DOI:
10.1039/C7GC03110F
(Paper)
Green Chem., 2018,
20, 170-182
Mechanistic studies of base-catalysed lignin depolymerisation in dimethyl carbonate†
Received
16th October 2017
, Accepted 24th November 2017
First published on 24th November 2017
Abstract
The depleting fossil reservoirs have stimulated global research initiatives on renewable lignin feedstocks as sustainable alternatives to petroleum-derived aromatics. Base-catalysed depolymerisation (BCD) is regarded as an economical and efficient approach for the valorisation of technical lignins. The major limiting factor encountered during this process is the re-condensation of the formed phenolic products, which results in lower monomer yields. To diminish these side reactions, we selected alkali earth metal catalysts in dimethyl carbonate (DMC) to produce methylated phenol derivatives as the final products. Herein, we demonstrate for the first time a base-promoted depolymerisation process affording low-molecular weight oils in high yields (52–67 wt%) wherein the employed bases are used in truly catalytic quantities (with catalyst loadings of around 5 mol%). The general applicability of this methodology was proved on four different lignin samples (1 Kraft, 3 organosolv) using caesium carbonate and lithium tert-butoxide as catalysts. The 2D NMR studies on the post-reaction lignin samples showed a similar degradation of the major lignin linkages for both bases. A difference in the reduction of phenolic moieties was revealed by quantitative 31P NMR analysis. Furthermore, GPC analysis demonstrated a significant shift towards lower mass fragments for the Cs2CO3-catalysed lignin degradation. A detailed GC-MS analysis for these samples identified a range of methoxy capped-monomeric degradation products. The scope of this reaction system was further expanded to lignocellulosic biomass such as milled beechwood chips, which notably showed similar product distributions. Based on the correlation of the experimental observations for extracted lignin samples and model compound studies, a mechanistic pathway for the Cs2CO3-catalysed system was suggested. DFT calculations provided reaction pathways for the observed cleavage products.
Introduction
Lignin is a renewable biopolymer rich in aromatics which represents a considerable portion (roughly 30% on a weight basis and 40% on an energy basis) of the lignocellulosic biomass.1 Despite being widely available as a waste product from the pulp and paper industry, lignin is by far the least utilised fraction in lignocellulose in comparison with cellulose and hemicelluloses.2 The structural diversity and complexity of this three-dimensional, amorphous lignin biopolymer contributes to its overall recalcitrance. This represents a major hindrance for obtaining high-value chemicals in an integrated biorefinery. Thus, lignin fractions are most often relegated to low-value uses and burnt to get energy.2,3 Since in the forthcoming years biorefineries will process considerable amounts of renewable feedstocks, it becomes important to find efficient valorisation routes and upgrade this readily available resource to make the overall process more economical.2,3a
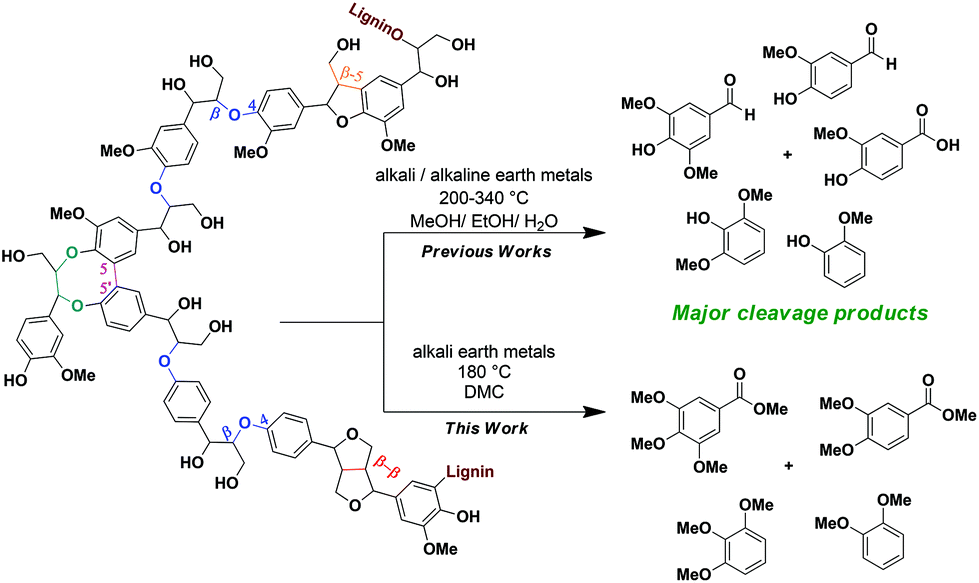
The lignin structure is composed of a randomly branched network of electron rich phenylpropanols (coumaryl, coniferyl, sinapyl alcohols) that are connected to each other through a variety of stable C–C and ethereal C–O linkages.4 Amongst these, β-O-4 linkages (Scheme 1) are the most predominant alkyl–aryl ether interconnecting bonds.2–4 A controlled and selective lignin depolymerisation strategy targeting these β-O-4 linkages is a key way for producing a variety of high-value, low molecular weight aromatics.5 Numerous publications in recent years have been devoted to such strategies involving oxidative,5,6 reductive,7 redox-neutral,5c,8 acid-9 and base-catalysed10,11 degradation of lignin β-O-4 model compounds and extracted lignin samples. Most of the studies in the literature on BCD highlight the advantages of using cheap and readily available reagents (NaOH, KOH, Ca(OH)2, LiOH, liq. NH3etc.).10 Lately, in order to make processes more economically viable, there have been attempts to perform the degradation reactions using solid bases that are frequently combined with other metal catalysts.11 Along these lines a recent article by Chaudhary and Dhepe compared the efficiency for lignin depolymerisation using a variety of heterogeneous bases.11g The highest yields of the low molecular weight products (monomers to trimers, 51%) were attained using NaX zeolites at 250 °C with a reaction time of 1 h. It is noteworthy, however, that the base was employed in stoichiometric amounts and a significantly decreased yield of low molecular weight products (monomers to trimers) was observed (51% to 34%) in the first recycling run.11g
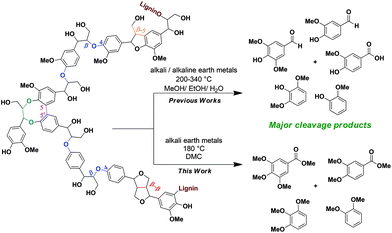 |
| | Scheme 1 Base-catalysed cleavage of lignin in DMC. | |
The use of stoichiometric amounts of base in BCD relative to monomer units present in lignin is a common theme in the literature.10b,c,j,k,m,q The catalyst loadings are generally given in wt% relative to the weight of solvent and not lignin.10b–l,n,q For example, one standard reaction condition entails 10% [w(lignin)/w(solvent)] lignin loading and 2% to 4% [w(base)/w(solvent)] catalyst loading using a base such as NaOH.10c,i–k,q The catalyst loadings relative to lignin are therefore usually between 20 wt% and 40 wt%. The majority of BCD reactions require elevated temperatures from 200 °C to 340 °C and are often accompanied by high pressures. Furthermore, these conditions promote secondary reactions between the reactive phenolic and aldehyde/ketone intermediates, hence leading to an overall decrease in monomer yields. To obtain higher yields of the monomer rich oil fractions under higher temperature and pressure conditions, Lercher and Toledano independently reported boric acid and phenols as suitable capping agents.10j,l Alternatively, we have exploited the versatile chemical nature of DMC at elevated temperatures (>90 °C) in the presence of bases, and reported a BCD strategy for a range of lignin β-O-4 model compounds obtaining methoxy-capped phenolic and benzoic acid derivatives as products.10p Recently, Barrett et al. also demonstrated the intricate role of a MeOH/DMC solvent system which helped in increasing the aromatic yields by O-methylation of the phenolic intermediates for their Cu20PMO-catalysed reductive lignin disassembly pathway.7i
Predicting the reaction behaviour of real lignin based on the results from lignin model compound studies is often not reliable because they frequently differ from each other.4b,c Considering these implications, the current study focuses on the BCD of four lignin polymers, aiming to increase the yields of the oil fractions with a higher composition of methoxy-capped low molecular weight products. After having established that the same types of products are formed from lignin as in model compound-based studies, efforts were put in studying the mechanistic aspects of the reaction. We herein propose reaction pathways leading to the formation of the observed capped-methoxy-aromatics.
Results and discussion
Characterisation of lignins
In this study, four lignin samples, namely three organosolv lignins, isolated from cherry (OS-C), oak (OS-O), and beechwood (OS-BW), and commercially available Kraft lignin from Sigma Aldrich (370959) were evaluated under our BCD reaction conditions in DMC. Since the isolation process of a lignin sample significantly influences the physical and chemical properties of the polymer including the identity and ratio of the chemical linkages, the lignin samples were characterised by 2D-HSQC NMR, GPC and 31P NMR [after derivatisation with the phosphorylating agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP)] measurements prior to use.
These methods were used to determine the abundance of β-O-4, β-5 and β–β linkages as well as to quantify the total amounts of aliphatic hydroxyl groups present in the individual lignin samples. The data from these experiments are outlined in Tables 1 and 2.
Table 1 Characterisation of lignin samples by GPC and 2D-HSQC NMR
| Lignin source |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (Da) a (Da) |
S, G, Hb (%) |
β-O-4-OHc |
Linkagesb (per 100 C9 units) |
| β-O-4-ORd |
β-5 |
β–β |
|
Determined by GPC (DMF/LiCl) using a RI detector with respect to polystyrene standards.
Determined by 2D-HSQC NMR by comparing the aliphatic and aromatic signal intensities (ESI 2.2 a).
Amounts of β-O-4 linkages with free α-OH.
