
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general synthesis of aromatic and heteroaromatic lipoxin B4 analogues†
Benjamin
Owen
 and
Patrick J.
Guiry
*
and
Patrick J.
Guiry
*
Centre for Synthesis and Chemical Biology, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: p.guiry@ucd.ie
First published on 19th September 2023
Abstract
Lipoxins are an important class of pro-resolving mediators that play a crucial role in the resolution of inflammation. Thus, the synthesis of more chemically and metabolically stable synthetic lipoxin analogues is an area of significant interest. Whereas synthetic analogues of lipoxin A4 (LXA4) have been well studied, analogues of lipoxin B4 (LXB4) have been the focus of considerably less attention. Herein we report the asymmetric synthesis of a focused library of LXB4 mimetics in which the triene core of the molecule has been replaced with different aromatic and heteroaromatic rings. The synthesis of each of these analogues was achieved by a general strategy in which the key steps were a Suzuki cross coupling between a common upper chain fragment and an aromatic lower chain, followed by a stereoselective ketone reduction.
Introduction
Specialized pro-resolving mediators (SPMs) are a class of endogenously derived lipid mediators that are known to play a crucial role in the resolution of the innate inflammatory response.1,2 The two lipoxins, lipoxin A4 (1) and lipoxin B4 (2) (Fig. 1a), are important members of this class of compounds and both have been shown to stimulate a range of pro-resolving bioactions such as slowing the recruitment of polymorphonuclear neutrophils (PMNs) to the site of inflammation, while accelerating the recruitment of monocytes and stimulating the phagocytosis of apoptotic PMNs.3–5 However, a major drawback that limits the therapeutic use of these compounds is their chemical instability and rapid enzymatic degradation in vivo. As a result, there has been a lot of interest in designing synthetic lipoxin analogues that are more metabolically stable, easier to handle, and more synthetically accessible.6,7 Alongside oxidation of a hydroxy group to the corresponding ketone, one of the major pathways of enzymatic degradation for both LXA4 and LXB4 is known to be reduction of one of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in the triene core to form the metabolites 13,14-dihydro-LXA4 and 6,7-dihydro-LXB4 respectively.8 Inspired by this observation, we have previously reported the synthesis and biological evaluation of a number of different aromatic analogues of LXA4 in which the triene core of the molecule has been replaced with a more chemically stable aromatic or heteroaromatic ring (Fig. 1b). The first analogue synthesized, benzo-LXA4 (3), was shown to stimulate an increase in the phagocytosis of apoptotic PMNs comparable to native LXA4.9 Since then, we have demonstrated the favourable anti-inflammatory properties of LXA4 mimetics containing a heterocyclic core such as pyrido-LXA4 (4),10 quinoxalino-LXA4 (5)11 and imidazolo-LXA4 (6) (Fig. 1c).12 However, despite the progress made in the development of synthetic analogues of LXA4, the synthesis and evaluation of stable analogues of its isomer LXB4 has been the focus of considerably less attention. Although the biological properties of LXA4 and LXB4 are similar, they are not identical. For example, LXB4 has been shown to have consistently more potent neuroprotective effects on retinal ganglion cells than LXA4 both in vitro and in vivo.13 Furthermore, it is clear that LXA4 and LXB4 operate through completely different signalling pathways.14 LXA4 is known to bind to FPR2 (also known as the ALX receptor) expressed in the cell membranes of human PMNs and other immune cells.15 In contrast, no receptor for LXB4 has so far been identified. As a result, the mechanism of action of LXB4 is less well understood and research into the therapeutic benefits of LXB4 and its analogues has been somewhat neglected in comparison to LXA4. We initially reported a stereoselective synthesis of benzo-LXB4 (7a) alongside benzo-LXA4 (3) and showed that 7a was also able to stimulate phagocytosis of apoptotic PMNs.9 Since then, however, the only other examples of LXB4 analogues have been reported by Nubbemeyer and co-workers who developed a synthesis of analogues based on a cycloheptatriene core.16,17
