
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable synthesis of amino acids by catalytic fixation of molecular dinitrogen and carbon dioxide†
Manuel
Rivas
a,
Luís J.
del Valle
ab,
Pau
Turon
 *ac,
Carlos
Alemán
*ab and
Jordi
Puiggalí
*ab
*ac,
Carlos
Alemán
*ab and
Jordi
Puiggalí
*ab
aDepartament d'Enginyeria Química, EEBE, Universitat Politècnica de Catalunya, C/Eduard Maristany, 10-14, 08019, Barcelona, Spain. E-mail: pau.turon@bbraun.com; carlos.aleman@upc.edu; jordi.puiggali@upc.edu
bBarcelona Research Center for Multiscale Science and Engineering, Universitat Politècnica de Catalunya, C/Eduard Maristany, 10-14, Ed. C, 08019, Barcelona, Spain
cB. Braun Surgical, S.A. Carretera de Terrasa 121, 08191 Rubí, Barcelona, Spain
First published on 18th December 2017
Abstract
The industrial process of nitrogen fixation is complex and results in a huge economic and environmental impact. It requires a catalyst and high temperature and pressure to induce the rupture of the strong N–N bond and subsequent hydrogenation. On the other hand, carbon dioxide removal from the atmosphere has become a priority objective due to the high amount of global carbon dioxide emissions (i.e. 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 million tons in 2015). In this work, we fix nitrogen from N2 and carbon from CO2 and CH4 to obtain both glycine and alanine (D/L racemic mixture), the two simplest amino acids. The synthesis, catalyzed by polarized hydroxyapatite under UV light irradiation and conducted in an inert reaction chamber, starts from a simple gas mixture containing N2, CO2, CH4 and H2O and uses mild reaction conditions. At atmospheric pressure and 95 °C, the glycine and alanine molar yields with respect to CH4 or CO2 are about 1.9% and 1.6%, respectively, but they grow to 3.4% and 2.4%, when the pressure increases to 6 bar and the temperature is maintained at 95 °C. Besides, the minimum temperature required for the successful production of detectable amounts of amino acids is 75 °C. Accordingly, an artificial photosynthetic process has been developed by using an electrophotocatalyst based on hydroxyapatite thermally and electrically stimulated and coated with zirconyl chloride and a phosphonate. The synthesis of amino acids by direct fixation of nitrogen and carbon from gas mixtures opens new avenues regarding the nitrogen fixation for industrial purposes and the recycling of carbon dioxide.
200 million tons in 2015). In this work, we fix nitrogen from N2 and carbon from CO2 and CH4 to obtain both glycine and alanine (D/L racemic mixture), the two simplest amino acids. The synthesis, catalyzed by polarized hydroxyapatite under UV light irradiation and conducted in an inert reaction chamber, starts from a simple gas mixture containing N2, CO2, CH4 and H2O and uses mild reaction conditions. At atmospheric pressure and 95 °C, the glycine and alanine molar yields with respect to CH4 or CO2 are about 1.9% and 1.6%, respectively, but they grow to 3.4% and 2.4%, when the pressure increases to 6 bar and the temperature is maintained at 95 °C. Besides, the minimum temperature required for the successful production of detectable amounts of amino acids is 75 °C. Accordingly, an artificial photosynthetic process has been developed by using an electrophotocatalyst based on hydroxyapatite thermally and electrically stimulated and coated with zirconyl chloride and a phosphonate. The synthesis of amino acids by direct fixation of nitrogen and carbon from gas mixtures opens new avenues regarding the nitrogen fixation for industrial purposes and the recycling of carbon dioxide.
Introduction
Nitrogen fixation is an extremely relevant process from an industrial perspective.1 It is difficult to overestimate the impact of the Haber–Bosch process that yields ammonia and further results in more than 450 million tons of fertilizers per year, using more than 1% of the global energy supply.2 Moreover, the synthesis of ammonia from nitrogen is constantly under research and only in the last decade new catalysts based on ruthenium and zirconium have been developed.3 However, the environmental impact of the Haber–Bosch process is not negligible as natural gas or coal is used as an energy source (i.e. 1.87 tons of CO2 are released per ton of ammonia produced and globally 245 million tons of CO2 were released by the ammonia industry in 2010, equivalent to about 50% of the UK CO2 emissions).4 Thus, the outstanding benefits of nitrogen fixation are shadowed by their impact on environmental contamination. Furthermore, the catalytic reduction of dinitrogen under mild conditions has been achieved by using molybdenum catalysts. Shilov, in the 1980s, obtained a mixture of hydrazine and ammonia5 and Schrock and co-workers6,7 converted dinitrogen into ammonia with excellent yield using protons and electrons. Very recently, Kuriyama et al.8 reported the catalytic reduction of molecular dinitrogen into ammonia and hydrazine by using iron complexes bearing an anionic ligand as catalysts under mild reaction conditions. However, to our knowledge, the nitrogen fixation into amino acids (AAs) using hydroxyapatite (HAp) has not been reported yet.On the other hand, CO2 recycling is an absolute necessity for our society knowing that its accumulation in the atmosphere is now approaching 1 Tera ton.9 Significant steps have been taken towards the utilization of CO2 in order to convert it into valuable chemicals (150 Mt urea, 100 Mt methanol, 70 Mt salicylic acid, 9.7 Mt formaldehyde and 0.7 Mt formic acid are the most produced).10–12 In the last few years, catalytic reactions via carbon dioxide fixation have gained a prominent role as representative green processes with enhanced sustainability.13–16 However, by learning from nature, photosynthesis as performed by living organisms is the carbon fixation process par excellence. Efforts to mimic it synthetically have been elusive through time. The development of photosynthetic processes requires significant advances in new materials for light harvesting and the development of fast, stable, and efficient electrocatalysts.17,18
In this work, we introduce a new catalyst based on permanently polarized crystalline hydroxyapatite (p-cHAp) with enhanced electrical and photochemical properties that allows the coupling of nitrogen and carbon fixation processes. Such a catalyst family opens an interesting field of research where simple gas mixtures that usually do not react among themselves are combined to yield basic organic molecules, such as AAs, one of the main building blocks of life. More specifically, in this work we prove that glycine (Gly) and alanine (Ala) are produced at atmospheric pressure through an artificial photosynthetic nitrogen and carbon fixation reaction, starting from a weakly reducing atmosphere (N2, H2O, CO2 and CH4) and using UV radiation as a source of energy. This reaction represents a very simple alternative to the costly chemical and enzymatic processes used to produce commercially Gly and Ala.