Amounts of β-O-4 linkages with capped α-OH units.
|
| OS-O |
2610 |
85, 14, trace |
27 |
6 |
3 |
5 |
| OS-BW |
3103 |
73, 27, trace |
20 |
4 |
3.2 |
5.1 |
| OS-C |
15220 |
86, 14, trace |
37 |
1.4 |
2 |
6 |
| Aldrich-Kraft |
7106 |
Trace, 100, trace |
5 |
0 |
1 |
1 |
Table 2 Quantitative 31P NMR measurements of the aliphatic, phenolic, and carboxylic hydroxyl groups in lignin samplesa
| Lignin source |
OH (mmol g−1) |
| Aliphatic OH |
5-Substituted OH |
Guaiacyl OH (G) |
p-Hydroxyphenyl OH (H) |
Total phenolic OH |
COOH |
|
Lignin samples phosphitylated with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (ESI 2.2.b).
|
| OS-O |
2.37 |
1.06 |
0.56 |
0.05 |
1.66 |
0.12 |
| OS-BW |
2.38 |
1.24 |
0.69 |
0.01 |
1.94 |
0.01 |
| OS-C |
1.90 |
1.70 |
0.88 |
0.03 |
2.63 |
0.07 |
| Aldrich-Kraft |
1.79 |
0.90 |
1.77 |
0.17 |
2.84 |
0.20 |
Lignin degradation studies
The lignin samples were subjected to BCD studies using 10 wt% [w(base)/w(lignin)] of either of the two bases (Cs2CO3 or LiOt-Bu) at 180 °C for 8 h (Cs2CO3) or 12 h (LiOt-Bu). Depending on the lignin source this corresponded to a catalyst loading of 5 to 6 mol% for Cs2CO3 and 23 to 26 mol% for LiOt-Bu. The resulting reaction mixtures were analysed by 2D-HSQC NMR, quantitative 31P NMR and GPC. Lower molecular weight fractions (oil) were separated from the residual lignins by solvent extraction using ethyl acetate and analysed by GC-MS to identify and individually quantify the capped-aromatic monomer products.
2D NMR studies
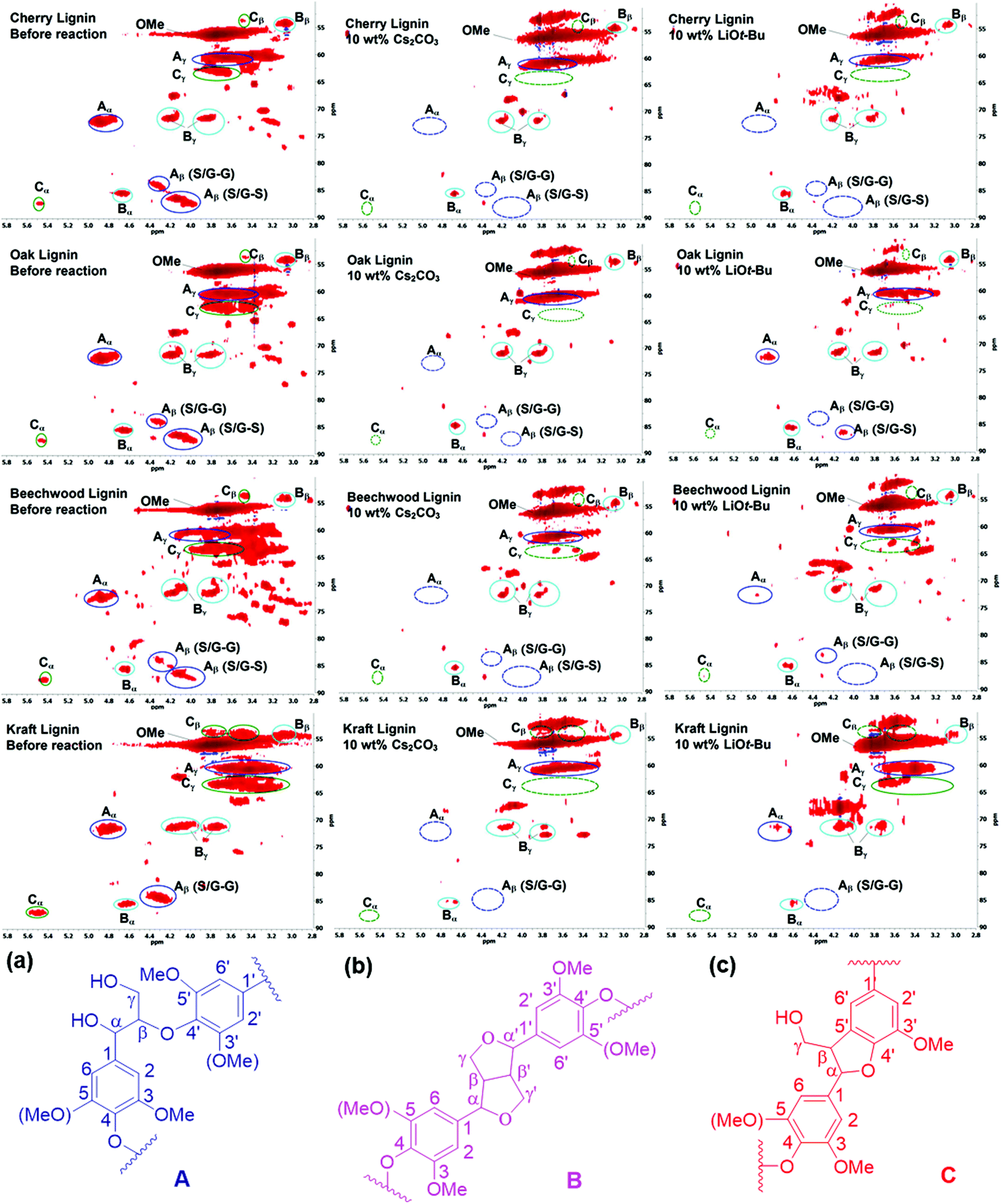
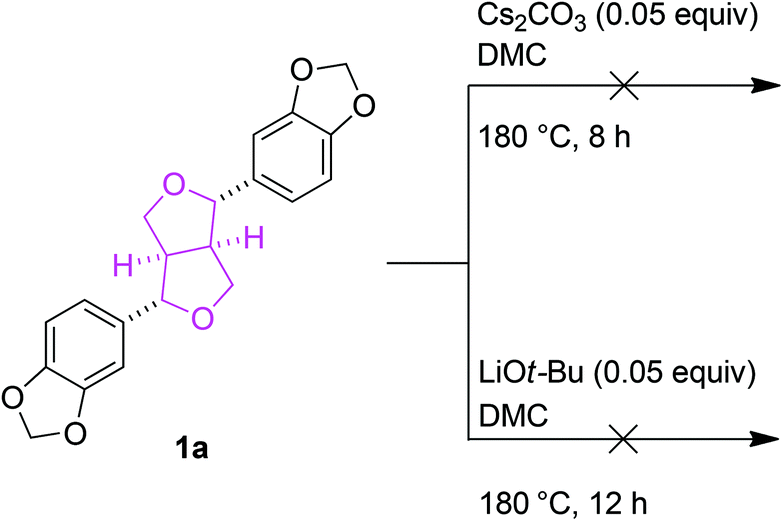
Preliminary studies to determine the changes in lignin structures mainly focused on their two-dimensional 1H–13C correlation NMR spectroscopy (HSQC) before and after the BCD reactions. In particular, the aliphatic oxygenated side chain regions (δC/δH 50.0–90.0/2.5–5.8) of the HSQC spectra showed a major decrease in the regions corresponding to the β-aryl-ether (β-O-4) and phenylcoumaran (β-5) bonds. The resinol (β–β) linkages seemed to remain unaltered for both the Cs2CO3 and LiOt-Bu-catalysed BCD processes in all four lignin samples (Fig. 1). A follow-up reaction using model compound 1a further confirmed the low reactivity of the resinol (β–β) linkages for our reaction system (Scheme 2). This could in turn be attributed to the lack of aliphatic hydroxyl groups, which were later proven during the mechanistic studies to have a considerable influence on the overall degradation pathway.
