
Synergistic effects in Fe nanoparticles doped with ppm levels of (Pd + Ni). A new catalyst for sustainable nitro group reductions†
Haobo
Pang
a,
Fabrice
Gallou
 b,
Hyuntae
Sohn
c,
Jeffrey
Camacho-Bunquin
c,
Massimiliano
Delferro
c and
Bruce H.
Lipshutz
*a
b,
Hyuntae
Sohn
c,
Jeffrey
Camacho-Bunquin
c,
Massimiliano
Delferro
c and
Bruce H.
Lipshutz
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: lipshutz@chem.ucsb.edu
bNovartis Pharma AG, Basel, Switzerland
cChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 8th December 2017
Abstract
A remarkable synergistic effect has been uncovered between ppm levels of Pd and Ni embedded within iron nanoparticles that leads to mild and selective catalytic reductions of nitro-containing aromatics and heteroaromatics in water at room temperature. NaBH4 serves as the source of inexpensive hydride. Broad substrate scope is documented, along with several other features including: low catalyst loading, low residual metal in the products, and recycling of the catalyst and reaction medium, highlight the green nature of this new technology.
Introduction
Bimetallic catalysis is a blossoming area of synthetic organic chemistry, potentially offering tremendous opportunities that augment traditional metal-catalyzed processes.1 As recently reviewed by Pye and Mankad,2 both mixed metal and multi-valent, single metal catalysts have led to exciting C–C and C–X couplings, while selected oxidations using related species have also been highlighted this year as well by Parmeggiani, Matassini, and Cardona.3 Missing from these discussions are applications of synergistic effects to multiple metal-catalyzed processes in general, and to nitro group reductions, in particular. Development of a highly efficient new technology that not only takes advantage of two transition metals working in harmony towards a common synthetic goal, but also adheres to many of the 12 Principles of Green Chemistry4 would be of special merit. In this report we describe a newly discovered synergistic relationship between two group 10 metals. That is, very clean reduction of aromatic and heteroaromatic nitro groups to the corresponding primary amines takes place in water at room temperature when ppm levels of both Pd and Ni are embedded within iron nanoparticles.Selective reduction of nitro groups represents an essential methodology, given the importance of this functional group within intermediates for fine chemicals, agrochemicals, pharmaceuticals, dyes, and polymers.5 In general, reductions of nitroarenes tend to be either metal-free or metal-based.6 The latter category tends to rely on precious metals such as Pd,7 Au,8 Ru,9 Pt10 and In,11 or non-noble metals such as Co,12 Ni,13 Mo,14 Zn,15 Ti16 and Fe.17 Although hydrogenation has been the most widely used approach, issues such as the need for high pressure equipment, flammable hydrogen gas, and relatively high loadings of expensive metals that oftentimes can localize as residual metal in the product remain. Environmental concerns stemming in large measure from organic waste that is mainly attributed to organic solvents18 can add to the unattractiveness of such traditional approaches.
In previous work from our group,19 it was found that commercial sources of FeCl3 augmented with only 80 parts per million (ppm) Pd, upon processing into nanoparticles (NPs), could be used to catalyze the reduction of nitroarenes at room temperature. Only two weight percent of our designer surfactant TPGS-750-M in water was needed to generate nanomicelles that serve as the reaction medium, with NaBH4 as the stoichiometric source of hydride. Due to the extremely low palladium loading, we endeavoured to further enhance the activity of this catalyst (Fe/ppm Pd NPs), especially with highly functionalized substrates. Therefore, a far more reactive and functional group tolerant catalyst was sought that would minimize reaction times, of particular interest to reactions run at scale.
