DOI:
10.1039/C7GC02627G
(Paper)
Green Chem., 2018,
20, 266-273
Stable and reusable nanoscale Fe2O3-catalyzed aerobic oxidation process for the selective synthesis of nitriles and primary amides†
Received
28th August 2017
, Accepted 27th November 2017
First published on 28th November 2017
Abstract
The sustainable introduction of nitrogen moieties in the form of nitrile or amide groups in functionalized molecules is of fundamental interest because nitrogen-containing motifs are found in a large number of life science molecules, natural products and materials. Hence, the synthesis and functionalization of nitriles and amides from easily available starting materials using cost-effective catalysts and green reagents is highly desired. In this regard, herein we report the nanoscale iron oxide-catalyzed environmentally benign synthesis of nitriles and primary amides from aldehydes and aqueous ammonia in the presence of 1 bar O2 or air. Under mild reaction conditions, this iron-catalyzed aerobic oxidation process proceeds to synthesise functionalized and structurally diverse aromatic, aliphatic and heterocyclic nitriles. Additionally, applying this iron-based protocol, primary amides have also been prepared in a water medium.
1. Introduction
The development of iron-catalyzed reactions for modern state-of-the-art organic synthesis is an important goal of chemical research.1,2 Remarkably, the high abundance, lower price and lower toxicity of iron make it an ideal metal for catalysis applications,1,2 and it can be a suitable candidate for replacing existing precious metal-based catalysts. Compared to homogeneous iron complexes, heterogeneous catalysts based on nanostructured iron materials are more practical and cost-efficient catalytic systems due to their ease of separation, reusability and stability.1d,2a,3 In order to realize more environmentally benign syntheses, iron catalyzed reactions should exploit the use of green and renewable reagents under mild reaction conditions.1c,d,4–7 Clearly, iron-catalyzed syntheses utilizing atmospheric O2 or air1c,d,4–7 and ammonia8 that demonstrate exquisite selectivity represent a more sustainable process that is crucial for the advancement of academic research and industrial production. Therefore, the development of iron catalyzed aerobic oxidation reactions is highly desired for the production of valuable chemicals, especially nitriles and amides. It is worth noting that nitriles and amides represent major building blocks and central intermediates for the synthesis of advanced chemicals, life science molecules and materials.9–13 Furthermore, these structural motifs have been found to be integral parts of bio-active molecules and natural products.13–17
While simple nitriles have been prepared by industrial ammoxidation processes under harsh conditions (>300 °C in the gas phase),18 functionalized and structurally diverse nitriles still rely on toxic cyanation reactions.19 The limitations of ammoxidation and drawbacks of cyanation inspired the search for alternative sustainable routes for the synthesis of nitriles from suitable starting materials. In this regard, the catalytic oxidative conversion of alcohols20 or aldehydes20a,21 using ammonia represents a green method to produce various nitriles.
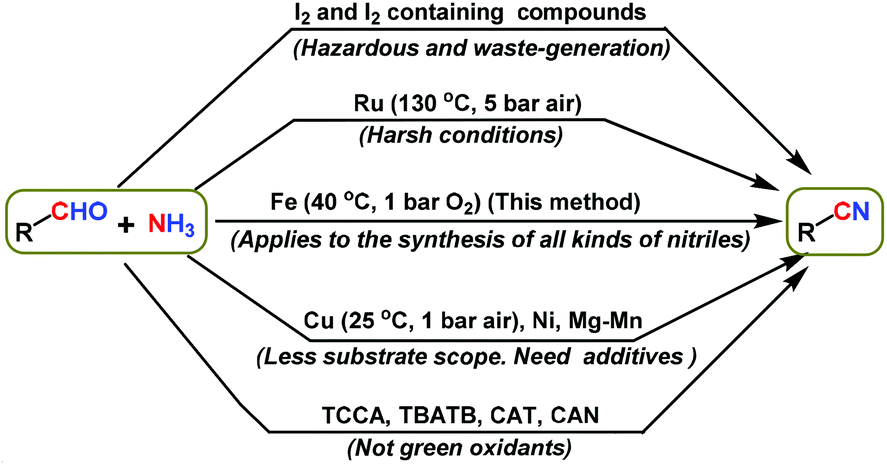
Complementary to alcohols are aldehydes, which are readily available fine and bulk chemicals, and they serve as suitable starting materials for the preparation of various chemicals such as amines,22 carboxylic acids,23 alcohols,24 esters25 and heterocycles,26 as well as for C–C bond forming reactions that enable the preparation of an array of interesting molecules24a–c,27. In particular, the recently reported Fe-23a and Cu-23b catalyzed oxidative conversions of aldehydes to carboxylic acids are quite interesting. Regarding the synthesis of nitriles from aldehydes, few methods have been reported using different nitrogen sources, such as hexamethyldisilazane (HMDS),28 hydroxylamine,29 NaN3,30 K3[Fe(CN)6]31 and ammonia,20a,21,32 using different catalysts. Unfortunately, except ammonia all of these reagents are either toxic or produce stoichiometric amounts of waste. Remarkable ammonia is considered to be a green and abundant nitrogen source for the preparation of nitrogen containing compounds. With regards to the utilization of ammonia for the synthesis of nitriles from aldehydes, various oxidants and catalysts have already been reported.20a,b,21,32–34 Among these, iodine and iodine-containing compounds were commonly employed.32 However these reagents are not only sensitive and hazardous, but also produce significant amounts of waste. Hence these reagents are not suitable from a green chemistry perspective. Next, Ru-based catalysts were used under harsh conditions (130 °C and 6 bar air).20a,b In addition to ruthenium, Cu,21 Ni,33a and Mg–Mn33b based systems are also used to catalyze the reaction of aldehydes and ammonia to obtain nitriles. Among these, the Cu/TEMPO/bipyridin catalyst system works under mild reaction conditions (25 °C and 1 bar O2).21a Even though this catalyst works under mild reaction conditions, it is a homogeneous catalyst and cannot be recycled.21a In addition, the use of NaOH additive and narrower substrate scope have hindered its practical utility for the sustainable synthesis of nitriles.21a Furthermore, other oxidants/catalysts such as trichloroisocyanuric acid (TCCA),34a tetrabutylammonium tribromide (TBATB),34b chloramine-T (CAT)34c and ceric ammonium nitrate (CAN)34d have also been used for the preparation of nitriles from aldehydes and ammonia. Again these compounds led to the formation of a significant amount of waste and hence they are not considered as green oxidants. Overall, most of these methodologies have disadvantages regarding the use of toxic and waste generating reagents, poor selectivity or narrower substrate scope for the synthesis of functionalized and structurally diverse nitriles. Hence, greener and more efficient methodologies are required for the synthesis of advanced nitriles from aldehydes using heterogeneous iron-based catalysts under mild reaction conditions. In this regard, herein we report the application of a reusable iron oxide catalyst for the synthesis of a variety of nitriles from aldehydes and aqueous ammonia under mild reaction conditions. Compared to previously reported catalysts (Fig. 1), our iron-based catalyst is more advantageous and allows the synthesis of functionalized and structurally diverse nitriles. Notably, the synthetic utility and practical applicability of this catalyst have been demonstrated by performing gram-scale syntheses and recycling the catalyst. In addition to the synthesis of nitriles, a greener protocol for the preparation of amides is also demonstrated using iron-based catalysts, starting from aldehydes and ammonia in a water medium.