Results and discussion
The catalyst design arises from the properties induced by the thermally stimulated polarization process to cHAp (discussed below). Specifically, p-cHAp exhibits electrical and electrochemical properties, both associated with charge transport phenomena, that remain practically unaltered with time (i.e. p-cHAp is a permanent polarization mineral). In order to take advantage of p-cHAp properties, other materials were necessary to complete the whole catalytic system. Thus, the requirements of the catalyst were, on one hand, a material able to transform an energy source, as for example UV radiation, into electron/hole pairs and, on the other hand, materials to bring in contact the gases used as reactants with the catalyst (i.e. adsorb the gases), which was essential to conduct the reactions. Obviously, the success of the catalyst was also based on the fact that it could avoid unfavourable synergies among the materials when they were combined. After evaluating different options, two materials, which correspond to those presented in this work, were selected: zirconia and a phosphonic acid with chelating properties. The details about the roles played by each component of the catalytic system are given throughout the whole work.Synthesis of amino acids at atmospheric pressure from an electrophotocatalyst based on polarized hydroxyapatite
The new catalytic system, hereafter denoted as p-cHAp/Phos-ZC-Phos, is based on p-cHAp that is, subsequently, coated with 3 layers. One of them, the intermediate coating layer, is made of zirconyl chloride (ZC), which probably hydrolyzes into zirconia, and two of them are made of amino tris(methylene phosphonic acid) (Phos). p-cHAp is a mineral with electrical and electrochemical activity obtained by applying a thermally and electrically stimulated polarization process to crystalline hydroxyapatite (cHAp). More specifically, p-cHAp is prepared using a three-step process: (1) cHAp is obtained by adding (NH4)2HPO4 in de-ionized water to Ca(NO3)2 in ethanol, and applying hydrothermal conditions to the resulting suspension for 24 h; (2) sintered cHAp is achieved by heating the cHAp synthesized in step 1 at 1000 °C for 2 h in air; and (3) p-cHAp was produced by sandwiching discs of sintered cHAp (step 2) between stainless steel plates and polarizing for 1 h with a constant DC field of 3 kV cm−1 at 1000 °C. The bulk resistance of p-HAp (∼105 Ω cm2) is significantly lower than that of the as-prepared HAp (∼108 Ω cm2), indicating that the ionic conductivity increases by three orders of magnitude when the thermally stimulated polarization process is applied. Moreover, the ionic conductivity is preserved for a long time (i.e. the bulk resistance increases by only 4% after 3 months). Similarly, the electrochemical activity of p-HAp is ∼150% higher than that of the as-prepared HAp, which evidences that the polarization treatment facilitates the diffusion of ions through the mineral matrix. On the other hand, the adsorption capacity of phosphonates onto p-cHAp is around four times higher than that onto cHAp, ensuring an efficient contact between the p-cHAp and the Phos-ZC-Phos 3-layer (Fig. 1a). Besides, ZC and Phos layers have been used to adsorb the gaseous reactants, essentially N2 and CO2, facilitating their reactivity. The details about the preparation of the catalyst and its components are provided in the ESI.†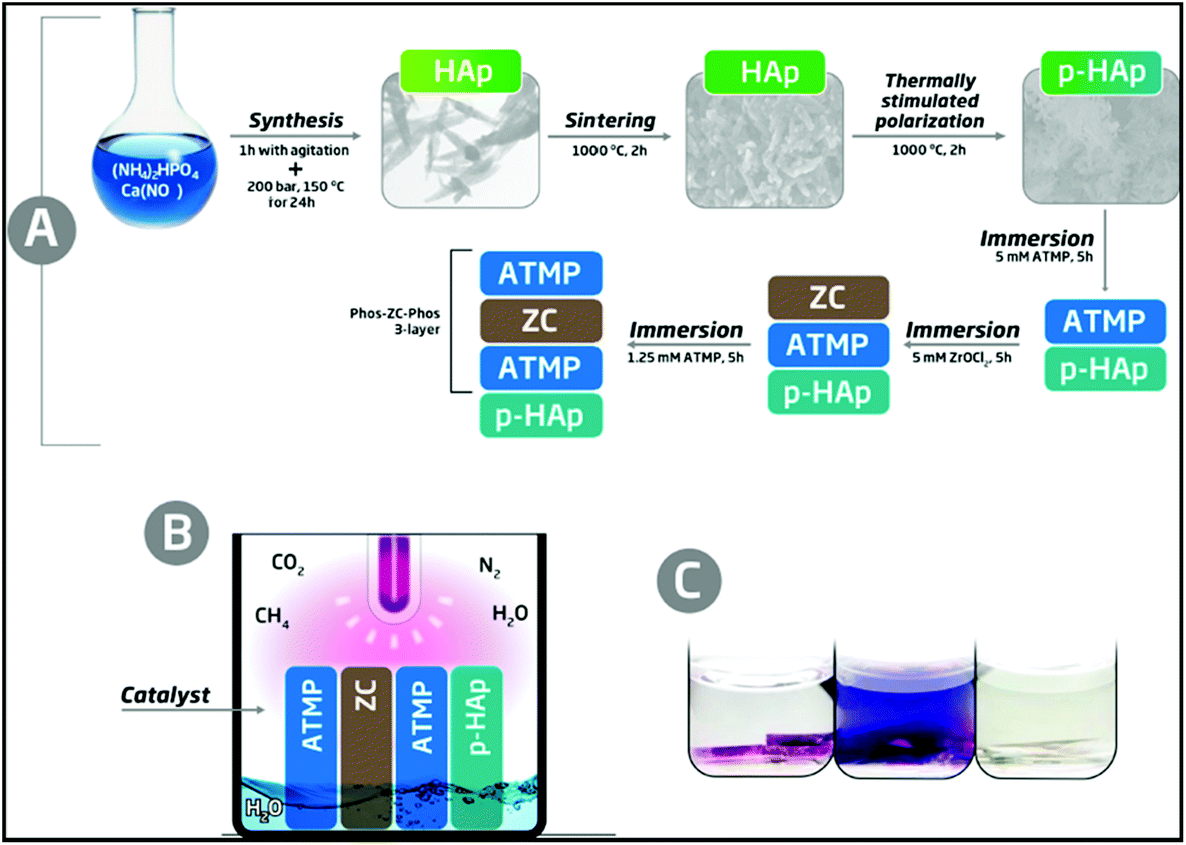 | ||
| Fig. 1 Schemes describing (a) the preparation of the p-cHAp/Phos-ZC-Phos catalytic system and (b) the reaction medium used to produce AAs. The details of the reactor are provided in the ESI.† (c) Representative results of the ninhydrin test for a positive reaction before stirring (left) and after stirring (middle) and a negative reaction (right). | ||
The synthesis of AAs was carried out in an inert reaction chamber under a weakly reducing atmosphere constituted of N2 (0.33 bar), CO2 (0.33 bar), CH4 (0.33 bar) and liquid H2O, using an UV lamp directly irradiating the catalyst and gas mixture (Fig. 1b). The formation of primary amines adsorbed onto the solid substrate was shown by positive ninhydrin tests through the development of purple spots inside the catalyst recovered after the reaction (Fig. 1c, left). Amine compounds were well dissolved in an acetone solution after vigorous stirring (Fig. 1c, middle), contrasting with the uncolored solid and solution (Fig. 1c, right) observed under many other assayed reaction conditions and catalytic systems (see below).