Results and discussion
Our initial study began with replacement of the Pd present in the NPs with varying amounts of Ni salts, with best results obtained using Ni(NO3)2·6H2O (Fig. 1). Reagent optimization studies revealed a dependence on two key variables in addition to the nickel(II) source: (1) the equivalents of MeMgCl used to form the NPs, and (2) the amount of NaBH4 used for catalyst activation (see the ESI, Tables S1 to S5†). Using 4-chloronitrobenzene as a model substrate, reaction with Fe/ppm Ni NPs was surprisingly slower (12 h) than the corresponding reaction using Fe/ppm Pd NPs (2 h; Table 1). Remarkably, preparing the same NPs containing both 80 ppm Pd, 1600 ppm Ni and 2 mol% Fe led to complete conversion in 15 minutes. Additional examples, as illustrated in Table 2, attest to this same trend. Fe NPs prepared from pure FeCl3 in the absence of both Pd and Ni led to a very slow reaction (19% product after 14 h). | ||
| Fig. 1 Generation and appearance of iron nanoparticles continuing ppm levels of Pd and Ni. | ||
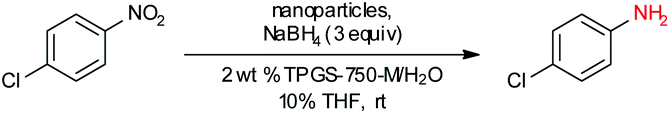
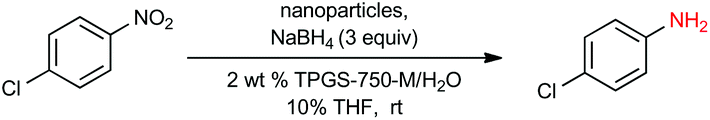

Following reduction with MeMgCl, the mixture was quenched with pentanes containing traces of water, and then triturated by additional pentanes to afford khaki-colored Fe/ppm Pd + Ni NPs (Fig. 1). The nanoparticles could be used directly upon activation with NaBH4 (Fig. 2), or isolated and stored in solid form, without loss of activity, for several months at room temperature and without protection from air. Should these NPs turn from yellow-brown to orange upon exposure to air over time, there is no loss in activity since the NaBH4 in the pot quickly converts them back to their active form. This process could be further streamlined by taking advantage of the recently disclosed co-solvent effect,20 in these cases using 10% THF. As revealed by each of the five additional examples illustrated in Table 2, the rates of reduction could be increased significantly.
 | ||
| Fig. 2 Generation (A) and appearance, after exposure to NaBH4; (B) of iron nanoparticles containing ppm levels of Pd and Ni. | ||