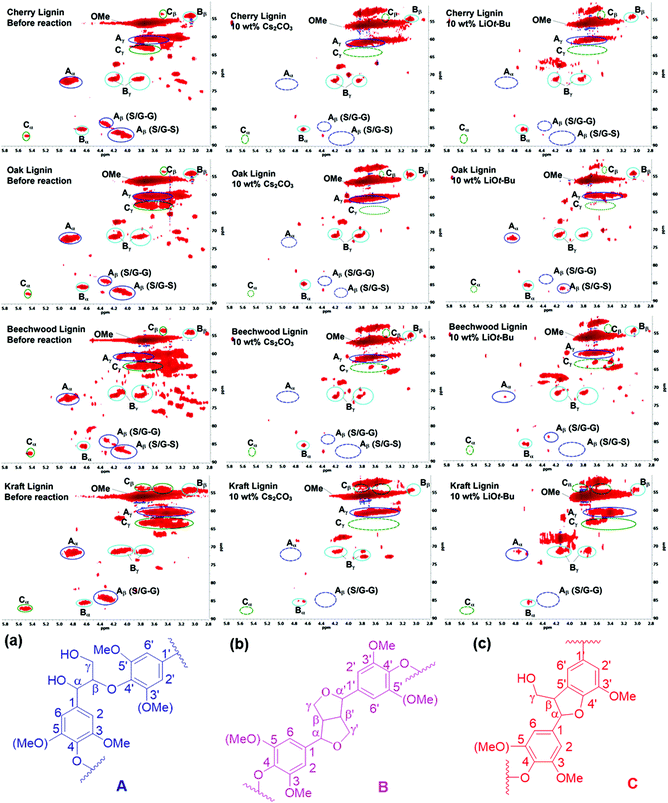 |
| | Fig. 1 2D-HSQC NMR spectra (in DMSO-d6) of organosolv, cherry, oak, beechwood and Aldrich-Kraft lignin; before the reaction; after the reaction with Cs2CO3 at 180 °C; after the reaction with LiOt-Bu at 180 °C; (a) β-O-4′ aryl ether linkages; (b) resinol substructures formed by β–β′, α-O-γ′, and γ-O-α′ linkages; (c) phenylcoumaran substructures formed by β-5′ and α-O-4′ linkages. | |
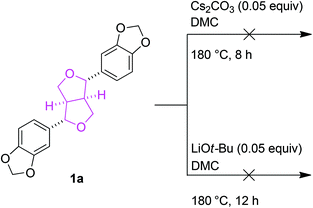 |
| | Scheme 2 Reactivity of β–β model compound 1a in the BCD reactions. | |
Quantitative 31P NMR studies
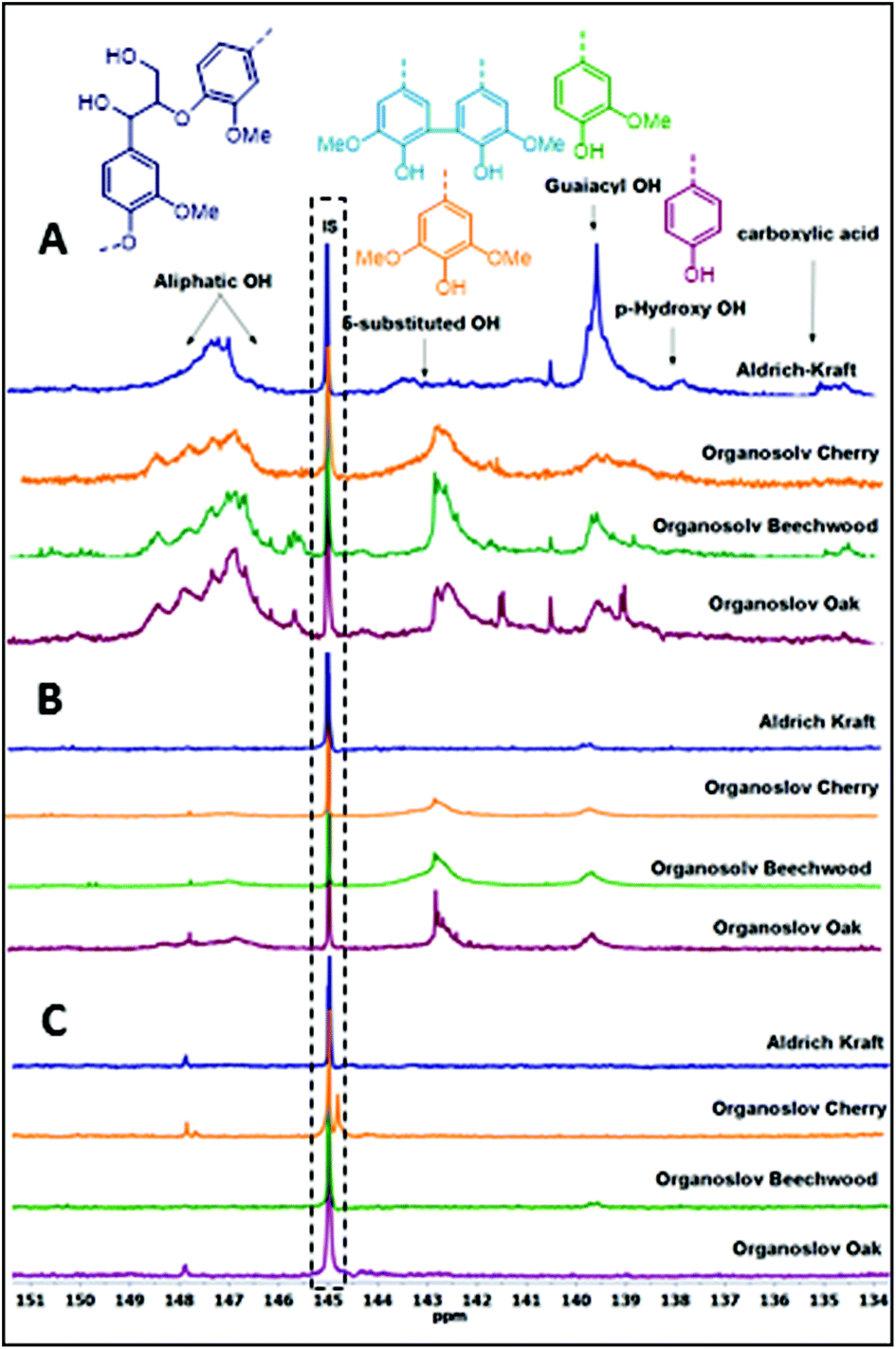
The degradation of the lignin linkages as observed in the 2D-NMR studies prompted us to further investigate its implications for the functional groups still present in the lignin structures. Thus, a series of product analyses were carried out using quantitative 31P NMR spectroscopy in the presence of a known amount of an internal standard (Fig. 2, Table 3).12 As shown in Fig. 2, for both bases an overall reduction in the amounts of aliphatic, phenolic, and carboxylic –OHs in the lignin samples was observed. However, quantification studies revealed that the Cs2CO3-catalysed reactions underwent a complete removal of these functional groups (Table 3 and Table S1†). One likely reason for the decrease in aliphatic –OH groups can be credited to the transesterification reaction with DMC catalysed by both the employed bases, as shown in Scheme 3. Reports in the literature suggest that the various phenolic-OH groups can undergo base promoted methylation.7h,13 Additionally, the cleavage of the interconnecting bonds could also result in decreased peak intensities in the defined regions of the spectra. Therefore, follow-up experiments focused on determining any shifts in the average molecular weights of the residual lignin mixtures after BCD reactions.
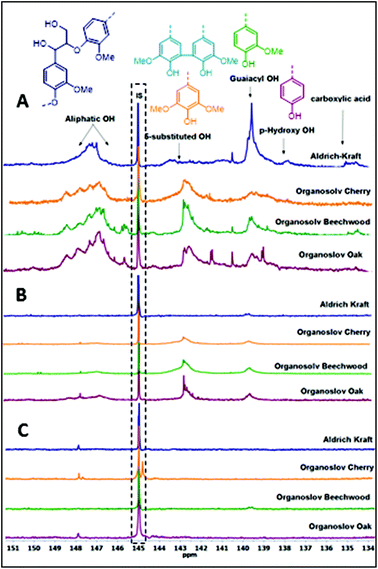 |
| | Fig. 2 Quantitative 31P NMR spectra of lignins; (A) before the reaction; (B) after the reaction with LiOt-Bu; (C) after the reaction with Cs2CO3. | |
 |
| | Scheme 3 Base-catalysed degradation in lignin β-O-4 model compounds. | |
Table 3 Reduction in hydroxyl groups of lignins quantified by 31P NMR using cyclohexanol as an internal standard
| Base |
% reduction in –OH |
| Lignin |
Aliphatic –OH |
5-Substituted OH |
Guaiacyl OH |
| LiOt-Bu |
OS-O |
84 |
48 |
73 |
| OS-BW |
89 |
68 |
74 |
| OS-C |
92 |
64 |
74 |
| Kraft |
96 |
93 |
93 |
| Cs2CO3 |
OS-O |
98 |
98 |
97 |
| OS-BW |
99 |
97 |
98 |
| OS-C |
97 |
98 |
97 |
| Kraft |
99 |
99 |
94 |
Gel permeation chromatography analysis
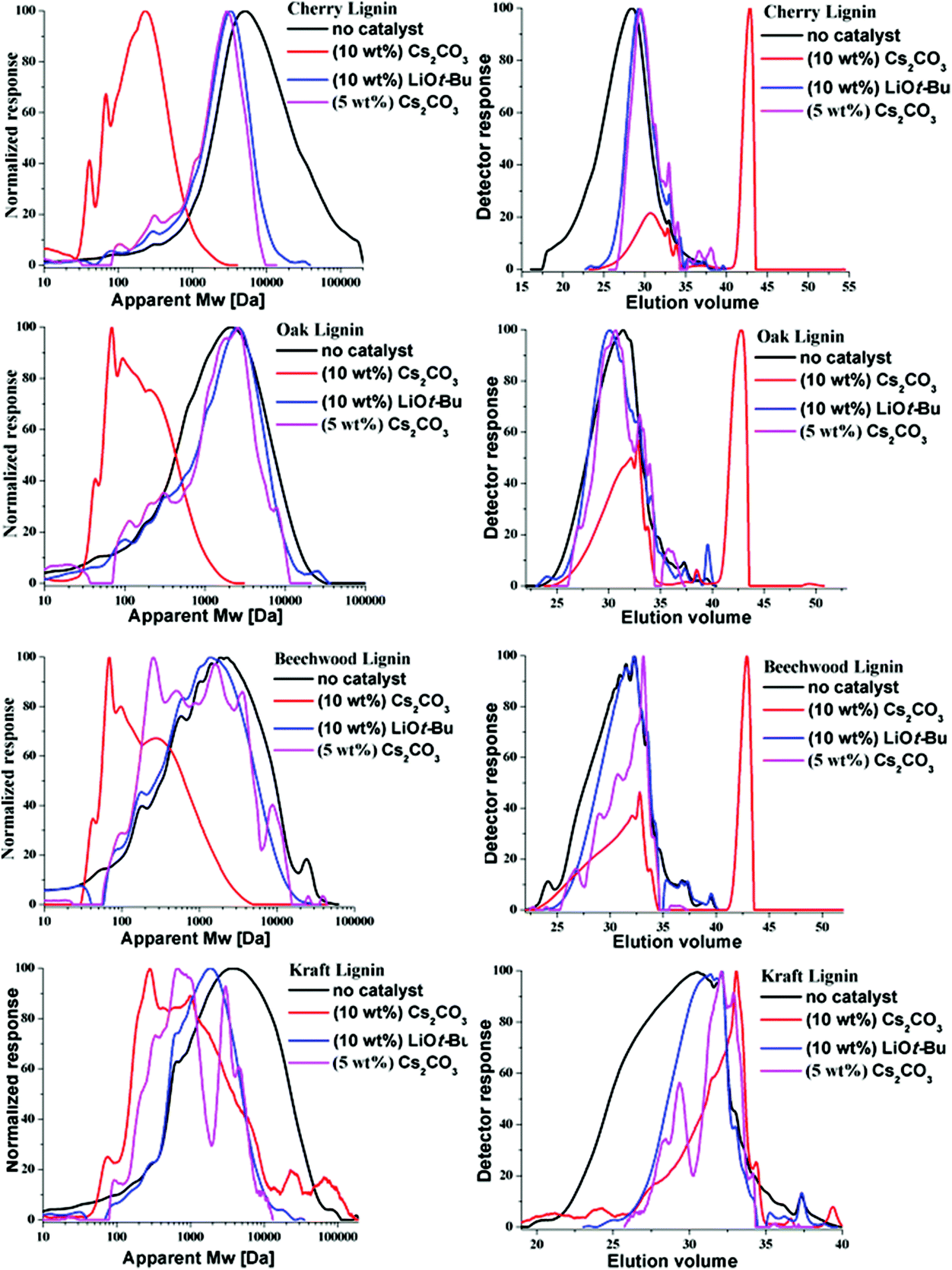
The relative change in the weight average molecular weight (Mw) of the lignin samples preceding the BCD was determined by GPC analysis. Fig. 3 depicts the mass distribution chromatograms and the elugrams for the lignins both before and after the BCD with 10 wt% [w(base)/w(lignin)] of Cs2CO3 and LiOt-Bu under the given reaction conditions. As shown in Table 4, there was a stark decrease in the Mw values for all of the samples treated with Cs2CO3 (10 wt%, Fig. 3, red lines), in comparison with the corresponding LiOt-Bu-catalysed reactions (Fig. 3, blue lines). For the organosolv lignins, the decrease in mass was more significant in comparison with the Kraft lignin. This can be attributed to the rather condensed structure of Kraft lignin with less alkyl–aryl ether linkages resulting from the more invasive pre-treatment conditions. Furthermore, reactions with reduced amounts of Cs2CO3 (5 wt%) proved to be less effective as observed in the GPC (pink lines), thereby emphasising the effect of the base concentration on the product outcome. The influence of the base loading did not become apparent through previous HSQC measurements considering that the degradation of the alkyl–aryl ether linkages was already observed with 5 wt% of Cs2CO3 and 1.2 wt% of LiOt-Bu,10p illustrating the importance of various analytical techniques for the characterisation of lignin depolymerisation.
