
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew biobased non-ionic hyperbranched polymers as environmentally friendly antibacterial additives for biopolymers†
Carlos R.
Arza
a,
Sedef
İlk
b,
Deniz
Demircan
a and
Baozhong
Zhang
 *a
*a
aLund University, Centre for Analysis and Synthesis, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: baozhong.zhang@chem.lu.se
bÖmer Halisdemir University, Central Research Laboratory, TR-51240, Niğde, Turkey
First published on 1st February 2018
Abstract
The aim of this research was to develop new biobased non-ionic polymeric additives with significant bacterial inhibition and low leaching potential, so that they can be used to produce biopolymer materials for various applications such as biomedical devices, surgical textile, or food packaging. Two new non-ionic hyperbranched polymers (HBPs) were prepared by a facile solvent-free polymerization of an AB2-monomer derived from naturally existing molecular building blocks 2-phenylethanol, isatin, and anisole. The molecular structures and thermal properties of the obtained HBPs were characterized by GPC, NMR, FTIR, HRMS, MALDI-TOF, TGA and DSC analyses. Disk diffusion tests revealed that the two obtained HBPs showed more significant antibacterial activity against 9 different food and human pathogenic bacteria, compared with small molecular antibiotics. The maximal antibacterial effect of HBPs was achieved at 2 μg per disk (or 0.1 mg mL−1), which was significantly lower (∼1/15) compared to the linear antibacterial polymer chitosan. Such enhanced antibacterial properties can be attributed to the unique highly branched structures and effectively amplified functionalities of HBPs. Finally, the prepared HBPs were added into natural polymers cellulose and polyhydroxybutyrate (PHB), and the resulting biopolymer films showed no significant leakage after being merged in water for 5 days. This was in sharp contrast to the biopolymer films containing a small model compound, which leaked out significantly under the same conditions. To our knowledge, this is the first report on non-ionic bio-based dendritic macromolecules with significant bacteria inhibition and low leakage.
1. Introduction
Today, the continuously growing fossil consumption of plastics and the associated environmental challenges (e.g. plastic pollution, greenhouse gas emission, toxic additives) have gained enormous attention from society and academia. Currently, plastics derived from sustainable resources (biopolymers) are considered the most promising solution for such challenges.1,2 Among the large variety of biopolymers, those with good biocompatibility and biodegradability (e.g. PLA, PHAs, cellulose) are of particular importance for the development of food packaging materials, clinical textile, and biomedical devices.3–5 A particular challenge for biopolymers in such applications is bacterial contamination, which may cause severe food contamination or bacterial infections for patients.6,7 As such, there has been a strong demand from the society and industry for the development of new biopolymer materials with bacteria inhibition. The addition of small antibacterial agents (e.g. antibiotics, drugs, or metal nanoparticles) into biopolymers is an industrially adopted strategy for water sterilization and food preservation.8–10 However, such biopolymer materials can suffer from the leakage of small antibacterial agents, which can reduce the antibacterial effect and threaten the environment.11 Therefore, non-leachable green additives for biopolymer materials with a strong antibacterial effect are expected to greatly facilitate the development of biopolymers and benefit the society.Covalently grafting antibacterial functionalities onto polymers is a convenient strategy to achieve non-leachable antibacterial materials. Compared to the use of small molecular antibacterial agents, polymers with covalently grafted antibacterial functionalities have advantages like increased efficacy and selectivity, lower eco-toxicity, longer lifetime, and lower risk of skin permeation.12–14 Such antibacterial polymers have great potential in the production of sterile bandages, surgical clothing, food-contact plastic containers or films, or various anti-biofouling medical implants. In the past decade, the number of FDA-approved antibacterial polymers has increased significantly, which indicates the growing demand of such materials. The state of the art antibacterial polymers have been summarized in several recent reviews.15–19
Today, diversified synthetic and natural antibacterial polymers have been reported, of which the majority are positively charged polymers that can inhibit the bacterial growth by ionic interactions with anionic bacterial membranes.20–24 Anionic antibacterial polymers have also been reported, in which the bacterial inhibition was endowed by the cationic counter-ions of the polymers.25 However, ionic polymers are not ideal additives for biopolymers, because they usually suffer from poor miscibility with a non-ionic biopolymer matrix and they have relatively high water solubility and eco-toxicity.26 On the other hand, due to the lack of ionic interactions, non-ionic polymers need to have specific functionalities to interact with bacteria. It is known that toxic functionalities (e.g. chlorine, organotin) can endow polymers with antibacterial properties,16 but the use of such toxic groups are potentially dangerous for the environment and people's health. Meanwhile, non-harmful antibacterial functionalities are abundant in nature (e.g. isatin, anisole, curcumin, astaxanthin), and the incorporation of such naturally existing functional groups can provide antibacterial polymers. An example of this kind of polymer dates back to 1965, when Cornell and Donaruma reported the enhanced antibacterial effect of tropolone-containing polymers.27 More examples include aspirin-based non-ionic polyesters,28,29 curcumin-derived biopolymers,30,31 limonene-based polycarbonates,32 and astaxanthin-based polymers.33 Today, all the reported polymers with natural antibacterial functionalities have linear structures, with the functionalities being incorporated in the backbone or on the pendant groups of polymers. Meanwhile, the recent advances in the polymer synthesis allowed for the production of highly branched polymer structures by “dendronization” or “hypergrafting” strategies,34,35 which can conveniently amplify or modify the desired functionality.36 Such a new class of dendritic polymers are of great interest in the context of antibacterial polymers, because it is known that densely grafted positive charges on dendritic polymers can significantly enhance their interactions with the bacterial membrane.37–44 However, it is unclear whether the bacteria inhibition can be amplified by the localization of the non-ionic antibacterial functionalities of dendritic polymers. To date, non-ionic dendritic polymers have only been investigated as anti-viral drugs,45,46 and we are unaware of any investigation on antibacterial non-ionic dendritic polymers.
This work aims to prepare non-ionic dendritic polymers derived from bio-based resources, and to use such new polymers as non-leachable additives for matrix biopolymers to achieve desirable antibacterial properties. Such new antibacterial dendritic polymers are expected to have three main advantages: (1) an enhanced antibacterial effect by the amplified natural functional groups, (2) reduced toxicity and improved compatibility with matrix biopolymers due to the non-ionic nature of such polymers, and (3) eco-friendliness due to the low leaching potential. For this aim, 3 key bio-based building blocks (1–3, Scheme 1) were selected for the construction of bio-based dendritic functional polymers. 2-Phenylethanol (1) is widely present in essential oils,47 which can be produced by bio-fermentation of tobacco waste.48 Isatin (2) is well known for its biological activities in plants such as bacteria inhibition.49–54 Anisole (3) and its derivatives (e.g. anethole or butylated hydroxyanisole) are effective antioxidant and antimicrobial agents that can be used in the food industry.55 Anisole is also a naturally occurring molecule that has been recommended by academia and the pharmaceutical industry as a green solvent.56 Herein, we report on a facile and green synthetic protocol to prepare two new non-ionic dendritic polymers (specifically, hyperbranched polymers or HBPs57–59) from bio-based building blocks 1–3, and the evaluations of their thermal properties, bacteria inhibition, and leaching potential as additives for biopolymers (i.e. polyhydroxybutyrate and regenerated cellulose). Compared to small molecular agents (model compounds or commercial antibiotics), the new macromolecular green additives showed significantly enhanced antibacterial activities and negligible leakage in water.
