
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLactobacillus plantarum LC27 and Bifidobacterium longum LC67 mitigate alcoholic steatosis in mice by inhibiting LPS-mediated NF-κB activation through restoration of the disturbed gut microbiota
Won-Gyeong
Kim
a,
Hye In
Kim
a,
Eun Kyung
Kwon
b,
Myung Joo
Han
*a and
Dong-Hyun
Kim
 *b
*b
aDepartment of Life and Nanopharmaceutical Sciences and Department of Pharmacy, Kyung Hee University, 26, Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Korea. E-mail: mjhan@khu.ac.kr; Fax: +82-2-961-0260
bDepartment of Food and Nutrition, Kyung Hee University, 26, Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Korea. E-mail: dhkim@khu.ac.kr; Fax: +82-2-957-5030
First published on 16th July 2018
Abstract
Long-term exposure to ethanol simultaneously causes gastrointestinal inflammation, liver injury, and steatosis. In the present study, we investigated the effects of Bifidobacterium longum LC67, Lactobacillus plantarum LC27, and their mixture (LM) against ethanol-induced steatosis in mice. Exposure to ethanol caused liver damage: it increased ALT, AST, TG, TC, and lipopolysaccharide levels in the blood and induced NF-κB activation in the liver. Oral administration of LC27, LC67, or LM in mice reduced ethanol-induced ALT, AST, TG, and TC levels in the blood and liver. These also suppressed ethanol-induced NF-κB activation and α-smooth muscle actin expression in the liver and increased ethanol-suppressed AMPK activation. Treatment with LC27, LC67, or LM increased ethanol-suppressed alcohol dehydrogenase and acetaldehyde dehydrogenase activities in the liver, as well as tight junction protein expression in the liver and colon. Moreover, treatment with LC27, LC67, or LM restored the ethanol-disturbed gut microbiota composition, such as the increased population of Proteobacteria, and inhibited fecal and blood lipopolysaccharide levels. These inhibited NF-κB activation and increased tight junction protein expression in ethanol- or lipopolysaccharide-stimulated Caco-2 cells. These findings suggest that LC27, LC67, and LM can alleviate alcoholic steatosis by inhibiting LPS-mediated NF-κB activation through restoration of the disturbed gut microbiota.
1. Introduction
Exposure to ethanol remains a main risk factor in chronic liver disease.1 Alcoholic liver damage, ranging from simple hepatic steatosis to serious liver damage such as fibrosis and cirrhosis, results from chronic ethanol abuse or binge drinking.2 Chronic exposure to ethanol increases oxidative stress, endotoxin, and inflammatory cytokines in the blood and liver and disturbs the gut microbiota composition and gut membrane function, leading to liver damage including liver steatosis, fibrosis, and cirrhosis.3,4 Ethanol is metabolized to acetate via acetaldehyde and produces reactive oxygen species, which stimulates inflammation.5–7 Moreover, the intake of ethanol disturbs the gut microbiota composition and increases bacterial endotoxin production.8–10 Excessive exposure to endotoxins can cause inflammation in the gastrointestinal tract (e.g., colitis), and disruption in gut wall functions; this further accelerates the absorption of intestinal bacterial by-products, including endotoxins, into the blood.10,11 Excessive absorption of endotoxins such as lipopolysaccharide (LPS) into the blood causes inflammation in the liver, and deteriorates non-alcoholic and alcoholic liver diseases including steatosis, hepatitis, and hepatic fibrosis.10,12,13Lactic acid bacteria are frequently used in reducing the disease risks and increasing the quality of life of patients.14,15 Of these, lactobacilli and bifidobacteria suppress the proliferation of pathogens,16 increase the immune function of the host,17 attenuate metabolic diseases,18,19 and alleviate alcohol-induced liver damage.8,20,21Lactobacillus rhamnosus CCFM1107 attenuates liver injury in ethanol-treated mice by reducing oxidative stress and restoring intestinal microflora.20Lactobacillus rhamnosus GG alleviated liver injury in ethanol-treated mice by increasing miR122a expression.22Lactobacillus brevis SBC8803 attenuates liver damage in ethanol-treated mice by inhibiting proinflammatory cytokine expression.23Bifidobacterium longum LC67 and Lactobacillus plantarum LC27 attenuated acute ethanol-induced gastritis and liver injury in mice by inhibiting NF-κB activation and IL-8 expression.21 Some lactic acid bacteria including Lactobacillus rhamnosus GG attenuate ethanol-induced steatosis.24 However, the inhibitory mechanisms of bifidobacteria and lactobacilli including LC27 and LC67 against ethanol-induced steatosis have remained elusive.
Therefore, we examined the effects of LC27 and LC67, which inhibited NF-κB activation and TNF-α expression in ethanol-stimulated Kupffer cells and LPS-stimulated macrophages,21 against alcoholic steatosis in mice.