Analyses of the active NPs by transmission electron microscopy (TEM), using a copper grid, in 2 wt% TPGS-750-M aqueous solutions show spherical particles (Fig. 3). As seen previously,21 these metal NPs are associated with existing nanomicelles in aqueous solution due to the presence of (M)PEG within the surfactant. Energy Dispersive X-Ray (EDX) data (Fig. 4) for the same nanoparticles indicate that the ratio of Fe/Ni is 12.3, which is close to the theoretical ratio of 11.9 (see the detailed calculation in ESI 5b†). The presence of palladium at these low levels, as expected,19 was not observed.
 | ||
| Fig. 3 Catalyst characterization by cryo-TEM of Fe/ppm Pd + Ni NPs in aqueous TPGS-750-M after adding NaBH4. | ||
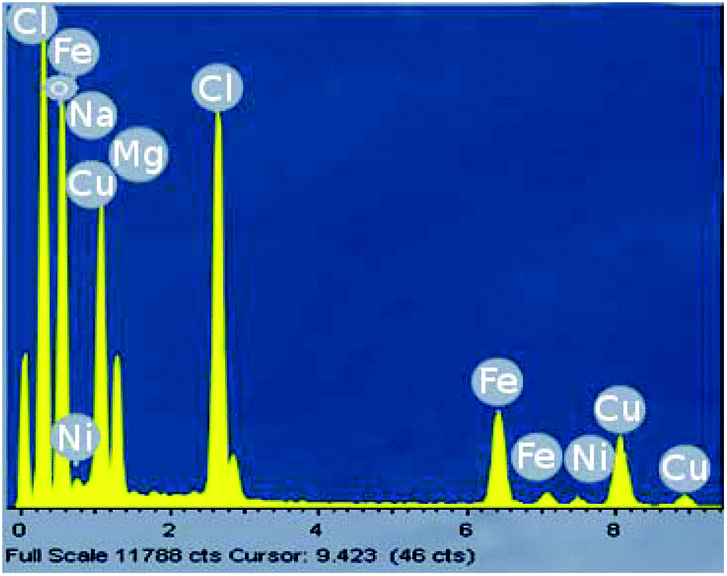 | ||
| Fig. 4 EDX for Fe/ppm Pd + Ni NPs in the presence of NaBH4. | ||
To assess the scope of this technology, an expansive study was undertaken examining several types of nitroarenes and -heteroarenes. Under standard conditions (i.e., 2 wt% TPGS-750-M/H2O, NaBH4 (3 equiv.), 10% THF co-solvent at room temperature), most reactions took place quite smoothly and in excellent yields (Table 3). Products containing substituents on the aromatic/hetroaromatic ring, including fluoro-(10), chloro-(1, 4), bromo-(2, 7, 10, 14), trifluoromethyl-(12, 17, 18), chlorodifluoromethoxy-(23), were formed without incident. Reduction in the presence of other functionality, such as ester, amide, sulfonyl-, thio-, hydrazinyl-, as well as C–C double bonds, likewise, occurred smoothly. Heterocycles including pyridine, isoxazole, quinoline, piperazine, piperidine, morpholine were all tolerated, strongly suggesting potential applications to targets in the pharmaceutical and agrochemical arenas. By contrast, reactions run under identical conditions but in pure THF were extremely slow, with 4-chloronitrobenzene affording the targeted product to the extent of only 27% after 16 hours.
| a Reaction conditions: Nitro compound (0.5 mmol), Fe nanoparticles (6 mg, 2% Fe, 80 ppm Pd, 1600 ppm Ni), NaBH4 (1.5 mmol), 2 wt% TPGS-750-M/H2O (1 mL), THF (0.1 mL). b Isolated yield. c Nitro compound (0.125 mmol), Fe nanoparticles (3 mg), NaBH4 (0.375 mmol), 2 wt% TPGS-750-M/H2O (1 mL), THF (0.1 mL). d DMSO (0.25 mL). e Nitro compound (0.125 mmol), Fe nanoparticles (3 mg), NaBH4 (0.375 mmol), 2 wt% TPGS-750-M/H2O (1 mL), THF (0.3 mL). f Nitro compound (0.25 mmol), Fe nanoparticles (3 mg), NaBH4 (0.75 mmol), TPGS-750-M/H2O (1 mL), THF (0.1 mL). g Nitro compound (0.25 mmol), Fe nanoparticles (3 mg), NaBH4 (0.75 mmol), TPGS-750-M/H2O (1 mL), CH3OH (0.25 mL). h Nitro compound (0.5 mmol), Fe nanoparticles (12 mg), NaBH4 (3 mmol), 2 wt% TPGS-750-M/H2O (1 mL), DCM (0.1 mL). i THF (0.25 mL). j Nitro compound (0.5 mmol), Fe nanoparticles (12 mg), KBH4 (3 mmol), 2 wt% TPGS-750-M/H2O (1 mL), THF (0.25 mL). k Nitro compound (0.125 mmol), Fe nanoparticles (1.5 mg), KBH4 (0.375 mmol), 2 wt% TPGS-750-M/H2O (1 mL), DMSO (0.25 mL). l Nitro compound (0.125 mmol), Fe nanoparticles (2.5 mg), KBH4 (0.75 mmol), 2 wt% TPGS-750-M/H2O 1 mL, DMSO 0.25 mL. m Nitro compound (0.25 mmol), Fe nanoparticles (2.5 mg), KBH4 (0.75 mmol), 2 wt% TPGS-750-M/H2O (1 mL), THF (0.25 mL). n Nitro compound (0.25 mmol), Fe nanoparticles (3 mg), NaBH4 (0.75 mmol), 2 wt% TPGS-750-M/H2O (1 mL), DCM (0.3 mL). o Nitro compound (0.125 mmol), Fe nanoparticles (1.5 mg), KBH4 (0.75 mmol), 2 wt% TPGS-750-M/H2O (1 mL), THF (0.25 mL). |
|---|
|
Several of the examples studied (Table 3) presented special challenges due to the nature of nitro compounds that can be highly crystalline, and/or flocculent solids and therefore, slow to gain entry to the inner micellar cores. Practical issues, such an adherence to the stir bar, or formation of insoluble gums, were readily solved by the presence of varying amounts of a co-solvent. While THF was the first co-solvent to be chosen (see products 14, 18, 20, 25, 27), varying percentage of other water-miscible co-solvents such as MeOH (see 16) and DMSO (see 13, 19, 22, 24) were found to also lead to clean overall primary amine formation. Lowering the global concentration from 0.5 to 0.25, or even 0.10 M also occasionally was beneficial (see 11, 14, 15, 16, 22, 24, 25, 26, 27, 28). Typically, NaBH4 was used to generate the Pd–H species, although on occasion, KBH4 led to a more effective reduction, as noted previously by Gallou and co-workers.22
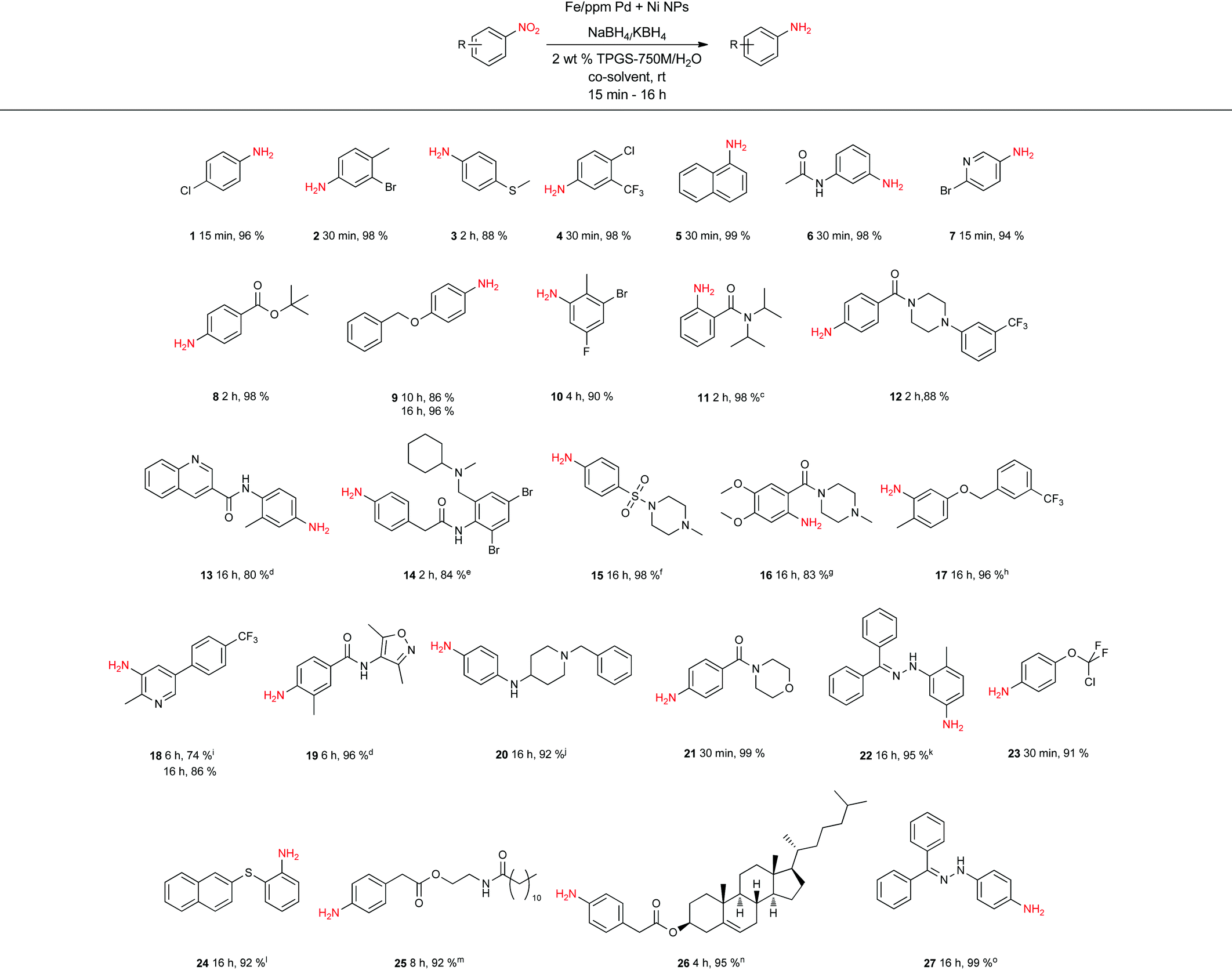
Given the increasing number of key synthetic transformations that can now be run under mild conditions in aqueous media using micellar technology,23 sequences can be planned that take advantage of the commonality of this reaction medium. As examples that include nitro group reductions, product 27 could be formed via this NP-mediated reduction, after which it is then converted to its Cbz derivative 28. Final cyclization via a Fischer indole synthesis to 29 proceeded in good overall yield (Scheme 1).