 | ||
| Scheme 1 Synthesis of AB2 monomer 4, HBPs 5 and 6, and a model compound 7. The biomass origins of the building blocks are geranium oil for phenylethanol (1), isatis flower for isatin (2), and anise seeds for anisole (3). | ||
2. Experimental section
2.1. Chemicals and materials
Cellulose (microcrystalline, powder), 2,5-dihydroxybenzoic acid (DHB) (>99%(HPLC)), N,N-dimethylacetamide (DMAc) (ReagentPlus, ≥99%), isatin, lithium chloride (LiCl) (BioXtra, ≥99.0%), 2-phenylethanol (≥99.0%), potassium carbonate (K2CO3) (puriss, p.a., ACS reagent, anhydrous, ≥99.0%), p-toluenesulfonyl chloride (≥98%), and trimethylamine (≥99%) were purchased from Sigma-Aldrich. Acetonitrile (MeCN) (HPLC grade) and chloroform (Analytical grade, stabilized with ethanol) were purchased from Scharlau. Dichloromethane (DCM) (Reag. Ph. Eur., ACS), NaHCO3 (ACS, Reag. Ph. Eur.), and MgSO4 were purchased from VWR Chemicals. Trifluoromethanesulfonic acid (TFSA) (99%) and anisole (99%) were purchased from Acros Organics. 2-Bromoethylbenzene (>97%) was purchased from Fluka. Methanol was purchased from Honeywell. Ethanol (99.7%) was purchased from Solveco. All chemicals and reagents were used as received. Poly(3-hydroxybutyrate) (PHB) powder was supplied by BIOMER (Germany), which was washed with 0.001 N aqueous HCl for 30 min, washed with deionized water, and dried at 50 °C under vacuum for 2 days before further use. The acid-treated PHB had a weight-average molar mass (Mw) of 620![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and a polydispersity index (PDI) of 2.0.
000 g mol−1 and a polydispersity index (PDI) of 2.0.
2.2. Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str.), 1727 (CO str.), 1613 (Ar C–C str.), 1470 (Ar C–CH in-plane bend. + Ar C–C str.), 1325 (Ar CCH in-plane bend. + C–N str. + Ar C–C str.), 1095 (Ar C–C str. + Ar C–CH in-plane bend. + Ar C–C str.), 751 (Ar C–H out-of-plane bend.). HRMS (ESI+) exact mass calcd for C16H14NO2 252.1025, found 252.1024.
O str.), 1608 (Ar C–C str.), 1507 (Ar C–CH in-plane bend. + Ar C–C str.), 1350 (Ar CCH in-plane bend. + C–N str. + Ar C–C str.), 1246 (ArO–C sym. str.), 1037 (ArO–C asym. str.), 745 (Ar C–H out-of-plane bend.). HRMS (ESI+) exact mass calcd for C30H28NO3 450.2069, found 450.2069.
O str.), 1727 (CO str.), 1613 (Ar C–C str.), 1470 (Ar C–CH in-plane bend. + Ar C–C str.), 1325 (Ar CCH in-plane bend. + C–N str. + Ar C–C str.), 1095 (Ar C–C str. + Ar C–CH in-plane bend. + Ar C–C str.), 751 (Ar C–H out-of-plane bend.). HRMS (ESI+) exact mass calcd for C16H14NO2 252.1025, found 252.1024.
O str.), 1608 (Ar C–C str.), 1507 (Ar C–CH in-plane bend. + Ar C–C str.), 1350 (Ar CCH in-plane bend. + C–N str. + Ar C–C str.), 1246 (ArO–C sym. str.), 1037 (ArO–C asym. str.), 745 (Ar C–H out-of-plane bend.). HRMS (ESI+) exact mass calcd for C30H28NO3 450.2069, found 450.2069.
2.3. Preparation of biopolymer films for leaching tests
Cellulose solution (4.3 wt%) was prepared according to a previously described procedure.60–62 A suspension of dry cellulose (10.1 g, 59.4 mmol) in DMAc (235 mL) was kept at 160 °C for 1 h under constant stirring. The volume of DMAc was reduced to ∼20 mL by vacuum distillation under nitrogen. The resulting slurry was cooled to 100 °C, and anhydrous LiCl (20.0 g) was added. The mixture was further cooled to room temperature with vigorous stirring to yield a clear, viscous cellulose solution. Films of cellulose with or without 5 wt% additive (small molecules or HBPs) were prepared by casting their respective solutions onto a Petri dish and dried at 60 °C for 3 days. PHB solutions with or without additives (95/5 PHB/additive in weight) were prepared by dissolution at 100 °C for 5 min in a sealed vessel, followed by 5 h standing at room temperature. PHB films were prepared by casting the corresponding PHB solutions in chloroform in a Petri dish at room temperature.For the evaluation of the leaching potential, the prepared biopolymer films (ca. 100 mg) were merged in distilled water: 10 mL water for cellulose films, and 3 mL water for PHB films. After 5 days, the UV-absorbance of the water phase was measured.
2.4. Analytical methods
Nuclear magnetic resonance (NMR) measurements were carried out on a Bruker DRX400 spectrometer at the proton frequency of 400.13 MHz and a carbon frequency of 100.61 MHz. Fourier transform infrared (FTIR) spectra were obtained with an attenuated total reflection (ATR) setup using a Bruker Alpha FT-IR spectrometer. Twenty-four scans were co-added using a resolution of 4 cm−1. Gel permeation chromatography (GPC) was carried out with three Shodex columns in series (KF-805, 2804, and 2802.5) and a refractive index (RI) detector (Viscotek Model 250). All measurements were carried out at room temperature at a concentration of 10 mg mL−1 using chloroform as the eluent, and at an elution rate of 1 mL min−1. Calibration was performed with four polystyrene standard samples (Mn = 650 kg mol−1 from Water Associates, 96 and 30 kg mol−1 from Polymer Laboratories, and 3180 g mol−1 from Agilent Technologies). Differential scanning calorimetry (DSC) measurements were performed using a TA Instruments DSC Q2000. The samples were studied with a heating rate of 10 °C min−1 under nitrogen with a purge rate of 50 mL min−1. The sequence consisted of a heating ramp from 40 to 300 °C and held at that temperature for 30 s, followed by a cooling ramp to −50 °C and held at that temperature for 3 min, and finally a heating ramp to 300 °C, which was employed to determine the glass transition temperature (Tg). Thermogravimetric analysis (TGA) was performed with a thermogravimetric analyser TA Instruments Q500 at a heating rate of 10 °C min−1 under nitrogen with a purge rate of 50 mL min−1. UV-visible absorption measurements were carried using a Varian Cary 1E UV-visible spectrometer in the wavelength range from 200 to 700 nm with a resolution of 1 nm, employing quartz cuvettes of 10 mm path length. High resolution mass spectrometry (HRMS) was performed by direct infusion on a Water Xero-G2 QTOF mass spectrometer using electrospray ionization. MALDI-TOF measurements were carried out using a 4700 Proteomics Analyzer (Applied Biosystems/MDSSCIEX, USA) in a positive reflector mode with DHB as the matrix.2.5. Antimicrobial bioassay
3. Results and discussion
3.1. Synthesis of HBPs
The synthesis started from a commercially available molecule 2-bromoethylbenzene, which can be conveniently prepared by a clean bromination in an aqueous medium of 2-phenylethanol (1).64 A biobased AB2-monomer (4) was obtained by a simple SN2 reaction of 2-bromoethylbenzene and 2 (Scheme 1), which yielded monomer 4 as orange needle-like crystals with 40% yield after crystallization in ethanol. The same monomer 4 could also be prepared by tosylation of 1 in a green solvent anisole, followed by in situ SN2 reaction with 2. However, this latter procedure produced monomer 4 with a lower yield (30%). Solvent-free Friedel–Crafts polymerization of monomer 4 at 50 °C for 72 h yielded a brown solid as the crude product, which was washed with hot methanol to afford pure HBP 5. It was noticed that similar polymerization was usually carried out at room temperature according to the literature.65 However, polymerization of monomer 4 at room temperature yielded only oligomers with low conversion (>10%) after 3 days. This can be attributed to the low reactivity of the methylated isatin and alkylated benzene of monomer 4, which can retard the Friedel–Crafts polymerization.65 Therefore, the reaction temperature was carefully investigated and it was found that 50 °C was the optimum temperature, and even higher temperature led to the formation of an insoluble gel. The anisole-functionalized HBP 6 was prepared from the same batch of reaction for the preparation of HBP 5. After the polymerization of monomer 4, half of the crude reaction mixture was quenched with methanol to produce HBP 5, and the other half was quenched with excess anisole followed by methanol. Such a procedure would allow for both HBP 5 and HBP 6 to have comparable molecular weights and degrees of branching. A model compound 7 with two anisole groups was synthesized by reacting excess anisole with 4 in TFSA, and the product was characterized by 1H- and 13C-NMR spectroscopy (ESI, Fig. S2 and S6†).3.2. Characterization of HBPs