2. Materials and methods
2.1. Materials
RPMI1640 and LPS purified from Escherichia coli O111:B4 were purchased from Sigma (St Louis, MO, USA). Antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Radioimmunoprecipitation assay (RIPA) lysis buffer, phosphatase inhibitor cocktail, and protease inhibitor cocktail were purchased from Invitrogen (Carlsbad, CA, USA). Enzyme-linked immunosorbent assay (ELISA) kits for cytokines were purchased from Ebioscience (San Diego, AC, USA). Triglyceride (TG), total cholesterol (TC), low-density lipoprotein (LDL) cholesterol, alanine aminotransferase (ALT) and aspartate transaminase (AST) assay kits were purchased from Asan Co. (Seoul, Korea).2.2. Preparation of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67
LC27 and LC67 were grown in general media for probiotics. Briefly, these probiotics were grown to the density of 2–4 × 109 CFU mL−1 and centrifuged to harvest cells. Collected bacterial cells (2 × 109 CFU per 0.2 mL) were suspended in phosphate buffered saline (PBS) or 1% glucose. Cells suspended in PBS were inactivated at 80 °C for 30 min and used for cell experiments. Cells suspended in 1% dextrose were used for in vivo experiment.2.3. Culture of Caco-2 cells
Caco-2 cells (Korean Cell Line Bank, Seoul, Korea) were cultured in RPMI 1640 (Sigma, St Louis, MO) containing 1% antibiotic–antimycotic and 10% fetal bovine serum (RAF) at 37 °C.25 For the assay of NF-κB activation and tight junction protein expression, cells (2 × 105 cells per mL) were treated with LPS (100 ng mL−1) or ethanol (50 μL mL−1) in the absence or presence of LC27 or LC67 (1 × 103 or 1 × 105 cells per mL) for 90 min (for NF-κB activation) or 20 h (for tight junction, iNOS, and COX-2 proteins). Proteins were measured by immunoblotting.2.4. Animals
Male ICR mice (21–23 g, 7 weeks old) were purchased from the Orient Co., Ltd, a branch of Charles River Laboratories (Gyeonggi-do, Korea). The mice were housed in a wire cage under controlled conditions (temperature, 20–22 °C; and humidity, 50 ± 10%) and fed standard laboratory chow and water. The experiment was ethically approved by the Committee for the Care and Use of Laboratory Animals of Kyung Hee University (IRB No, KHUASP(SE)-17-013) and performed in accordance with the Kyung Hee University Guidelines for Laboratory Animals Care and Usage.2.5. Induction of ethanol-induced steatosis in mice
Ethanol-induced steatosis was induced in mice according to the modified method of Chen et al.26 The mice were randomly divided into eight groups. Each group consisted of 7 mice. Briefly, the mice were sequentially fed Lieber-DeCarli (LB) diet (DYET#710260, Dyets, Inc., Bethlehem, PA. USA) containing 1% ethanol for one day, 2% ethanol for 4 days, and 5% ethanol for 10 days and gavaged 30% ethanol (5.0 g kg−1, dissolved in saline) on day 16. Test agents (group 2; vehicle [1% dextrose] alone; group 3, silymarin [100 mg kg−1]; group 4, 5 × 108 CFU of LC27 per mouse; group 5, 2 × 109 CFU of LC27 per mouse; group 6, 5 × 108 CFU of LC67 per mouse; group 7, 2 × 109 CFU of LC67 per mouse; or group 8, their mixture [LM, the mixture of 1 × 109 CFU of LC27 per mouse and 1 × 109 CFU of LC27]) were orally administered for 5 days (from day 7 after the initial treatment with 5% ethanol-containing LB diet). The final test agents were treated 1 h before the oral gavage of ethanol. The normal control group (group 1) was fed with normal LB diet (Dyet#710027) instead of the ethanol-containing LB diet and 1% glucose instead of test agents (suspended in 1% glucose). The mice were sacrificed 15 h after the final administration of test agents, and whole-blood samples were immediately withdrawn from the carotid artery. Serum was prepared by centrifugation (10 min, 250g) and TG, TC, LDL cholesterol, HDL cholesterol, ALT, AST, and γ-GTP levels were determined according to the methods of Kwon et al.21 Liver and colon tissues were removed for histological examination, ELISA, and immunoblotting.2.6. Histological examination
For the histological examination, the livers were fixed in 4% paraformaldehyde, frozen in optimal cutting temperature solution, cut into 10 μm sections using a cryostat (Leica, Nussloch, Germany), stained with hematoxylin–eosin or oil red O, and observed under a light microscope.2.7. Tissue preparation
Excised liver and colon tissues of the mice were perfused with ice-cold perfusion solution (0.15 M KCl, 2 mM EDTA, pH 7.4), homogenized in 50 mM Tris–HCl buffer (pH 7.4), and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and 4 °C for 30 min21 The resulting supernatants were used for ELISA and immunoblotting.
000g and 4 °C for 30 min21 The resulting supernatants were used for ELISA and immunoblotting.
2.8. ELISA and immunoblotting
For ELISA, the supernatant of tissues was transferred to 96-well ELISA plates. Tumor necrosis factor (TNF)-α concentrations were determined, using commercial ELISA kits.21,25 For immunoblotting, the supernatant of tissues (20 μg protein) was separated by 10% SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidenedifluoride membranes, and then hybridized overnight, at 4 °C with 1:1000 diluted AMP-activated protein kinase (AMPK), p-AMPK, inducible NO synthase (iNOS), p65, p-p65, and β-actin antibodies. The membrane was then incubated with 1:2000 diluted anti-rabbit or anti-mouse immunoglobulin G secondary antibody for 1 h at room temperature and washed 3 times with phosphate buffered saline containing 0.1% Tween 20 for 10 min. Protein bands were visualized with an enhanced chemiluminescence reagent.
2.9. Determination of malondialdehyde and nitric oxide
Malondialdehyde levels of tissues were assayed according to the method of Lee et al.27 The reaction solution containing 8.1% sodium dodecyl sulfate, 20% acetate buffer (pH 3.5), and thiobarbituric acid was mixed well with 0.2 mL tissue supernatant for 3 min, incubated at 95 °C for 60 min, cooled on ice for 5 min, and extracted with 1 mL distilled water and 2.5 mL of an n-butanol:pyridine mixture (15:1, v/v). Supernatants were measured at 532 nm. Nitrite levels were measured in the tissue supernatant according to the method of Lee et al.27
2.10. Determination of blood and colonic fluid LPS
LPS levels in sera and feces were determined using diazo-coupled LAL assays according to the manufacturer's protocol.28 Briefly, feces collected from the mice (100 mg) were placed in 50 mL of PBS in a pyrogen-free tube, sonicated for 1 h on ice, and centrifuged at 400g for 10 min. Supernatants were collected and sterilized by filtration through a 0.45 μm filter, followed by re-filtration through a 0.22 μm filter. The fecal supernatant and sera (5 μL) were diluted 1:10 in pyrogen-free water and heated at 70 °C for 10 min. LPS levels in the heated fecal and sera samples were assayed using the LAL Assay kit.