 |
| | Fig. 1 The synthesis of nitriles from aldehydes and ammonia using different catalysts/oxidants: a comparison of our method with previously reported methods. | |
2. Results and discussion
2.1. Preparation of nanoscale Fe2O3-based (Fe2O3-N/C) catalysts
In 2013, we developed nanoscale Fe2O3-based particles activated by nitrogen doped graphene layers for catalysis applications (Fe2O3-N/C).2a Inspired by these materials and based on our objective to develop iron-based sustainable chemical processes, herein we demonstrate the synthesis of all kinds of aromatic, heterocyclic and aliphatic nitrile as well as primary amides starting from aldehydes and aqueous ammonia using 1 bar of molecular oxygen or air. Typically, this Fe2O3-based catalyst has been prepared (Fig. 2) by impregnation of an in situ generated Fe(II)(1,10-phenanthroline)3(OAc)2 complex on commercial carbon (Vulcan XC 72R) and subsequent pyrolysis at 800 °C for 2 h in an argon atmosphere (Fe-phen/C-800; phen = 1,10-phenanthroline).2a The detailed preparation and characterization of this material by transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR) spectroscopy, and Mössbauer spectroscopy has been reported in our previously published paper2a (see also ESI†). The Fe-phen/C-800 catalyst was characterized by the formation of nanoscale Fe2O3 particles (2–5 and 20–80 nm), which are surrounded by 3–5 nitrogen doped graphene layers (Fig. S1A†). In contrast to the active material, the inactive material, Fe(OAc)2/C-800, prepared without phenanthroline contained much larger and well facetted Fe2O3 particles with a size of 100–800 nm. In this case there was no formation of graphene layers and hence these particles are not surrounded by graphene layers.
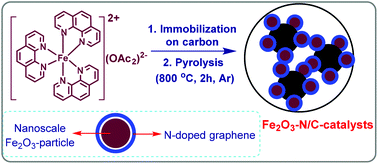 |
| | Fig. 2 The preparation of nitrogen doped graphene activated nanoscale Fe2O3-particles supported on carbon. | |
The X-ray photoelectron spectra for the N 1s electrons (Fig. S3A†) of the Fe2O3-N/C (Fe-phen/C-800) catalyst reveal that there are three N 1s peaks that occur at 398.3 eV, 399.3 eV and 401.0 eV. The first two peaks are assigned to the pyridinic nitrogens and Fe–N centers respectively. The peak observed at 401.0 eV is assigned to nitrogen in a grapheme-like structure. In the XPS for the Fe 2p electrons, two peaks corresponding to Fe species in the near-surface region were observed (Fig. S3B†). The first (major) appeared at 712.4 eV and the second (minor) at 709.9 eV. The first peak is due to the presence of Fe3+ species (more likely Fe2O3) while the second peak is assigned to Fe2+ species. Fe2+ species are in a minor proportion compared to Fe3+ species. By XPS analysis, in the Fe-phen/C-800 catalyst both the Fe3+ (major) and Fe2+ (minor) species are present on the surface of the material. However, the bulk composition is somewhat different to the surface composition. In the bulk, we can see the presence of mainly the Fe3+ species (in the form of Fe2O3). The presence of Fe2O3 in the bulk composition was confirmed by EPR spectroscopy and Mössbauer spectral analysis (Fig. S4 and S5; see ESI† for more details). Based on the reactivity and detailed spectroscopic characterization of the active (Fe-phen/C-800) and inactive (Fe(OAc)/C-800) catalysts, the active sites of the catalyst consist mainly of nanoscale Fe2O3 particles, which are surrounded by the nitrogen species of the graphene layers. Both the nano-sized particles and the Fe–N interactions are responsible for the remarkable activity of this catalyst. Thus, the active sites appear to be Fe–Nx centers that govern the catalytic activities.
2.2. Reaction design for the synthesis of nitriles
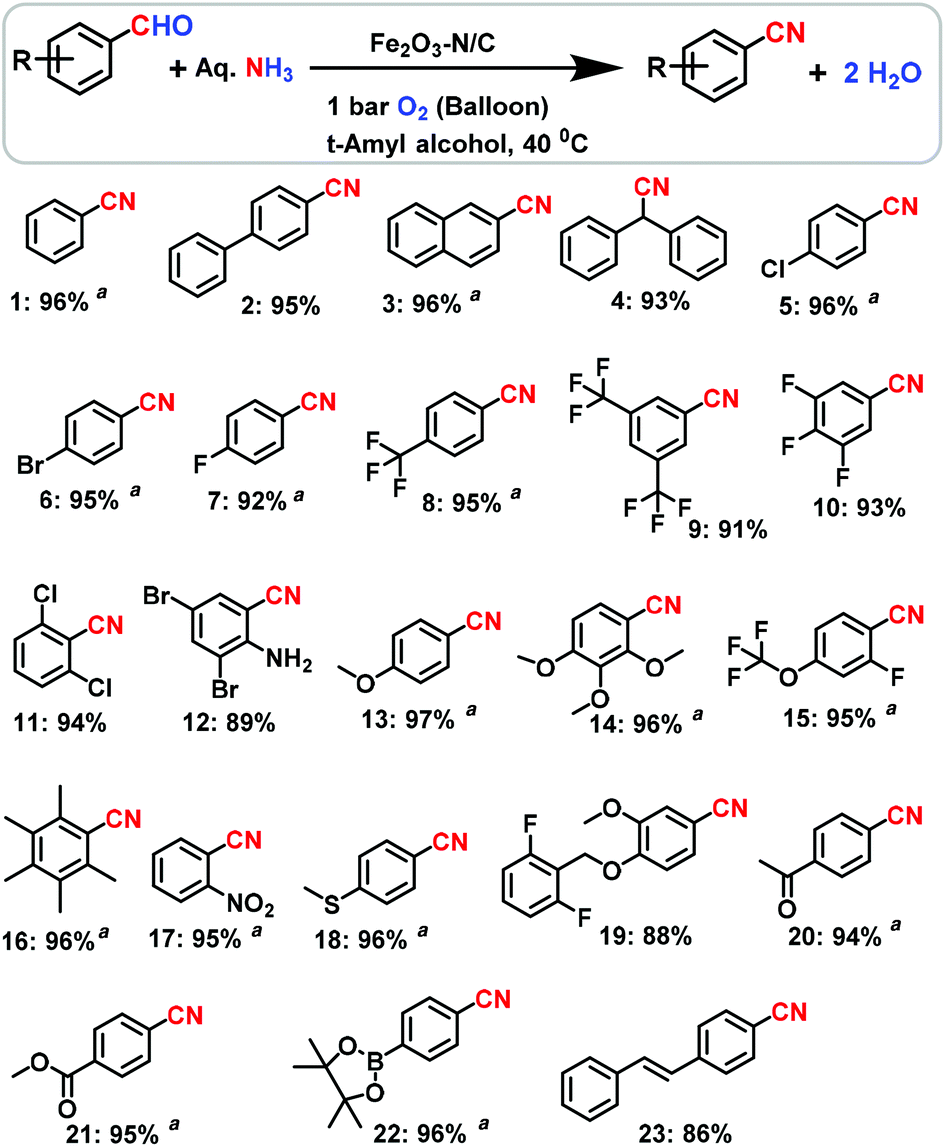
Using our Fe-phen/C-800 catalyst (Fe2O3-N/C), we started to investigate the preparation of nitriles from suitable starting materials using aqueous ammonia in the presence of 1 bar molecular oxygen at a mild temperature (<50 °C) in conventional apparatus (in a glass vial using a balloon), without the use of any special equipment or an autoclave. Under these conditions, first we tested benzyl alcohol at 40 °C with 1 bar O2 in the presence of the Fe2O3-N/C catalyst and found that no reaction occurred to produce the desired benzonitrile. However, to our surprise the reaction of benzaldehyde selectively gave benzonitrile in a 96% yield (Table 1, entry 6). Obviously, the homogeneous catalyst