C double bonds in the triene core to form the metabolites 13,14-dihydro-LXA4 and 6,7-dihydro-LXB4 respectively.8 Inspired by this observation, we have previously reported the synthesis and biological evaluation of a number of different aromatic analogues of LXA4 in which the triene core of the molecule has been replaced with a more chemically stable aromatic or heteroaromatic ring (Fig. 1b). The first analogue synthesized, benzo-LXA4 (3), was shown to stimulate an increase in the phagocytosis of apoptotic PMNs comparable to native LXA4.9 Since then, we have demonstrated the favourable anti-inflammatory properties of LXA4 mimetics containing a heterocyclic core such as pyrido-LXA4 (4),10 quinoxalino-LXA4 (5)11 and imidazolo-LXA4 (6) (Fig. 1c).12 However, despite the progress made in the development of synthetic analogues of LXA4, the synthesis and evaluation of stable analogues of its isomer LXB4 has been the focus of considerably less attention. Although the biological properties of LXA4 and LXB4 are similar, they are not identical. For example, LXB4 has been shown to have consistently more potent neuroprotective effects on retinal ganglion cells than LXA4 both in vitro and in vivo.13 Furthermore, it is clear that LXA4 and LXB4 operate through completely different signalling pathways.14 LXA4 is known to bind to FPR2 (also known as the ALX receptor) expressed in the cell membranes of human PMNs and other immune cells.15 In contrast, no receptor for LXB4 has so far been identified. As a result, the mechanism of action of LXB4 is less well understood and research into the therapeutic benefits of LXB4 and its analogues has been somewhat neglected in comparison to LXA4. We initially reported a stereoselective synthesis of benzo-LXB4 (7a) alongside benzo-LXA4 (3) and showed that 7a was also able to stimulate phagocytosis of apoptotic PMNs.9 Since then, however, the only other examples of LXB4 analogues have been reported by Nubbemeyer and co-workers who developed a synthesis of analogues based on a cycloheptatriene core.16,17
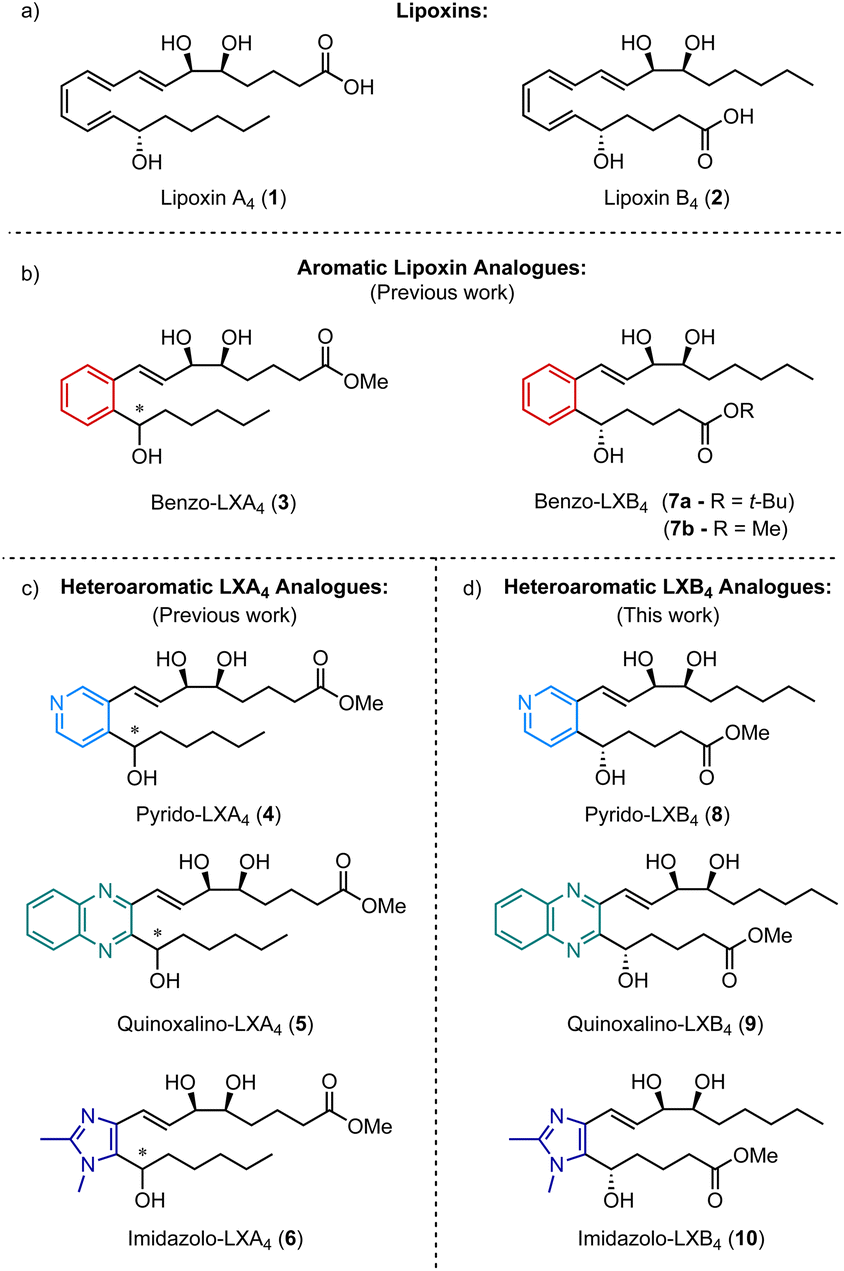 | ||
| Fig. 1 Overview of previously synthesized analogues of LXA4 and the corresponding LXB4 analogues synthesized in this work. | ||
Given our experience with heteroaromatic analogues of LXA4, we aimed to develop a general synthetic route that would allow for the efficient stereoselective synthesis of a focused library of LXB4 mimetics containing a variety of different aromatic and heteroaromatic cores. This would rapidly expand the number of LXB4 analogues accessible and hopefully lead to better understanding of LXB4 and the potential therapeutic applications of its stable synthetic analogues in the future. To demonstrate this, we selected four different LXB4 analogues as targets to be synthesized: benzo-LXB4 (7), pyrido-LXB4 (8), quinoxalino-LXB4 (9) and imidazolo-LXB4 (10) (Fig. 1d). Targets containing these particular heteroaromatic cores were chosen based on the findings of our SAR studies on heteroaromatic LXA4 mimetics. In particular, analogues containing a dimethylimidazole core12,18 and a quinoxaline core11 have been shown to be especially potent and effective inhibitors of LPS-induced NF-κB activity. The success of LXA4 analogues based on a dimethylimidazole core and a quinoxaline core encouraged us to also investigate the synthesis of LXB4 analogues containing these key heterocycles. A summary of the results of our previous SAR studies on synthetic LXA4 mimetics is shown in the ESI (ESI, Table S1†).