The molar masses of the obtained HBPs were measured by GPC as ∼2000–3000 g mol−1 (Table 1). Such values are in the range for most reported dendritic polymers to be blended with matrix biopolymers.66–69 According to the GPC results, the apparent molar mass of 6 (Mn = 2000) is lower than that of 5 (Mn = 3200). Such an apparent counter-intuitive result can be attributed to the fact that the anisole groups at the periphery of HBP 6 are softer and much less polar compared to the isatin groups in HBP 5. It is assumed that during the GPC measurements in chloroform, HBP 6 adopted a more “collapsed” conformation compared with HBP 5, so it showed a smaller hydrodynamic volume and lower apparent molar mass.| HBP | M n (g mol−1) | M w (g mol−1) | PDI | T g (°C) | T 5 (°C) | T 10 (°C) | T d (°C) | Char yield (%) |
|---|---|---|---|---|---|---|---|---|
| 5 | 3200 | 6800 | 2.13 | 220 | 355 | 435 | 498 | 57 |
| 6 | 2000 | 3400 | 1.70 | 205 | 370 | 392 | 405, 465 | 42 |
The chemical structures of HBPs were characterized by 1H-NMR spectroscopy (Fig. 1). First, the signals for monomer 4 were unambiguously assigned (Fig. 1a). The signals at 2.99 and 3.94 ppm (peaks 1 and 2, respectively) corresponded to the methylene protons (–CH2–). The four aromatic signals observed at 6.75, 7.08, 7.52, and 7.57 ppm corresponded to the four protons on the isatin group (peaks 3, 4, 8, 9, respectively). The signals for the phenyl groups (peaks 5, 6, 7) overlapped at 7.19–7.30 ppm. In the 1H-NMR spectrum of HBP 5 (Fig. 1b), all the sharp signals of monomer 4 disappeared, and only broad signals were observed, which indicated the formation of polymers. The broad signals at ∼4.00 and 2.90 ppm corresponded to the protons on the ethylene bridges (Fig. 1b). The new signal appeared at 3.47 ppm was assigned to the OCH3 protons of the linear structures caused by partial reaction (structure 5b in Scheme 2).70 These linear structures formed due to the partial reaction during the polymerization, which led to the formation of intermediate 5a with tertiary OH groups. After the polymerization, the unreacted tertiary OH groups (of 5a) were converted into OCH3 groups (of 5b) by methanol quenching. The partial reaction of monomer 4 was consistent with the observed low reactivity of monomer 4. Since the 1H-NMR signals for the OH groups of 5a and the OCH3 groups of 5b are expected to appear both at ∼3.4 ppm, we measured the 1H-NMR spectrum of 5 in CDCl3 with an added drop of D2O. As a result, the intensity of the signal at 3.47 ppm did not change after the D2O addition, which indicated that this signal at 3.47 ppm corresponded to the non-active methoxy groups (5b). The existence of linear structure 5b was corroborated by mass spectrometrical analyses (ESI, Fig. S16†). When HBP 5 was converted to 6, two new signals appeared at 6.80 and 3.71 ppm (Fig. 1c), which corresponded to the protons of the grafted anisole units. Meanwhile, Fig. 1c shows a small signal at 3.47 ppm (OCH3 protons on the linear structure of HBP 6b), which has a much lower intensity compared to that of HBP 5 (Fig. 1b). This is consistent with the fact that HBP 6 was prepared by quenching intermediate 5a with anisole and methanol so the resulting HBP 6 contained two types of linear structures 6a (anisole quenched) and 6b (methanol quenched) (highlighted in Scheme 2). The ratio of 6a to 6b was estimated as 4.0:6.0, by examining the reduced intensity of the 1H-NMR signal at 3.47 ppm (for details see Fig. S13†). The presence of the linear structural units in HBPs 5 and 6 indicated that the degree of branching (DB)71,72 of these polymers is not 100%, as some previously reported HBPs with isatin structures.73,74 We therefore evaluated the DB values of the new HBPs based on the 1H-NMR signal intensities corresponding to the linear, dendritic, and terminal units (ESI, Fig. S12†). As summarized in Table 1, the DB value of HBP 5 was calculated as 0.55 (calculations shown in the ESI†). Such DB values are consistent with the theoretically most probable value (DB = 0.5) for the polymerization of AB2-monomers with two equal “B” groups.71 This suggested that for the polymerization of monomer 4, the tertiary OH groups in intermediate 5a did not have higher reactivity compared to the carbonyl groups in the monomer, as reported for some other isatin-containing monomers.73,74 Our observation is consistent with the low reactivity of monomer 4, which had to be polymerized at elevated temperature (50 °C), while other more reactive isatin-containing AB2 monomers (e.g. monomers with alkoxy benzene) could be polymerized at room temperature. Since the focus of this work was to develop new eco-friendly HBP additives for biopolymers, no further synthetic investigation was made toward HBPs with varied DB values. Due to signal overlapping, it is difficult to directly calculate the DB of HBP 6. However, HBP 5 and HBP 6 were prepared from the same intermediate 5a, so we assume that the DB values of both HBPs are the same. MALDI-TOF analyses of HBP 6 confirmed the existence of polymers (oligomers) with different DB values (ESI, Fig. S17†).
 | ||
| Fig. 1 1H-NMR spectra of (a) monomer 4, (b) HBP 5, and (c) HBP 6. | ||
 | ||
| Scheme 2 Linear structural units (5a) caused by partial reaction of the monomer 4. Direct methanol quenching of 5a yielded HBP 5b, which was the structure of product HBP 5. Quenching 5a with anisole followed by methanol yielded two different linear structures 6a and 6b, which were both present in the product HBP 6. The ratio of 6a:6b was estimated by 1H NMR spectroscopic analyses as 4.0:6.0. | ||
The chemical structures of HBPs were further characterized by 13C-NMR spectroscopy (Fig. 2). For monomer 4, the signals observed at 33.74 and 41.88 ppm corresponded to the carbons of the ethylene bridges (Fig. 2a). For both HBPs, the two corresponding ethylene “bridge” carbons appeared at 33.43 and 41.85 ppm. For HBP 5, two new signals at 61.85 and 50.94 ppm were observed (Fig. 2b), which corresponded to quaternary carbons of the 3,3′-diphenyloxindoles and the OCH3 groups due to partial reaction, respectively. For HBP 6, two carbon signals observed at 61.90 and 61.10 ppm corresponded to the quaternary carbons of 3,3′-diphenyloxindole structures at the “interior” and “periphery” of the HBP 6, respectively. In addition, a new signal at 55.33 ppm also appeared, corresponding to the OCH3 groups in the “periphery”. It was noted that for HBP 6, no signal was observed corresponding to the OCH3 groups (6b, Scheme 2), which was consistent with the reduced amount of the methanol-quenched linear structure 6b according to the previously discussed 1H-NMR results.
 | ||
| Fig. 2 Aliphatic region of the 13C-NMR spectra of (a) monomer 4, (b) HBP 5, and (c) HBP 6. Full spectra are provided in the ESI.† | ||
The conversion of HBP 5 into 6 was confirmed by FTIR spectroscopy (Fig. 3). For HBP 5, the symmetric stretching of the two carbonyl groups 1 and 2 (Fig. 3A) on the isatin units were discerned at 1737 and 1712 cm−1, respectively.75 For HBP 6, the band at 1737 cm−1 disappeared, and only a broad band at ∼1708 cm−1 was observed, which indicated that the terminal isatin groups of 5 were all consumed. Furthermore, in Fig. 3B, two new absorption bands at 1248 and 1024 cm−1 were observed (marked with 3 in Fig. 3B), corresponding to the symmetric and asymmetric C–O stretching of anisole moieties, respectively. This confirmed the formation of HBP 6 with anisole structures.