After 72 h at 95 °C, the clean synthesis of both Gly and Ala is demonstrated by the NMR spectra displayed in Fig. 2. The 1H NMR spectrum of the samples obtained by dissolving the catalyst and products of the reaction (Fig. 2a) shows the presence of the catalyst signal corresponding to the Phos methylene group (doublet at 3.79–3.76 ppm) and the signals corresponding to the newly produced AAs such as methylene protons (singlet at 3.65 ppm) of Gly and both methine (quadruplet at 3.91–3.85 ppm) and methyl (doublet at 1.54–1.52 ppm) groups of Ala. The same compounds are also observed in the solid-state 13C NMR spectrum (Fig. 2b), where only peaks assigned to the Phos (54.34 and 53.00 ppm), Gly (171.95 and 41.26 ppm) and Ala (175.25, 50.25 and 16.01 ppm) units are detected. The 31P NMR spectrum (Fig. 2c) shows the presence of the p-HAp (7.21 ppm) and Phos (−0.03 ppm) peaks, but additional signals related to the products coming from the decomposition of the catalyst were not detected. With respect to CH4 or CO2, the Gly and Ala molar yields at 95 °C after 72 h are 1.9% and 1.6%, respectively (Table 1). Instead, after 24 h at 95 °C no trace of Gly and Ala is detected by NMR. Furthermore, the formation of AAs is unsuccessful without the sustained exposure to the UV radiation, which appears to be a fundamental issue to generate radicals (e.g. ˙CH3 and ˙OH) needed for further reaction intermediates towards the final yielding of Ala and Gly.
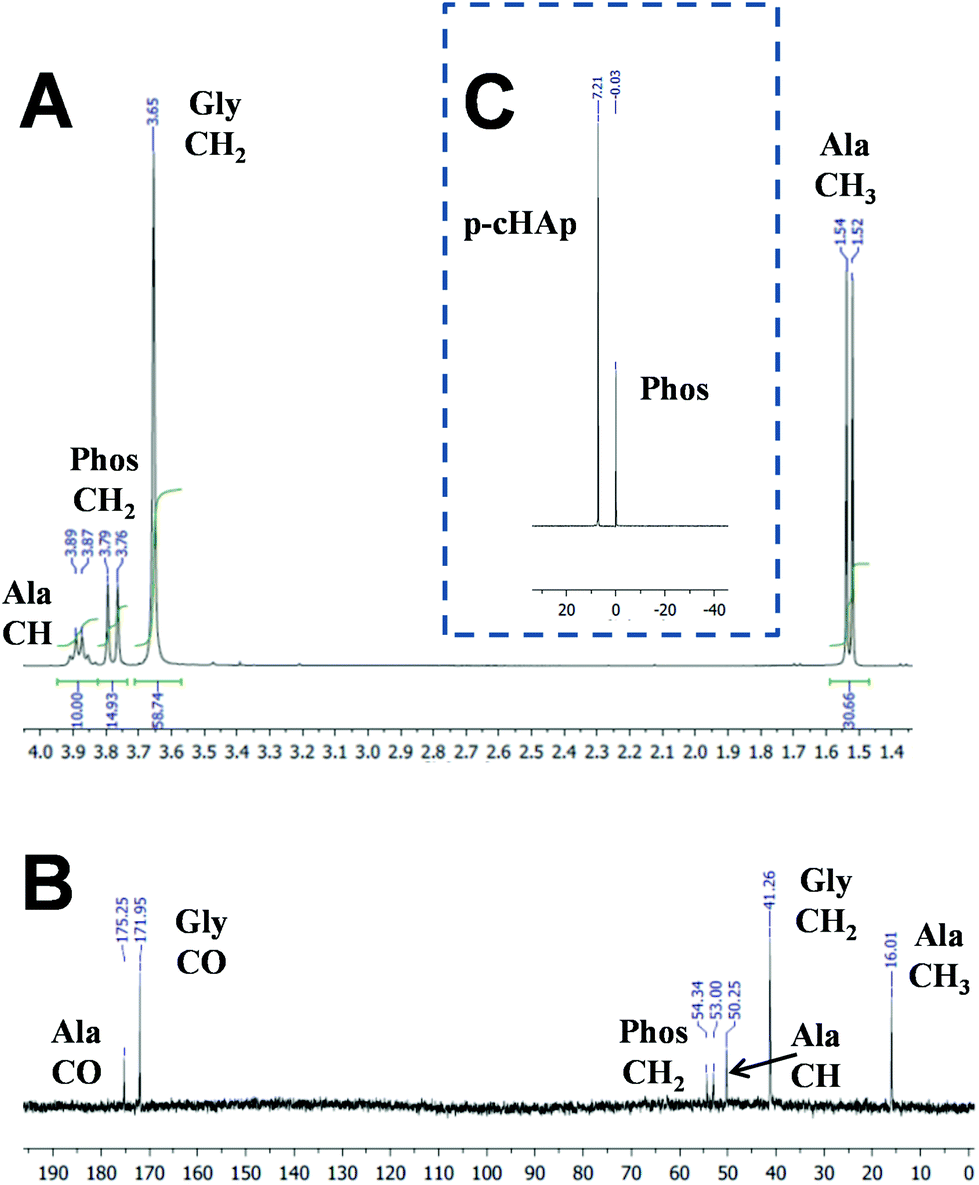 | ||
| Fig. 2 For samples obtained after the reaction (95 °C and 24 h) using a chamber pressure of 1 bar (i.e. 0.33 bar of each feeding reaction gas): (a) 1H NMR spectrum of the solution obtained after the extraction of the AAs from the catalyst by dissolving the sample in deuterated water containing 100 mM of HCl and 50 mM of NaCl; and the solid state (b) 13C and (c) 31P NMR spectra of the catalyst with the synthesized AAs. | ||
| Experimental conditionsa | Yield | |||
|---|---|---|---|---|
| Mineral | P (atm) | Time | Gly | Ala |
| a Temperature was kept fixed at 95 °C in all cases. b P N2 = 0.33 bar; PCH4 = 0.33 bar; PCO2 = 0.33 bar. c P N2 = 2 bar; PCH4 = 2 bar; PCO2 = 2 bar. d P N2 = 1 bar; PCH4 = 2 bar; PCO2 = 2 bar. e P N2 = 3 bar; PCH4 = 2 bar; PCO2 = 2 bar. | ||||
| p-cHAp | 1b | 24 h | — | — |
| p-cHAp | 1b | 72 h | 1.9% (6.7 mg cm−2) | 1.6% (3.3 mg cm−2) |
| p-cHAp | 6c | 24 h | 1.9% (6.6 mg cm−2) | 1.3% (2.7 mg cm−2) |
| p-cHAp | 6c | 72 h | 3.4% (11.9 mg cm−2) | 2.4% (5.0 mg cm−2) |
| p-aHAp | 6c | 72 h | 0.9% (3.2 mg cm−2) | 0.7% (1.3 mg cm−2) |
| p-cHAp | 5d | 24 h | 1.5% (5.3 mg cm−2) | 0.9% (1.9 mg cm−2) |
| p-cHAp | 7e | 24 h | 2.4% (8.3 mg cm−2) | 1.6% (3.3 mg cm−2) |
Photoemission spectroscopy (XPS) analyses show that the amines in AAs come from the molecular nitrogen and not from a hypothetical decomposition of the Phos. The N 1s spectra registered for different representative samples (Fig. 3a) indicate that the peak at 399.5 eV, which is ascribed to the C–N of Phos, is observed with practically the same intensity when both negative and positive reactions (i.e. without and with sustained exposure to UV radiation) are monitored. However, only in the latter case bands at lower binding energies appear due to the formation of deprotonated and protonated amino groups (401.2 and 404.5 eV, respectively).19 Furthermore, the amount of atomic nitrogen increased from 0% to 2.75–2.97% when the Phos-ZC-Phos 3-layer coating was added to the p-cHAp substrate. The nitrogen percentage, after the positive reaction, was increased up to 6.2%, which corroborates the fact that the formed AAs remain adsorbed into the catalysts.
 | ||