Results and discussion
For all four of the target analogues, we proposed a general retrosynthetic analysis that would allow us to vary the heterocyclic core in a modular fashion without needing to redesign the synthesis each time (Scheme 1). The key steps of this general strategy were anticipated to be (i) a Suzuki coupling between a boronic ester “upper chain” and an aromatic δ-keto ester “lower chain”, (ii) a stereoselective ketone reduction, and (iii) an acetonide deprotection of the 1,2-diol. Upper chain 11 would thus serve as a common fragment in the synthesis of each of the target analogues, while the synthesis of the lower chain would be slightly different each time in order to successfully vary the aromatic or heterocyclic core.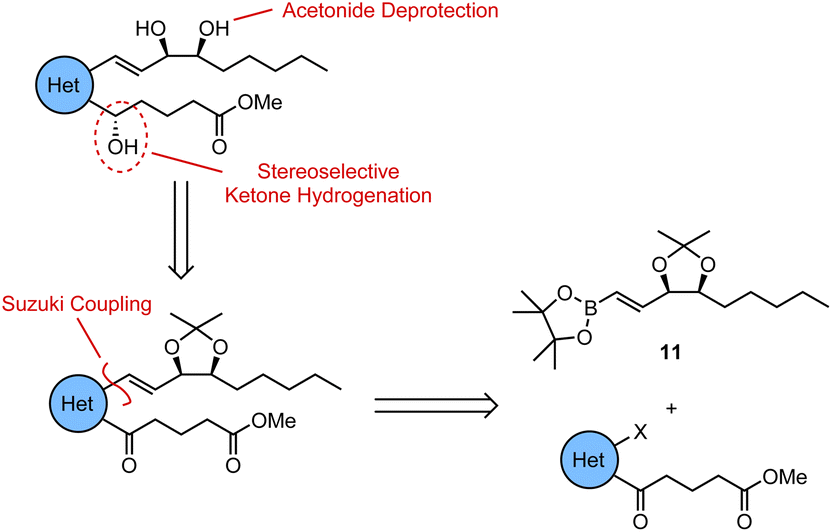 | ||
| Scheme 1 Proposed general retrosynthetic analysis for the target (hetero)aromatic LXB4 analogues. | ||
To begin with, the synthesis of upper chain 11 was achieved via a chiral pool strategy starting from 2-deoxy-D-ribose (12) (Scheme 2). Following acetonide protection of the 1,2-diol, a Wittig olefination was carried out between 13 and the ylide generated in situ by deprotonation of propyl phosphonium bromide with NaHMDS. This afforded alkene 14 in 53% yield exclusively as the Z-geometric isomer. However, the E/Z-selectivity of the Wittig reaction was inconsequential as alkene 14 was subsequently reduced to the corresponding alkane 15 in 97% yield by H2 gas and Pd/C. To complete the synthesis, alcohol 15 was first oxidized to aldehyde 16 in 74% yield using catalytic TEMPO, and PIDA as the co-oxidant. After that, the key vinyl boronic ester functionality was successfully installed via a chromium(II)-mediated Takai olefination with (dichloromethyl)boronic ester 17. This afforded upper chain 11 in 69% yield with only the desired E-geometric isomer being detectable by 1H-NMR spectroscopic analysis.
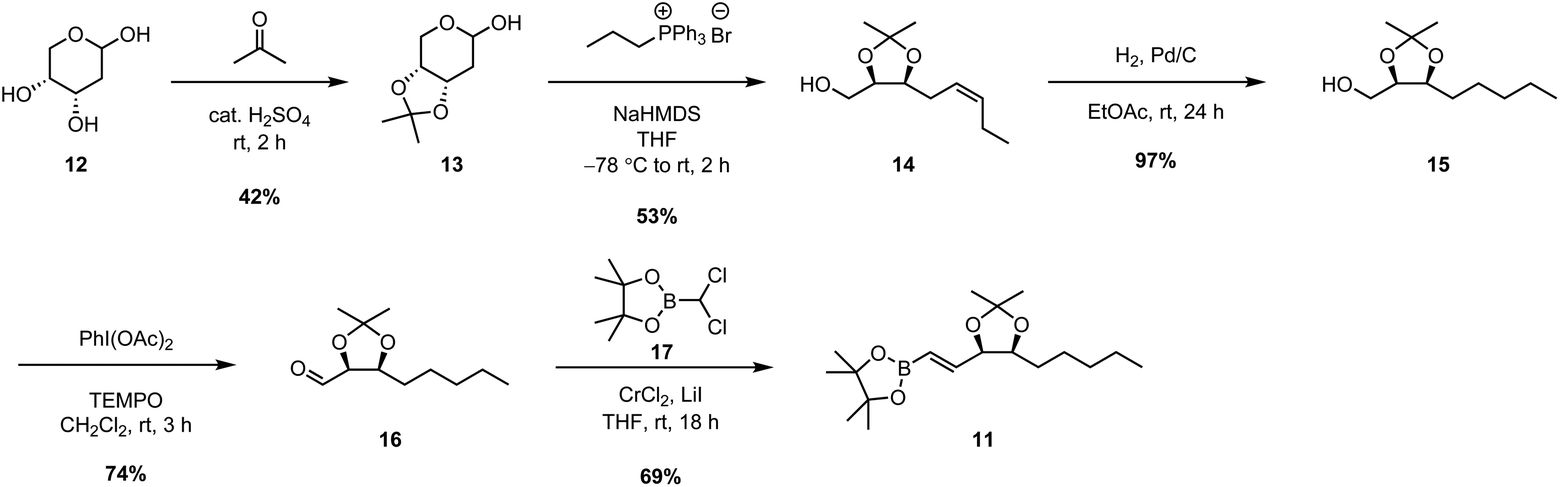 | ||
| Scheme 2 Synthesis of upper chain boronic ester 11. | ||
With the upper chain coupling partner in hand, we then turned our attention to completing the synthesis of our first target analogue, benzo-LXB4 (7b). We have previously reported the synthesis of this particular compound, albeit as the tert-butyl ester 7a, by employing a somewhat different strategy involving a Heck coupling as the key retrosynthetic disconnection.9 This work therefore represents an alternative approach to the target analogue. The δ-keto ester lower chain 20 was prepared in a single step from 1,2-dibromobenzene (18) using turbo-Grignard methodology developed by Knochel and co-workers.19 1,2-Dibromobenzene was first treated with a solution of i-PrMgCl·LiCl to induce Br/Mg exchange, and the resulting ArMgCl·LiCl species was then transmetallated to copper by addition of CuCN·2LiCl and subsequently trapped with acid chloride 19 to afford lower chain δ-keto ester 20 in 73% yield (Scheme 3). A Suzuki coupling was then attempted by heating the two coupling partners, 11 and 20, under microwave-irradiation in the presence of Pd(PPh3)4 and K2CO3. Pleasingly, these microwave-assisted conditions furnished the desired couple product 21 in an excellent 81% yield after only 1 hour at 125 °C. After this, we then sought to carry out a stereoselective reduction of the ketone via a ruthenium-catalyzed Noyori hydrogenation. Due to its ability to tolerate a large number of different heterocyclic ketones,20 we hoped this catalytic system would be the most general and reliable for the synthesis of our LXB4 analogues. The reduction of 21 was thus carried out under 20 bar of H2 gas in the presence of the catalyst, RuCl2[(R)-DM-BINAP][(R)-DAIPEN] which afforded the desired (S)-alcohol 22 in 38% yield after 24 h.
