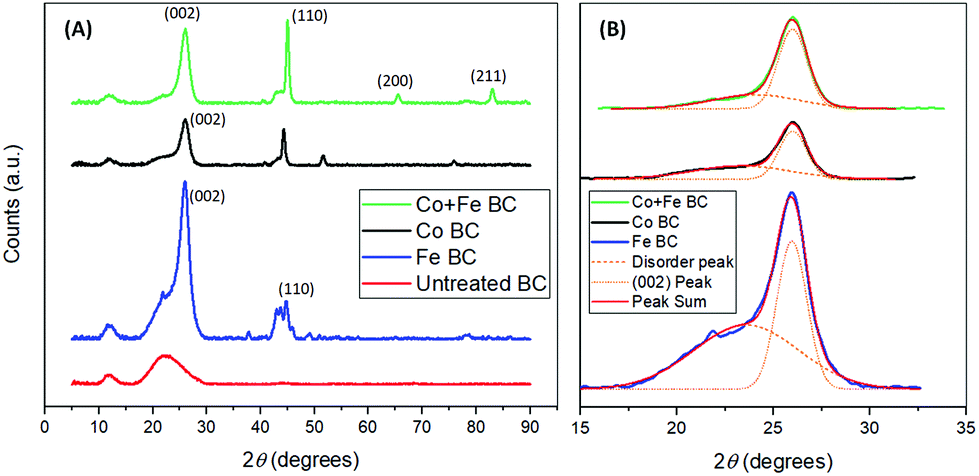 | ||
| Fig. 5 X-ray diffractograms of the biocarbons prepared at 900 °C (A) and related curve fitting to determine graphitic content (B). | ||
| Raman ID/IG | XRD disorder/(002) |
L
ca (nm) |
d
002b (nm) |
n | g | |
|---|---|---|---|---|---|---|
|
a Scherrer equation Lc = kλ/βcosθ.
b Bragg's law d002 = nλ/2sinθ.
c
n = Lc/d002.
d
g = (0.344 − d002)/0.0086.
|
||||||
| Co + Fe BC | 0.70 | 0.53 | 9.21 | 0.342 | 26.9 | 0.233 |
| Co BC | 1.33 | 0.98 | 9.70 | 0.342 | 28.4 | 0.233 |
| Fe BC | 1.18 | 1.52 | 8.91 | 0.343 | 26.0 | 0.116 |
| Untreated BC | 5.52 | N/A | N/A | |||
HRTEM was used to validate the potential existence of nanoparticles in the system treated with both iron(III) nitrate and cobalt(II) nitrate. Fig. 6(A and B) shows dark, dense masses of roughly 30 nm dispersed throughout the lighter carbon structure. These nanoparticles are likely FeCo alloy nanoparticles, based on the diffraction peaks and size calculations from XRD. In Fig. 6(C and D) it appears that even along the edge of the carbon phases, the alloy nanoparticles are surrounded by carbon layers, described as a “core–shell structure”.52 This may be due to the nanoparticles providing a site for carbon to begin stacking, eventually promoting the outward growth of graphite layers.57 The confirmation of the presence of FeCo nanoparticles not only allows for understanding the catalytic pyrolysis mechanism, but proceeds to open new routes to the development of multifunctional materials. The unique magnetic properties of FeCo nanoparticles, which possess both high Curie temperature and saturation magnetization, make them very promising nanomaterials for a myriad of applications.58 Their magnetic properties are an important asset that could offer better understanding of the graphitization process. Indeed, the TGA thermogram shown in Fig. S3 (ESI†) represents the same scan of Co + Fe BC presented in Fig. 1, however, this time the electromagnet positioned below the pan containing the sample is active. Herein, if a magnetic substance is generated during the pyrolysis, the measured weight during the scan should be perturbed. We can indeed observe a sharp discontinuity appearing at 488 °C which could be associated with the generation of FeCo magnetic nanoparticles. This temperature is in good agreement with those found in the literature for obtaining this kind of alloy.59 Furthermore, this temperature matches with the reduction of the metal species by H2 as an example. The reductive gases generated through the biomass pyrolysis should occur below 600 °C, as described for pure iron in the chemical reaction drawn above. A small perturbation was also noticed at higher temperatures, in between 600 and 900 °C. This could be associated with the catalyzed sequential generation of graphite which is supposed to start to grow in between these temperatures. Now we have sufficient information to put forth a proposal of