| Fig. 3 (A) N 1s high-resolution XPS spectra for (1) p-cHAp, (2) p-cHAp/Phos-ZC-Phos, and (3) p-cHAp/Phos-ZC-Phos after the negative reaction (i.e. without exposure to UV radiation) and (4) p-cHAp/Phos-ZC-Phos after the positive reaction (24 h at 95 °C). SEM micrographs before (B) and after (C) successful reaction using the p-HAp/Phos-ZC-Phos catalytic system (24 h at 95 °C). The reactions were performed at 1 bar. Variation of the Gly/Phos (○), Ala/Phos (□) (Gly + Ala)/Phos (◆) and Gly/Ala (▲) ratios versus (D) time for reactions performed at 95 °C and (E) temperature for reactions performed for 24 h using the p-cHAp/Phos-ZC-Phos catalytic system prepared as displayed in Fig. 1a. The reactions were performed at 6 bar. | ||
The p-cHAp/Phos-ZC-Phos catalyst exhibits a rough and relatively irregular surface morphology (Fig. 3b), which changed after the reaction due to the sporadic formation of micrometric prismatic crystals with the hexagonal basal plane parallel to the disk surface (Fig. 3c). Although the ninhydrin test reflects the presence of AAs adsorbed inside the catalyst, micrographs also demonstrate the growth of AA crystals on the surface of the catalytic system. This behavior is consistent with the capacity of organophosphonate films for inducing the crystallization of oriented molecular sieves as proved by the growth of stable, vertically-oriented and one dimensional aluminum phosphate crystals.20,21 On the other hand, the formation of AAs has also been proven by FTIR spectroscopy, which shows intense absorption bands in the amine region of the corresponding spectra, and characteristic X-ray diffraction patterns. Finally, chiral high-performance liquid chromatography (HPLC) analyses were carried out to quantify the ratio of D- and L-Ala adsorbed into the catalysts. As was expected, after dissolution of the catalysts after the reaction in a 0.1 M HCl solution with 50 mM NaCl, a racemic D-Ala:L-Ala mixture was determined.
The effect of varying the pressure and the temperature
In order to increase the rate of the reaction, the chamber pressure was increased up to 6 bar by introducing 2 bar of each feeding reaction gas, while the temperature was kept fixed at 95 °C. After 24 h, the molar yields, expressed with respect to CH4 or CO2, are 1.9% and 1.3% for Gly and Ala, respectively (Table 1), which are pretty similar to those achieved at atmospheric pressure after 72 h. Moreover, at high pressure Gly and Ala molar yields increase to 3.4% and 2.4%, respectively, after 96 h. Amazingly, the reported molar yields of Gly and Ala, based on the same carbonaceous species, by the action of electric discharges on a mixture of CH4, N2, water and traces of ammonia were only 0.26% and 0.71%, respectively, increasing to 1.55% when the sum of the 10 produced AAs was considered.22The effects of both the reaction time and the temperature in this catalytic process were studied considering a chamber pressure of 6 bar. The variation of the Gly/Phos and Ala/Phos ratios, which were determined from the areas of signals corresponding to CH2 (Gly and Phos) and CH (Ala) protons in the 1H NMR spectra, against the reaction time (from 2 to 96 h) reflects that Gly is produced first (Fig. 3a), while Ala is subsequently derived from this simple AA. Thus, the Gly/Ala ratio decreases from 5.4 to 2.2, while a continuous increase of the Gly/Phos ratio with the reaction time is nevertheless observed (i.e. from 0.8 to 4.5). On the other hand, the minimum temperature required for the successful production of detectable amounts of Gly and Ala after 24 hours is 75 °C (Fig. 3b). The Ala/Phos ratio increases progressively with the reaction temperature while the Gly/Phos ratio decreases at 105 °C due to the conversion of Gly into Ala, even though the (Gly + Ala)/Phos ratio still increases.