2.11. Quantitative real time-polymerase chain reaction (qPCR)
Real-time PCR for Firmicutes, Bacteroidetes, Actinobacteria, and δ/γ-Proteobacteria was performed with 100 ng total DNA isolated from the colon fluid using a Takara thermal cycler and SYBR premix reagents.29 The thermal cycling conditions were as follows: 95 °C for 30 s, followed by 35 cycles of denaturation and amplification at 95 °C for 5 s and 63 °C for 30 s, respectively. Gene expression levels were calculated relative to bacterial rRNA, using Microsoft Excel. Primers were used as follows: 16 s rRNA forward, 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG GTG CCA GCM GCC GCG GTA A-3′ and reverse, 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGG ACT ACH VGG GTW TCT AAT-3′; Firmicutes forward, 5′-GGA GYA TGT GGT TTA ATT CGA AGC A-3′ and reverse, 5′-AGC TGA CGA CAA CCA TGC AC-3′; Bacteroidetes forward, 5′-AAC GCG AAA AAC CTT ACC TAC C-3′ and reverse, 5′-TGC CCT TTC GTA GCA ACT AGT G-3′; Actinobacteria forward, 5′-TGT AGC GGT GGA ATG CGC-3′ and reverse, 5′-AAT TAA GCC ACA TGC TCC GCT-3′; δ/γ-Proteobacteria forward, 5′-GCT AAC GCA TTA AGT RYC CCG-3′ and reverse, 5′-GCC ATG CRG CAC CTG TCT-3′.2.12. Statistical analysis
Data are indicated as mean ± SD. Significant differences were analyzed using one-way ANOVA followed by Duncan's multiple range test (P < 0.05).3. Results
3.1. Effects of LC27 and LC67 on ethanol-induced steatosis in mice
To understand whether LC27 and LC67 could attenuate ethanol-induced steatosis, we induced steatosis in mice by sequential feeding of LB diet containing 1–5% ethanol and gavaging ethanol and investigated the effects of LC27, LC67, or LM against ethanol-induced steatosis. Ethanol intake significantly increased TG, TC, and LDL-cholesterol levels, as well as ALT, AST, and γ-GTP levels in the blood (Fig. 1). Furthermore, ethanol treatment increased TNF-α and LPS levels in the blood. The oral administration of LC27, LC67, or LM