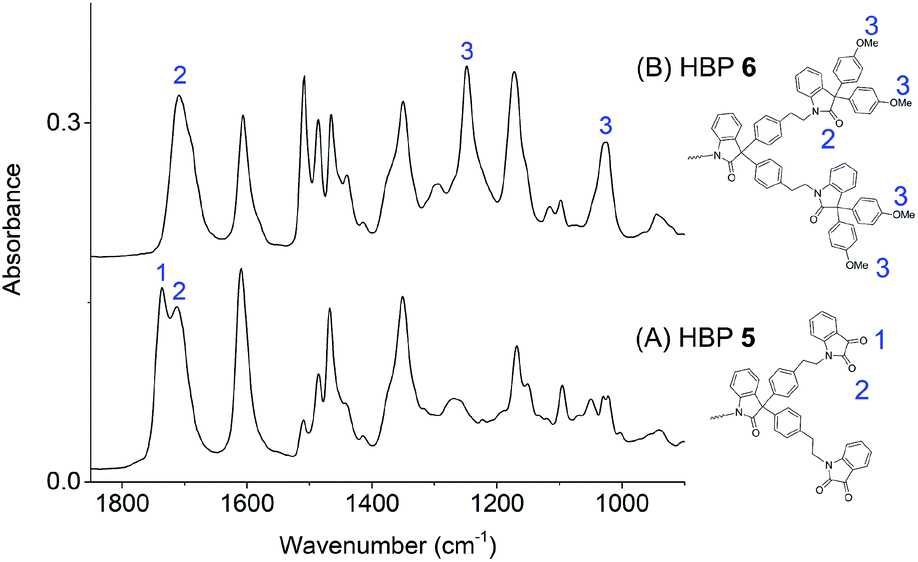 | ||
| Fig. 3 FTIR spectra of HBPs 5 (A) and 6 (B). | ||
Thermal properties of HBPs were characterized by DSC and TGA analyses. As shown in Fig. 4a, HBPs 5 and 6 both showed high glass transition temperatures (Tg = 220 and 205 °C, respectively). Such a high Tg was due to their highly rigid molecular structures, which was consistent with the previously reported polymers based on isatin or other 1,2-quinones.73,74,76 It was also noted that HBP 5 showed higher Tg compared with 6, which was ascribed to the strong polar interactions of the isatin units in 5, as well as the increased free volume of 6 caused by the flexible methoxy end-groups.77 HBPs 5 and 6 are both amorphous without a melting endotherm, which is clearly different from the small molecules 4 and 7 (melting points of 105 and 144 °C, respectively). Furthermore, according to the TGA data (Fig. 5 and Table 1), HBPs 5 and 6 are thermally more stable than monomer 4 and model compound 7, and their thermal stability is comparable to that of other high performance HBPs published before.78 It was noted that HBP 6 was thermally less stable compared with 5, which was attributed to the degradation of anisole end-groups. The high char yields of 5 and 6 were also noticed (57% and 42%, respectively), which was consistent with other reported rigid polymers containing high carbon contents.79,80 Such high char yields may provide an additional advantage for these HBPs as additives for biopolymers in terms of flame retardation, because high char yields can lower the formation of flammable gases upon heating. Such a phenomenon has been widely observed in rigid aromatic polymers with heterocyclic rings (e.g. polyimide, polybenzoxazole, polybenzimidazole, polybenzothiazoles, or polybenzoxazoles), which can be used as halogen-free flame-retardant polymer materials.81
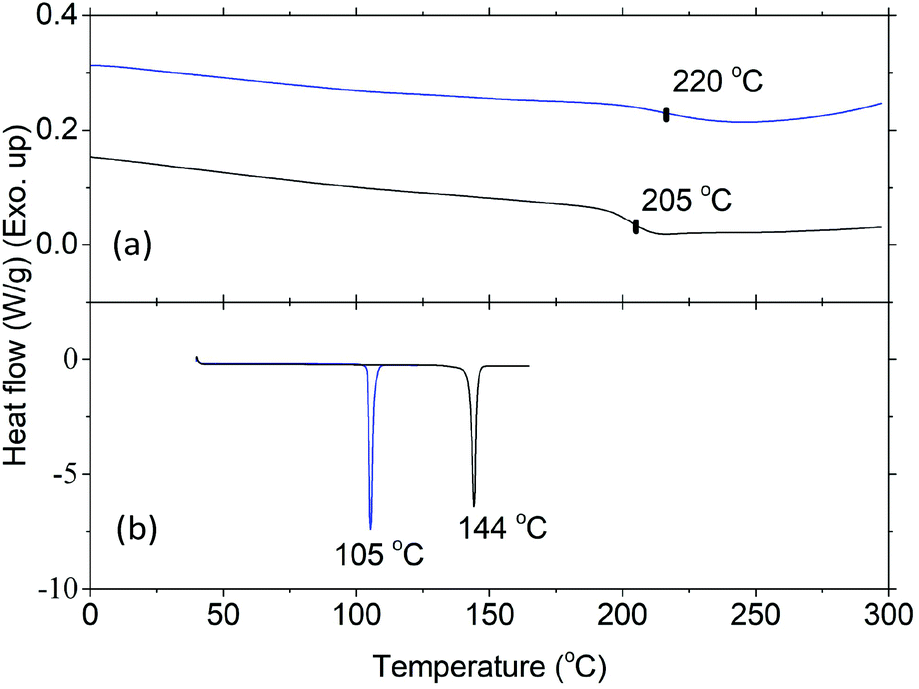 | ||
| Fig. 4 DSC curves of (a) 5 (blue line) and 6 (black line) (second heating), and (b) 4 (blue line), and 7 (black line) (first heating). | ||
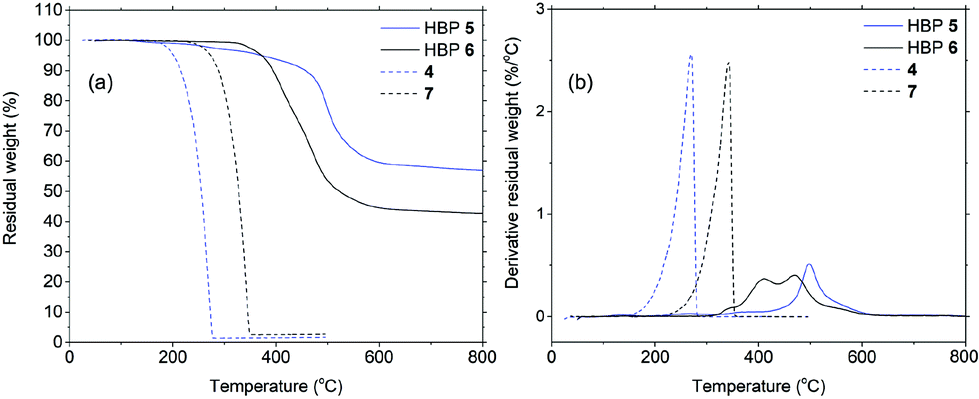 | ||
| Fig. 5 Residual weight (a) and its derivative (b) as a function of temperature for HBP 5 (blue solid), HBP 6 (black solid), 4 (blue dashed), and 7 (black dashed). | ||
3.3. Leakage of HBPs from biopolymer films
Plastic materials containing small molecular additives (e.g. antibacterial agents) often suffer from the leakage of harmful chemicals into the environment. Although it is generally accepted (or hypothesized) that macromolecular additives can have lower leakage, there has been surprisingly little effort in the evaluation of such leakage. We in this study evaluated the leakage of HBPs from two biopolymer films based on regenerated cellulose and PHB matrices. As shown in Fig. 6, after 5 days merging in distilled water, the UV-vis spectra of biopolymers containing 5% HBP 5 did not show an observable absorbance corresponding to HBP 5. In contrast, under the same conditions, the biopolymer films containing small molecule 4 showed a significant UV-vis absorbance of 4 in the water phase, which indicated significant leaching after 5 days. The leakage of the additives in the biopolymer films was also evaluated by the appearance of the films. For cellulose films (ESI, Fig. S9†), it was observed that the film containing 5% HBP 5 did not change significantly, and the aqueous phase remained colourless. However, the cellulose film containing 5% monomer 4 changed significantly after being merged in water for 5 days. Discolouration of the film and yellow colouration of the solution were clearly observed. PHB films (ESI, Fig. S10 and S11†) did not change the appearance as significantly as cellulose films did, possibly due to their lower hydrophilicity that could retard the leakage in aqueous medium. Finally, the leakage of monomer 4 into the aqueous phase was quantitatively determined by UV-vis measurements according to the Lambert–Beer law (ESI, Fig. S18†). We found that 4.7 and 2.5% of monomer 4 was leaked into the aqueous media from cellulose and PHB films, respectively. The same quantification method showed that the leakage of HBP 5 was effectively zero for both films.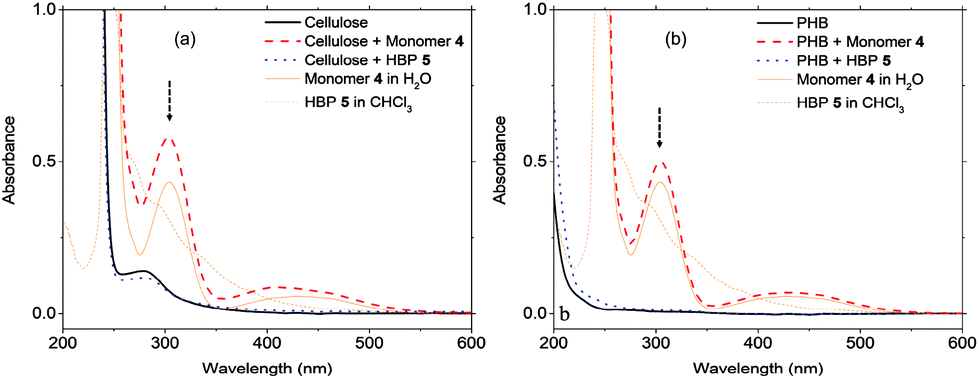 | ||