Influence of changes in the catalytic system
Substitution of p-cHAp by p-aHAp, which is obtained by replacing cHAp by amorphous hydroxyapatite (aHAp) and by applying the same thermally and electrically stimulated polarization process, did not affect the production of AAs, even though the yield of Gly and Ala decreases to 0.92% and 0.65%, respectively (Table 1). The latter has been attributed to the partial thermal decomposition of aHAp during the sintering process, which gives rise to the formation of a β-tricalcium phosphate23 causing a reduction of the electrochemical activity with respect to the samples obtained using cHAp. Conversely, negative results were obtained when p-cHAp was replaced by sintered s-cHAp (i.e. non-polarized), polarized silicates or aluminosilicates (mica), proving that the HAp and its electrochemical activity play an important role in the catalytic mechanism.For the sake of completeness, the effectiveness of the substitution of the Phos-ZC-Phos 3-layer coating by two possible combinations of 2-layer coatings (Phos-ZC and ZC-Phos), by Phos monolayers or by a Phos/ZC mixture was also assayed. In all cases, negative results were obtained demonstrating the importance of the 3-layered architecture, and thus discarding a process based on the photodecomposition of Phos. In addition, both the capability of incorporating water molecules24 and the effective role of metal/phosphonate compounds as chelating ligands25 are noteworthy due to the presence of free phosphonic acid groups. The results obtained after introducing the above-mentioned changes in the 3-layered p-HAp catalyst are summarized in Fig. 4.
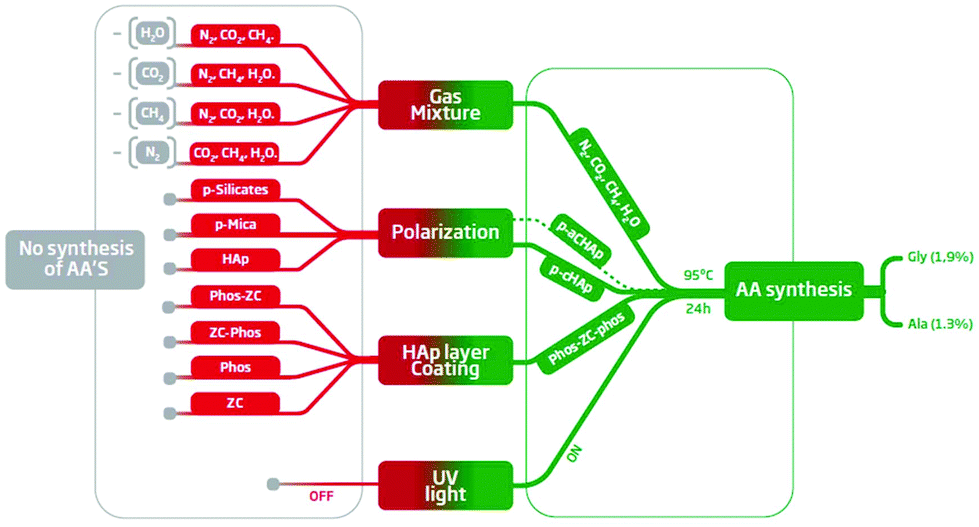 | ||
| Fig. 4 Summary of the results obtained using different experimental conditions: green and red boxes refer to the successful and unsuccessful production of AAs, respectively. The details about the 16 different experimental conditions are provided in the ESI.† | ||
Molecular nitrogen and carbon dioxide fixation
Different experiments (Table S5†) have also been performed in order to determine the reactants that are essential to obtain AAs under the defined experimental conditions (UV radiation, 6 bar, 95 °C, and 24 h). The results indicate that N2, CH4, CO2 and H2O were all necessary, because the removal of any of them led to negative ninhydrin tests as well as an absence of the AA peaks in the corresponding NMR spectra (Fig. 4). As was expected, the yield of AAs systematically correlates with the pressure of N2 in the reaction chamber (Table 1).These observations support the idea that the nitrogen source for the AAs is N2 and not Phos. Furthermore, the experiments performed without incorporating the coating to the catalyst failed to produce AAs (Table S5†), whilst XPS measurements evidenced that N2 was adsorbed by the Phos layers of the coating (Table S3†). Altogether, these results support that the catalyst coating facilitates both the adsorption and further reactivity of N2. This point is particularly relevant since most of the reported AA syntheses are based on ammoniacal solutions as the nitrogen source. It is well known that N2 is fixed by means of some specific bacteria by the action of enzymes that have metal-sulfide active sites.26 Numerous efforts have been made to develop metal-based catalysts to coordinate and activate N2 through the formation of metal–dinitrogen complexes with limited success.27 The splitting of the N–N bond in bridging dinitrogen complexes was first achieved by Cummins, who showed how three-coordinate molybdenum amido-complexes react with dinitrogen at −35 °C to yield an unstable dinitrogen bridging complex that, subsequently, breaks down into two molecules of the nitride-complex.28 Although some additional progress has been made in this area, the fixation of atmospheric N2 remains a challenge because of the limited reactivity and the harsh conditions required to convert dinitrogen into useful nitrogen-containing organic compounds. The p-cHAp/Phos-ZC-Phos catalyst overcomes such drawbacks rendering nitrogen containing compounds under relatively mild reaction conditions.
Regarding carbon fixation, experiments performed without CH4 in the initial mixture failed to produce AAs and, therefore, prove its role as a carbon source for the methyl group of Ala and for the methylene group of Gly (i.e. it again discards a synthetic route based on the decomposition of Phos). On the other hand, the carboxylic groups of the two AAs would appear to come from CO2. The transformation of CO2 into organic moieties under UV irradiation, mimicking photosynthesis, represents a very interesting approach for the minimization of CO2 emissions and for developing sustainable industrial processes. Thus, the catalytic process reported in this work fulfils the very ambitious goals of artificial photosynthesis: to use earth-abundant and inexpensive materials converting them into highly appreciated organic compounds (i.e. AAs) of industrial and environmental significance. Although zirconium phosphonates are able to adsorb CO2,29 HAp has shown a noticeable capacity to incorporate carbonate and hydrogen carbonate from atmospheric CO2 when dissolved in water23,30 facilitating the formation of carboxylic groups. In this work, XPS experiments prove that both the p-cHAp substrate and the ZC layer are responsible for the CO2 adsorption onto the catalyst, while Phos layers do not contribute.