suppressed ethanol-induced TG, TC, LDL-cholesterol, ALT, AST, γ-GTP, TNF-α, and LPS levels while their treatments increased HDL-cholesterol levels. The oral administration of LC67 at a dose of 2 × 109 CFU per mouse most potently reduced ALT, AST, γ-GTP, and LPS levels. Its inhibitory effect is comparable to those treated with silymarin (50 mg kg−1), a positive agent. When LC27 and LC67 were combined at 1:1, 3:1, and 9:1 ratios, their effects were not different.
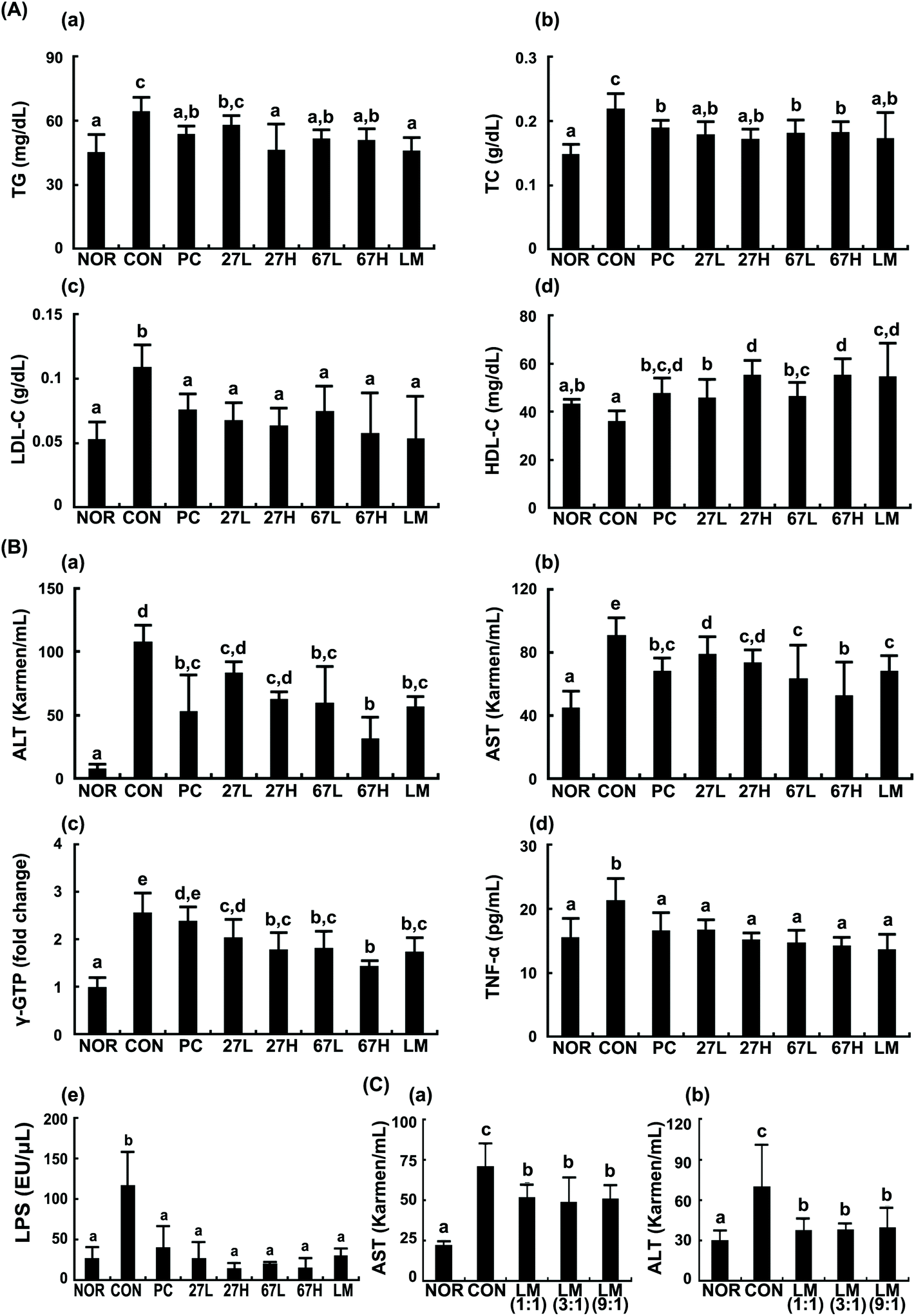 | ||
| Fig. 1 Effects of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 on the ethanol-induced steatosis and liver damage in mice. (A) Effects on the triglyceride (TG, a), total cholesterol (TC, b), LDL-cholesterol (LDL-C, c), and HDL-cholesterol (HDL-C, d) levels in the blood. (B) Effects on the AST (a), AST (b), γ-GTP (c), TNF-α (d), and LPS € levels in the blood. (C) Effects of LC27 and LC67 mixtures (1:1, 3:1, and 9:1) on AST (a) and ALT (b) levels. Test agents (CON, vehicle alone; PC, 50 mg kg−1 of silymarin; 27L, 5 × 108 CFU per mouse of LC27; 27H, 2 × 109 CFU per mouse of LC27; 67L, 5 × 108 CFU per mouse of LC27; 67H, 2 × 109 CFU per mouse of LC27; LM, the (1:1, 3:1, or 9:1) mixtures of LC27 and LC67 [2 × 109 CFU per mouse]) were orally administered once a day for 5 days. The mice were sacrificed 15 h after the final administration of test agents. The normal control group (NOR) was treated with saline instead of ethanol and 1% dextrose instead of test agents. All data are represented as mean ± SD (n = 7). Means with the same letters are not significantly different (p < 0.05). | ||
Treatment with ethanol also increased TG, TC, and LDL-cholesterol levels and reduced ADH and ALDH activities in the liver (Fig. 2). Treatment with LC27, LC67, or LM suppressed ethanol-induced TG, TC, LDL-cholesterol, ALT, and AST levels and increased ethanol-suppressed ADH and ALDH activities. The oral administration of LC27 at a dose of 2 × 109 CFU