a scenario describing the events occurring during the pyrolysis. The metal species undergo different chelations throughout the pyrolysis process, providing a catalysis effect at each step. First the iron and cobalt species could catalyze the cascade of dehydration reactions allowing the ligno-cellulose depolymerization. Then they could consume the syngas released by the pyrolysis biomass, catalyzing the carbonization process. Eventually around 500 °C, after reduction of the iron and cobalt species, the FeCo alloy is generated that permits the catalysis of the graphite production. A last point to highlight can be related to the catalytic mechanism responsible for the graphite growing at the FeCo surface. The corroborate results of HRTEM with the literature can provide substantial information about a possible mechanism. Indeed, if we look carefully at Fig. 6(E and F) we can observe a parallelism in between both FeCo and the graphite lattice. This epitaxial relationship between the (110) plane of FeCo and the (002) plane of graphite argues in favor of a growing graphite monocrystal orientation provided by the FeCo substrate.59 Furthermore, the locally darker zone observed in the interface between FeCo and the graphene layers is a well-known phenomenon generally observed during the inspection of strained-layer epitaxies. It is a result of strong mechanical stress between the alloy and the carbon phases, explained by metal–carbon covalent bonds. Further discussion about the mechanism of graphite growing at the FeCo surface can be found in ref. 60. The authors confronted the bulk and surface diffusion model with the carbide migration model proposed by Schaper et al.61
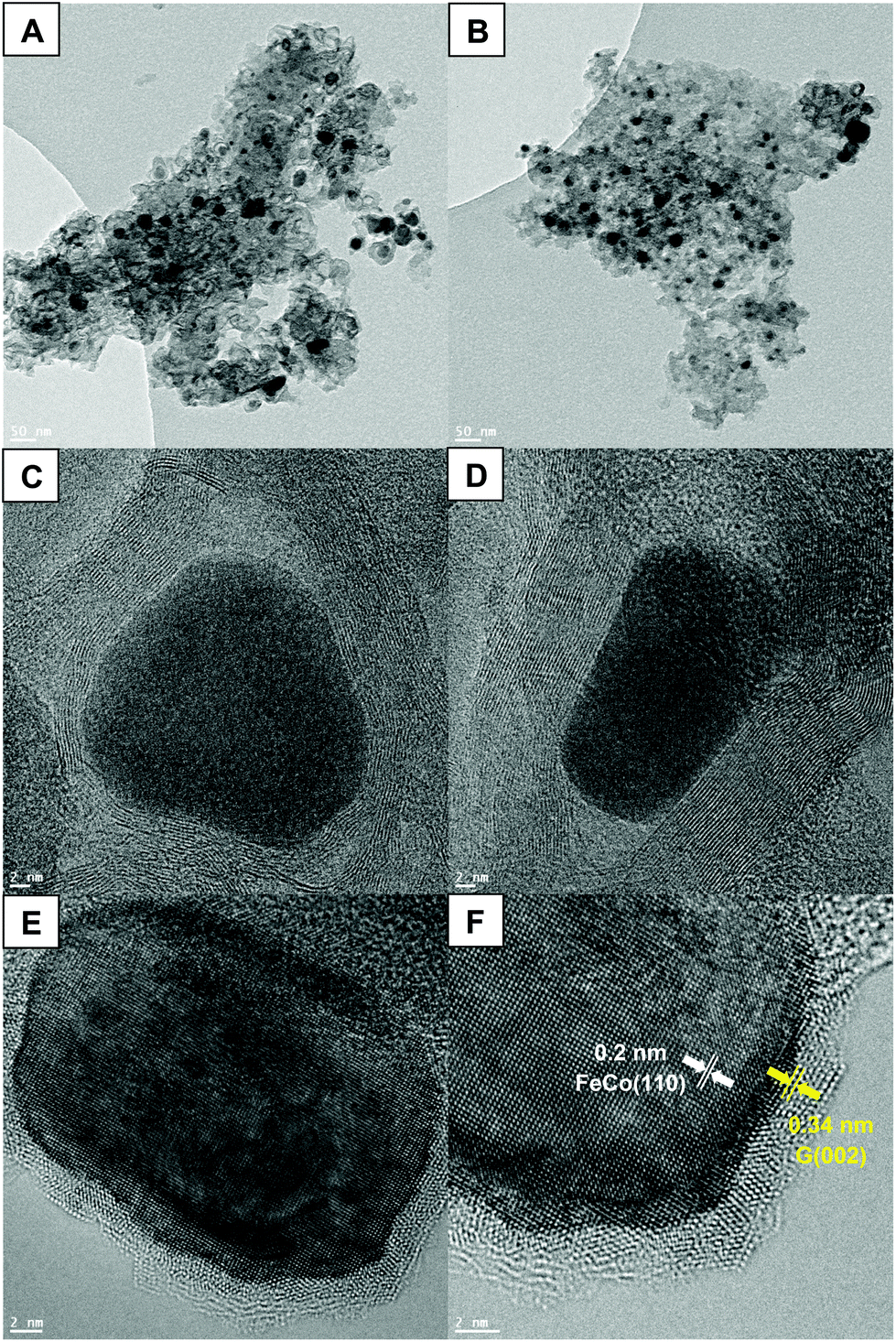 | ||
| Fig. 6 HRTEM micrographs of Co + Fe BC: dense FeCo alloy nanoparticles are more contrasted than the carbon phase (A) and (B). FeCo nanoparticles with their graphitic shell (C) and (D). Observation of FeCo nanoparticles/graphite interface and assignation of FeCo alloy face-centred-cubic structure FeCo(110) as well as the stacked graphene layers of graphite G(002). | ||
FeCo nanoparticles have a sensitive character toward oxidation making the preparation and utilization of these nanoparticles challenging. One of the best strategies is to use graphitic carbon or graphene to prevent this oxidation. The addition of this carbon shell further increases the potential of these nanomaterials by giving new properties such as high optical absorbance in the near-infrared region.62 Another very interesting application for these types of core–shell FeCo-graphite nanoparticles is related to the catalysis field:63 it has been shown that these nanoparticles are very efficient in the catalysis of reduction reactions.64 This last application is particularly relevant for further developing our benchmark study. Indeed, the natural porosity of biocarbon coupled with the FeCo graphite nanoparticles makes this composite a very promising starting material for heterogeneous catalysis.