systems based on iron acetate or iron acetate/phenanthroline were found to be inactive (Table 1, entries 1 and 2). Similarly, the unpyrolyzed Fe-phen@C material, pyrolyzed simple iron acetate on carbon (Fe(OAc)2@C-800) and bulk Fe2O3 were also not active (Table 1, entries 3–5). After getting excellent results using the Fe2O3-N/C catalyst, and establishing a set of mild reaction conditions, we explored the applicability of this methodology for the preparation of nitriles from aldehydes and aqueous ammonia. As seen from Schemes 1–3, structurally diverse and functionalized aromatic, heterocyclic and challenging aliphatic nitriles have been synthesized from various aldehydes.
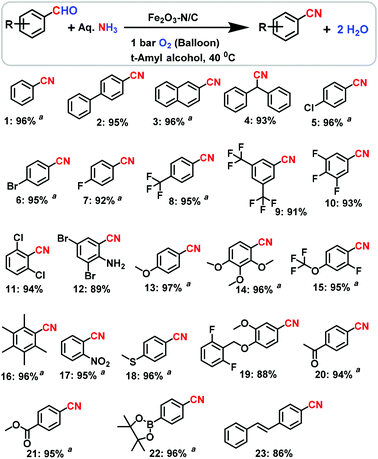 |
| | Scheme 1 The Fe2O3-N/C-catalyzed synthesis of structurally diverse and functionalized benzonitriles. Reaction conditions: 0.5 mmol aldehyde, 200–300 μL aq. NH3 (28–30% NH3 basis), 30 mg catalyst (3 mol% Fe), 1 bar O2 (balloon), 2 mL t-amyl alcohol, 40 °C, 24 h. Isolated yields are shown. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yields were determined by GC using 100 μL n-hexadecane as standard. Yields were determined by GC using 100 μL n-hexadecane as standard. | |
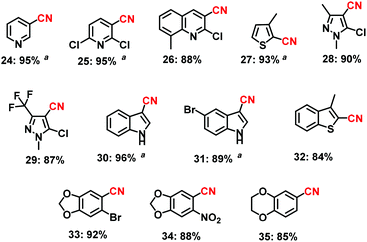 |
| | Scheme 2 The preparation of cyano-heterocycles using the Fe2O3-N/C catalyst. Reaction conditions: 0.5 mmol aldehyde, 200–300 μL aq. NH3 (28–30% NH3 basis), 30 mg catalyst (3 mol% Fe), 1 bar O2 (balloon), 2 mL t-amyl alcohol, 40 °C, 24 h. Isolated yields are shown. aYields were determined by GC using 100 μL n-hexadecane as standard. | |
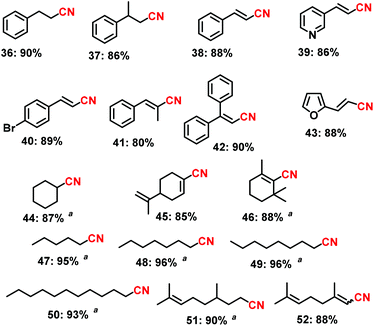 |
| | Scheme 3 The synthesis of challenging aliphatic nitriles using the Fe2O3-N/C catalyst. Reaction conditions: 0.5 mmol aldehyde, 200–300 μL aq. NH3 (28–30% NH3 basis), 30 mg catalyst (3 mol% Fe), 1 bar O2 (balloon), 2 mL t-amyl alcohol, 40 °C, 24 h. Isolated yields are shown. aYields were determined by GC by using 100 μL n-hexadecane as standard. | |
Table 1 The preparation of benzonitrile from benzaldehyde and aqueous ammonia using iron catalysts: a comparison of the catalytic activity
|
|
| Entry |
Catalyst |
Yield of benzonitrile (%) |
| Carbon = Vulcan XC72R, phen = phenanthroline. Materials were pyrolyzed at 800 °C for 2h under argon and bulk Fe2O3 was obtained commercially (>96% Fe basis). Homogeneous catalysis reaction conditions: 0.5 mmol benzaldehyde, 300 μL aq. NH3 (28–30% NH3 basis), 0.015 mmol Fe(OAc)2, 0.045 mmol phenanthroline, 1 bar O2 (balloon), 2 mL t-amyl alcohol, 40 °C, 24 h. GC yields determined using 100 μL n-hexadecane as standard. Heterogeneous catalysis reaction conditions: 0.5 mmol benzaldehyde, 300 μL aq. NH3 (28–30% NH3 basis), weight of catalyst corresponds to 3 mol% Fe, 1 bar O2 (balloon), 2 mL t-amyl alcohol, 40 °C, 24 h. GC yields determined using 100 μL n-hexadecane as standard. |
| 1a |
Fe(OAc)2 |
<1 |
| 2a |
Fe(OAc)2 + phen |
<1 |
| 3b |
Fe-phen@C |
<1 |
| 4b |
Fe(OAc)2@C-800 |
<3 |
| 5b |
Bulk Fe2O3 |
<1 |
| 6b |
Fe-phen@C-800 |
96 |
2.3. Synthesis of structurally diverse aryl nitriles
Aryl nitriles, which are an important motif found in a majority of nitrile containing life science molecules, especially in the central nervous system (CNS), cardiovascular and anti-HIV drugs,14 were obtained in good to excellent yields (Scheme 1). Interestingly, halogenated benzonitriles, which encounter selectivity problems when made by typical cyanation reactions, were prepared in 89–97% yields (Scheme 1). The resulting halogenated benzonitriles serve as precursors for agrochemicals, pesticides and engineering materials. As an example, 2,6-dichlorobenzonitrile (DCBN), which is a herbicide and a key intermediate in the preparation of various potential pesticides and the engineering of highly thermally resistant plastics, was obtained in a 94% yield (Scheme 1; product 11). This particular reaction is of high commercial significance and is also a very challenging topic in the field of heterogeneous catalysis under gas phase conditions due to steric hindrance (i.e. due to the substitution of two deactivating chlorine atoms in the ortho positions). The yield of DCBN achieved in the present study is much higher than that of state of the art catalysts (YDCBN = ca. 80% only). Also, functionalized benzaldehydes underwent selective reactions to produce challenging benzonitriles in yields of up to 97% (Scheme 1).