| Fig. 6 UV-visible absorbance spectra of the aqueous phase after (a) cellulose films and (b) PHB films were merged in deionized water for 5 days. The arrows indicate the maximum absorbance at 304 nm used for quantitative analyses. | ||
3.4. Antibacterial activity
The antibacterial activity of HBPs was evaluated by simple disk diffusion assay (images shown in the ESI, Fig. S14†). The tested bacteria include six Gram-negative G(−) bacteria (Proteus microbilis, Pseudomonas aeruginosa, Proteus vulgaris, Enterobacter aerogenes, and Salmonella typhmurium, and Escherichia coli) and three Gram-positive G(+) bacteria (Staphylococcus aureus, Bacillus thuringiensis and Streptococcus mutans). First, the effect of the sample loading was evaluated. In the range of 1–20 μg per disk (Fig. 7a and b), both HBPs showed a larger zone of inhibition with lower sample loading. The strongest antibacterial activity was observed when 2 μg HBP was loaded per disk. Further reduction of the sample loading (0.5–0.05 μg per disk) led to a decreased antibacterial activity (ESI, Fig. S15†). The existence of a maximal antibacterial effect at 2 μg per disk for the obtained HBPs was clearly different from the behaviour of small molecular antibiotics, which usually have a positive correlation between the loading amount of antibiotics and the antibacterial effects.82,83 It was noted that the maximal antibacterial effect was reported for linear polymers with a much higher concentration. For example, chitosan was reported to exhibit the maximum antibacterial effect at 1.5 mg mL−1 (corresponding to 30 μg per disk),84 which was 15 times higher than our new HBPs. Such a powerful antibacterial effect of HBPs could be ascribed to the dendritic structures and high local concentration of the functional groups of the HBPs.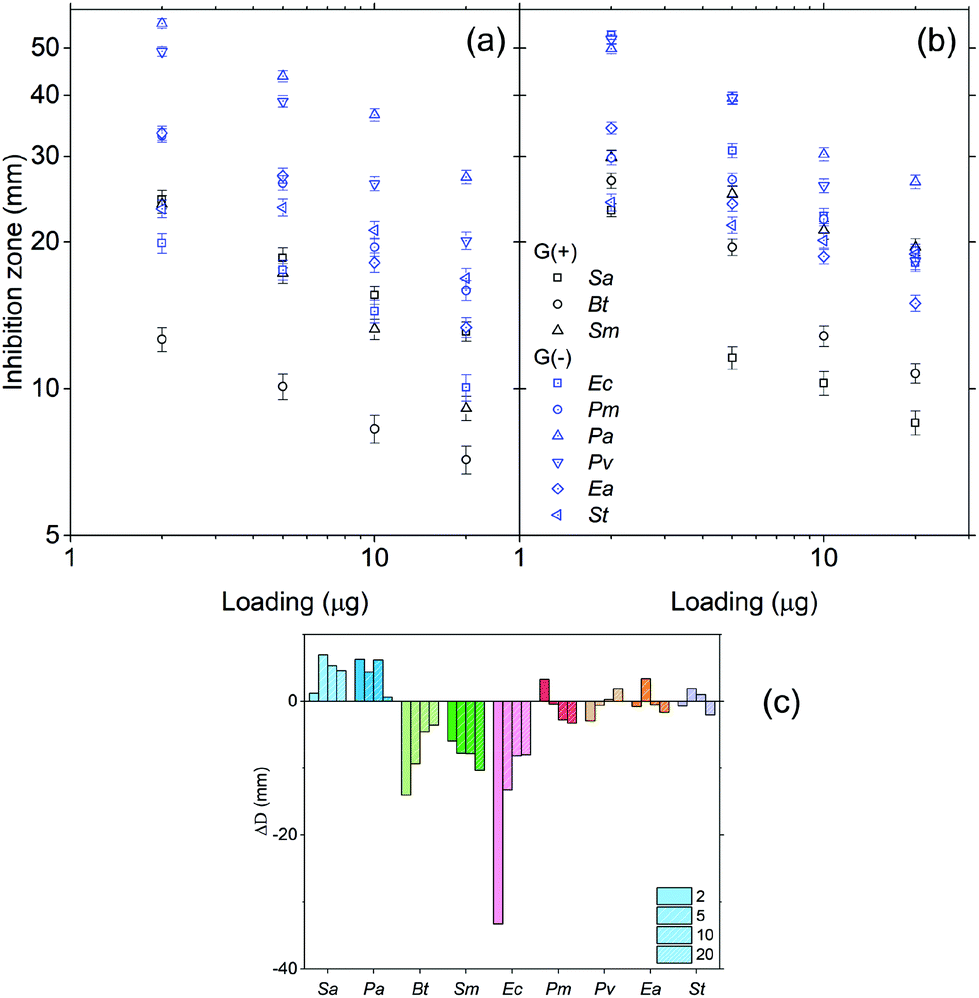 | ||
| Fig. 7 Zone of inhibition of (a) 5 and (b) 6, as a function of the loading amount (μg per disk), and (c) the differences in the zones of inhibition (ΔD = D5 − D6). D5 and D6 are the diameters of the inhibition zones of HBPs 5 and 6, respectively. | ||
Furthermore, the different antibacterial effects of two HBPs were compared by calculating the difference in the zones of inhibitions (ΔD) of HBPs 5 and 6. ΔD values for each bacterium at four loading amounts (2–20 μg per disk) are shown in Fig. 7c. Interestingly, the 9 tested bacteria exhibited 3 types of ΔD values. For two bacteria Sa and Pa, clear positive ΔD were observed, which indicated that HBP 5 was more effective to inhibit these two bacteria. For three bacteria Bt, Sm and Ec, large negative ΔD values were observed, which indicated that HBP 6 was significantly more effective for these three bacteria. For the rest of the 4 bacteria (Pm, Pv, Ea, St), ΔD values were effectively zero within the experimental error range, which indicated that there was no significant difference between the antibacterial effects of the 2 HBPs. It was noted that ΔD values showed no apparent correlation with whether a bacterium is G(+) or G(−). Such different antibacterial behaviour of the two HBPs could be ascribed to the higher polarity of 5 compared with 6 (the dipole moments of isatin and anisole are 5.65 and 1.38 D, respectively),85,86 which could significantly influence the interactions between HBPs and bacterial cell membranes. In addition, the rigidity of the HBPs may also play a role in their interactions with bacteria.