per mouse most potently decreased TG, TC, and LDL-cholesterol levels and increased HDL-cholesterol levels. Moreover, ethanol intake induced the expression of α-smooth muscle actin (α-SMA), which plays an important role in fibroblast contractility,31 while treatment with LC27, LC67, or LM suppressed ethanol-induced α-SMA expression.
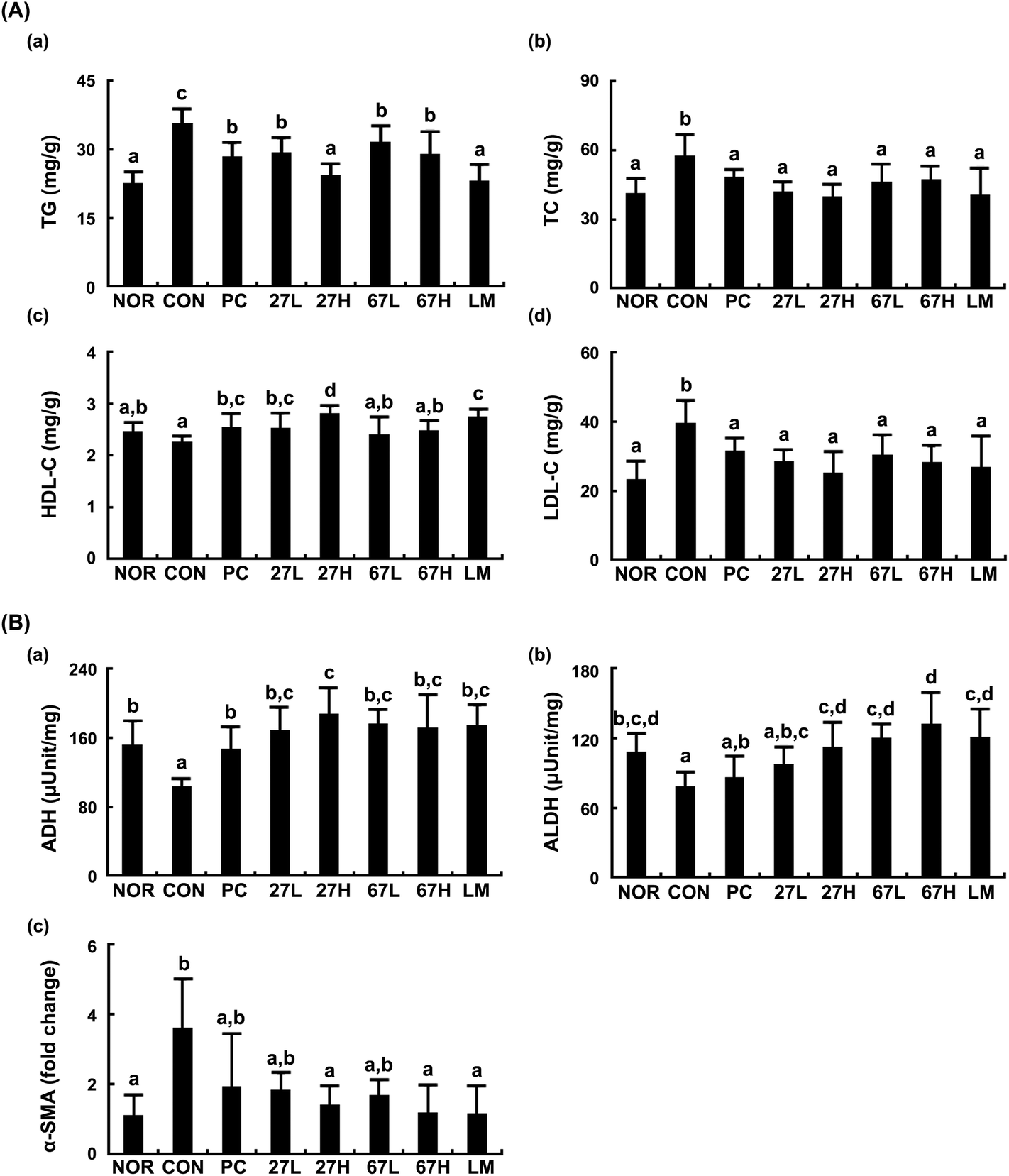 | ||
| Fig. 2 Effects of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 on the ethanol-induced steatosis and liver damage in mice. (A) Effects on the TG (a), TC (b), HDL-C (c), and LDL-C (d) levels in the liver. (B) Effects on the ADH (a) and ALDH activities (b) in the liver. (C) Effects on the α-smooth muscle actin (α-SMA), assessed by qPCR. Test agents (CON, vehicle alone; PC, 50 mg kg−1 of silymarin; 27L, 5 × 108 CFU per mouse of LC27; 27H, 2 × 109 CFU per mouse of LC27; 67L, 5 × 108 CFU per mouse of LC27; 67H, 2 × 109 CFU per mouse of LC27; LM, the (1:1 mixture of LC27 and LC67 [2 × 109 CFU per mouse]) were orally administered once a day for 5 days. The mice were sacrificed 15 h after the final administration of test agents. The normal control group (NOR) was treated with saline instead of ethanol and 1% dextrose instead of test agents. All data are represented as mean ± SD (n = 7). Means with the same letters are not significantly different (p < 0.05). | ||
Ethanol intake increased myeloperoxidase activity and TNF-α, NO, and malondialdehyde levels in the liver (Fig. 3). Histological examination showed that ethanol treatment accelerated fat accumulation in the liver. However, the oral administration of LC27, LC67, or LM significantly suppressed ethanol-induced TNF-α, NO, and malondialdehyde levels, myeloperoxidase activity, and fat accumulation. Particularly, the oral administration of LC27 at a dose of 2 × 109 CFU per mouse most potently inhibited myeloperoxidase activity. Ethanol intake induced iNOS, and COX-2 expression and NF-κB activation in the liver