Finally, water also displays a fundamental role in the synthesis of AAs (Fig. 4). In fact, water molecules do not appear as an element source of AAs, but they have a significant influence on the mechanism contributing to the formation of ˙OH radicals and dissolving CO2. Furthermore, hydration of Phos layers is also expected to play a major role in ionic mobility through such Phos layers.31
Mechanism
Similar syntheses of AAs (mainly Gly and Ala) have been demonstrated when ammoniacal solutions saturated with methane were exposed to near-UV irradiation (λ < 405 nm) and even natural sunlight in the presence of the catalyst platinized TiO2 powder.32,33 The total yield of AAs after 72 h of irradiation with a xenon lamp was 472 nmol and after 312 h of exposure to sunlight was 2140 nmol, which represented a very low conversion (molar yield: <0.01%).32 The reaction mechanism proposed in this case was based on the electron/hole (e−/h+) pair produced when TiO2 was irradiated with light having a higher energy of its band gap. However, the results were negative when CO2 was employed as a carbon source instead of CH4. The present work reveals an alternative way by obtaining organic nitrogen from molecular nitrogen instead of ammonia.Our first hypothesis for the reaction mechanism of the processes presented in this work is the occurrence of water-splitting photocatalysis on the surface of p-cHAp, in a way that resembles the one on TiO2 electrodes earlier reported by Fujishima and Honda.34 Thus, the reaction is not successful without water, which is consistent with the fact that water plays an important role in the mechanism. The hydroxyl radical (˙OH) photoproduction on those mineral surfaces is a well-known process that can be described as follows.35 The photon energy is adsorbed and electrons are excited to the conduction band, leaving holes in the valence band (h+). These electron–hole pairs travel to the surface of the substrate to participate in water splitting. Thus, the transfer of electrons from water molecules to valence band holes forms ˙OH (eqn (1)), whereas in the corresponding reductive reaction the transfer of electrons to O2 molecules forms superoxide radical anions (O2˙−).
| H2O + h+ → {˙}OH + H+ | (1) |
Because of the electrical properties of p-cHAp, we have hypothesized a similar process for the water photolysis on this mineral, regardless of the possible variations in the intermediate stages of the photooxidation and photoreduction reactions. It is worth noting that p-cHAp presents vacancies that originate from the transformation of hydrogen phosphate into phosphate during the thermally stimulated polarization process, as proved by NMR experiments. Therefore, its catalytic role should be comparable to that of activated TiO2 surfaces. Furthermore, Nishikawa36 demonstrated that, after heat treatment, HAp becomes electro-conductive by UV irradiation, leading to the formation of stable but very reactive O2˙− by altering its own PO43− groups. These features together with the electrochemical activity of p-cHAp, in combination with the Phos-ZC-Phos layers could produce an electrochemical capacitor,37 explaining the electrophotocatalytic activation of the p-cHAp/Phos-ZC-Phos system. Accordingly, the mobility of O2˙−, which may produce ˙OH in an aqueous environment, H+ and vacancies could be the driving forces of this electrochemical mechanism. The high electrochemical activity of p-cHAp in comparison with the as-prepared and sintered cHAp is proved in Fig. 5, which presents the voltammetric charge per surface unit (Q) against the number of consecutive oxidation–reduction cycles. As can be seen, Q is significantly higher for p-HAp than for the two non-polarized samples, such a difference increasing with the number of redox processes.
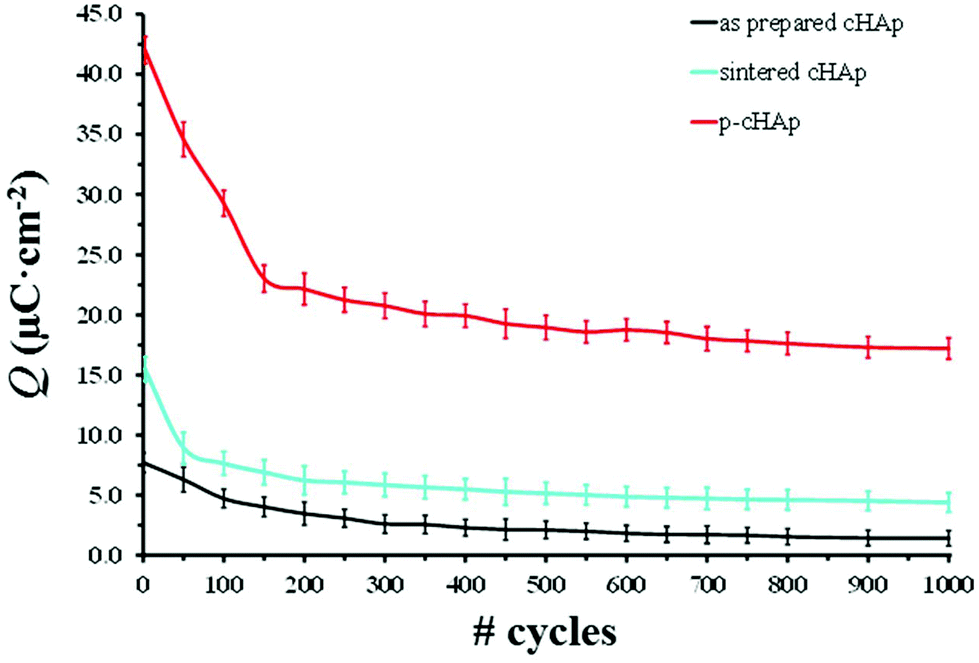 | ||
| Fig. 5 Variation of the electroactivity (voltammetric charge stored for reversible exchange in a redox cycle) of the as-prepared cHAp, sintered cHAp and p-HAp. In all cases, electroactivity measurements are expressed as voltammetric charge per surface unit (Q). | ||
As the hydroxyl radicals are very oxidative in nature, we suggest several possible reaction steps that could lead to the production of carbonaceous intermediates required for the production of Gly and Ala:
| CH4 + {˙}OH → {˙}CH3 + H2O | (2) |
| ˙CH3 + {˙}OH → {:}CH2 + H2O | (3) |
On the other hand, the one electron reduction of CO2 into its radical anion, CO2˙−, is proposed as a net electron transfer process possible at the interface between p-cHAp and Phos-ZC-Phos:
| CO2 + e− → CO2˙− | (4) |
Although this process is expected to be disfavored, because of both the thermodynamic stability of CO2 and the energy required for a stereochemical change in the geometry from a linear to a bent configuration, it has frequently been observed on HAp surfaces.23,30 This step is consistent with the analysis of volatile compounds, which revealed the formation of small amounts of aldehydes. Thus, around 600 μg m−3 of acetaldehyde and 90 μg m−3 of formaldehyde were detected after 16 h of reaction at 95 °C.
Besides, a number of mechanisms have been reported to explain the electrochemical transformation of N2 into NH3 through different steps that involve the N–N cleavage and the N protonation onto the catalyst surface.38 Within this context, such a transformation could follow associative or dissociative mechanisms. In the associative mechanism, N2 molecules adsorbed onto the catalysts are protonated sequentially without breaking the N–N bond until the first NH3 molecule is produced, as occurs in the reaction of N2-fixing by enzymes;39 while in the dissociative mechanism, the N2 molecules dissociate immediately and nitrogen radicals are protonated as they are in the Haber–Bosch process.40 After this the resulting NH3 may be easily oxidized by ˙OH to form ˙NH2.41 Then, Gly may be produced in the successive free-radical reactions involving ˙NH2, CO2˙− and ˙CH3 and the intermediate species. Finally, the transformation of Gly into Ala is possibly due to the α-H–(CαH) electrochemical removal and subsequent reaction with ˙CH3.