2.4. Synthesis of heterocyclic nitriles
Next, cyano-substituted heterocycles and aryl nitriles bearing heterocyclic backbones were synthesized in 84–96% yields (Scheme 2). In general, heterocyclic nitriles have been found to be key starting materials for the preparation of various active subunits of medicinal and biological molecules.
2.5. Synthesis of challenging aliphatic nitriles
In subsequent efforts, various aliphatic aldehydes, that are difficult to react, were chosen as potential substrates to produce their corresponding nitriles in good to excellent yields (up to 96%, Scheme 3). More interestingly, the allylic nitriles, which are important starting compounds for allylic amines, were obtained without further oxidation of their olefinic group (Scheme 3).
2.6. Green synthesis of primary amides
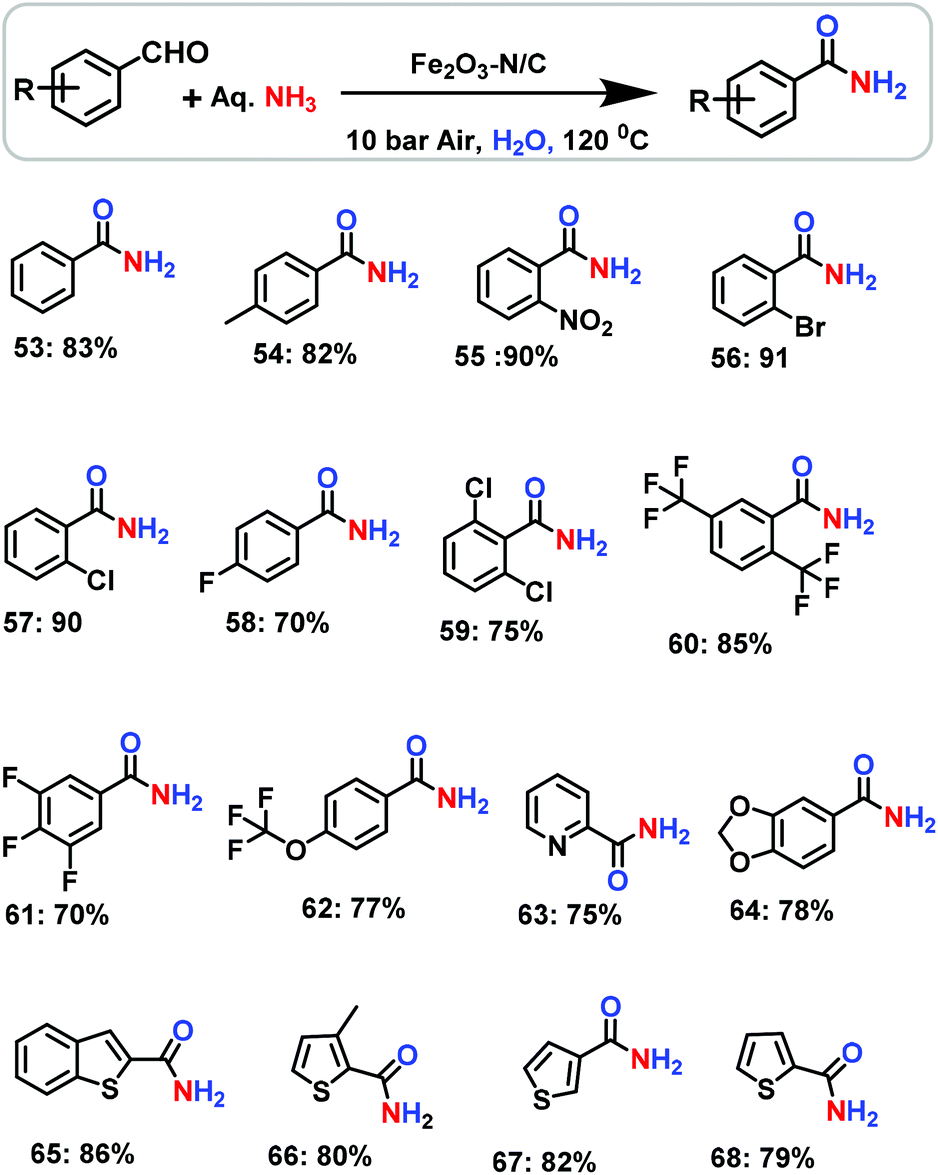
The creation of amide functionalities35–37 is highly important in chemistry, medicine and biology because they play a major role in the amplification and configuration of life science molecules, peptides, proteins and materials.13,16,17 In general, primary amides have been prepared by the reaction of carboxylic acids or their activated derivatives, such as acid chlorides and anhydrides, with ammonia. Nevertheless for the synthesis of this important class of amides, more environmentally benign procedures are required. In addition to alcohols,37a,b the synthesis of primary amides from aldehydes and ammonia has also been carried out using manganese oxide,37a Rh,38a KMnO438b and iodine.38c However, the use of iron-based heterogeneous catalysts for the synthesis of primary amides is obviously more advantageous.