In order to assess the effect of the dendritic structures of HBPs, the antibacterial effects of two small molecules with the same functional groups (4 and 7) were measured and compared with that of HBPs. As shown in Fig. 8, for all the bacteria tested, 4 and 7 exhibited significantly lower antibacterial effects compared with those of HBP 5 and 6, respectively. The enhanced antibacterial behaviour of HBPs can be attributed to the dendritic structures and high local concentration of the functional groups,87,88 which may intensify the interactions of the bioactive functionalities with the bacterial membranes.
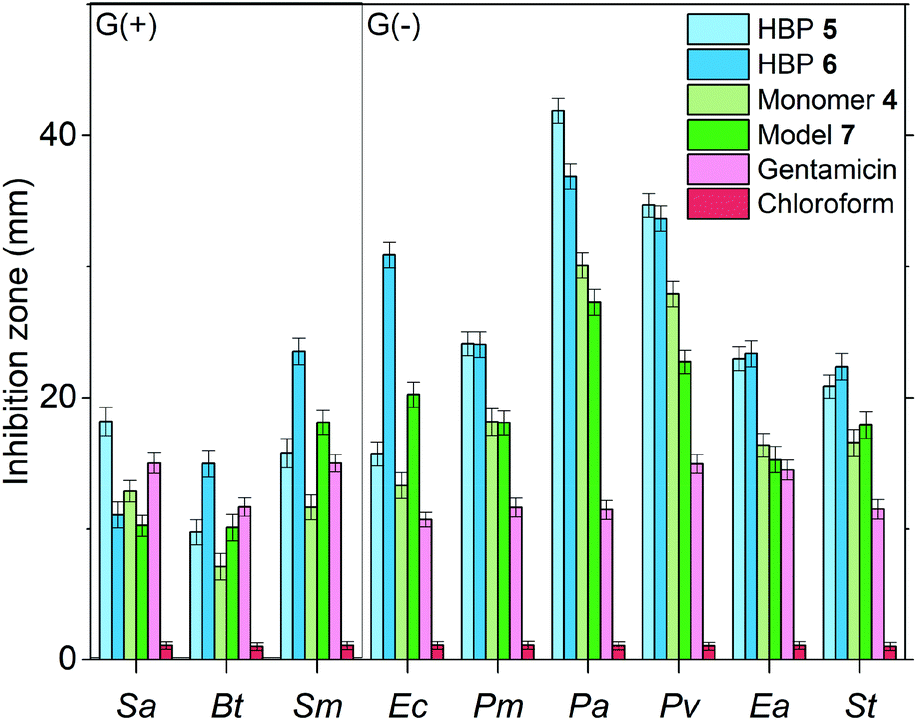 | ||
| Fig. 8 Comparison of the antibacterial effects of HBPs and small molecular antibiotics (4, 7 and gentamicin) at the loading level of 10 μg per disk. In the negative control experiments, pure chloroform was used to treat the samples. | ||
Furthermore, we compared the antibacterial effects of the two HBPs (5, 6) and two small model molecules (4, 7) with that of a commercial antibiotic (gentamicin). For consistency, we compared the antibacterial effects for all the tested molecules or polymers at 10 μg per disk (Fig. 8). For the six tested G(−) bacteria, gentamicin was always less effective than the two small molecules (4 and 7) and the two HBPs (5 and 6). This is attributed to the great antibacterial effects of the isatin or anisole groups, compared to that of gentamicin. For the tested three G(+) bacteria, the results were more complex, and the difference between the antibacterial effects of gentamicin and small molecules (4 and 7) are less significant. For bacteria Sa and Bt, gentamicin is slightly more effective than 4 and 7. For bacterium Sm, gentamicin is less effective than compound 7, but more effective than monomer 4. Comparison between gentamicin and HBPs suggested that for Sa gentamicin was more effective than HBP 6, but less effective than HBP 5. For Bt, gentamicin is more effective than HBP 5, but less effective than HBP 6. For Sm, gentamicin is significantly less effective than HBP 6, but similar to HBP 5. In general, the new non-ionic HBPs showed more significant inhibition against G(−) bacteria compared to G(+) bacteria. This characteristic makes the new HBPs particularly attractive, due to the strong demand from the society and medical units searching for new effective antibiotics against drug-resistant G(−) bacteria.89–91 It should be mentioned that in Fig. 8 the antibacterial properties of different compounds were compared at the same loading level (10 μg per disk). This loading level was the one at which gentamicin showed the highest antibacterial effect. However, this loading level was not the one at which HBPs are most effective. As shown in Fig. 7a and b, more significant antibacterial effects of HBPs could be achieved at a lower loading level. A comparison of the antibacterial effects of HBPs and gentamicin at their most effective loading levels (2 μg per disk for HBPs, and 10 μg per disk for gentamicin) is presented in Fig. 9, from which significantly stronger antibacterial effects of HBPs were observed, compared with that of gentamicin.