Conclusion
The synergistic catalytic effect of both iron(III) and cobalt(II) nitrates on the pyrolysis of Miscanthus grass has been highlighted. The utilization of both metal species has shown to be much more efficient for producing more ordered graphitic carbon than the iron and cobalt catalysts used separately. After observing the catalytic effect of the iron and cobalt on the ligno-cellulosic biomass degradation, the in situ production of new metal species revealed by XRD induced a profound impact on the level of biocarbon graphitization. After confrontation and discussion of different measures of the graphitization based on the Raman and XRD data exploitation, it has been proposed that the highest level of graphitization has been reached due to the creation of FeCo alloy nanoparticles. The direct perspective of this work can be related to the heat treatment. Indeed, for modeling purposes, we have been interested in temperatures below 1000 °C, however the yield of graphite could be improved by increasing the pyrolysis temperature. In a more general context, these findings open the door for a wide variety of uses for renewable graphitized biocarbon, and allow for the creation of cost-competitive, environmentally friendly, smart materials for catalysis, electronics and composites. As a tangible example, the creation of FeCo/graphite composite has been already shown to have a great potential for catalysis and biological applications due to its particular magnetic properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the Natural Sciences and Engineering Research Council (NSERC), the Canada Discovery Grants (Project # 401111, 400320 and NSERC- CRD Project # 401190) and the Ontario Ministry of Research, Innovation and Science (MRIS) Ontario Research Fund, Research Excellence Program Round-7 (ORF-RE07) (Project # 052644 and 052665). The authors would like to deeply thank Dr James F. Britten and Victoria Jarvis, from McMaster University, for their assistance in XRD measurements.References
- Y. Zhan and G. Zhang, Mater. Des., 2006, 27, 79–84 CrossRef.
- R. N. Bhowmik, Composites, Part B, 2012, 43, 503–509 CrossRef.
- M. Terrones, A. R. Botello-Méndez, J. Campos-Delgado, F. López-Urías, Y. I. Vega-Cantú, F. J. Rodríguez-Macías, A. L. Elías, E. Muñoz-Sandoval, A. G. Cano-Márquez, J.-C. Charlier and H. Terrones, Nano Today, 2010, 5, 351–372 CrossRef.
- W.-J. Liu, H. Jiang and H.-Q. Yu, Chem. Rev., 2015, 115, 12251–12285 CrossRef PubMed.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- M. Wissler, J. Power Sources, 2006, 156, 142–150 CrossRef.
- E. Behazin, M. Misra and A. K. Mohanty, Polym. Test., 2017, 61, 364–372 CrossRef.
- K. Qian, A. Kumar, H. Zhang, D. Bellmer and R. Huhnke, Renewable Sustainable Energy Rev., 2015, 42, 1055–1064 CrossRef.
- N. Z. Muradov and T. N. Veziroğlu, Int. J. Hydrogen Energy, 2008, 33, 6804–6839 CrossRef.