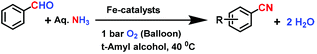
After having successfully demonstrated the preparation of important nitriles, we have further extended the potential scope of the Fe2O3-N/C catalyst for the direct and one-pot synthesis of primary amides from benzaldehyde and aqueous ammonia. Interestingly, this catalyst was also observed to be active and selective for the preparation of even primary amides (Scheme 4). However, to achieve sufficient activity and selectivity, the reaction temperature and pressure had to be raised to a certain extent and the reaction conditions needed to be adjusted accordingly. In a water medium, we were able to convert aldehydes into primary amides using aqueous ammonia and air in a yield of up to 91% (Scheme 4).
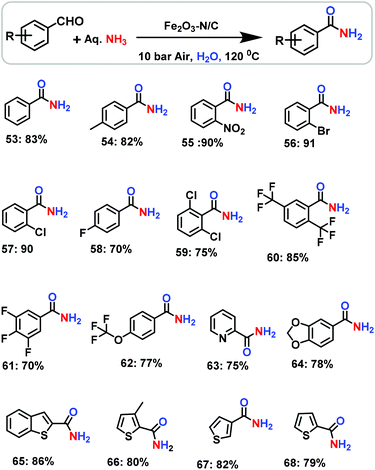 |
| | Scheme 4 The nanoscale Fe2O3-catalysed synthesis of amides from aldehydes and aqueous ammonia. Reaction conditions: 0.5 mmol aldehyde, 100 μL aq. NH3 (28–30% NH3 basis), 50 mg catalyst (5.0 mol% Fe), 10 bar air, 3 mL H2O, 120 °C, 24 h. Isolated yields are shown. | |
The reaction pathway for the formation of nitriles and amides is presented in Scheme 5. In the first step, the formation of the intermediate, a primary imine was formed from an aldehyde and ammonia. In the presence of the Fe2O3-N/C catalyst, this unstable imine underwent oxidation to produce a nitrile. In the final step, in presence of water and ammonia, the formed nitrile was hydrolyzed to produce a primary amide. The production of the amide from the nitrile requires a higher temperature to proceed under forcing conditions. In order to confirm the formation mechanism of the amide, we performed the reaction of benzonitrile in water at 120 °C in the presence of aq. ammonia and a catalyst. In this case, the formation of benzamide in an 80% yield was observed. This result clearly evidenced that water is required for the formation of amides from aldehydes and ammonia. Due to the involvement of a two step reaction, the obtained yields of the amides are somewhat lower than those of the nitriles. In this case the unreactive nitrile was observed as the other product.
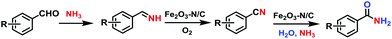 |
| | Scheme 5 The reaction pathway for the Fe2O3-N/C-catalyzed synthesis of nitriles and amides from aldehydes and ammonia. | |
2.7. Scale up studies for gram scale synthesis
In order to demonstrate the synthetic and practical utility of this approach using our iron-catalyzed synthetic protocol, we also performed the gram scale (i.e. 1 to 5 g) reactions. In the interest of scale up, a few grams of some selected aldehydes were converted to their corresponding nitriles under identical reaction conditions. The yields of the desired nitriles obtained from these tests are very much comparable and quite consistent with those obtained from the small (0.5 mmol) scale tests (Scheme 6).
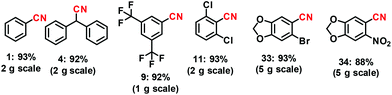 |
| | Scheme 6 Demonstrating the gram scale reactions for the synthesis of some selected nitriles over the Fe2O3-N/C catalyst. Reaction conditions: 2–5 g substrate, the weight of the catalyst corresponds to 3 mol% Fe, 200 μL aq. NH3 for every 0.5 mmol substrate, 40 °C, 10–50 mL t-amyl alcohol, 1 bar O2 (balloon), 24–30 h. Isolated yields are shown. | |
2.8. Stability and recyclability of the Fe2O3-N/C catalyst
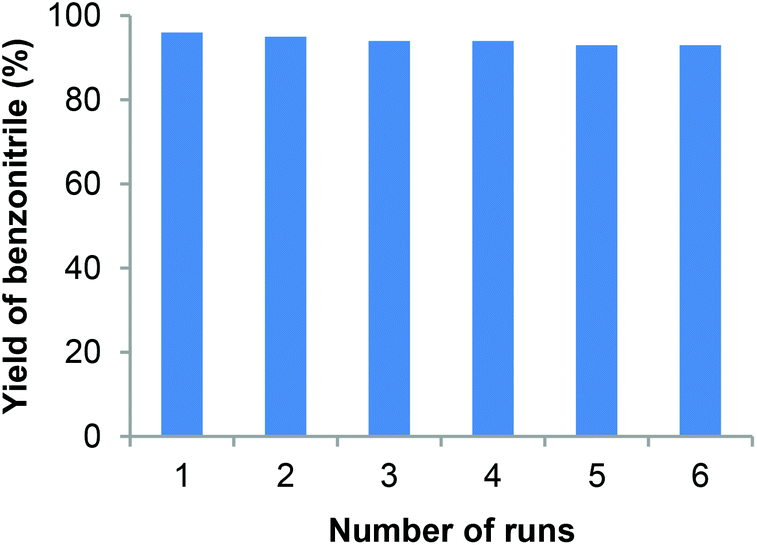
In general, the stability, recyclability and reusability features of any catalyst are very important for the advancement of sustainable industrial processes. Noticeably, our Fe2O3-N@C catalyst is highly stable and can be conveniently recycled up to 5 times without any significant loss of catalytic activity and selectivity (Fig. 3). The recycled catalyst was subjected to ICP analysis and it was found that there was no leaching of iron in the recycled catalyst.