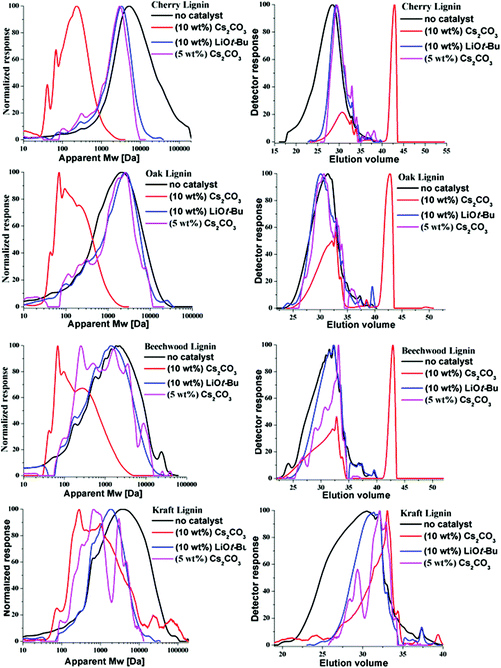 |
| | Fig. 3 GPC measurements for the cherry, oak, beechwood and Kraft lignin samples: before the reaction (no catalyst); after the reaction with Cs2CO3 (10 wt%, 0.030 mmol), 8 h, 180 °C; after the reaction with LiOt-Bu (10 wt%, 0.125 mmol), 12 h, 180 °C; after the reaction with Cs2CO3 (5 wt%, 0.015 mmol), 8 h, 180 °C. Left: Mass distribution with respect to polystyrene standard calibration; right: elugram. | |
Table 4 Average molecular weight change before and after the reactions
| Lignin |
M
w (Da) |
| Before the reaction |
5 wt% Cs2CO3 |
10 wt% Cs2CO3 |
10 wt% LiOt-Bu |
| Organosolv-cherry |
15220 |
2436 |
253 |
3235 |
| Organosolv-oak |
2610 |
2022 |
212 |
2506 |
| Organosolv-beechwood |
3103 |
2008 |
383 |
1898 |
| Aldrich-Kraft |
7106 |
2192 |
1696 |
5790 |
GC-MS analysis
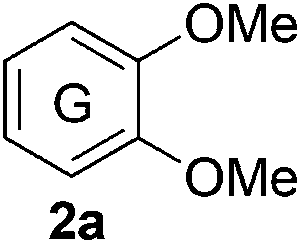
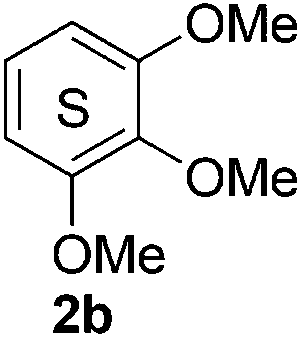
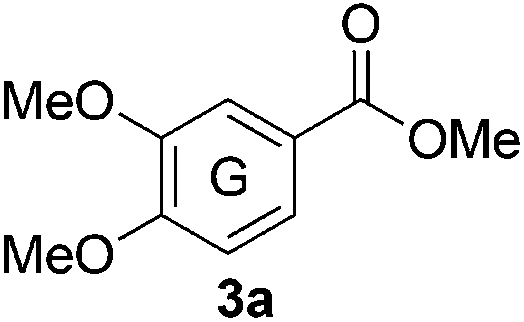
Based on the GPC results, reactions catalysed by 10 wt% of Cs2CO3 showed the highest reduction in molecular weight with a Mw value in the range of aromatic monomers and oligomers (200–1900 Da) for the four lignin samples (Table 4). The identification and quantification of these products were thus sought to be done by GC-MS analysis of the low-molecular weight oil fractions (52–67%) [w(oil)/w(initial lignin)] obtained after their separation from the ethyl acetate insoluble residue of the reaction mixture (for further details, see ESI section 2.4†). A correlation was observed between the yields of the low-molecular weight oil fractions and the general solubility of the lignin sample in the reaction solvent DMC. Organosolv cherry lignin had the lowest solubility in DMC and accordingly afforded the lowest low-molecular weight oil fraction (52 wt%). Table 5 summarises the quantification results of the major mono-aromatic cleavage products (2a,b and 3a,b) that we expected to find based on our model-based studies (Scheme 3). The product mixtures from oak, cherry, and beechwood lignin contained the capped syringaldehyde ester (3b) in major quantities while the capped guaiacyl ester yields remained within the expected product ratio (2a,b/3a,b), corresponding well to the amount of S and G units in the lignin starting material (compare Tables 1 and 5). Because the Kraft lignin from Sigma Aldrich (#370959) almost exclusively contains G units, the S containing units were only observed in trace quantities. Additionally, various capped liner esters were observed in the oil fraction of the lignin samples (see the ESI†). Considering that the cost of the reagents can be a limiting factor for reaction scale up with regard to biorefinery application, we also performed the BCD reaction with a widely available Kraft lignin and KOH as the base (Table 5, last column). To our delight, similar selectivities for the aromatic products were observed with comparable yields to the Cs2CO3-catalysed reactions.
Table 5 Quantification of selected methoxy-capped aromatic monomers identified in the oils obtained after the depolymerisation of lignin samples (OS-OL, OS-CL, OS-BWL, KL = 100 mg) at 180 °C for 8 h with 10 wt% [w(base)/w(lignin)] of Cs2CO3 in DMC (5 mL)
| Capped monomers |
OS-OL |
OS-CL |
OS-BWL |
KL |
KLa |
| wt% of starting material |
|
Reaction with 10 wt% [w(base)/w(lignin)] of KOH.
|
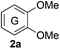
|
0.03 |
0.02 |
0.05 |
0.3 |
0.2 |
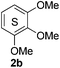
|
0.2 |
— |
0.1 |
0.05 |
— |
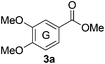
|
0.6 |
0.9 |
1.1 |
1.8 |
1.5 |
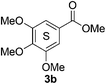
|
3.4 |
4.0 |
3.2 |
— |
— |
| Total (2 + 3) |
4.23 |
4.92 |
4.45 |
2.15 |
1.7 |
Depolymerisation of beechwood chips
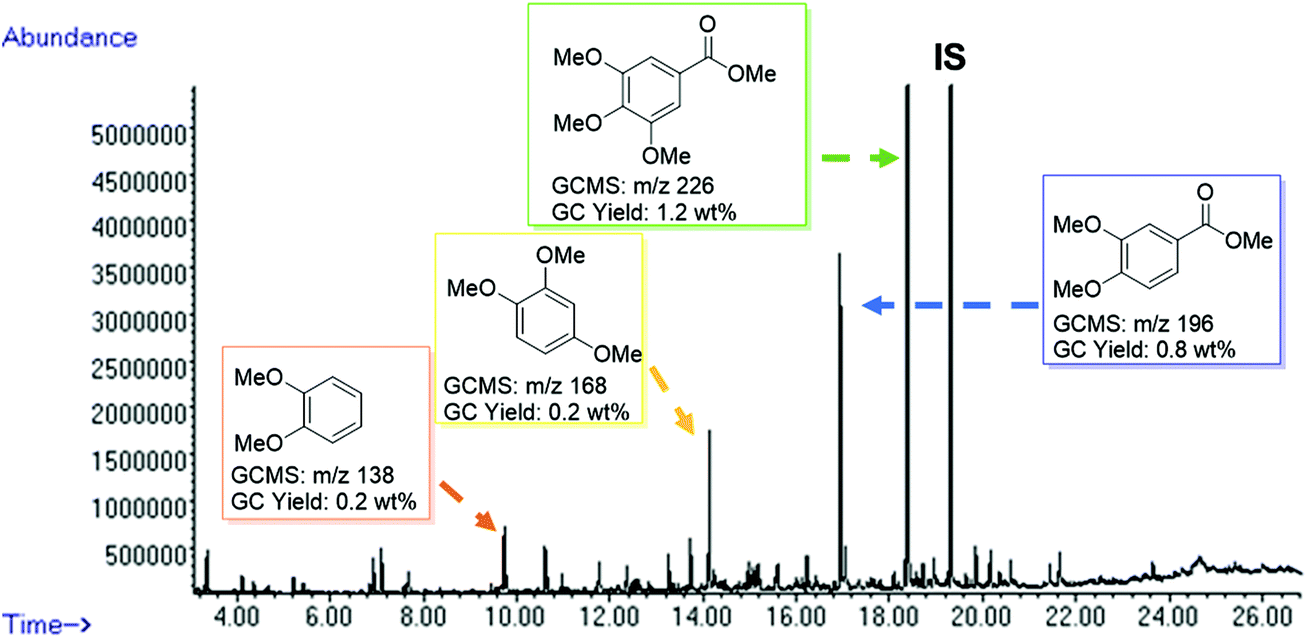
After the successful degradation of extracted lignin samples, we decided to test the direct applicability of our BCD system on wood. The reaction was performed using a sample of pre-milled beechwood chips (100 mg) with Cs2CO3 (10 wt%) in DMC. Pleasingly, an oil fraction of 40 wt% was recovered while affording a deep brown residue in 60 wt%. The oil fraction was then subjected to GC-MS analysis (Fig. 4) and gratifyingly we could identify methoxy-capped monoaromatic products with a similar product distribution as observed for the extracted lignin samples. A control reaction carried out in the absence of the base did not afford any cleavage products thereby ruling out any influence of the pre-milling conditions on the product formation (see ESI Fig. S9†).
