
Liensinine perchlorate inhibits colorectal cancer tumorigenesis by inducing mitochondrial dysfunction and apoptosis†
 *a
and
*a
and
Abstract
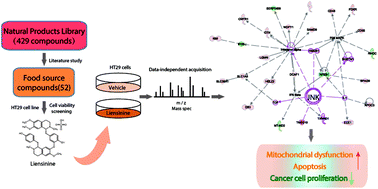
Scope: Colorectal cancer (CRC) is one of the most common cancers worldwide with poor survival and limited therapeutic options, and there is an urgent need to develop novel therapeutic agents with good treatment efficiency and low toxicity. This study aims to examine the anticancer bioactivity of liensinine, a constituent of Nelumbo nucifera Gaertn, in CRC and investigate the action mechanisms involved. Methods and results: Liensinine was found to induce apoptosis and exert a significant inhibitory effect on the proliferation and colony-forming ability of CRC cells in a dose-dependent manner without any observed cytotoxicity on normal colorectal epithelial cells. Mechanistically, our data from quantitative proteomics, western blot analysis and flow cytometry analyses demonstrated that exposure of CRC cells to liensinine caused cell cycle arrest, mitochondrial dysfunction and apoptosis, accompanied by the activation of the JNK signaling pathway. Furthermore, animal experiments showed that liensinine markedly suppressed the growth of CRC tumor xenografts in nude mice by reducing the Ki-67 proliferation index, but did not damage the vital organs of the animals. Conclusion: This study demonstrated for the first time that liensinine, a food-source natural product, could be a novel therapeutic strategy for treating CRC without obvious side effects.
 Please wait while we load your content...
Please wait while we load your content...