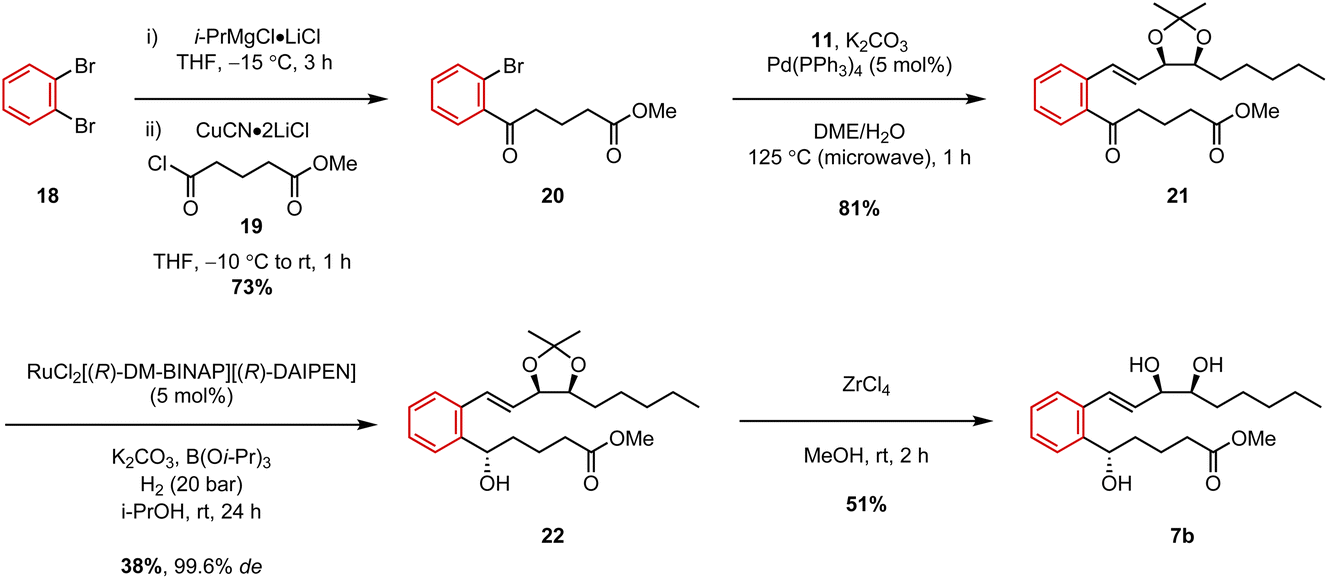 | ||
| Scheme 3 Synthesis of benzo-LXB4 (7b). | ||
Although the isolated yield was somewhat moderate, the diastereoselectivity was excellent with 22 being obtained in 99.6% de (99.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 dr) as determined by SFC analysis. We also observed that it was important to use K2CO3 as the base instead of KOt-Bu in order to avoid transesterification of the methyl ester with the i-PrOH solvent. The stereochemistry of the product was tentatively assigned as (S)-based on the transition state model associated with the (R,R)-Ru catalyst,21 but this assignment was not confirmed. Nevertheless, we have previously confirmed predictions made by the transition state model on multiple separate occasions by using both X-ray crystallography and Mosher's ester analysis, so we place high confidence in our stereochemical assignment of 22.11,12,18 Finally, to complete the synthesis, the acetonide protecting group was removed using ZrCl4 in MeOH and the target analogue 7b was obtained in 51% yield.
:0.2 dr) as determined by SFC analysis. We also observed that it was important to use K2CO3 as the base instead of KOt-Bu in order to avoid transesterification of the methyl ester with the i-PrOH solvent. The stereochemistry of the product was tentatively assigned as (S)-based on the transition state model associated with the (R,R)-Ru catalyst,21 but this assignment was not confirmed. Nevertheless, we have previously confirmed predictions made by the transition state model on multiple separate occasions by using both X-ray crystallography and Mosher's ester analysis, so we place high confidence in our stereochemical assignment of 22.11,12,18 Finally, to complete the synthesis, the acetonide protecting group was removed using ZrCl4 in MeOH and the target analogue 7b was obtained in 51% yield.
The successful synthesis of benzo-analogue 7b confirmed the viability of our synthetic strategy, so in order to further explore the generality of the route, we then turned our attention to synthesizing examples of heteroaromatic analogues of LXB4. Pyrido-LXB4 (8) was thus chosen as our first target heteroaromatic analogue (Scheme 4). The synthesis of pyridine lower chain ketone 26 was achieved in two straightforward steps. The first step involved a LiTMP-mediated deprotonation of 3-bromopyridine (23) at −85 °C followed by trapping of the resulting lithiated species with aldehyde 24. Provided that the temperature of the reaction was maintained below −80 °C, clean conversion to the desired product was observed and alcohol 25 was isolated in 60% yield following column chromatography.22 The second step was a DMP-mediated oxidation of the secondary alcohol which afforded lower chain ketone 26 in 78% yield. With both coupling partners in hand, we were pleased to find that the same conditions used to synthesize the benzo-analogue 7b could be extended to the synthesis of pyrido-analogue 8 with little difficulty. The microwave-assisted Suzuki coupling between lower chain 26 and upper chain 11 afforded coupled product 27 in an excellent 78% yield. After that, the stereoselective Noyori hydrogenation conditions converted ketone 27 to (S)-alcohol 28 in 57% yield. Once again, the stereoselectivity was excellent with 28 being obtained in 99.6% de as confirmed by SFC analysis. Finally, the ZrCl4/MeOH acetonide deprotection conditions afforded the target analogue, pyrido-LXB4 (8), in 62% yield. The synthesis of a second heteroaromatic target, quinoxalino-LXB4 (9) also proceeded as planned (Scheme 5). The quinoxaline lower chain 30 was prepared in one step from 2-chloroquinoxaline (29) and aldehyde 24via a cross-dehydrogenative coupling using conditions reported by Antonchick and co-workers.23 After that, the microwave-assisted Suzuki coupling between 11 and 30 furnished coupled ketone 31 in 75% yield, and the subsequent stereoselective Noyori hydrogenation led to the successful formation of (S)-alcohol 32 in 62% yield and excellent 99.6% de. However, a slight complication arose in the final step. It was found that ZrCl4/MeOH could not be used to remove the acetonide protecting group as these conditions led to decomposition of the starting material. Camphorsulfonic acid (CSA) in MeOH was therefore used instead to afford the target analogue, quinoxalino-LXB4 (9) in 81% yield.