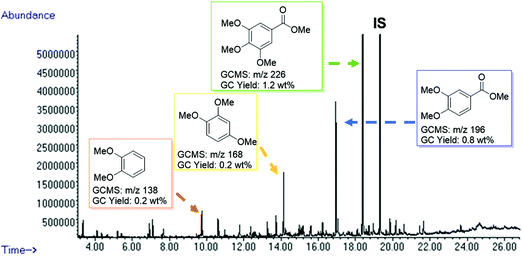 |
| | Fig. 4 GC-FID trace of the product mixture obtained from the depolymerisation of milled beechwood chips (100 mg) using Cs2CO3 (10 wt%) in DMC (5 mL) at 180 °C for 8 h. Identification of the major peaks is based on the GC-MS data (full peak overview in Table S6†). Quantification is based on the standardization of individual products, using n-octadecane as an internal standard. | |
Model compound studies
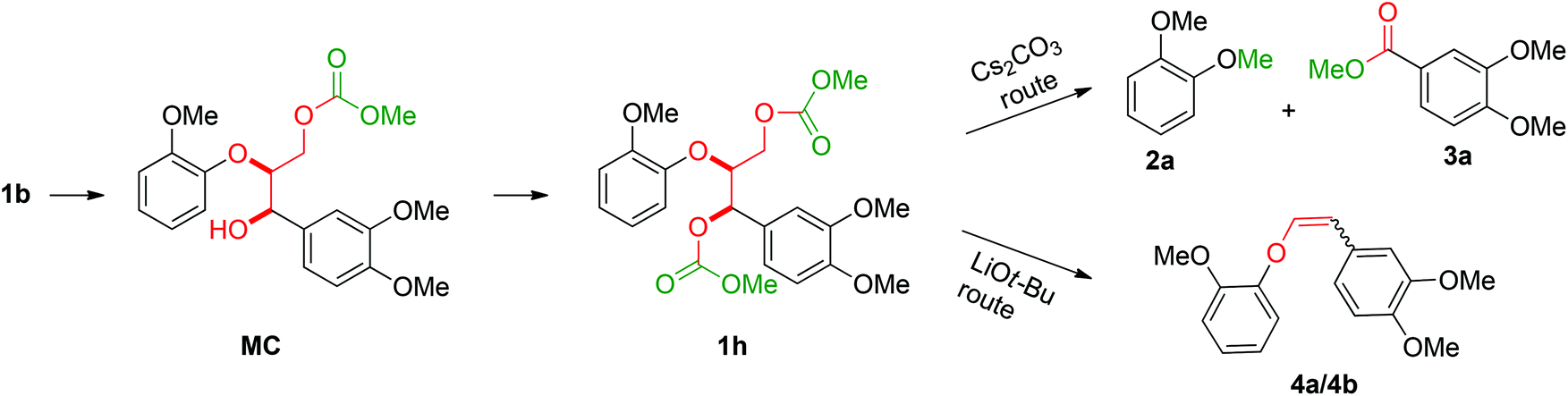
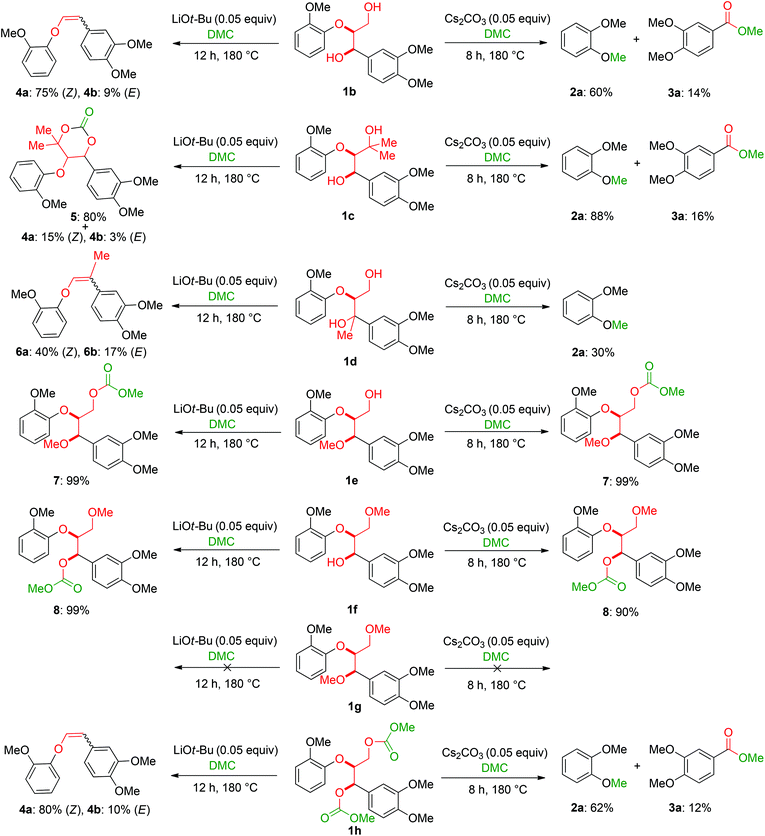
The obtained results for the BCD on lignin samples and wood gave rise to the question which reaction pathway is involved in the cleavage of the β-O-4 bonds and the subsequent capping of products. To answer this question, a range of lignin β-O-4 model compounds were investigated having some of their pivotal sites such as the α-proton, γ-proton, or either one or both the aliphatic hydroxyls substituted by methyl or methoxy groups (Scheme 3). The idea was to compare the reactivity of these models (1c–h) with the unsubstituted model compound 1b. Model compound 1c which has both of its γ-protons replaced by methyl groups resulted in increased veratrole (2a) and dimethoxybenzoate (3a) yields for the Cs2CO3-catalysed reaction. However, model 1d containing the substituted α-proton formed a complex mixture of products with the overall yields of 2a decreasing by half (30%). This loss in yield suggests a possible involvement of the α-proton in the cleavage reactions for 1b. In contrast, the reaction of both 1c and 1d with LiOt-Bu resulted in an overall decrease in the yields of the corresponding alkenes 4 and 6 possibly due to the formation of an additional cyclic product such as 5. Furthermore, substituting either one of the two aliphatic hydroxyls such as in models 1e and 1f restricted the reactions for both bases to stop at the monocarboxylated products 7 and 8, thereby highlighting the necessity of both the positions to be free to initiate a successful cleavage and elimination reaction. This was further confirmed by model 1g that remained unreactive for both base catalysts. Based on our observations on model compound studies (Scheme 4), the formation of dicarboxylated product 1h starting from model 1b in DMC was speculated to be a common intermediate for the pathways involving both Cs2CO3 and LiOt-Bu. Indeed, individual reactions of 1h with these bases furnished comparable results as with 1b affording slightly increased yields (Schemes 3 and 4).
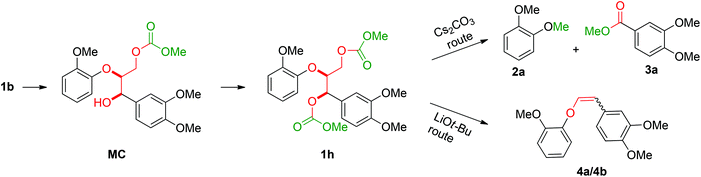 |
| | Scheme 4 Reaction of the β-O-4 model linkage under BCD conditions showing the reaction sequence and the point of divergence in reactivity for Cs2CO3 and LiOt-Bu. | |
Reaction profile study
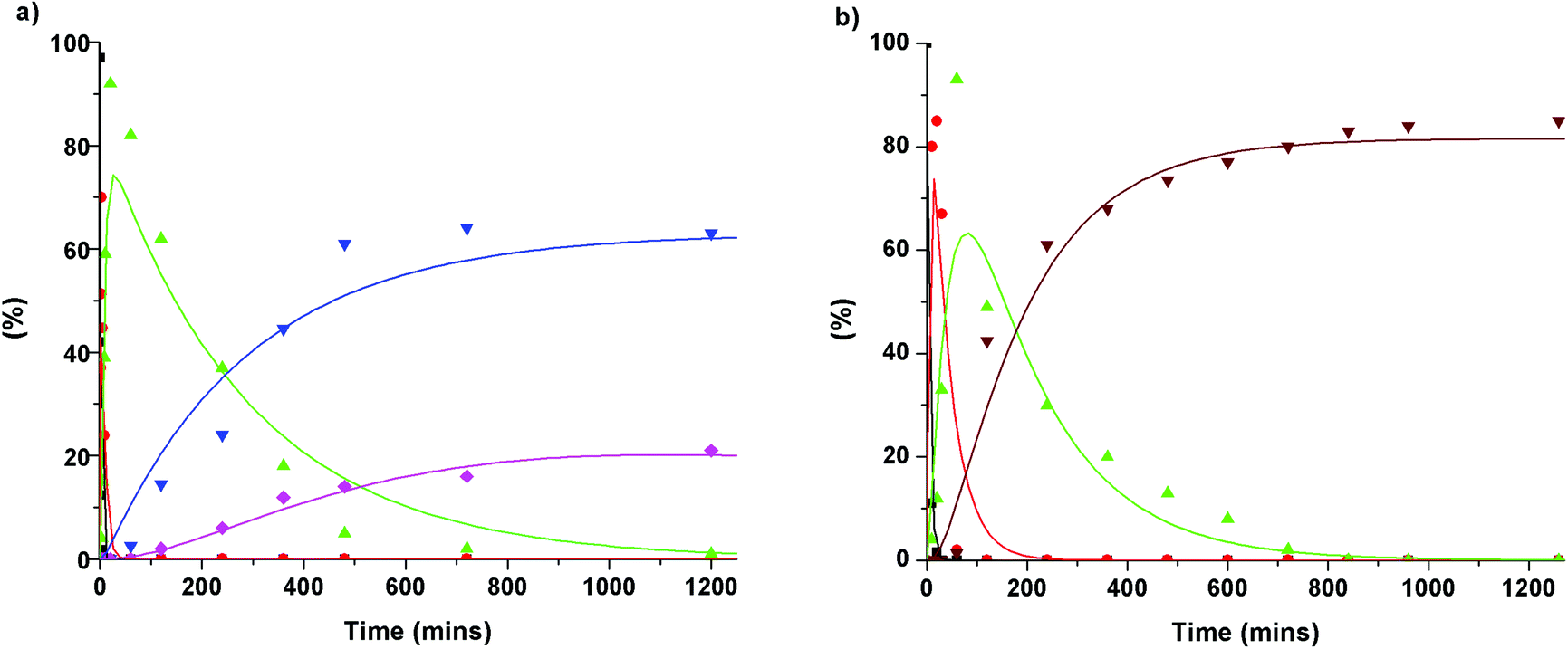
To gain further insight into the degradation of the β-O-4 linkage under the Cs2CO3- and LiOt-Bu-catalysed reaction conditions, the consumption of 1b and the formation of stable intermediates and products were monitored by HPLC analysis (Fig. 5, see ESI section 4†). From this analysis, a series of reaction pathways were proposed, and the rate analysis provided the curve fits shown in the corresponding figures.