 | ||
| Scheme 1 1-Pot nitro group reduction followed by amine protection and Fischer indole synthesis. | ||
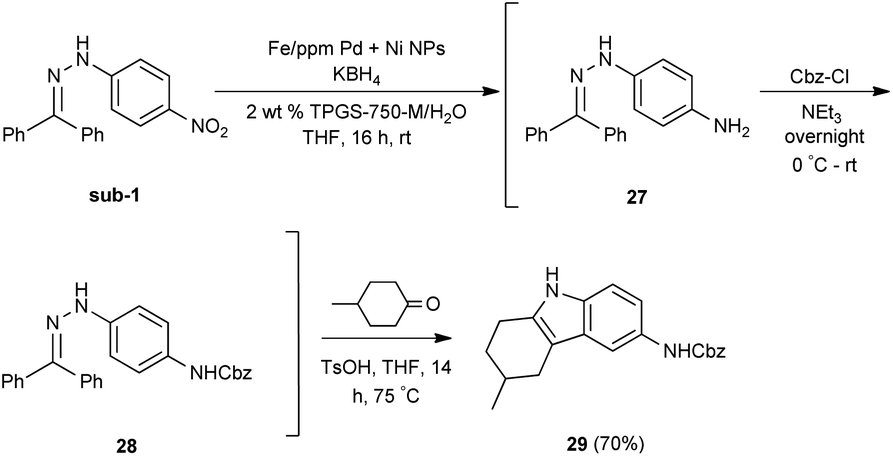
An alternative, representative 1-pot sequence, illustrated Scheme 2, converts 4-chloronitorbenzene initially to its aniline derivative. Subsequent SNAr addition leads to adduct 30, and then a Suzuki–Miyaura coupling affords biaryl 31, all done in water between rt and 45 °C in high overall yield.
 | ||
| Scheme 2 1-Pot, 3-step sequence involving NO2 group reduction /SNAr/Suzuki–Miyaura coupling, in water. | ||
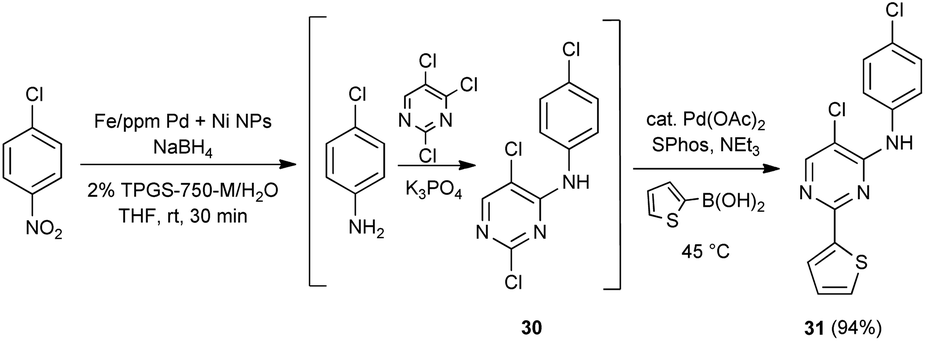
As is characteristic of designer surfactant technology in water, “in flask” recycling of an aqueous reaction mixture is straight forward.18 Thus, following reduction of a nitroaromatic (Scheme 3), extraction of the resulting substituted aniline upon addition of minimal amounts of a single organic solvent (e.g., MTBE) afforded the desired product, subject to purification. The E Factor24 associated with this process based on solvent usage as a measure of “greenness” is <4, which is quite low and reflects the limited solvent needed to remove the product from the reaction mixture. As THF remains partly in the aqueous phase, no additional co-solvent is needed with each recycle. Addition of fresh NPs, however, was required to ensure full conversion after an extractive workup. Yields for each of the four recycles studied remained high.