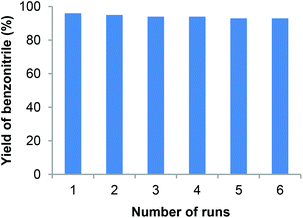 |
| | Fig. 3 Recyclability of the Fe2O3-N/C catalyst for the synthesis of benzonitrile. Reaction conditions: 1 mmol benzalydehyde, 60 mg catalyst (3.2 mol% Fe), 400 μL aq. NH3 (28–30% NH3 basis), 1 bar O2 (balloon), 4 mL t-amyl alcohol, 40 °C, 24 h. Yields were determined by GC using n-hexadecane as standard. | |
The stability of the catalyst was also tested by performing kinetic studies (Fig. S6†). The effect of ammonia concentration, reaction time, amount of catalyst, concentration of substrate (benzaldehyde) and pressure of O2 on the yield of benzonitrile was studied (Fig. S6†). Noticeably, the catalyst is stable and exhibited similar catalytic activity in the presence of an excess of ammonia (1 mL), with a prolonged reaction time and at a high pressure of O2 (5 bar). As seen from the kinetic plots (Fig. S6†), on increasing the reaction time, catalyst amount and ammonia concentration, the yield of benzonitrile increased and the highest yield was observed for the reaction that took 24 h with 30 mg of catalyst, in the presence of 200 μL aq. ammonia and one bar O2.
3. Experimental section
3.1. Procedure for the preparation of the Fe2O3-N/C (Fe-phen/C-800) catalyst (1 g)2a
Appropriate amounts of Fe(OAc)2 and 1,10-phenanthroline corresponding to 3 wt% of Fe in a 1:3 molar ratio of Fe to phenathroline were stirred in ethanol for 30 minutes at room temperature. Then, carbon powder (VULCAN® XC72R, Cabot Corporation Prod. Code XVC72R, CAS no. 1333-86-4) was added and the whole reaction mixture was stirred at 60 °C for 12–15 hours. The reaction mixture was cooled to room temperature and the ethanol was removed in vacuo. The solid material obtained was dried at 60 °C for 12 hours, after which it was ground to a fine powder. Then, the powdered material was pyrolyzed at 800 °C for 2 hours under an argon atmosphere and then cooled to room temperature.
Elemental analysis of Fe2O3-N/C (pyrolyzed Fe-phen/C at 800 °C) (wt%): C = 91.1, H = 0.19, N = 2.69, Fe = 2.95.
3.2. General procedure for the preparation of nitriles
An 8 mL oven dried glass vial was charged with a magnetic stirrer bar and 0.5 mmol of the appropriate aldehyde and then 2 mL t-amyl alcohol was added. Next, 30 mg Fe2O3-N/C (i.e. Fe-phen/C-800) catalyst (3 mol% Fe) was added followed by the addition of 200–300 μL aq. NH3 (28–30% NH3 basis). The glass vial was fitted with a septum and screw cap. Then a balloon containing 1 bar O2 was connected to the reaction vial via a needle and the reaction was allowed to proceed for 24 h at 40 °C under continuous stirring. After completion of the reaction, the reaction vial was cooled down to room temperature and the excess O2 was gradually discharged. Then, the catalyst was filtered-off, and washed with ethyl acetate. The solvent from the filtrate containing the reaction products and unreacted substrates (if any) was removed in vacuo and the target nitrile was purified by column chromatography. In the case of determining the yields by GC, 100 μL n-hexadecane was added to the reaction vial containing the products, and diluted with ethyl acetate. Then, this was filtered through a plug of silica and the filtrate containing product was analysed by GC (HP-6890, column HP-5 30 m × 250 mm × 0.25 μm) equipped with a FID. The conversion and yields were determined on the basis of the GC area counts using pre-calibrations. All products were analyzed by GC, GC-MS and NMR spectroscopy analysis.
3.3. General procedure for the preparation of amides
A magnetic stirrer bar and the appropriate aldehydes were transferred to an 8 mL glass vial and then 3 mL H2O was added. Later on, 50 mg Fe2O3-N/C (i.e. Fe-phen/C-800) catalyst (5 mol% Fe) was added followed by the addition of 100 μL aq. NH3. Then, the vial was fitted with a septum, cap and needle. The reaction vials (i.e. 8 vials in each test) were placed into a 300 mL autoclave (PARR Instrument Company, USA) and then the autoclave was pressurized with 10 bar of air. The autoclave was placed into an aluminium block and the temperature of the aluminium block was set to be 120 °C inside the autoclave. The reactions were allowed to progress under continuous stirring for the required time at 120 °C. In this set-up the temperature of the aluminium block was set to more than 120 °C (130–140 °C) in order to attain exactly 120 °C inside the autoclave. After completion of the reaction, the excess air that remained in the autoclave was slowly depressurized and then the samples were removed from the autoclave. Afterwards, the catalyst was filtered-off and washed with methanol. The solvent from the filtrate containing the reaction products was removed in vacuo and the corresponding amide was purified by column chromatography. All products were analyzed by GC-MS and NMR spectroscopy analysis.
3.4. Procedure for the gram scale reactions
The reactions were performed similarly to the procedure mentioned in section 3.2, in a 100 or 300 mL glass fitted autoclave with the following conditions; 2–5 g aldehyde, a corresponding 3 mol% Fe, 200 μL aq. NH3 for every 0.5 mmol substrate, 40 °C, 10–50 mL t-amyl alcohol, 24–30 h. After completion of the reaction, the autoclave was cooled to room temperature and the remaining air was discharged. The catalyst from the reaction mixture was filtered off and washed with ethyl acetate. The solvent from the filtrate containing the reaction products was removed in vacuo. The corresponding nitrile was purified by column chromatography (silica and an n-hexane–ethyl acetate mixture). All products were analyzed by GC-MS and NMR spectroscopy analysis.
3.5. Procedure for recycling the catalyst
The recycling of the catalyst was carried out for the synthesis of benzonitrile using the same procedure given in section 3.2 under the following reaction conditions: 1 mmol benzaldehyde, 60 mg Fe2O3-N/C (Fe-phen/C-800) (3% Fe), 400 μL aq. NH3, 1 bar O2, 4 mL t-amyl alcohol, 40 °C, 24 h; yields were determined by GC using 100 μL n-hexadecane as standard. In each run, after the reaction, the catalyst was separated by centrifugation, washed thoroughly with ethyl acetate and dried under vacuum. Then, the dried catalyst was used further, without any purification or re-activation. The recycled catalyst and centrifugate containing the reaction products were subjected to ICP analysis. This analysis showed that the content of Fe in the recycled catalysts had not decreased (2.94) and also Fe was not detected in the centrifugate.
4. Conclusions
In conclusion, we have demonstrated a green method for the preparation of nitriles and primary amides starting from various easily accessible aldehydes by making use of a combination of an earth abundant Fe-based nano-catalyst and green reagents such as atmospheric O2 or air and inexpensive aqueous ammonia. Under operationally simple and mild reaction conditions, this iron-based catalyst is highly reactive and selective for the synthesis of a series of functionalized and structurally diverse aromatic, heterocyclic and aliphatic nitriles. Additionally, this aerobic green oxidative method has been applied in the preparation of primary amides. Moreover, the synthetic utility and practical applicability of this method were demonstrated by up scaling the reactions to gram scales (1–5 g scale) and recycling of the catalyst.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Prof. Matthias Beller, the Director of the Leibniz Institute for Catalysis for his helpful discussions. The European Research Council for financial support and the State of Mecklenburg-Vorpommern for general support are gratefully acknowledged. We also thank the analytical staff of LIKAT for their excellent service.