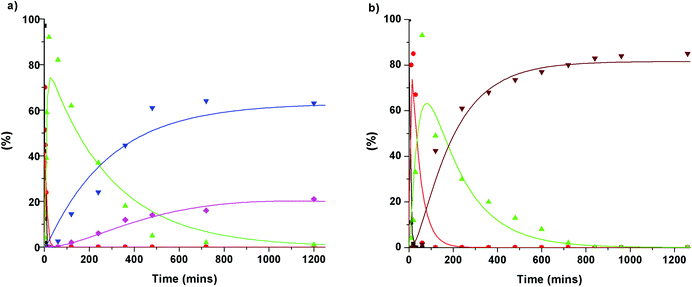 |
| | Fig. 5 Reaction profiles: (a) Cs2CO3 at 180 °C in DMC with (β-O-4) model compound 1b. (b) LiOt-Bu at 180 °C in DMC with (β-O-4) model compound 1b. Dots show experimental data points, whereas the line is a modeled reaction. (a) Black: Dilignol (1b); red: monocarboxylated dilignol (MC); green: dicarboxylated dilignol (1h); blue: veratrole (2a); pink: 3,4-dimethoxybenzoate (3a). (b) Black: Dilignol (1b); red: monocarboxylated dilignol (MC); green: dicarboxylated dilignol (Ih); brown: cis/trans alkenes (4a/4b). | |
The initial reactions were found to be the same for both Cs2CO3 and LiOt-Bu. Carboxylation of the γ-hydroxyl group occurred first (reaction rates of MC = 0.69 min−1 for Cs2CO3 and 0.2 min−1 for LiOt-Bu) followed by a second carboxylation of the α-hydroxyl group resulting in common intermediate 1h (reaction rates of 1h = 0.15 min−1 for Cs2CO3 and 0.025 min−1 for LiOt-Bu). Following the formation of 1h, the reaction pathways diverge. The reaction with Cs2CO3 promotes the cleavage of the C–O bond resulting in veratrole (2a). The production of 2a, however, does not correlate directly with the accruement of 3,4-dimethoxybenzoate (3a). A probable intermediate M is speculated to be formed which undergoes a subsequent reaction to 3a. The detailed reaction pathway leading to M is discussed later during the computational analysis.
Upon the formation of 1h in the reaction with LiOt-Bu, the alkenes 4a and 4b are formed without any observable intermediates (the rate of formation of 4a/4b = 0.005 min−1).
Computational study
Based on the studies above, quantum chemical calculations were considered necessary to find a reasonable mechanism rationalising the observed selectivities for the obtained cleavage products. Here again dilignol 1b was chosen as a representative substrate and caesium methanolate (CsOMe) as a model base considering the depletion of Cs2CO3 (0.05 equiv.) over the course of the reaction resulting in a methanolate as a by-product.
Thus, after an initiation period mainly methanolates acted as bases in the reaction. The investigated reaction pathways were limited to intermediates and transition states without explicit coordination of the caesium ions. The main reason for this approximation is the dramatically increased number of different structures and possible oligomers that would have to be taken into consideration for each step. Secondly, the PCM solvation model would be inaccurate for the description of the solvated species in this system preventing a reliable comparison of structures with different coordination of the caesium cation. Also, based on Pearson's HSAB principle, it can be expected that the uncoordinated intermediates and transition states provide a good correlation for the reactions with a caesium base owing to its weak coordination to the deprotonated oxygen atoms of the substrate.14
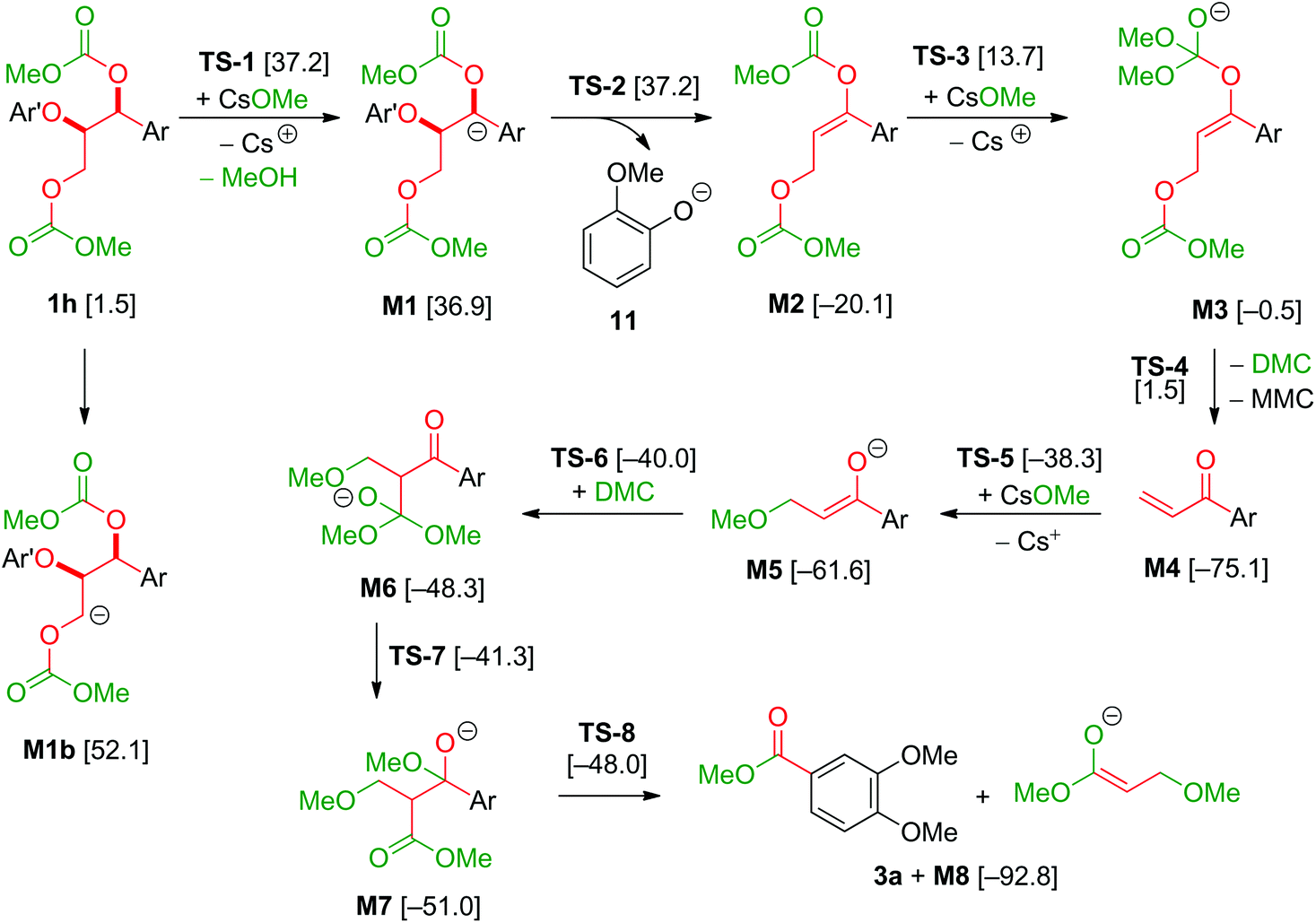
The model compound studies (Scheme 3) suggest the involvement of both the α- and γ-protons in the formation of veratrole (2a) and 3,4-dimethoxybenzoate (3a). The decreased yield of 2a for model compound 1d specifically indicates that these reaction products are formed after a deprotonation at the α-position. This deprotonation reaction, however, most likely occurs on the dicarboxylated dilignol 1h since, in contrast to 1b, it has no free hydroxyl groups, which are more acidic than the aliphatic hydrogen atoms. Moreover, the experimental results already indicated 1h as an intermediate in the formation of 2a and 3a (see Scheme 3; the reaction of 1h with Cs2CO3 and Fig. 5a).
The deprotonation at the α-carbon of 1h results in intermediate M1 (Scheme 5). This intermediate is favoured with a Gibbs energy difference of 15.2 kcal mol−1 over M1b which is deprotonated at the γ-carbon. The energy of M1 is 36.9 kcal mol−1 relative to 1b. The transition state of the deprotonation (TS-1) has a Gibbs energy of 37.2 kcal mol−1 relative to 1b. The elimination of guaiacolate (11) from M1 proceeds without a significant barrier and gives enol ether M2. The formation of veratrole from 1d (Scheme 3) can be attributed to a deprotonation at the γ-position. However, the lower acidity of the hydrogen at the γ-position compared to the one at the α-position explains the low yield of veratrole (2a). Furthermore, the absence of 3a in the product spectrum of 1d can be explained by the formation of a different intermediate after the elimination process. Additionally, a similar product inhibition for the formation of veratrole by o-methoxy groups was also observed in our previous study.10p The overall change of the selectivity thus indicates that the steric hindrance at the α-carbon might be preventing the deprotonation.