and suppressed the expression of tight junction proteins occludin and caludin-1 and the activation of AMPK, which stimulates hepatic fatty acid oxidation and inhibits cholesterol synthesis, lipogenesis, and triglyceride synthesis,30 while treatment with LC27, LC67, or LM suppressed iNOS, COX-2 expression and NF-κB activation and increased AMPK activation (Fig. 4). Moreover, treatment with LC27, LC67, or LM suppressed ethanol-induced α-SMA expression.
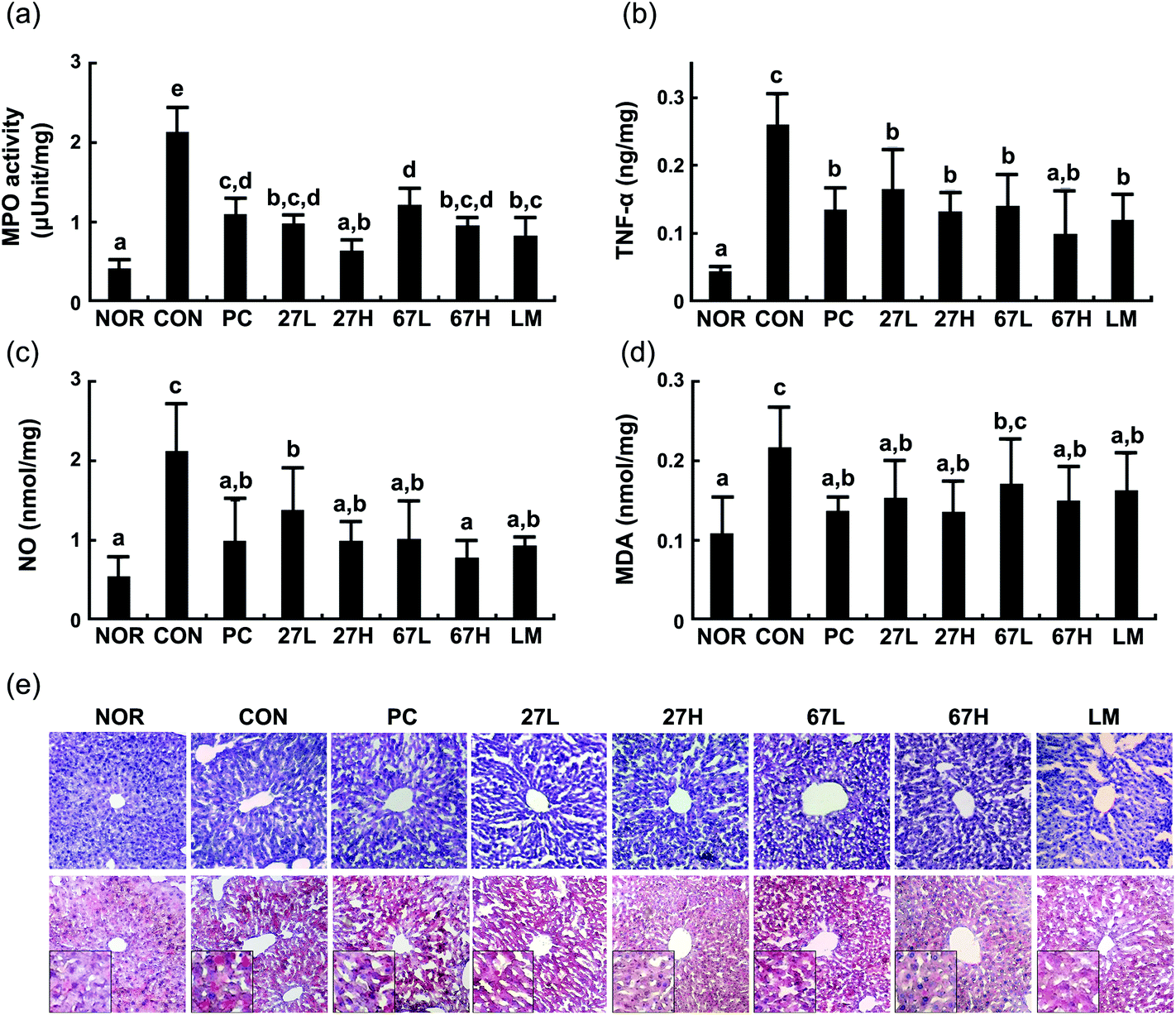 | ||
| Fig. 3 Effects of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 on the myeloperoxidase activity (a), TNF-α (b), NO (c), malondialdehyde (MDA, d) levels, and histological examination (e) in the liver of the mice with ethanol-induced steatosis. Test agents (CON), vehicle alone; PC, 50 mg kg−1 of silymarin; 27L, 5 × 108 CFU per mouse of LC27; 27H, 2 × 109 CFU per mouse of LC27; 67L, 5 × 108 CFU per mouse of LC27; 67H, 2 × 109 CFU per mouse of LC27; LM, the (1:1 mixture of LC27 and LC67 [2 × 109 CFU per mouse]) were orally administered once a day for 5 days. The mice were sacrificed 15 h after the final administration of test agents. The normal control group (NOR) was treated with saline instead of ethanol and 1% dextrose instead of test agents. All data are represented as mean ± SD (n = 7). Means with the same letters are not significantly different (p < 0.05). | ||
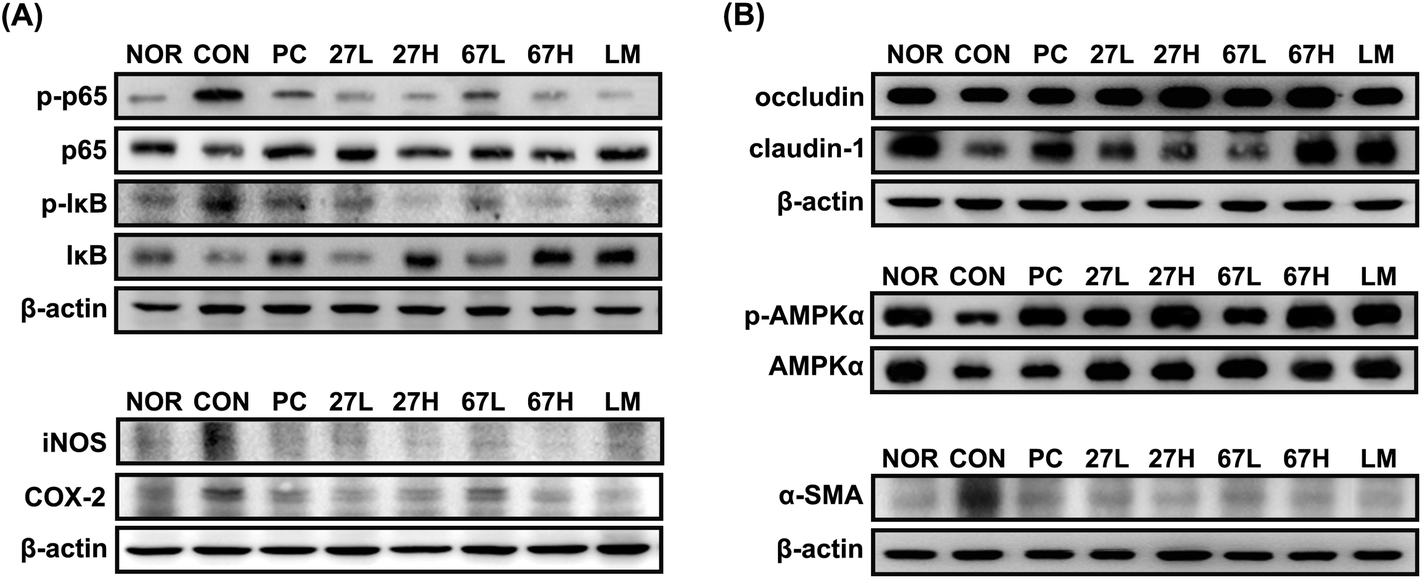 | ||
| Fig. 4 Effects of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 on the NF-κB activation and expression of iNOS, and COX-2 (A), phosphorylation of AMPK, and expression of tight junction proteins (occludin and claudin-1) and α-SMA (B) in the liver of mice with ethanol-induced steatosis. Test agents (CON), vehicle alone; PC, 50 mg kg−1 of silymarin; 27L, 5 × 108 CFU per mouse of LC27; 27H, 2 × 109 CFU per mouse of LC27; 67L, 5 × 108 CFU per mouse of LC27; 67H, 2 × 109 CFU per mouse of LC27; LM, the (1:1 mixture of LC27 