Conclusions
In summary, AAs are produced by using a new process based on electrophotosynthesis which uses a simple gas mixture. The process is able to fix nitrogen and carbon dioxide into organic species under mild reaction conditions, becoming an efficient and environmentally friendly synthesis. The newly developed electrophotocatalyst enhances the catalytic properties of cHAp to the extent that artificial photosynthesis is achieved, thus opening a new era in the treatment of contaminated atmospheres and nitrogen fixation research.Experimental methods
Preparation of polarized hydroxyapatite
The synthesis and characterization of aHAp and cHAp are described in detail in the ESI.† The sintering of aHAp, cHAp, montmorillonite and mica powders was carried out by heating them at 1000 °C for 2 h under air. After this, the samples were uniaxially pressed at 620 MPa for 10 min. Discs of 100 mm diameter, 1.7 mm thickness and sufficient mechanical consistence were finally obtained for all sintered materials (s-aHAp, s-cHAp, s-Nanofil 757 and s-LM).In order to get thermally and electrically stimulated minerals, preliminary assays were performed with s-cHAp discs using DC voltages that ranged from 250 to 2000 V. For this purpose, s-cHAp discs were sandwiched between stainless steel (AISI 304) plates, heated in a furnace to 1000 °C in air and, simultaneously, polarized for 1 h under the application of a constant DC voltage of 500 V (3 kV cm−1). Subsequently, the samples were cooled to room temperature, maintaining the DC voltage. The best results, in terms of both the mechanical consistence and maximum adsorption capacity of phosphates and phosphonates, were obtained at 500 V. After this, the discs of s-cHAp, s-aHAp, s-Nanofil 757 or s-LM were polarized using the same procedure for 1 h under the application of a constant DC voltage of 500 V, and the resultant polarized systems are denoted as p-aHAp, p-cHAp, p-N757 and p-LM, respectively.
Characterization of polarized hydroxyapatite
p-cHAp samples were characterized by cyclic voltammetry (CV), wide angle X-ray diffraction (WAXD) and FTIR spectroscopy as detailed and discussed in the ESI.†Deposition of phosphonate and zirconyl chloride layers
3-Layered systems consisting of the successive deposition of ATMP, ZC and ATMP layers (Phos-ZC-Phos) onto a mineral substrate (i.e. s-cHAp, s-aHAp, s-Nanofil 757 or s-LM before and after being subjected to the thermally stimulated polarization process) were obtained by immersion in the corresponding aqueous solutions at room temperature for 5 h. The detailed experimental conditions are described in the ESI.†After different trials to evaluate the influence of the content of ZC (detailed in the ESI†) all the results displayed in this work, including the main text, correspond to the ZC layer deposited from a 5 mM solution (unless another concentration is explicitly indicated).
Synthesis of amino acids
A high pressure stainless steel reactor was designed ad hoc and employed to perform the synthesis of amino acids (AAs). The designed reactor, which is discussed in the ESI,† was equipped with three independent inlet valves for N2, CH4, and CO2 and an outlet valve to recover the gaseous reaction products. A UV lamp (GPH265T5L/4, 253.7 nm) was also placed in the middle of the reactor to irradiate the catalyst directly, the lamp being protected by a UV transparent quartz tube. All surfaces were coated with a thin film of a perfluorinated polymer in order to avoid any contact between the reaction medium and the reactor surfaces, in this way discarding other catalytic effects.Regarding the temperature range, the reactions were performed within 75–105 °C for reaction times between 2 and 96 h. The catalyst samples weighed approximately 150 mg and 0.5 mL of de-ionized liquid water were initially incorporated into the reaction chamber, except for assessing the water effect. The chamber was extensively purged with the first selected gas in order to eliminate the initial air content (i.e. N2 or CO2). After this, in order to reach the target pressure with the right mixture, each selected gas was introduced to increase the reaction chamber pressure (measured at room temperature) as described in the ESI.† The yields of AAs were determined by using the areas of 1H NMR signals corresponding to CH2 (Gly) and CH3 (Ala) protons, as is described in the ESI.†
Measurements
The synthesis of AAs was routinely verified by the ninhydrin (2,2-dihydroxyindane-1,3-dione) detection test for primary amines. On the other hand, quantitative analyses were performed by NMR spectroscopy and X-ray photoelectron spectroscopy (XPS). Morphological studies were performed by scanning electron microscopy (SEM). Analysis of volatile organic compounds has been carried out by gas chromatography. The details about the equipment and experimental conditions for such analyses are provided in the ESI.†Conflicts of interest
Authors declare that the technology reported in this paper has been patented by B Braun Surgical S.A. and UPC (EP16382381, EP16382524, PCT/EP2017/069437).Acknowledgements
The authors acknowledge MINECO-FEDER (MAT2015-69367-R and MAT2015-69547-R) for financial support. The support for the research of C. A. was received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. This work is integrated within a wider research project supported by B. Braun Surgical S.A., UPC, ICS and ICFO. Special thanks to Dr J. F. Julián, Dr J. Navinés, Mrs Anna M. Rodríguez, Mrs Cristina Manjón, Dr E. Rodríguez and Mr Paul Wakely for their valuable contributions and to Mr A. Rodríguez for his contribution to the design of Fig. 1 and 4 and to Mr A. Lacasa for the design of the graphical abstract.References
- Editorial comment, The Nitrogen Fix, Nature, 2013, 501, 6–6 CrossRef.
- B. E. Smith, Nitrogenase reveals its inner secrets, Science, 2002, 297, 1654–1655 CrossRef CAS PubMed.
- K. C. Macleod and P. L. Holland, Recent developments in the homogeneous reduction of dinitrogen by molybdenum and iron, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- R. Lan, J. T. S. Irvine and S. Tao, Synthesis of ammonia directly from air and water at ambient temperature and pressure, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- A. E. Shilov, Catalytic reduction of molecular nitrogen in solutions, Russ. Chem. Bull. Int. Ed., 2003, 52, 2555–2562 CrossRef CAS.
- D. V. Yandulov and R. R. Schrock, Catalytic reduction of dinitrogen to ammonia at a single molybdenum center, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- R. R. Schrock, Reduction of dinitrogen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17087–17087 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii and K. Yoshizawa, Catalytic transformation of dinitrogen into ammonia and hydrazine by iron-dinitrogen complexes bearing pincer ligand, Nat. Commun., 2016, 7, 12181 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Synthesis and characterization of zwitterionic carbon dioxide fixing reagents, Int. J. Greenhouse Gas Control, 2010, 4, 452–458 CrossRef CAS.