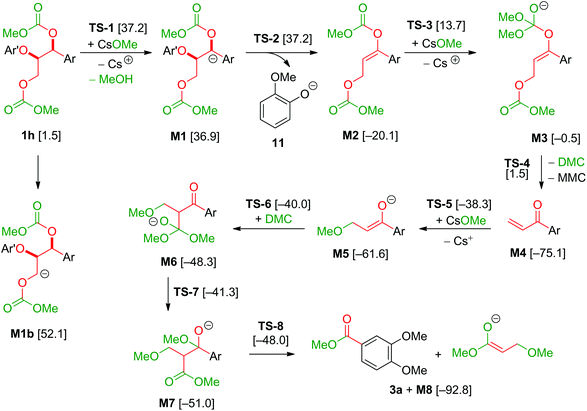 |
| | Scheme 5 Reaction pathway from dicarboxylated dilignol 1h to 3,4-dimethoxybenzoate (3a); Ar: 3,4-dimethoxyphenyl; Ar′: 2-methoxyphenyl; MMC: monomethoxy carbonate; [Gibbs energies relative to 1b in kcal mol−1]. | |
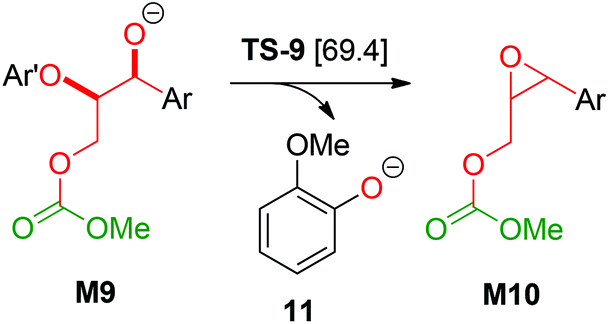
Based on a recent computational study by Yu,15 we also investigated a possible elimination of guaiacolate from the monocarboxylated structure M9 resulting in an oxirane intermediate (see Scheme 6). However, the Gibbs energy of the transition state TS-9 of this reaction is 69.4 kcal mol−1 relative to 1b and 32.2 kcal mol−1 relative to TS-1. Therefore, Yu's mechanism is highly disfavoured in our reaction system.
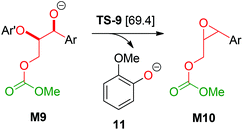 |
| | Scheme 6 Elimination of 11 according to Yu et al.;15 Ar: 3,4-dimethoxyphenyl; Ar′: 2-methoxyphenyl; [relative Gibbs energies to 1b in kcal mol−1]. | |
The final step in the formation of 2a is the methylation of 11 by dimethyl carbonate. The relative Gibbs energy of the transition state of the methylation under Walden inversion at the methyl group is 24.5 kcal mol−1 relative to 11.
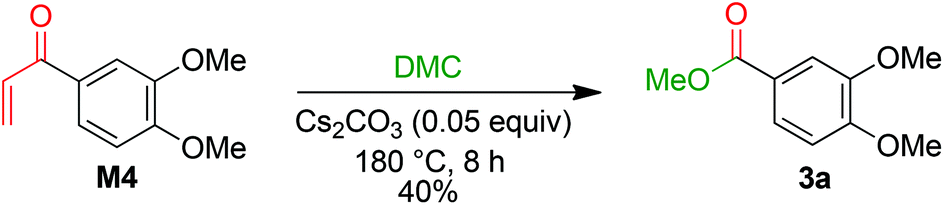
The formation of 3a is a result of the decomposition of M2. The addition of a methanolate to the methoxy carbonate group is endergonic. The resulting intermediate M3 has a Gibbs energy of 19.6 kcal mol−1 relative to M2. The transition state of this step has an energy of 33.8 kcal mol−1 compared to M2. Elimination of dimethyl carbonate from M3 results in a simultaneous elimination of monomethoxy carbonate (MMC, decomposing into MeOH and CO2). The transition state TS-4 of this step is only 2.0 kcal mol−1 higher in energy than M3. Moreover, the reaction is highly exergonic since the Gibbs energy of the vinyl ketone M4 is 74.6 kcal mol−1 lower than that of M3 and the energy is 55.0 kcal mol−1 lower than that of M2. The vinyl ketone M4 can react with a methanolate to give the enolate M5. The addition reaction is endergonic with an energy difference of 13.9 kcal mol−1 and the corresponding transition state TS-5 having an energy of 37.2 kcal mol−1. Addition of dimethyl carbonate to the α-carbon of M5 in a Claisen condensation-type reaction (TS-6) results in intermediate M6. Instead of an elimination of a methanolate, a transfer of a methoxy group of the dimethyl carbonate group to the carbonyl group is likely to occur. The resulting intermediate M7 resembles the intermediate of a Claisen condensation between 3,4-dimethoxybenzoate and the enolate M8. Hence, a retro-Claisen condensation-type step (TS-8) yields the experimentally observed product 3a. The formation of 3a and M8 is highly exergonic with an energy difference of 92.8 kcal mol−1 relative to 4a and 17.7 kcal mol−1 relative to M4. TS-5 is the transition state with the highest Gibbs energy (35.1 kcal mol−1 relative to M4) between M4 and 3a. To support the proposed reaction pathway, we investigated a crucial intermediate: the vinyl ketone M4 (Scheme 7). M4 was synthesised (see ESI 5†) and used as a starting material in a reaction under the same conditions as shown for 1b in Scheme 3.
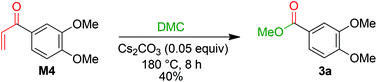 |
| | Scheme 7 Reaction of M4 with Cs2CO3 in DMC. | |
The formation of 3a in 40% yield when employing M4 as a starting material confirms that there is a reaction pathway between M4 and 3a. This is also in accordance with the proposed formation of an intermediate M while studying the reaction profile for the decomposition of 1b with caesium carbonate to 3a (ESI section 4.3;†M = M4).
Conclusion
We have developed a base-catalysed depolymerisation strategy for various lignin samples including organosolv and Kraft lignin in dimethyl carbonate using alkaline earth metal catalysts. For the first time, a truly catalytic base-catalysed depolymerisation process (catalyst loadings of around 5 mol%) is demonstrated affording low-molecular weight oils. For all lignin samples, the low-molecular weight oil and the ethyl acetate insoluble fractions were quantified and characterised using HSQC 2D-NMR, 31P NMR, GPC, and GC-MS measurements. The oil fractions for the Cs2CO3-catalysed lignin degradation were found to be within the range of 52–67 wt% of the starting lignin material and contained a range of methoxy capped aromatic monomers. Furthermore, it was shown that for potential future biorefinery applications the base can be changed to readily available KOH affording similar monomer yields. The methodology with the caesium salt was also effective for the direct degradation of milled beechwood chips thereby further expanding the scope to a more robust lignocellulosic starting material. These results are well in agreement with our model compound studies and hence prove the system's efficiency and applicability to the complex lignin polymer. In our quest to understand the different reaction pathways involved in the degradation using LiOt-Bu and Cs2CO3 in dimethyl carbonate, we further studied a variety of lignin model compounds (1a–1h), which demonstrated the importance of the free aliphatic hydroxyl groups and the proton at the α-carbon of the lignin β-O-4 linkage in the depolymerisation reaction. Monitoring the reaction of 1b for both bases helped us to elucidate the overall reaction pathway and the point of divergence for the bases that leads to the different product selectivities. This was further confirmed by the quantum chemical calculations performed for the Cs2CO3-catalysed cleavage reaction on model compound 1b. Ultimately, we could provide a rational reaction pathway for our BCD system in dimethyl carbonate based upon our extensive studies on both the model compound and real lignin samples.16
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the European Commission (SuBiCat Initial Training Network, Call FP7-PEOPLE-2013-ITN, grant no. 607044). S. D., S. S. M. S., and C. W. L. thank the international training network SuBiCat for their predoctoral stipends. J. M. is grateful to the NRW Graduate School BrenaRo and the German Academic Exchange Service (DAAD) for predoctoral stipends. J. E. thanks the Deutsche Forschungsgemeinschaft through the International Research Training Group Seleca (IGRK 1628) and the IT Center of RWTH Aachen University for providing the computational resources. We thank Dr N. Anders (RWTH Aachen University) for the GPC measurements, T. Rinesch (RWTH Aachen University) for proofreading the manuscript, and Dr C. Räuber (RWTH Aachen University) for helpful discussions during the NMR investigations. Prof. Dr Paul C. J. Kamer, Prof. Dr Andrew D. Smith, and Prof. Dr Nick Westwood (all from the University of St Andrews, Scotland) and Dr R. Rinaldi (formerly MPI für Kohlenforschung, Mülheim a.d.R.) are acknowledged for their helpful discussions and for supplying oak, cherry and beechwood lignin samples, respectively.
Notes and references
-
(a) A. Sakakibari, Wood Sci. Technol., 1980, 14, 89–100 CrossRef;
(b)
R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as Feedstock for a bioenergy and bioproducts industry: the technical feasibility of a billion-ton annual supply, U. S. Department of Energy, 2005 Search PubMed.
-
(a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed;
(b) A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed;
(c) M. D. Karkas, B. S. Matsuura, T. M. Monos, G. Magallanes and C. R. Stephenson, Org. Biomol. Chem., 2016, 14, 1853–1914 RSC;
(d) S. Dabral, M. Turberg, A. Wanninger, C. Bolm and J. G. Hernández, Molecules, 2017, 22, 146 CrossRef PubMed.
-
(a) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed;
(b) D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
-
(a) P. J. Deuss, K. Barta and J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 RSC;
(b) C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC;
(c) R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed;
(d) U. Weißbach, S. Dabral, L. Konnert, C. Bolm and J. G. Hernández, Beilstein J. Org. Chem., 2017, 13, 1788–1795 CrossRef PubMed;
(e) S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- Selected oxidative degradation approaches via benzylic alcohol oxidation:
(a) A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC;
(b) A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed;
(c) J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1122 CrossRef CAS PubMed;
(d) A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed;
(e) K. Walsh, H. F. Sneddon and C. J. Moody, Org. Lett., 2014, 16, 5224–5227 CrossRef CAS PubMed;
(f) M. Dawange, M. V. Galkin and J. S. M. Samec, ChemCatChem, 2015, 7, 401–404 CrossRef CAS;
(g) C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed;
(h) R. Zhu, B. Wang, M. Cui, J. Deng, X. Li, Y. Ma and Y. Fu, Green Chem., 2016, 18, 2029–2036 RSC;
(i) S. Dabral, J. G. Hernández, P. C. J. Kamer and C. Bolm, ChemSusChem, 2017, 10, 2707–2713 CrossRef CAS PubMed.