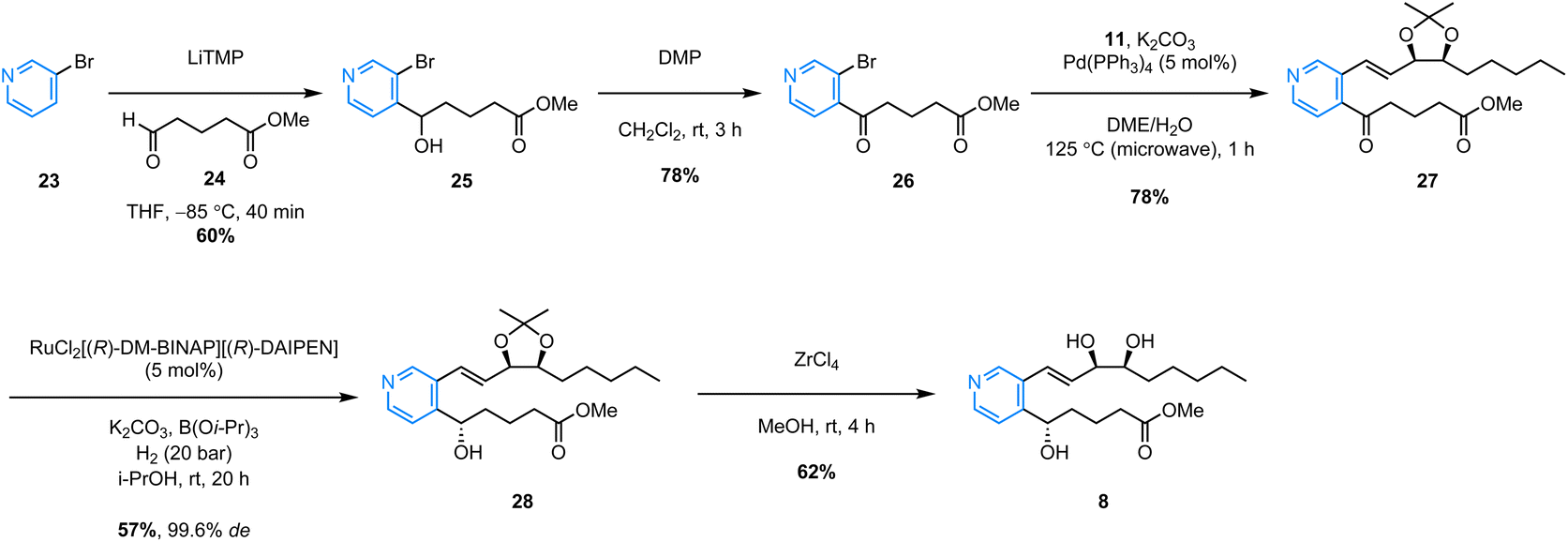 | ||
| Scheme 4 Synthesis of pyrido-LXB4 (8). | ||
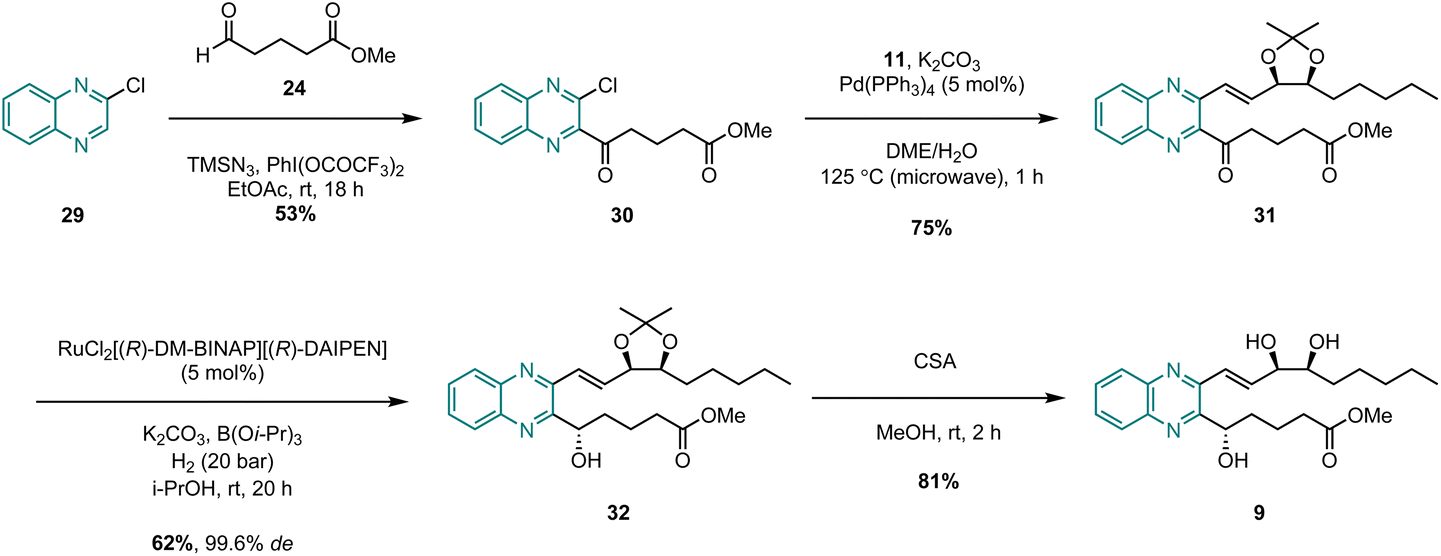 | ||
| Scheme 5 Synthesis of quinoxalino-LXB4 (9). | ||
The synthesis of a fourth and final LXB4 analogue based on a dimethylimidazole core was then investigated (Scheme 6). Imidazole lower chain 34 was prepared in a single step via addition of i-PrMgCl·LiCl to dibromoimidazole 33 followed by trapping of the resulting magnesiated intermediate with acid chloride 19. This led to regiospecific acylation at C-5 with no trace of any C-4 acylated product observed. However, the isolated yield of 34 was somewhat low due to competing protonation of the magnesiated intermediate. Protonation was typically observed in an equal ratio to acylation even when the reaction was conducted under an inert atmosphere with anhydrous THF. This proved difficult to avoid, presumably due to the presence of acidic α-protons in acid chloride 19. Nevertheless, despite the low yield, we believed the ability to rapidly access 34 in one step was still preferable to developing an alternative multi-step route. δ-Keto ester 34 was thus carried forward to the subsequent microwave-assisted Suzuki coupling with upper chain 11 to give the desired coupled product 35 in 68% yield. However, when a stereoselective Noyori hydrogenation of 35 was attempted in the presence of the (R,R)-Ru catalyst and 20 bar of H2 gas, no reaction was observed after 24 h. This was surprising because we had previously used these conditions to successfully carry out asymmetric reductions of related imidazole substrates.11,12,18
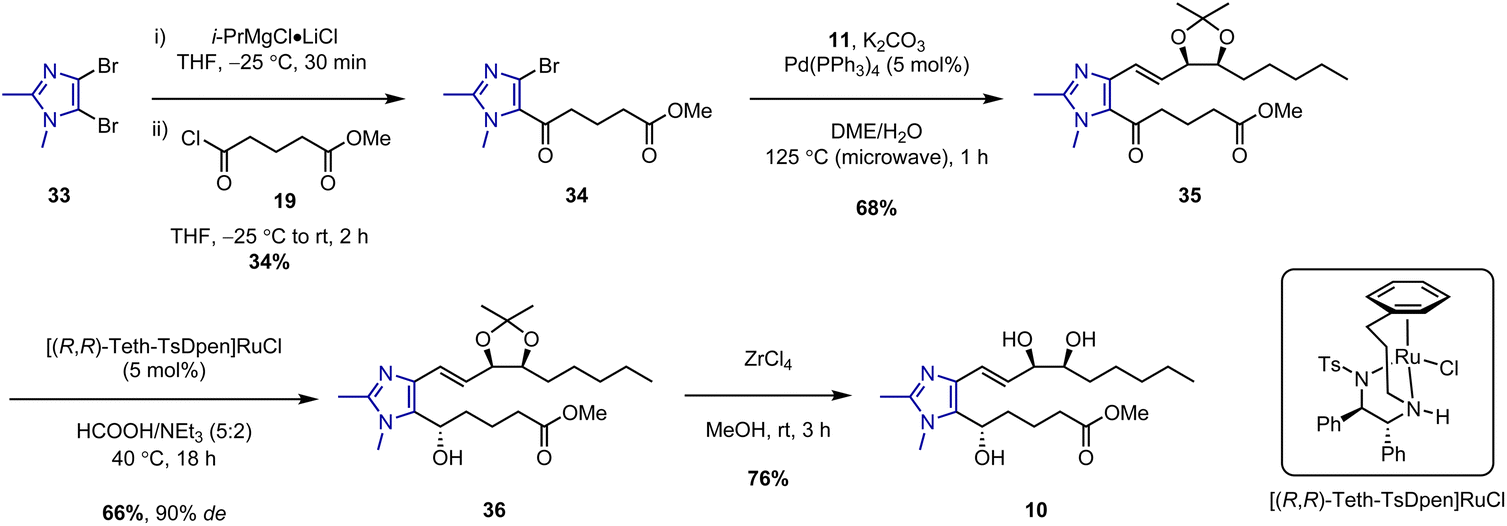 | ||
| Scheme 6 Synthesis of imidazolo-LXB4 (10). | ||
We therefore decided to use uncoupled ketone 34 as a model substrate to screen for suitable alternative stereoselective reduction conditions (Table 1). As with coupled ketone 35, lower chain 34 was also unreactive under the asymmetric Noyori hydrogenation conditions regardless of whether K2CO3 or KOt-Bu was used as the base (Table 1, entries 1 and 2). A number of other asymmetric reduction conditions were attempted, but all were unsuccessful (Table 1, entries 3–5).
|
|
|||
|---|---|---|---|
| Entry | Conditions | Yield | er |