 | ||
| Scheme 3 Studies on recycling and determination of E factor. | ||
Another important aspect of this protocol concerns the amounts of residual metal to be expected in the product from reductions of nitroarenes. Established guidelines for allowable levels of transition metal impurities vary as a function of levels of toxicity.25 Residual Fe is considered relatively innocuous (limit: 1300 ppm), while limits for both Ni (25 ppm) and especially Pd (10 ppm) are far more rigorous.19 Metal impurities that exceed these levels require additional time, effort, and cost to have them removed or at least, reduced to allowable levels.
Analyses were conducted on several products resulting from Fe/ppm Pd + Ni NP-catalyzed nitro group reductions by inductively coupled plasma mass spectrometry (ICP-MS). Results (see ESI†) showed residual palladium to be below 10 ppm, while residual nickel was below 1 ppm. Residual iron was also measured and found to be below 5 ppm, implying in the composite that release of any metal from these NPs is minimal.
The role played by the added Ni in enhancing reactivity of the in situ-formed palladium hydride can be initially ascertained via XAS experiments on the activated catalysts, Fe/ppm Pd and Fe/ppm Pd + Ni. On the basis of EXAFS data, a higher shell scattering feature attributed to Pd–Pd interactions and the presence of Pd oligomers was observed for the Fe/ppm Pd system. Such a feature was not observed in the presence of Ni (see ESI†), indicating that Ni dilutes the palladium content on the surface of these NPs, leading to increased Pd dispersity and thereby, increasing the reactivity of the isolated palladium atoms.26 More definitive imaging techniques such as aberration-corrected HAADF STEM would be needed to establish Pd site isolation in the presence of Ni, and these experiments are presently under consideration. That a base metal such as nickel can be used to enhance the reactivity of a precious metal within a nanoparticle catalyst, leading to greater activity notwithstanding the ppm levels involved, is an exciting discovery that may have a considerable number of applications to other types of precious metal catalysis.
Conclusion
In summary, a new catalyst useful for nitro group reductions in water has been uncovered that contains ppm levels of both Pd and Ni that work in harmony to afford greater reactivity than that observed using related catalysts based on either individual metal. The process is safe, takes place in water under mild conditions, and allows for facile in-flask product isolation as well as recycling of the entire aqueous reaction mixture. Very low levels of residual metals are found in the isolated products, further enhancing the attractiveness of this environmentally responsible technology.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support provided by Novartis and the NSF (SusChEM 1566212) is warmly acknowledged with thanks. Work at Argonne National Laboratory is supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences and Geosciences under Contract DE-AC02-06CH11357. Use of the Advanced Photon Source at Argonne National Laboratory is supported by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. MRCAT operations are supported by the Department of Energy and the MRCAT member institutions.Notes and references
- (a) S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC; (b) M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC.
- C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030 RSC.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- (a) A. M. Birch, S. Groombridge, R. Law, A. G. Leach, C. D. Mee and C. Schramm, J. Med. Chem., 2012, 55, 3923 CrossRef CAS PubMed; (b) R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS; (c) H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210 CrossRef CAS.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2016 DOI:10.1021/acs.oprd.6b00205.
- (a) M. M. Dell'Anna, S. Intini, G. Romanazzi, A. Rizzuti, C. Leonelli, F. Piccinni and P. Mastrorilli, J. Mol. Catal. A: Chem., 2014, 395, 307 CrossRef; (b) A. Chinnappan and H. Kim, RSC Adv., 2013, 3, 3399 RSC; (c) O. Verho, K. P. J. Gustafson, A. Nagendiran, C.-W. Tai and J.-E. Bäckvall, ChemCatChem, 2014, 6, 3153 CrossRef CAS.
- L. He, L.-C. Wang, H. Sun, J. Ni, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2009, 48, 9538 CrossRef CAS PubMed.