References
- For selected reviews and books on iron catalysis:
(a) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed;
(b) C. Bolm, Nat. Chem., 2009, 1, 420 CrossRef CAS PubMed;
(c) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed;
(d) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed;
(e) K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS PubMed;
(f)
B. Plietker, in Iron Catalysis in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- For selected examples of iron catalysis:
(a) R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed;
(b) J. R. Ludwig, P. M. Zimmerman, J. B. Gianino and C. S. Schindler, Nature, 2016, 533, 374–379 CrossRef CAS PubMed;
(c) K. Adams, A. K. Ball, J. Birkett, L. Brown, B. Chappell, D. M. Gill, P. K. T. Lo, N. J. Patmore, C. R. Rice, J. Ryan, P. Raubo and J. B. Sweeney, Nat. Chem., 2017, 9, 396–401 CrossRef CAS PubMed;
(d) R. K. Sharma, S. Dutta, S. Sharma, R. Zboril, R. S. Varma and M. B. Gawande, Green Chem., 2016, 18, 3184–3209 RSC.
- For selected examples and reviews on iron nanocatalysis:
(a) R. V. Jagadeesh, T. Stemmler, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Nat. Protoc., 2015, 10, 548–557 CrossRef CAS PubMed;
(b) H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. l. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef PubMed;
(c) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
-
(a) L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed;
(b) W. N. Oloo and L. Que Jr., Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed.
-
S. S. Stahl and S. J. Lippard, Dioxygen and Alkane Activation by Iron-Containing Enzymes in Iron Metabolism: Inorganic Biochemistry and Regulatory Mechanisms, Wiley, 2008 Search PubMed.
- X. Jiang, J. Zhang and S. Ma, J. Am. Chem. Soc., 2016, 138, 8344–8347 CrossRef CAS PubMed.
- M. O. Ratnikov, X. Xu and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 9475–9479 CrossRef CAS PubMed.
- J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86–95 CrossRef CAS PubMed.
-
(a)
R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed;
(b)
A. J. Aatiadi in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and R. Rappaport, Wiley, 2010 Search PubMed.
- M. Movassaghi and M. D. Hill, Nat. Protoc., 2007, 2, 2018–2023 CrossRef CAS PubMed.
- J. Yang, M. R. Karver, W. Li, S. Sahu and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 5222–5225 CrossRef CAS PubMed.
- T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef CAS PubMed.
-
G. Arthur, The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000 Search PubMed.
- F. F. Fraser, Y. Lihua, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef PubMed.
- S. T. Murphy, H. L. Case, E. Ellsworth, S. Hagen, M. Huband, T. Joannides, C. Limberakis, K. R. Marotti, A. M. Ottolini, M. Rauckhorst, J. Starr, M. Stier, C. Taylor, T. Zhu, A. Blaser, W. A. Denny, G. L. Lu, J. B. Smaill and F. Rivault, Bioorg. Med. Chem. Lett., 2007, 17, 2150–2155 CrossRef CAS PubMed.
- A. Graul and J. Castaner, Drugs Future, 1997, 22, 956–968 CrossRef CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
-
(a) A. Martin and V. N. Kalevaru, ChemCatChem., 2010, 2, 1504–1522 CrossRef CAS;
(b)
F. Cavani, G. Centi and P. Marion, Metal Oxide Catalysis, ed. S. D. Jackson and J. S. J. Hargreaves, Wiley-VCH Verlag GmbH, 2009 Search PubMed;
(c)
R. K. Grasselli and M. A. Tenhover, Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 3489–3517 Search PubMed;
(d) A. Martin and B. Lücke, Catal. Today, 2000, 57, 61–70 CrossRef CAS.
-
(a) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC;
(b) M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem. – Eur. J., 2003, 9, 1828–1836 CrossRef CAS PubMed.
-
(a) T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286–6288 CrossRef CAS PubMed;
(b) T. Oishi, K. Yamaguchi and N. Mizuno, Top. Catal., 2010, 53, 479–486 CrossRef CAS;
(c) W. Yin, C. Wang and H. Huang, Org. Lett., 2013, 15, 1850–1853 CrossRef CAS PubMed;
(d) R. V. Jagadeesh, T. Stemmler, A.-E. Surkus, M. Bauer, M.-M. Pohl, J. Radnik, K. Junge, H. Junge, A. Brückner and M. Beller, Nat. Protoc., 2015, 10, 916–926 CrossRef PubMed;
(e) R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CAS;
(f) S. Shang, W. Dai, L. Wang, Y. Lv and S. Gao, Chem. Commun., 2017, 53, 1048–1051 RSC.
-
(a) L. M. Dornan, Q. Cao, J. C. A. Flanagan, J. J. Crawford, M. J. Cook and M. J. Muldoon, Chem. Commun., 2013, 49, 6030–6032 RSC;
(b) M. Bhardwaj, M. Kour and S. Paul, RSC Adv., 2016, 6, 99604–99614 RSC.
-
(a) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS;
(b) H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef CAS;
(c) O.-Y. Lee, K.-L. Law and D. Yang, Org. Lett., 2009, 11, 3302–3305 CrossRef CAS PubMed.
-
(a) H. Yu, S. Ru, G. Dai, Y. Zhai, H. Lin, S. Han and Y. Wei, Angew. Chem., Int. Ed., 2017, 56, 3867–3871 CrossRef CAS PubMed;
(b) M. Liu and C.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806–10810 CrossRef CAS PubMed.
-
(a) H. Wang, X.-J. Dai and C.-J. Li, Nat. Chem., 2017, 9, 374–378 CrossRef CAS PubMed;
(b) L. Pu, Acc. Chem. Res., 2014, 47, 1523–1535 CrossRef CAS PubMed;
(c) F. Meng, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 11304–11307 CrossRef CAS PubMed;
(d) S. Fleischer, S. Zhou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 5120–5124 CrossRef CAS PubMed;
(e) X. Tan, G. Wang, Z. Zhu, C. Ren, J. Zhou, H. Lv, X. Zhang, L. Wa Chung, L. Zhang and X. Zhang, Org. Lett., 2016, 18, 1518–1521 CrossRef CAS PubMed.