- For selected articles see:
(a) S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611–5618 CrossRef CAS PubMed;
(b) S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed;
(c) J. Zakzeski, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2011, 13, 671–680 RSC;
(d) B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. Pete Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS;
(e) S. K. Hanson, R. Wu and L. A. Pete Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed;
(f) B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730–2733 CrossRef CAS PubMed;
(g) J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS;
(h) B. Sedai and T. Baker, Adv. Synth. Catal., 2014, 356, 3563–3574 CrossRef CAS;
(i) J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ChemSusChem, 2015, 8, 2106–2113 CrossRef CAS PubMed;
(j) J. Mottweiler, T. Rinesch, C. Besson, J. Buendia and C. Bolm, Green Chem., 2015, 17, 5001–5008 RSC;
(k) T. Rinesch, J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ACS Sustainable Chem. Eng., 2017, 5, 9818–9825 CrossRef CAS.
- For selected articles see:
(a) N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed;
(b) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed;
(c) A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS;
(d) J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed;
(e) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed;
(f) J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed;
(g) Z. Strassberger, A. H. Alberts, M. J. Louwerse, S. Tanase and G. Rothenberg, Green Chem., 2013, 15, 768–774 RSC;
(h) T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttämaa, F. Ribeiro, J. T. Miller and M. M. AbuOmar, Chem. Sci., 2013, 4, 806–813 RSC;
(i) J. A. Barrett, Y. Gao, C. M. Bernt, M. Chui, A. T. Tran, M. B. Foston and P. C. Ford, ACS Sustainable Chem. Eng., 2016, 4, 6877–6886 CrossRef CAS.
- For selected articles see:
(a) M. Nagy, K. David, G. J. P. Britovsek and A. J. Ragauskas, Holzforschung, 2009, 63, 513–520 CrossRef CAS;
(b) J. M. Nichols, L. M. Bishop, R. J. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed;
(c) T. vom Stein, T. Weigand, C. Merkens, J. Klankermayer and W. Leitner, ChemCatChem, 2013, 5, 439–441 CrossRef CAS;
(d) D. Weickmann and B. Plietker, ChemCatChem, 2013, 5, 2170–2173 CrossRef CAS;
(e) R. G. Harms, I. I. E. Markovits, M. Drees, W. A. Herrmann, M. Cokoja and F. E. Kìhn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed;
(f) T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef CAS PubMed.
- For selected recent articles see:
(a) S. Jia, B. J. Cox, X. Guo, Z. C. Zhang and J. G. Ekerdt, ChemSusChem, 2010, 3, 1078–1084 CrossRef CAS PubMed;
(b) M. R. Sturgeon, S. Kim, K. Lawrence, R. S. Paton, S. C. Chmely, M. Nimlos, T. D. Foust and G. T. Beckham, ACS Sustainable Chem. Eng., 2014, 2, 472–485 CrossRef CAS;
(c) T. Yokoyama, J. Wood Chem. Technol., 2014, 35, 27–42 CrossRef CAS;
(d) R. G. Harms, I. I. E. Markovits, M. Drees, W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed;
(e) A. Kaiho, M. Kogo, R. Sakai, K. Saito and T. Watanabe, Green Chem., 2015, 17, 2780–2783 RSC;
(f) P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed;
(g) M. Scott, P. J. Deuss, J. G. de Vries, M. H. G. Prechtl and K. Barta, Catal. Sci. Technol., 2016, 6, 1882–1891 RSC;
(h) C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Z. Slawin, P. C. J. Kamer, J. G. de Vries, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef CAS PubMed.
- For homogeneous base-catalysed degradation of lignin model compounds and lignin see:
(a) R. W. Thring, Biomass Bioenergy, 1994, 7, 125–130 CrossRef CAS;
(b) J. E. Miller, I. Evans, A. Littlewolf and D. E. Trudell, Fuel, 1999, 78, 1363–1366 CrossRef CAS;
(c)
J. S. Shabtal, W. W. Zmierczak and E. Chornet, Process for conversion of lignin to reformulated, partially oxygenated gasoline, U. S. PatentUS6172272B1, Jan. 9, 2001 Search PubMed;
(d)
J. S. Shabtal, W. W. Zmierczak, E. Chornet and D. Johnson, Process for converting lignins into a high octane blending component, U. S. PatentUS2003/0115792A1, June 26, 2003 Search PubMed;
(e)
J. S. Shabtal, W. W. Zmierczak, E. Chornet and D. Johnson, Process for converting lignins into a high octane additive, U. S. PatentUS2003/0100807A1, May 29, 2003 Search PubMed;
(f) S. Karagöz, T. Bhaskar, A. Muto and Y. Sakata, Fuel, 2004, 83, 2293–2299 CrossRef;
(g) A. Vigneault, D. K. Johnson and E. Chornet, Can. J. Chem. Eng., 2007, 85, 906–916 CrossRef CAS;
(h) S. Nenkova, T. Vasileva and K. Stanulov, Chem. Nat. Compd., 2008, 44, 182–185 CrossRef CAS;
(i) J. M. Lavoie, W. Baré and M. Bilodeau, Bioresour. Technol., 2011, 102, 4917–4920 CrossRef CAS PubMed;
(j) V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed;
(k) R. Beauchet, F. Monteil-Rivera and J. M. Lavoie, Bioresour. Technol., 2012, 121, 328–334 CrossRef CAS PubMed;
(l) A. Toledano, L. Serrano and J. Labidi, J. Chem. Technol. Biotechnol., 2012, 87, 1593–1599 CrossRef CAS;
(m) T. Kleine, J. Buendia and C. Bolm, Green Chem., 2013, 15, 160–166 RSC;
(n) A. Toledano, L. Serrano and J. Labidi, Fuel, 2014, 116, 617–624 CrossRef CAS;
(o) E. M. Karp, B. S. Donohoe, M. H. O'Brien, P. N. Ciesielski, A. Mittal, M. J. Biddy and G. T. Beckham, ACS Sustainable Chem. Eng., 2014, 2, 1481–1491 CrossRef CAS;
(p) S. Dabral, J. Mottweiler, T. Rinesch and C. Bolm, Green Chem., 2015, 17, 4908–4917 RSC;
(q) R. Katahira, A. Mittal, K. Mc Kinney, X. Chen, M. P. Tucker, D. K. Johnson and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 1474–1486 CrossRef CAS;
(r) A. Rodriguez, D. Salvachúa, R. Katahira, B. A. Black, N. S. Cleveland, M. Reed, H. Smith, E. E. K. Baidoo, J. D. Keasling, B. A. Simmons, G. T. Beckham and J. M. Gladden, ACS Sustainable Chem. Eng., 2017, 5, 8171–8180 CrossRef CAS;
(s) Z. Strassberger, P. Prinsen, F. vam der Klis, D. S. van Es, S. Tanase and G. Rothenberger, Green Chem., 2015, 17, 325–334 RSC.
- For heterogeneous base-catalysed degradation of lignin model compounds and lignin see:
(a) G. S. Macala, T. D. Matson, C. L. Johnson, R. S. Lewis, A. V. Iretskii and P. C. Ford, ChemSusChem, 2009, 2, 215–217 CrossRef CAS PubMed;
(b) Z. Yuan, S. Cheng, M. Leitch and C. C. Xu, Bioresour. Technol., 2010, 101, 9308–9313 CrossRef CAS PubMed;
(c) M. R. Sturgeon, M. H. O'Brien, P. N. Ciesielski, R. Katahira, J. S. Kruger, S. C. Chmely, J. Hamlin, K. Lawrence, G. B. Hunsinger, T. D. Foust, R. M. Baldwin, M. J. Biddy and G. T. Beckham, Green Chem., 2014, 16, 824–835 RSC;
(d) K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC;
(e) J. Long, Q. Zhang, T. Wang, X. Zhang, Y. Xu and L. Ma, Bioresour. Technol., 2014, 154, 10–17 CrossRef CAS PubMed;
(f) J. S. Kruger, N. S. Cleveland, S. Zhang, R. Katahira, B. A. Black, G. M. Chupka, T. Lammens, P. G. Hamilton, M. J. Biddy and G. T. Beckham, ACS Catal., 2016, 6, 1316–1328 CrossRef CAS;
(g) R. Chaudhary and P. L. Dhepe, Green Chem., 2017, 19, 778–788 RSC.
-
(a) D. S. Argyropoulos, Res. Chem. Intermed., 1995, 21, 373–395 CrossRef CAS;
(b) Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 RSC;
(c) M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 CrossRef CAS;
(d) S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
-
(a) S. Ouk, S. Thiebaud, E. Borredon, P. Legars and L. Lecomte, Tetrahedron Lett., 2002, 43, 2661–2663 CrossRef CAS;
(b) K. M. Deshmukh, Z. S. Qureshi, K. P. Dhake and B. M. Bhanage, Catal. Commun., 2010, 12, 207–211 CrossRef CAS;
(c) J. N. G. Stanley, M. Selva, A. F. Masters, T. Maschmeyerb and A. Perosa, Green Chem., 2013, 15, 3195–3204 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- T. Lankau and C. H. Yu, Green Chem., 2016, 18, 1590–1596 RSC.
- For two very instructive reviews on the use of DMC, which were published during the evaluation of this manuscript, see:
(a) P. Tundo, M. Musolino and F. Aricò, Green Chem. 10.1039/c7gc01764b;
(b) G. Fiorani, A. Perosa and M. Selva, Green Chem. 10.1039/c7gc02118f.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc03110f |
| ‡ Present address: School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, United Kingdom. |
| § Present address: Instituto de Tecnologia Química (UPV-CSIC), Av de los Naranjos s/n, Valencia, Spain. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Julien
Engel‡
a,
Julien
Engel‡