- S. G. Oh, V. Mishra, J. K. Cho, B.-J. Kim, H. S. Kim, Y.-W. Suh, H. Lee, H. S. Park and Y. J. Kim, Catal. Commun., 2014, 43, 79 CrossRef CAS.
- (a) E. H. Boymans, P. T. Witte and D. Vogt, Catal. Sci. Technol., 2015, 5, 176 RSC; (b) H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed; (c) B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- N. Sakai, S. Asama, T. Konakahara and Y. Ogiwara, Synthesis, 2015, 47, 3179 CrossRef CAS.
- (a) F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M.-M. Pohl, J. Radnik, A.-E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537 CrossRef CAS PubMed; (b) R. K. Rai, A. Mahata, S. Mukhopadhyay, S. Gupta, P.-Z. Li, K. T. Nguyen, Y. Zhao, B. Pathak and S. K. Singh, Inorg. Chem., 2014, 53, 2904 CrossRef CAS PubMed; (c) R. V. Jagadeesh, D. Banerjee, P. B. Arockiam, H. Junge, K. Junge, M.-M. Pohl, J. Radnik, A. Bruckner and M. Beller, Green Chem., 2015, 17, 898 RSC.
- (a) R. J. Kalbasi, A. A. Nourbakhsh and F. Babaknezhad, Catal. Commun., 2011, 12, 955 CrossRef CAS; (b) O. Mazaheri and R. J. Kalbasi, RSC Adv., 2015, 5, 34398 RSC; (c) A. Chinnappan and H. Kim, RSC Adv., 2013, 3, 3399 RSC.
- E. Pedrajas, I. Sorribes, A. L. Gushchin, Y. A. Laricheva, K. Junge, M. Beller and R. Llusar, ChemCatChem, 2017, 9, 1128 CrossRef CAS.
- S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98 CrossRef CAS PubMed.
- M. L. Di Gioia, A. Leggio, I. F. Guarino, V. Leotta, E. Romio and A. Liguori, Tetrahedron Lett., 2015, 56, 5341 CrossRef CAS.
- (a) A. J. MacNair, M.-M. Tran, J. E. Nelson, G. U. Sloan, A. Ironmonger and S. P. Thomas, Org. Biomol. Chem., 2014, 12, 5082 RSC; (b) R. V. Jagadeesh, G. Wienhofer, F. A. Westerhaus, A.-E. Surkus, M.-M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972 RSC; (c) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC.
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10911 CrossRef CAS.
- J. Feng, S. Handa, F. Gallou and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 8979 CrossRef CAS PubMed.
- C. M. Gabriel, N. R. Lee, F. Bigorne, P. Klumphu, M. Parmentier, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 194 CrossRef CAS PubMed.
- E. D. Slack, C. M. Gabriel and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 14051 CrossRef CAS PubMed.
- (a) C. M. Gabriel, M. Parmentier, C. Riegert, M. Lanz, S. Handa, B. H. Lipshutz and F. Gallou, Org. Process Res. Dev., 2017, 21, 247 CrossRef CAS; (b) See also M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2016 DOI:10.1021/acs.oprd.6b00205.
- (a) B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 RSC; (b) B. H. Lipshutz, Johnson Matthey Technol. Rev., 2017, 61, 196 CrossRef.
- (a) R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007, pp. 1–47 Search PubMed; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- Committee For Medicinal Products For Human Use (CHMP), Guideline On The Specification Limits For Residues Of Metal Catalysts Or Metal Reagents, European Medicines Agency, London, 2008 Search PubMed.
- (a) J. Qu, X. Zhou, F. Xu, X.-Q. Gong and S. C. E. Tsang, J. Phys. Chem. C, 2014, 118, 24452 CrossRef CAS; (b) G. Liu, L. Zeng, Z.-J. Zhao, H. Tian, T. Wu and J. Gong, ACS Catal., 2016, 6, 2158 CrossRef CAS; (c) L. Shi, G.-M. Deng, W.-C. Li, S. Miao, Q.-N. Wang, W.-P. Zhang and A.-H. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994 CrossRef CAS PubMed; (d) B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02991h |
| This journal is © The Royal Society of Chemistry 2018 |