and LC67 [2 × 109 CFU per mouse]) were orally administered once a day for 5 days. The mice were sacrificed 15 h after the final administration of test agents. Proteins were analyzed by immunoblotting. The normal control group (NOR) was treated with saline instead of ethanol and 1% dextrose instead of test agents. | ||
3.2. Effects of LC27 and LC67 on ethanol-induced gastrointestinal (GI) inflammation in mice
Next, we investigated whether LC27, LC67, or LM could attenuate ethanol-induced gastrointestinal inflammation. Ethanol intake significantly induced gastrointestinal inflammation: it increased the myeloperoxidase activity in the stomach, small intestine, and colon, as well as distinctive signs of hemorrhagic gastritis, like the previous report26 (Fig. 5). These pathologies were not observed in the normal control group treated with the vehicle alone. The oral administration of LC27, LC67, or LM significantly suppressed the ethanol-induced GI inflammation: they suppressed the myeloperoxidase activity in the stomach, duodenum, and colon, as well as the distinctive signs of colitis. Treatment with LC27 (2 × 109 CFU per mouse) most potently inhibited the myeloperoxidase activity in the GI tract, as observed in the liver. Moreover, treatment with LC27, LC67, or LM suppressed ethanol-induced NF-κB activation and TNF-α expression, as well as increased the ethanol-suppressed expression of tight junction proteins.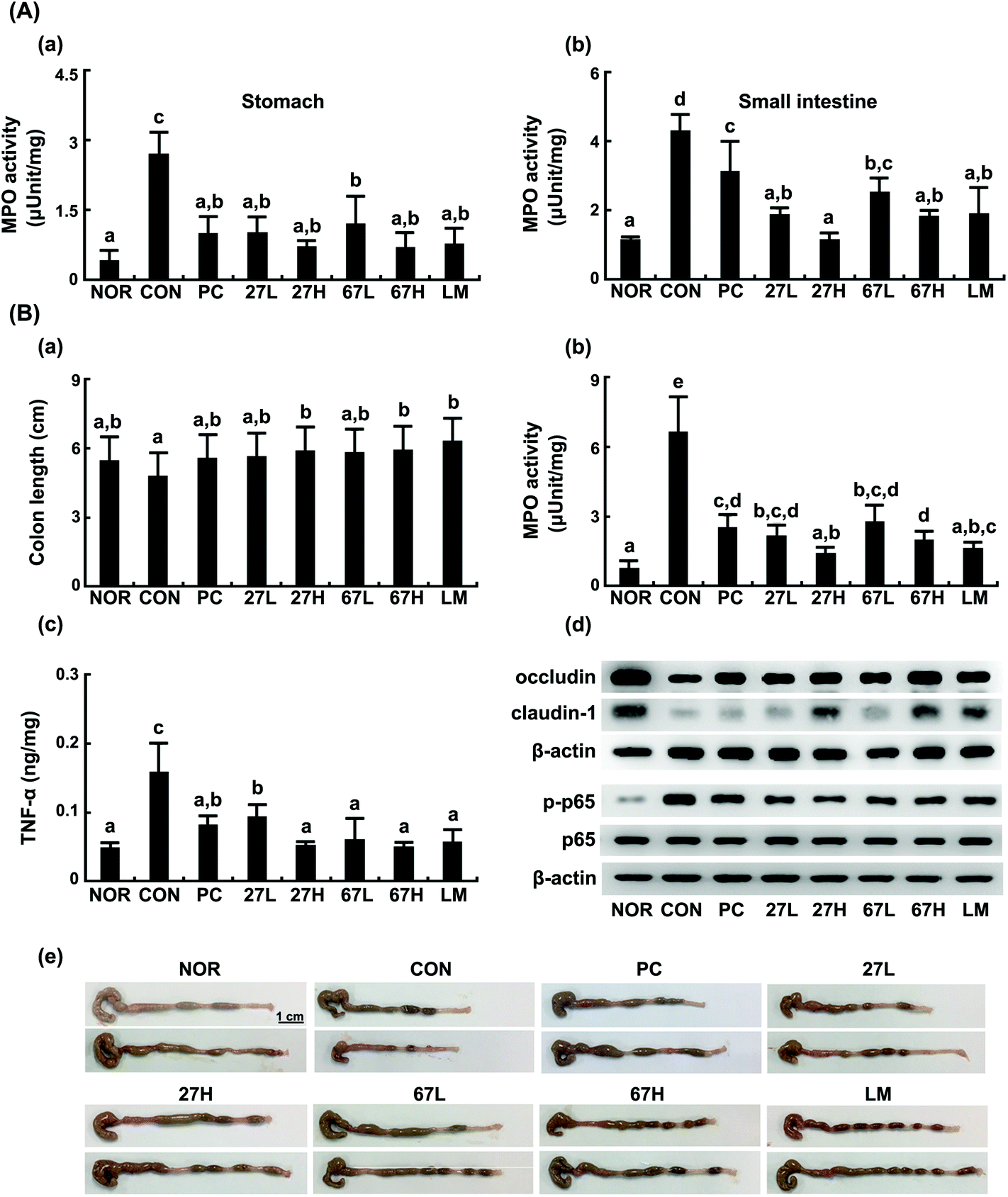 | ||
| Fig. 5 Anti-colitic effects of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 in mice with ethanol-induced steatosis. (A) Effects on the myeloperoxidase activities in the stomach (a) and small intestine (b). (B) Effects on the colon length (a), myeloperoxidase activity (b), TNF-α expression (c), tight junction protein expression and NF-κB activation (d), and photograph (e). Test agents (CON), vehicle alone; PC, 50 mg kg−1 of silymarin; 27L, 5 × 108 CFU per mouse of LC27; 27H, 