-
(a) E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef CAS PubMed;
(b) B. E. Marki and K. A. Scheidt, Org. Lett., 2008, 10, 4331–4334 CrossRef PubMed;
(c) R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, Org. Lett., 2015, 17, 3290–3293 CrossRef CAS PubMed;
(d) B. R. Travis, M. Sivakumar, G. O. Hollist and B. Borhan, Org. Lett., 2003, 5, 1031–1034 CrossRef CAS PubMed;
(e) S. D. Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef PubMed;
(f) R. Lerebours and C. Wolf, J. Am. Chem. Soc., 2006, 128, 13052–13053 CrossRef CAS PubMed.
-
(a) D. W. Piotrowski, A. S. Kamlet, A.-M. R. Dechert-Schmitt, J. Yan, T. A. Brandt, J. Xiao, L. Wei and M. T. Barrila, J. Am. Chem. Soc., 2016, 138, 4818–4823 CrossRef CAS PubMed;
(b) D. Mahesh, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2015, 80, 1644–1650 CrossRef CAS PubMed;
(c) V. Zuliani, G. Cocconcelli, M. Fantini, C. Ghiron and M. Rivara, J. Org. Chem., 2007, 72, 4551–4553 CrossRef CAS PubMed;
(d) S. Hati, P. K. Dutta, S. Dutta, P. Munshi and S. Sen, Org. Lett., 2016, 18, 3090–3093 CrossRef CAS PubMed.
-
(a) Y. Bernhard, B. Thomson, V. Ferey and M. Sauthier, Angew. Chem., Int. Ed., 2017, 56, 7460–7464 CrossRef CAS PubMed;
(b) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed;
(c)
V. Andrushko and N. Andrushko, Stereoselective Synthesis by Bond Formation, Stereoselective Synthesis of Drugs and Natural Products, Wiley VCH, Germany, 2013 Search PubMed.
-
(a) C. B. Kelly, K. M. Lambert, M. A. Mercadante, J. M. Ovian, W. F. Bailey and N. E. Leadbeater, Angew. Chem., Int. Ed., 2015, 54, 4241–4245 CrossRef CAS PubMed;
(b) C. Fang, M. Li, X. Hu, W. Mo, B. Hu, N. Sun, L. Jin and Z. Shen, Adv. Synth. Catal., 2016, 358, 1157–1163 CrossRef CAS.
-
(a) J. K. Augustine, R. N. Atta, B. K. Ramappa and C. Boodappa, Synlett, 2009, 3378–3382 CrossRef CAS;
(b) J.-L. Zhu, F.-Y. Lee, J.-D. Wu, C.-W. Kuo and K.-S. Shia, Synlett, 2007, 1317–1319 CrossRef CAS;
(c) E. Wang and G. Lin, Tetrahedron Lett., 1998, 39, 4047–4050 CrossRef CAS;
(d) M. A. Alia and T. Punniyamurthya, Adv. Synth. Catal., 2010, 352, 288–292 CrossRef.
- B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2012, 77, 5364–5370 CrossRef CAS PubMed.
- Q. Wu, Y. Luo, A. Lei and J. You, J. Am. Chem. Soc., 2016, 138, 2885–2888 CrossRef CAS PubMed.
-
(a) S. Talukdar, J.-L. Hsu, T.-C. Chou and J.-M. Fang, Tetrahedron Lett., 2001, 42, 1103–1105 CrossRef CAS;
(b) A. C. Lele, N. H. P. Desai and M. S. Degani, J. Pharm. Res., 2014, 7, 934–936 Search PubMed;
(c) A. G. Choghamarani, M. A. Zolfigol, M. Hajjami and S. Sardari, Synth. Commun., 2013, 43, 52–58 CrossRef;
(d) V. N. Telvekar, K. N. Patel, H. S. Kundaikar and H. K. Chaudhari, Tetrahedron Lett., 2008, 49, 2213–2215 CrossRef CAS;
(e) V. N. Telvekar, R. A. Rane and T. V. Namjoshi, Synth. Commun, 2010, 40, 494–497 CrossRef CAS;
(f) N. D. Arote, D. S. Bhalerao and K. G. Akamanchi, Tetrahedron Lett., 2007, 48, 3651–3653 CrossRef CAS;
(g) S. Bag, N. R. Tawari and M. S. Degani, ARKIVOC, 2009, xiv, 118–123 Search PubMed.
-
(a) S. Yamazaki and Y. Yamazaki, Chem. Lett., 1990, 571–574 CrossRef CAS;
(b) G. Lai, N. K. Bhamare and W. K. Anderson, Synlett, 2001, 230–231 CrossRef CAS.
-
(a) H. Veisi, Synthesis, 2010, 2631–2635 CrossRef CAS;
(b) Y.-Z. Zhu and C. Cai, Monatsh. Chem., 2010, 141, 637–639 CrossRef CAS;
(c) Y.-Z. Zhu, X.-Q. Zhang, F. Liu, H.-M. Gu and H.-L. Zhu, Synth. Commun., 2013, 43, 2943–2948 CrossRef CAS;
(d) L. Wang, C. Shen, H.-P. Wang, W.-Y. Zhou, F.-A. Sun, M.-Y. He and Q. Chen, J. Chem. Res., 2012, 36, 460–462 CrossRef CAS.
-
(a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed;
(b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
-
(a)
M. B. Smith, Compendium of Organic Synthetic Methods, Wiley, New York, 2001, p. 9 Search PubMed;
(b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
-
(a) K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544–547 CrossRef CAS PubMed;
(b) J. F. Soule, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550–18553 CrossRef CAS PubMed;
(c) C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528–6532 CrossRef CAS PubMed;
(d) F.-L. Zhang, Y.-F. Wang, G. H. Lonca, X. Zhu and S. Chiba, Angew. Chem., Int. Ed., 2014, 53, 4390–4394 CrossRef CAS PubMed;
(e) J. Zhang, Y. Liu, S. Chiba and T.-P. Loh, Chem. Commun., 2013, 49, 11439–11441 RSC;
(f) S. L. Zultanski, J. Zhao and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 6416–6419 CrossRef CAS PubMed;
(g) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
-
(a) H. Peng, L. Xu, H. Wu, K. Zhang and P. Wu, Chem. Commun., 2013, 49, 2709 RSC;
(b) D. Antoniak, A. Sakowicz, R. Loska and M. Mąkosza, Synlett, 2015, 26, 84–86 CAS;
(c) J.-J. Shie and J.-M. Fang, J. Org. Chem., 2003, 68, 1158–1160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02627g |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a