- M. Ahmad, A. U. Rajapaksha, J. E. Lim, M. Zhang, N. Bolan, D. Mohan, M. Vithanage, S. S. Lee and Y. S. Ok, Chemosphere, 2014, 99, 19–33 CrossRef PubMed.
- S. C. Peterson, M. A. Jackson, S. Kim and D. E. Palmquist, Powder Technol., 2012, 228, 115–120 CrossRef.
- R. Azargohar and A. K. Dalai, Microporous Mesoporous Mater., 2008, 110, 413–421 CrossRef.
- E. Thompson, A. E. Danks, L. Bourgeois and Z. Schnepp, Green Chem., 2015, 17, 551–556 RSC.
- A. Anstey, S. Vivekanandhan, A. Rodriguez-Uribe, M. Misra and A. K. Mohanty, Sci. Total Environ., 2016, 550, 241–247 CrossRef PubMed.
- M. J. Ahmed, M. A. Islam, M. Asif and B. H. Hameed, Bioresour. Technol., 2017, 243, 778–784 CrossRef PubMed.
- E. A. Heaton, F. G. Dohleman and S. P. Long, Glob. Chang. Biol., 2008, 14, 2000–2014 CrossRef.
- R. Muthuraj, M. Misra and A. K. Mohanty, RSC Adv., 2017, 7, 27538–27548 RSC.
- J. S. McDonald-Wharry, M. Manley-Harris and K. L. Pickering, Energy Fuels, 2016, 30, 7811–7826 CrossRef.
- G. A. Zickler, B. Smarsly, N. Gierlinger, H. Peterlik and O. Paris, Carbon, 2006, 44, 3239–3246 CrossRef.
- S. P. Jones, C. C. Fain and D. D. Edie, Carbon, 1997, 35, 1533–1543 CrossRef.
- F. J. Maldonado-Hódar, C. Moreno-Castilla, J. Rivera-Utrilla, Y. Hanzawa and Y. Yamada, Langmuir, 2000, 16, 4367–4373 CrossRef.
- C. P. Deck and K. Vecchio, Carbon, 2006, 44, 267–275 CrossRef.
- M. Demir, Z. Kahveci, B. Aksoy, N. K. R. Palapati, A. Subramanian, H. T. Cullinan, H. M. El-Kaderi, C. T. Harris and R. B. Gupta, Ind. Eng. Chem. Res., 2015, 54, 10731–10739 CrossRef.
- N. M. Rodriguez, A. Chambers and R. T. K. Baker, Langmuir, 1995, 11, 3862–3866 CrossRef.
- A. Nzihou, B. Stanmore and P. Sharrock, Energy, 2013, 58, 305–317 CrossRef.
- X. Fu, Y. Liu, X. Cao, J. Jin, Q. Liu and J. Zhang, Appl. Catal., B, 2013, 130, 143–151 CrossRef.
- W.-J. Liu, W.-W. Li, H. Jiang and H.-Q. Yu, Chem. Rev., 2017, 117, 6367–6398 CrossRef PubMed.
- F. Shafizadeh, J. Anal. Appl. Pyrolysis, 1982, 3, 283–305 CrossRef.
- Z. Liu, F. Zhang, S. K. Hoekman, T. Liu, C. Gai and N. Peng, ACS Sustainable Chem. Eng., 2016, 4, 3261–3267 CrossRef.
- M. V. Gil, D. Casal, C. Pevida, J. J. Pis and F. Rubiera, Bioresour. Technol., 2010, 101, 5601–5608 CrossRef PubMed.
- G. Zheng and J. A. Koziński, Fuel, 2000, 79, 181–192 CrossRef.
- N. Worasuwannarak, T. Sonobe and W. Tanthapanichakoon, J. Anal. Appl. Pyrolysis, 2007, 78, 265–271 CrossRef.
- C. H. Chia, B. Gong, S. D. Joseph, C. E. Marjo, P. Munroe and A. M. Rich, Vib. Spectrosc., 2012, 62, 248–257 CrossRef.
- M. Schwanninger, J. C. Rodrigues, H. Pereira and B. Hinterstoisser, Vib. Spectrosc., 2004, 36, 23–40 CrossRef.
- E. Smidt and K. Meissl, Waste Manage., 2007, 27, 268–276 CrossRef PubMed.
- X. Li, J.-i. Hayashi and C.-Z. Li, Fuel, 2006, 85, 1700–1707 CrossRef.
- E. Behazin, M. Misra and A. K. Mohanty, ACS Omega, 2017, 2, 2191–2199 CrossRef.