| a Transesterification to the i-propyl ester observed. b Major enantiomer was the unexpected (R)-enantiomer. | |||
| 1 | RuCl2[(R)-DM-BINAP][(R)-DAIPEN] (5 mol%) | No reaction | n/a |
| K2CO3, B(Oi-Pr)3, i-PrOH, rt, 24 h, H2 (20 bar) | |||
| 2 | RuCl2[(R)-DM-BINAP][(R)-DAIPEN] (5 mol%) | No producta | n/a |
| KOt-Bu, B(Oi-Pr)3, i-PrOH, rt, 24 h, H2 (20 bar) | |||
| 3 | (−)-DIP-chloride, Et2O, −20 °C, 72 h | No reaction | n/a |
| 4 | (R)-CBS cat. (10 mol%), BH3·THF, THF, rt, 24 h | No reaction | n/a |
| 5 | RuCl[p-cymene][(S,S)-TsDpen] (5 mol%) | Trace | n/a |
| HCOOH/NEt3 (5:2), rt, 4 days |
|||
| 6 | [(S,S)-Teth-TsDpen]RuCl (5 mol%) | 46% | 22:78b |
| HCOOH/NEt3 (5:2), 40 °C, 24 h |
|||
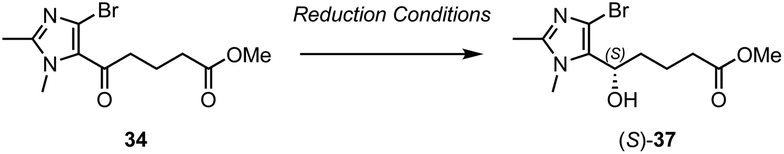
Eventually, a breakthrough was made when tethered transfer hydrogenation catalyst, [Teth-TsDpen]RuCl was identified. This catalytic system has been extensively studied by Wills and co-workers,24 and we were encouraged by their reports of its application to the reduction of similar imidazole-containing substrates.25 We thus reacted 34 with [(S,S)-Teth-TsDpen]RuCl in the presence of an azeotropic mixture of formic acid and triethylamine, and to our delight, complete conversion to alcohol 37 was observed after 24 h (Table 1, entry 6). However, despite this being the only catalytic system to result in the successful formation of the alcohol product, the enantioselectivity of the reaction was somewhat low (78:22 er). Additionally Mosher's ester analysis (ESI, Fig. S1 and S2†) was used to confirm that the major product was actually (R)-37, which is the opposite enantiomer to that predicted by the transition state model associated with ruthenium/(η6-arene)(TsDpen) catalysts of this type. The selectivity of the transfer hydrogenation is believed to be driven by a stabilizing C–H/π-intereaction in the transition state between an electron-deficient C–H bond on the η6-arene ligand and the aromatic substituent of the ketone.26,27 However, in this case, it is possible that the dimethyl-imidazole substituent is not sufficiently electron rich and too sterically encumbered to make this C–H/π-interaction favourable. Therefore, this would mean that the selectivity is instead driven by the avoidance of unfavourable steric interactions between the η6-arene ligand and the bulky imidazole substituent.