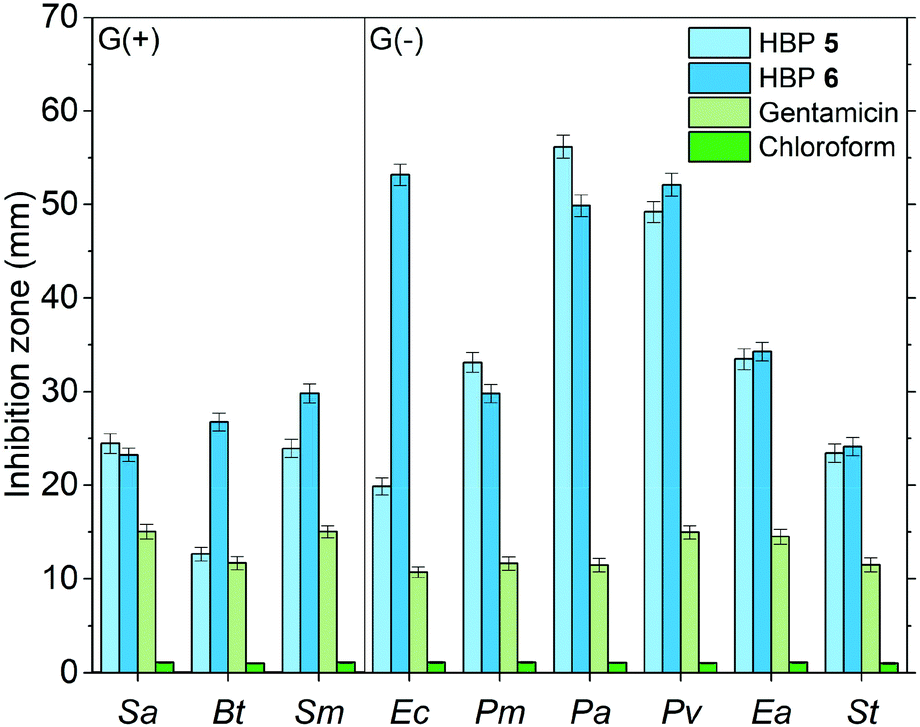 | ||
| Fig. 9 Comparison of the antibacterial effects of HBPs and gentamicin at their most effective loading amounts (2 μg per disk for HBPs, 10 μg per disk for gentamicin). In the negative control experiments, pure chloroform was used to treat the samples. | ||
4. Conclusions
Isatin and anisole based hyperbranched polymers (HBPs) have been prepared by a facile solvent-free polymerization with an acidic catalyst. Both HBPs are non-ionic, and they have rigid structures, high glass transition temperatures (>200 °C), and good thermal stability. The antibacterial effect of the new green HBPs was much more powerful compared with the small molecular antibiotic gentamicin, and the maximal bacterial inhibition of the HBPs was achieved at very low concentrations (loading amount per disk). Such an enhanced antibacterial effect can be attributed to the unique dendritic structures and densely packed functionalities of HBPs, which may enhance the adsorption of HBPs on the bacteria cell membrane and thus change the permeability and fluidity of the lipid bilayer. In addition, the biological activity of isatin or anisole groups may also be enhanced by the dendritic structures. Furthermore, the two HBPs have different sensitivities for the tested bacteria, which can be attributed to the different polarity and flexibility of isatin and the anisole terminal groups. In addition, the obtained green HBPs were blended with biopolymers PHB and cellulose and cast into films. The leakage tests of HBPs from the biopolymer films indicated that such macromolecular additives are non-leachable in water and thus environmentally friendly. The new green HBP additives are expected to have great potential in the development of bio-based polymers or plastics that can be used under bacteria-free conditions, such as biomedical applications or food packaging.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the ÅForsk Foundation (No. 16-479), the Crafoord Foundation (No. 20160774), and the Royal Physiographic Society in Lund. We thank Sofia Essén and Katja Bernfur for the help in mass spectrometry.References
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- M. Niaounakis, Biopolymers: applications and trends, Elsevier Inc., Oxford, 2015 Search PubMed.
- P. Appendini and J. H. Hotchkiss, Innovative Food Sci. Emerging Technol., 2002, 3, 113–126 CrossRef CAS.
- Shahid-ul-Islam, M. Shahid and F. Mohammad, Ind. Eng. Chem. Res., 2013, 52, 5245–5260 CrossRef CAS.
- E.-S. Park, H.-J. Lee, H. Y. Park, M.-N. Kim, K.-H. Chung and J.-S. Yoon, J. Appl. Polym. Sci., 2001, 80, 728–736 CrossRef CAS.
- M. B. Patel, S. A. Patel, A. Ray and R. M. Patel, J. Appl. Polym. Sci., 2003, 89, 895–900 CrossRef CAS.
- G. Mauriello, E. De Luca, A. La Storia, F. Villani and D. Ercolini, Lett. Appl. Microbiol., 2005, 41, 464–469 CrossRef CAS PubMed.
- T. J. Athauda, R. R. Ozer, J. M. Chalker, S. Bajpai, M. K. Jha, C. Shapiro, P. M. Griffin, R. V. Tauxe and D. W. T. Au, RSC Adv., 2013, 3, 10662 RSC.
- Y.-M. Weng and J. H. Hotchkiss, Packag. Technol. Sci., 1993, 6, 123–128 CrossRef CAS.
- N. Hasegawa, H. Okamoto, M. Kato and A. Usuki, J. Appl. Polym. Sci., 2000, 78, 1918–1922 CrossRef CAS.
- M. Bankova, T. Petrova, N. Manolova and I. Rashkov, Eur. Polym. J., 1996, 32, 325–330 CrossRef CAS.
- A. Kanazawa, T. Ikeda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2873–2876 CrossRef CAS.
- A. Kanazawa, T. Ikeda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1997–2001 CrossRef CAS.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. DeGrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- K.-S. Huang, C.-H. Yang, S.-L. Huang, C.-Y. Chen, Y.-Y. Lu and Y.-S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef PubMed.
- F. Hui and C. Debiemme-Chouvy, Biomacromolecules, 2013, 14, 585–601 CrossRef CAS PubMed.
- D. Raafat and H.-G. Sahl, Microb. Biotechnol., 2009, 2, 186–201 CrossRef CAS PubMed.
- F. Ferrero, M. Periolatto and S. Ferrario, J. Cleaner Prod., 2015, 96, 244–252 CrossRef CAS.
- V. W. L. Ng, J. P. K. Tan, J. Leong, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef CAS.
- N. D. Koromilas, G. C. Lainioti, G. Vasilopoulos, A. Vantarakis and J. K. Kallitsis, Polym. Chem., 2016, 7, 3562–3575 RSC.
- Y. Jiao, L. Niu, S. Ma, J. Li, F. Tay and J. Chen, Prog. Polym. Sci., 2017, 7, 53–90 CrossRef.
- E.-R. Kenawy and Y. R. Abdel-Fattah, Macromol. Biosci., 2002, 2, 261–266 CrossRef CAS.
- R. Costa, J. L. Pereira, J. Gomes, F. Gonçalves, D. Hunkeler and M. G. Rasteiro, Chemosphere, 2014, 112, 177–184 CrossRef CAS PubMed.
- R. J. Cornell and L. G. Donaruma, J. Med. Chem., 1965, 8, 388–390 CrossRef CAS PubMed.
- L. Erdmann and K. E. Uhrich, Biomaterials, 2000, 21, 1941–1946 CrossRef CAS PubMed.
- R. Jabara, N. Chronos and K. Robinson, Catheter. Cardiovasc. Interv., 2008, 72, 186–194 CrossRef PubMed.
- N. Shpaisman, L. Sheihet, J. Bushman, J. Winters and J. Kohn, Biomacromolecules, 2012, 13, 2279–2286 CrossRef CAS PubMed.
- H. Tang, C. J. Murphy, B. Zhang, Y. Shen, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Biomaterials, 2010, 31, 7139–7149 CrossRef CAS PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- S. Weintraub, T. Shpigel, L. G. Harris, R. Schuster, E. C. Lewis and D. Y. Lewitus, Polym. Chem., 2017, 8, 4182–4189 RSC.
- J. I. Paez, M. Martinelli, V. Brunetti and M. C. Strumia, Polymers, 2012, 4, 355–395 CrossRef.
- C. Schüll, H. Rabbel, F. Schmid and H. Frey, Macromolecules, 2013, 46, 5823–5830 CrossRef.
- D. Tomalia and J. Frechet, Dendrimers and other dendritic polymers, John Wiley & Sons, Ltd., Chichester, 2001 Search PubMed.
- C. Z. Chen and S. L. Cooper, Biomaterials, 2002, 23, 3359–3368 CrossRef CAS PubMed.
- P. Ortega, J. L. Copa-Patiño, M. A. Muñoz-Fernandez, J. Soliveri, R. Gomez and F. J. de la Mata, Org. Biomol. Chem., 2008, 6, 3264 CAS.
- C. Abid and S. Chattopadhyay, J. Appl. Polym. Sci., 2010, 116, 1640–1649 CAS.
- S. Charles, N. Vasanthan, D. Kwon and G. Sekosan, Tetrahedron Lett., 2012, 53, 6670–6675 CrossRef CAS PubMed.
- D. Gangadharan, N. Dhandhala and D. Dixit, J. Appl. Polym. Sci., 2012, 124, 1384–1391 CrossRef CAS.
- E. Fuentes-Paniagua, J. M. Hernández-Ros, M. Sánchez-Milla, M. A. Camero, M. Maly, J. Pérez-Serrano, J. L. Copa-Patiño, J. Sánchez-Nieves, J. Soliveri, R. Gómez and F. Javier de la Mata, RSC Adv., 2014, 4, 1256–1265 RSC.