- E. Alper and O. Y. Orhan, CO2 utilization: Developments in conversion processes, Petroleum, 2017, 3, 109–126 CrossRef.
- O. Ola, M. M. Maroto-Valer and S. Mackintosh, Turning CO2 into valuable chemicals, Energy Procedia, 2013, 37, 6704–6709 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock opportunities and challenges, Dalton Trans., 2007, 2975–2992 RSC.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Efficient, selective and sustainable catalysis of carbon dioxide, Green Chem., 2017, 19, 3707–3728 RSC.
- B. Zhao and Y. Su, Process effect of microalgal-carbon dioxide fixation and biomass production: A review, Renewable Sustainable Energy Rev., 2014, 31, 121–132 CrossRef CAS.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Glycerol-, pentaerythritol- and trimetylolpropane based polyurethanes and their cellulose carbonate composites prepared via the non-isocyanate route with catalytic carbon dioxide fixation, Green Chem., 2013, 15, 934–942 RSC.
- R. A. Molla, Md. A. Iqubal, K. Ghosh and S. M. Islam, A route for direct transformation of aryl halides to benzyl alcohols via carbon dioxide fixation reaction catalyzed by a (Pd@N-GMC) palladium nanoparticle encapsulated nitrogen doped mesoporous carbon material, Green Chem., 2016, 18, 4649–4656 RSC.
- A. Bar-Even, E. Noor, N. E. Lewis and R. Milo, Design and analysis of synthetic carbon fixation pathways, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8889–8894 CrossRef CAS PubMed.
- M. R. Dubois and D. L. Dubois, Development of molecular electrocatalysts for CO2 reduction and H2 production/oxidation, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef PubMed.
- G. Tzvetkov and F. P. Netzer, Synchrotron X-ray photoemission study of soft X-ray processed ultrathin glycine-water ice films, J. Chem. Phys., 2011, 134, 204704 CrossRef PubMed.
- S. Feng and T. Bein, Growth of oriented molecular sieve crystals on organophosphonate films, Nature, 1994, 368, 834–836 CrossRef CAS.
- S. Feng and T. Bein, Vertical aluminophosphate molecular sieve crystals grown at inorganic-organic interfaces, Science, 1994, 265, 1839–1841 CAS.
- D. Ring, Y. Wolman, N. Friedman and S. L. Miller, Prebiotic synthesis of hydrophobic and protein amino acids, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 765–768 CrossRef CAS.
- O. Bertran, L. J. del Valle, G. Revilla-López, M. Rivas, G. Chaves, M. T. Casas, J. Casanovas, P. Turon, J. Puiggalí and C. Alemán, Synergistic approach to elucidate the incorporation of magnesium ions into hydroxyapatite, Chem. – Eur. J., 2015, 21, 2537–2546 CrossRef CAS PubMed.
- B. Shah and U. Chudasam, Application of zirconium phosphonate-A novel hybrid material as an ion exchanger, Desalin. Water Treat., 2012, 38, 227–235 Search PubMed.
- C. S. Griffith, M. De Los Reyes, N. Scales, J. V. Hanna and V. Luca, Hybrid inorganic-organic adsorbents. Part 1: Synthesis and characterization of mesoporous zirconium titanate frameworks containing coordinating organic functionalities, ACS Appl. Mater. Interfaces, 2010, 2, 3436–3446 CAS.
- R. N. F. Thorneley, New light on nitrogenase, Nature, 1992, 360, 553–560 CrossRef.
- J. G. Andino, S. Mazumder, K. Pal and K. G. Caulton, New approaches to functionalizing metal-coordinates N2, Angew. Chem., Int. Ed., 2013, 52, 4726–4732 CrossRef CAS PubMed.
- C. E. Laplaza, M. J. A. Johnson, J. C. Peters, A. L. Odom, E. Kim, C. C. Cummins, G. N. George and I. J. Pickering, Dinitrogen cleavage by three-coordinate molybdenum(III) complexes: Mechanistic and structural data, J. Am. Chem. Soc., 1996, 118, 8623–8638 CrossRef CAS.
- X.-Z. Lin, Z.-Z. Yang, L.-N. He and Z.-Y. Yuan, Mesoporous zirconium phosphonates as efficient catalysts for chemical CO2 fixation, Green Chem., 2015, 17, 795–798 RSC.
- O. Bertran, L. J. del Valle, G. Revilla-López, G. Chaves, L. Cardús, M. T. Casas, J. Casanovas, P. Turon, J. Puiggalí and C. Alemán, Mineralization of DNA into nanoparticles of hydroxyapatite, Dalton Trans., 2014, 43, 317–327 RSC.
- H. E. Katz and M. L. Schilling, Electrical properties of multilayers based on zirconium phosphate/phosphonate bonds, Chem. Mater., 1993, 5, 1162–1166 CrossRef CAS.
- H. Reiche and A. J. Bard, Heterogeneous photosynthetic production of amino acids from methane-ammonia-water at platinum/titanium dioxide. Implications in chemical evolution, J. Am. Chem. Soc., 1979, 101, 3127–3128 CrossRef CAS.
- W. W. Dunn, Y. Aikawa and A. J. Bard, Heterogeneous photosynthetic production of amino acids at Pt/TiO2 suspensions by near ultraviolet light, J. Am. Chem. Soc., 1981, 103, 6893–6897 CrossRef CAS.
- A. Fujishima and K. Honda, Electrochemical photolysis of water at a semiconductor electrode, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- H. Nishikawa, Surface changes and radical formation on hydroxyapatite by UV irradiation for inducing photocatalytic activation, J. Mol. Catal. A: Chem., 2003, 206, 331–338 CrossRef CAS.
- D. Aradilla, F. Estrany and C. Alemán, Symmetric supercapacitors based on multilayers of conducting polymers, J. Phys. Chem. C, 2011, 115, 8430–8438 CAS.
- S. Back and Y. Jung, On the mechanism of electrochemical ammonia synthesis on the Ru catalyst, Phys. Chem. Chem. Phys., 2016, 18, 9161–9166 RSC.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Climbing nitrogenase: Toward a mechanism of enzymatic nitrogen fixation, Acc. Chem. Res., 2009, 42, 609–619 CrossRef CAS PubMed.
- T. H. Rod, A. Logadottir and J. K. Nørskov, Ammonia synthesis at low temperatures, J. Chem. Phys., 2000, 112, 5343–5347 CrossRef CAS.
- L. Huang, L. Li, W. Dong, Y. Liu and H. Lou, Removal of ammonia by OH radical in aqueous phase, Environ. Sci. Technol., 2008, 42, 8070–8075 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02911j |
| This journal is © The Royal Society of Chemistry 2018 |