- D. M. Keown, X. Li, J.-i. Hayashi and C.-Z. Li, Fuel Process. Technol., 2008, 89, 1429–1435 CrossRef.
- J. McDonald-Wharry, M. Manley-Harris and K. Pickering, Carbon, 2013, 59, 383–405 CrossRef.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2004, 362, 2271–2288 CrossRef PubMed.
- M. M. Titirici, A. Thomas, S.-H. Yu, J.-O. Müller and M. Antonietti, Chem. Mater., 2007, 19, 4205–4212 CrossRef.
- D. Potoczna-Petru, Carbon, 1991, 29, 73–79 CrossRef.
- O. Frank, M. Mohr, J. Maultzsch, C. Thomsen, I. Riaz, R. Jalil, K. S. Novoselov, G. Tsoukleri, J. Parthenios, K. Papagelis, L. Kavan and C. Galiotis, ACS Nano, 2011, 5, 2231–2239 CrossRef PubMed.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, N. L. Speziali, A. Jorio and M. A. Pimenta, Carbon, 2008, 46, 272–275 CrossRef.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef.
- A. Gupta, G. Chen, P. Joshi, S. Tadigadapa and P. C. Eklund, Nano Lett., 2006, 6, 2667–2673 CrossRef PubMed.
- M. Pawlyta, J.-N. Rouzaud and S. Duber, Carbon, 2015, 84, 479–490 CrossRef.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef.
- X. L. Dong, Z. D. Zhang, Q. F. Xiao, X. G. Zhao, Y. C. Chuang, S. R. Jin, W. M. Sun, Z. J. Li, Z. X. Zheng and H. Yang, J. Mater. Sci., 1998, 33, 1915–1919 CrossRef.
- Z. Schnepp, A. E. Danks, M. J. Hollamby, B. R. Pauw, C. A. Murray and C. C. Tang, Chem. Mater., 2015, 27, 5094–5099 CrossRef.
- J. Tong, W. Wang, Q. Li, F. Liu, W. Ma, W. Li, B. Su, Z. Lei and L. Bo, Electrochim. Acta, 2017, 248, 388–396 CrossRef.
- A. Wu, X. Yang and H. Yang, Dalton Trans., 2013, 42, 4978–4984 RSC.
- G. S. Chaubey, C. Barcena, N. Poudyal, C. Rong, J. Gao, S. Sun and J. P. Liu, J. Am. Chem. Soc., 2007, 129, 7214–7215 CrossRef PubMed.
- A. G. Kolhatkar, I. Nekrashevich, D. Litvinov, R. C. Willson and T. R. Lee, Chem. Mater., 2013, 25, 1092–1097 CrossRef PubMed.
- A. Monshi, M. R. Foroughi and M. Reza Monshi, Modified Scherrer Equation to Estimate More Accurately Nano-Crystallite Size Using XRD, 2012 Search PubMed.
- V. Pillai and D. O. Shah, J. Magn. Magn. Mater., 1996, 163, 243–248 CrossRef.
- P. E. Anderson and N. M. Rodriguez, J. Mater. Res., 2011, 14, 2912–2921 CrossRef.
- M. Yamada, S.-j. Okumura and K. Takahashi, J. Phys. Chem. Lett., 2010, 1, 2042–2045 CrossRef.
- A. Elias, J. Rodriguez-Manzo, M. McCartney, D. Golberg, A. Zamudio, S. Baltazar, F. Lopez-Urias, E. Munoz-Sandoval, L. Gu and C. Tang, Nano Lett., 2005, 5, 467–472 CrossRef PubMed.
- J. A. Rodríguez-Manzo, M. Terrones, H. Terrones, H. W. Kroto, L. Sun and F. Banhart, Nat. Nanotechnol., 2007, 2, 307 CrossRef PubMed.
- A. K. Schaper, H. Hou, A. Greiner and F. Phillipp, J. Catal., 2004, 222, 250–254 CrossRef.
- W. S. Seo, J. H. Lee, X. Sun, Y. Suzuki, D. Mann, Z. Liu, M. Terashima, P. C. Yang, M. V. McConnell, D. G. Nishimura and H. Dai, Nat. Mater., 2006, 5, 971–976 CrossRef PubMed.
- L. Hu, R. Zhang, L. Wei, F. Zhang and Q. Chen, Nanoscale, 2015, 7, 450–454 RSC.
- X. Fu, Y. Liu, X. Cao, J. Jin, Q. Liu and J. Zhang, Appl. Catal., B, 2013, 130–131, 143–151 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C7GC03457A |
| This journal is © The Royal Society of Chemistry 2018 |