Based on the observation that [(S,S)-Teth-TsDpen]RuCl converted 34 preferentially to (R)-37, these transfer hydrogenation conditions were then repeated with coupled ketone 35 using the opposite hand of the catalyst, [(R,R)-Teth-TsDpen]RuCl, with the expectation that the desired (S)-epimer would be obtained as the major product. This reaction proceeded well, and the desired product 36 was obtained in 66% yield after 18 h. Pleasingly, only a single diastereomer was detectable in the 1H-NMR spectrum and SFC analysis confirmed that 36 was obtained in 90% de (95:5 dr). This showed much higher selectivity can be obtained when the stereoselective transfer hydrogenation is carried out on the more sterically hindered coupled ketone as opposed to the less sterically hindered uncoupled ketone 34. With 36 finally obtained in high diastereomeric purity, the synthesis of the target analogue could be completed. Acetonide deprotection of 36 was conducted using ZrCl4/MeOH to afford imidazolo-LXB4 (10) in 76% yield.
Conclusion
An efficient and modular synthesis of aromatic and heteroaromatic lipoxin B4 analogues has been developed, and a focused library of compounds was generated in order to demonstrate the robustness and generality of the synthetic strategy. In total, the asymmetric synthesis of four aromatic LXB4 analogues was successfully completed. This included a new synthesis of benzo-LXB4 as well as the first syntheses of LXB4 analogues containing a heterocyclic core. The heteroaromatic analogues that were successfully synthesized in this study were based on a pyridine, a dimethylimidazole and a quinoxaline heterocyclic core. However, the general principles of this synthetic strategy could, in theory, be extended to the synthesis of analogues containing a variety of other heterocyclic cores. We hope to report a detailed evaluation of the anti-inflammatory properties of each of the heteroaromatic LXB4 analogues synthesized in this study in due course. The results of these studies could then potentially form the basis of a larger study probing the structure–activity relationships of (hetero)aromatic mimetics of lipoxin B4 which would allow us to learn more about the important differences between lipoxin A4 and lipoxin B4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. O. is grateful for the award of an Irish Research Council Enterprise Partnership Scheme PhD. Scholarship (EPSPG/2019/529) with Enterprise Partner SK Biotek Ireland. The authors thank Dr Yannick Ortin (University College Dublin) for help with NMR spectroscopy and Dr Jimmy Muldoon (University College Dublin) for mass spectrometric analysis [supported by a Science Foundation Ireland Infrastructure Award (18/RI/5702)].References
- C. N. Serhan, Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology, Nature, 2014, 510(7503), 92–101, DOI:10.1038/nature13479.
- M. C. Basil and B. D. Levy, Specialized Pro-Resolving Mediators: Endogenous Regulators of Infection and Inflammation, Nat. Rev. Immunol., 2016, 16(1), 51–67, DOI:10.1038/nri.2015.4.
- B. McMahon and C. Godson, Lipoxins: Endogenous Regulators of Inflammation, Am. J. Physiol.: Renal Physiol., 2004, 286(2), F189–F201, DOI:10.1152/ajprenal.00224.2003.
- C. N. Serhan, Lipoxins and Aspirin-Triggered 15-Epi-Lipoxins Are the First Lipid Mediators of Endogenous Anti-Inflammation and Resolution, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2005, 73(3), 141–162, DOI:10.1016/j.plefa.2005.05.002.
- S. J. O'Meara, K. Rodgers and C. Godson, Lipoxins: Update and Impact of Endogenous pro-Resolution Lipid Mediators, in Reviews of Physiology, Biochemistry and Pharmacology, Springer, Berlin, Heidelberg, 2008, pp. 47–70. DOI:10.1007/112_2006_0606.
- C. D. Duffy and P. J. Guiry, Recent Advances in the Chemistry and Biology of Stable Synthetic Lipoxin Analogues, MedChemComm, 2010, 1(4), 249, 10.1039/c0md00136h.
- C. Godson, P. Guiry and E. Brennan, Lipoxin Mimetics and the Resolution of Inflammation, Annu. Rev. Pharmacol. Toxicol., 2023, 63(1), 429–448, DOI:10.1146/annurev-pharmtox-051921-085407.
- J. F. Maddox and C. N. Serhan, Lipoxin A4 and B4 Are Potent Stimuli for Human Monocyte Migration and Adhesion: Selective Inactivation by Dehydrogenation and Reduction, J. Exp. Med., 1996, 183(1), 137–146, DOI:10.1084/jem.183.1.137.
- T. P. O'Sullivan, K. S. A. Vallin, S. T. Ali Shah, J. Fakhry, P. Maderna, M. Scannell, A. L. F. Sampaio, M. Perretti, C. Godson and P. J. Guiry, Aromatic Lipoxin A4 and Lipoxin B4 Analogues Display Potent Biological Activities, J. Med. Chem., 2007, 50(24), 5894–5902, DOI:10.1021/jm060270d.
- C. D. Duffy, P. Maderna, C. McCarthy, C. E. Loscher, C. Godson and P. J. Guiry, Synthesis and Biological Evaluation of Pyridine-Containing Lipoxin A4 Analogues, ChemMedChem, 2010, 5(4), 517–522, DOI:10.1002/cmdc.200900533.