- H. Bakhshi and S. Agarwal, Polym. Chem., 2016, 7, 5322–5330 RSC.
- H. Bakhshi, S. Agarwal, T. Hayashi, N. Zhu, X. Ni, Z. Shen, F. Padella, H. Wang, J. Soliveri, R. Gómez and F. J. de la Mata, J. Mater. Chem. B, 2017, 5, 6827–6834 RSC.
- M. Witvrouw, V. Fikkert, W. Pluymers and B. Matthews, Molecular, 2000, 58, 1100–1108 CrossRef CAS.
- J. Rojo and R. Delgado, J. Antimicrob. Chemother., 2004, 54, 579–581 CrossRef CAS PubMed.
- B. T. Lingappa, M. Prasad, Y. Lingappa, D. F. Hunt and K. Biemann, Science, 1969, 163, 192–194 CAS.
- Q. Wang, Y. Song, Y. Jin, H. Liu and H. Zhang, Biocatal. Biotransformation, 2013, 31, 292–298 CrossRef CAS.
- J. Da Silva, S. Garden and A. Pinto, J. Braz. Chem., 2001, 12, 273–324 CrossRef CAS.
- A. Abadi, S. Abou-Seri and D. Abdel-Rahman, Eur. J. Med. Chem., 2006, 41, 296–305 CrossRef CAS PubMed.
- N. Sin, B. L. Venables, K. D. Combrink, H. B. Gulgeze, K.-L. Yu, R. L. Civiello, J. Thuring, X. A. Wang, Z. Yang, L. Zadjura, A. Marino, K. F. Kadow, C. W. Cianci, J. Clarke, E. V. Genovesi, I. Medina, L. Lamb, M. Krystal and N. A. Meanwell, Bioorg. Med. Chem. Lett., 2009, 19, 4857–4862 CrossRef CAS PubMed.
- P. Paira, A. Hazra, S. Kumar, R. Paira, K. B. Sahu, S. Naskar, P. Saha, S. Mondal, A. Maity, S. Banerjee and N. B. Mondal, Bioorg. Med. Chem. Lett., 2009, 19, 4786–4789 CrossRef CAS PubMed.
- A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, L. Varoli, M. A. Cremonini, G. Placucci, R. Cervellati and E. Greco, Eur. J. Med. Chem., 2009, 45, 1374–1378 CrossRef PubMed.
- G. Cerchiaro and A. Ferreira, J. Braz. Chem. Soc., 2006, 17, 1473–1485 CrossRef CAS.
- M. Raccach, J. Food Saf., 1984, 6, 141–170 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada, P. J. Dunn, W. Raverty, A. J. Hunt, J. H. Clark and P. Hosek, Green Chem., 2016, 18, 288–296 RSC.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- H. Chen and J. Kong, Polym. Chem., 2016, 7, 3643–3663 RSC.
- W. Wu, R. Tang, Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC.
- D. Demircan and B. Zhang, Carbohydr. Polym., 2017, 157, 1913–1921 CrossRef CAS PubMed.
- T. Heinze, K. Rahn, M. Jaspers and H. Berghmans, J. Appl. Polym. Sci., 1996, 60, 1891–1900 CrossRef CAS.
- D. Demircan, S. Ilk and B. Zhang, Biomacromolecules, 2017, 18, 3439–3446 CrossRef CAS PubMed.
- Clinical and Laboratory Standards Institute (CLSI), Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard - Eleventh Edition. CLSI document M02-A11, 2012 Search PubMed.
- J. N. Ashley, H. J. Barber, A. J. Ewins, G. Newberry and A. D. H. Self, J. Chem. Soc., 1942, 1942, 103–116 RSC.
- D. A. Klumpp, K. Y. Yeung, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1998, 63, 4481–4484 CrossRef CAS.
- S. Xu, R. Luo, L. Wu, K. Xu and G.-Q. Chen, J. Appl. Polym. Sci., 2006, 102, 3782–3790 CrossRef CAS.
- A. Javadi, Y. Srithep, S. Pilla, C. C. Clemons, S. Gong and L.-S. Turng, Polym. Eng. Sci., 2011, 51, 1815–1826 CAS.
- J.-F. Zhang and X. Sun, Polym. Int., 2004, 53, 716–722 CrossRef CAS.
- W. Zhang, Y. Zhang and Y. Chen, Iran. Polym. J., 2008, 17, 891–898 CAS.
- J.-Y. Chen, Z.-L. Xiang, F. Yu, B. F. Sels, Y. Fu, T. Sun, M. Smet and W. Dehaen, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2596–2603 CrossRef CAS.
- C. J. Hawker, R. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS.
- D. Hölter, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef.
- M. Smet, E. Schacht and W. Dehaen, Angew. Chem., Int. Ed., 2002, 41, 4547–4550 CrossRef CAS PubMed.
- Y. Fu, J. Chen, H. Xu, C. Van Oosterwijck, X. Zhang, W. Dehaen and M. Smet, Macromol. Rapid Commun., 2012, 33, 798–804 CrossRef CAS PubMed.
- T. Polat, F. Bulut, I. Arıcan, F. Kandemirli and G. Yildirim, J. Mol. Struct., 2015, 1101, 189–211 CrossRef CAS.
- M. G. Zolotukhin, L. Fomina, R. Salcedo, L. E. Sansores, H. M. Colquhoun and L. M. Khalilov, Macromolecules, 2004, 37, 5140–5141 CrossRef CAS.
- M. Rostagno, S. Shen, I. Ghiviriga and S. A. Miller, Polym. Chem., 2017, 8, 5049–5059 RSC.
- A. Ghosh, S. Banerjee and B. Voit, in Porous Carbons – Hyperbranched Polymers – Polymer Solvation, ed. T. E. Long, B. Voit and O. Okay, Springer International Publishing, Cham, 2015, pp. 27–124 Search PubMed.
- G. Duan, S. Jiang, S. Chen and H. Hou, J. Nanomater., 2010, 2010, 1–5 CrossRef.
- E. Ekıncı, S. Kötepe, A. Paşahan, T. Seç and T. Seçkın, Turk. J. Chem., 2006, 30, 277–285 Search PubMed.
- H. Zhang, R. J. Farris and P. R. Westmoreland, Macromolecules, 2003, 36, 3944–3954 CrossRef CAS.
- O. O. Olajuyigbe, J. Pharm. Allied Health Sci., 2012, 2, 12–20 CrossRef.
- C. N. Obi, Am. J. Microbiol. Res., 2014, 2, 178–181 CrossRef.
- R. C. Goy, S. T. B. Morais and O. B. G. Assis, Rev. Bras. Farmacogn., 2016, 26, 122–127 CrossRef CAS.
- V. Galasso and G. C. Pappalardo, J. Chem. Soc., Perkin Trans. 2, 1976, 574 RSC.
- G. Baysinger, L. I. Berger, R. N. Goldberg, H. V. Kehiaian, K. Kuchitsu, D. L. Roth and D. Zwillinger, CRC Handbook of Chemistry and Physics, Taylor & Francis Group, Boca Raton, 95th edn, 2014 Search PubMed.
- A. M. Noz-Bonilla and M. Fernández-García, Prog. Polym. Sci., 2011, 37, 281–339 CrossRef.
- Y. Segawa, T. Higashihara and M. Ueda, Polym. Chem., 2013, 4, 1746–1759 RSC.
- J. H. Rex, Nat. Rev. Microbiol., 2014, 12, 231–232 CrossRef CAS.
- A. N. Amin and D. Deruelle, Future Microbiol., 2015, 10, 1049–1062 CrossRef CAS PubMed.
- K. Bush, ACS Infect. Dis., 2015, 1, 509–511 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc03401f |
| This journal is © The Royal Society of Chemistry 2018 |