- M. de Gaetano, C. Tighe, K. Gahan, A. Zanetti, J. Chen, J. Newson, A. Cacace, M. Marai, A. Gaffney, E. Brennan, P. Kantharidis, M. E. Cooper, X. Leroy, M. Perretti, D. Gilroy, C. Godson and P. J. Guiry, Asymmetric Synthesis and Biological Screening of Quinoxaline-Containing Synthetic Lipoxin A4 Mimetics (QNX-SLXms), J. Med. Chem., 2021, 64(13), 9193–9216, DOI:10.1021/acs.jmedchem.1c00403.
- M. de Gaetano, E. Butler, K. Gahan, A. Zanetti, M. Marai, J. Chen, A. Cacace, E. Hams, C. Maingot, A. McLoughlin, E. Brennan, X. Leroy, C. E. Loscher, P. Fallon, M. Perretti, C. Godson and P. J. Guiry, Asymmetric Synthesis and Biological Evaluation of Imidazole- and Oxazole-Containing Synthetic Lipoxin A4 Mimetics (SLXms), Eur. J. Med. Chem., 2019, 162, 80–108, DOI:10.1016/j.ejmech.2018.10.049.
- C. Kim, I. Livne-Bar, K. Gronert and J. M. Sivak, Fair-Weather Friends: Evidence of Lipoxin Dysregulation in Neurodegeneration, Mol. Nutr. Food Res., 2020, 64(4), 1801076, DOI:10.1002/mnfr.201801076.
- M. Romano, J. F. Maddox and C. N. Serhan, Activation of Human Monocytes and the Acute Monocytic Leukemia Cell Line (THP-1) by Lipoxins Involves Unique Signaling Pathways for Lipoxin A4 versus Lipoxin B4: Evidence for Differential Ca2+ Mobilization, J. Immunol., 1996, 157(5), 2149–2154, DOI:10.4049/jimmunol.157.5.2149.
- O. Corminboeuf and X. Leroy, FPR2/ALXR Agonists and the Resolution of Inflammation, J. Med. Chem., 2015, 58(2), 537–559, DOI:10.1021/jm501051x.
- L. Trippe, A. Nava, A. Frank and U. Nubbemeyer, Synthesis of Enantiopure 6,11-Methylene Lipoxin B4 Methyl Ester, Eur. J. Org. Chem., 2021,(7), 1156–1167, DOI:10.1002/ejoc.202001591.
- L. Trippe, A. Nava, A. Frank, D. Schollmeyer and U. Nubbemeyer, Synthesis of Enantiopure ω-(4-Fluorophenyl)-6,11-Methylene Lipoxin B4 Methyl Ester, Synthesis, 2021,(20), 3760–3768, DOI:10.1055/a-1512-1763.
- B. Owen, M. de Gaetano, A. Gaffney, C. Godson and P. J. Guiry, Synthesis and Biological Evaluation of Bicyclo[1.1.1]Pentane-Containing Aromatic Lipoxin A4 Analogues, Org. Lett., 2022, 24(32), 6049–6053, DOI:10.1021/acs.orglett.2c02345.
- A. Krasovskiy and P. Knochel, A LiCl-Mediated Br/Mg Exchange Reaction for the Preparation of Functionalized Aryl- and Heteroarylmagnesium Compounds from Organic Bromides, Angew. Chem., Int. Ed., 2004, 43(25), 3333–3336, DOI:10.1002/anie.200454084.
- T. Ohkuma, M. Koizumi, M. Yoshida and R. Noyori, General Asymmetric Hydrogenation of Hetero-Aromatic Ketones, Org. Lett., 2000, 2(12), 1749–1751, DOI:10.1021/ol0000814.
- C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, Mechanism of Asymmetric Hydrogenation of Ketones Catalyzed by BINAP/1,2-Diamine–Ruthenium(II) Complexes, J. Am. Chem. Soc., 2003, 125(44), 13490–13503, DOI:10.1021/ja030272c.
- A. I. Subota, A. O. Lutsenko, B. V. Vashchenko, D. M. Volochnyuk, V. Levchenko, Y. V. Dmytriv, E. B. Rusanov, A. O. Gorlova, S. V. Ryabukhin and O. O. Grygorenko, Scalable and Straightforward Synthesis of All Isomeric (Cyclo)Alkylpiperidines, Eur. J. Org. Chem., 2019,(22), 3636–3648, DOI:10.1002/ejoc.201900450.
- K. Matcha and A. P. Antonchick, Metal-Free Cross-Dehydrogenative Coupling of Heterocycles with Aldehydes, Angew. Chem., Int. Ed., 2013, 52(7), 2082–2086, DOI:10.1002/anie.201208851.
- H. G. Nedden, A. Zanotti-Gerosa and M. Wills, The Development of Phosphine-Free “Tethered” Ruthenium(II) Catalysts for the Asymmetric Reduction of Ketones and Imines, Chem. Rec., 2016, 16(6), 2623–2643, DOI:10.1002/tcr.201600084.
- Y. Zheng, J. A. Martinez-Acosta, M. Khimji, L. C. A. Barbosa, G. J. Clarkson and M. Wills, Asymmetric Transfer Hydrogenation of Aryl Heteroaryl Ketones Using Noyori-Ikariya Catalysts, ChemCatChem, 2021, 13(20), 4384–4391, DOI:10.1002/cctc.202101027.
- R. Noyori, M. Yamakawa and S. Hashiguchi, Metal–Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds, J. Org. Chem., 2001, 66(24), 7931–7944, DOI:10.1021/jo010721w.
- A. Matsuoka, C. A. Sandoval, M. Uchiyama, R. Noyori and H. Naka, Why P-Cymene? Conformational Effect in Asymmetric Hydrogenation of Aromatic Ketones with a H6-Arene/Ruthenium(II) Catalyst, Chem. – Asian J., 2015, 10(1), 112–115, DOI:10.1002/asia.201402979.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and spectra of all compounds. See DOI: https://doi.org/10.1039/d3ob01076g |
| This journal is © The Royal Society of Chemistry 2023 |