2 × 109 CFU per mouse of LC27; 67L, 5 × 108 CFU per mouse of LC27; 67H, 2 × 109 CFU per mouse of LC27; LM, the (1:1 mixture of LC27 and LC67 [2 × 109 CFU per mouse]) were orally administered once a day for 5 days. The mice were sacrificed 15 h after the final administration of test agents. The normal control group (NOR) was treated with saline instead of ethanol and 1% dextrose instead of test agents. All data are represented as mean ± SD (n = 7). Means with the same letters are not significantly different (p < 0.05). | ||
3.3. Effects of LC27 and LC67 on the gut microbiota composition in mice with ethanol-induced steatosis
Ethanol intake disturbed the gut microbiota composition and increased the gut microbiota LPS production. In the present study, we also found that ethanol intake increased the populations of Firmicutes and Proteobacteria but reduced the populations of Bacteroidetes and Actinobacteria, resulting in increased ratios of Firmicutes/Bacteroidetes (B/F), Proteobacteria/Bacteroidetes (P/B), and Actinobacteria/Firmicutes (P/A) (Fig. 6). However, treatment with LC27, LC67, or LM significantly inhibited the ethanol-induced population of Proteobacteria, and increased the ethanol-suppressed populations of Bacteroidetes and Actinobacteria while the survival rate of orally administered probiotics in the GI tract could not be analyzed. The oral administration of LC27 most potently increased the Actinobacteria population. Moreover, they suppressed the ethanol-induced gut microbiota LPS levels.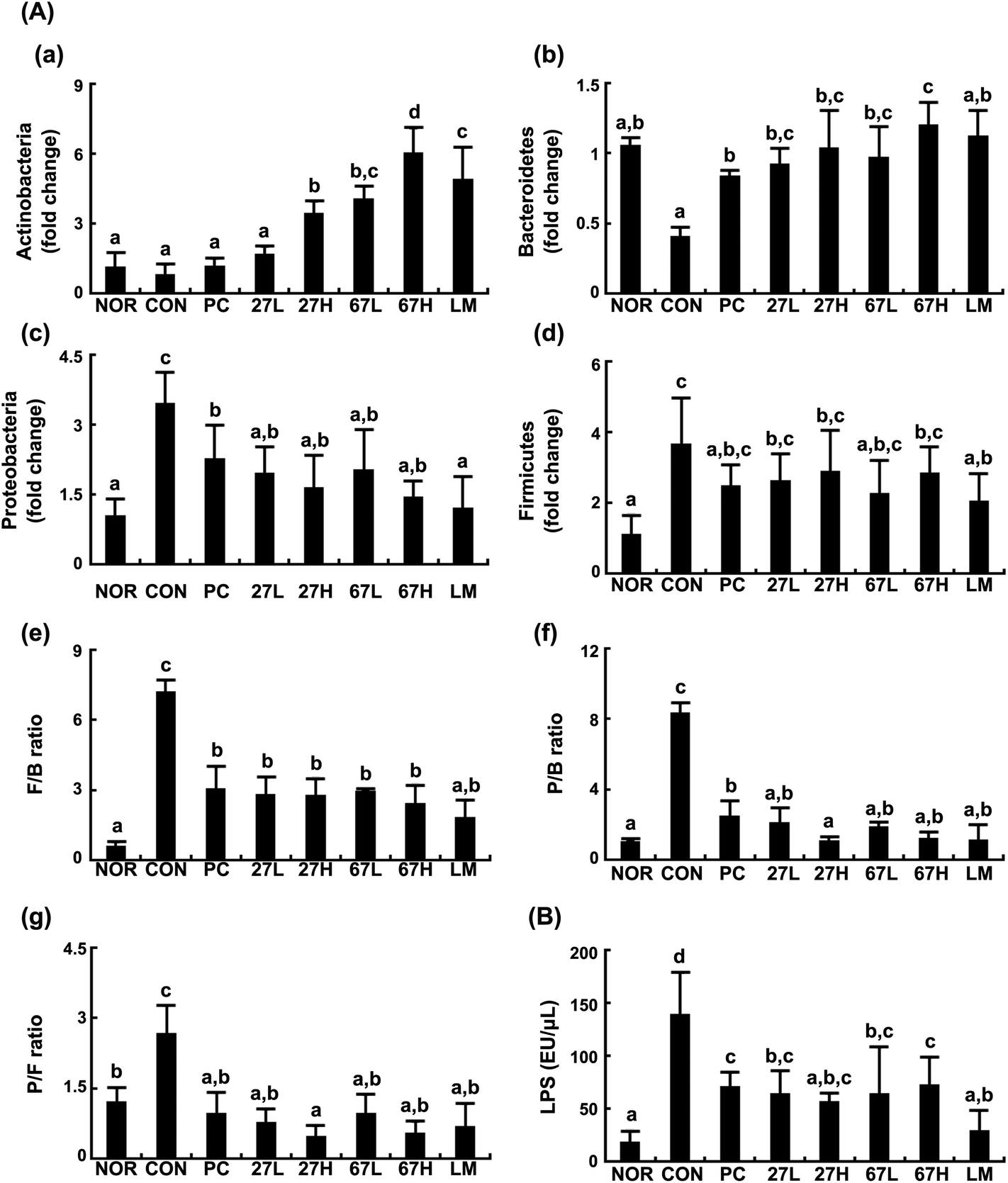 | ||
| Fig. 6 Effect of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 on the gut microbiota composition in mice with ethanol-induced steatosis. (A) Effects on the populations of Actinobacteria (a), Bacteroidetes (b), Proteobacteria (c), and Firmicutes (d) and ratios of Firmicutes to Bacteroidetes population (F/B, e), Proteobacteria to Bacteroidetes population (P/B, f), and Proteobacteria to Firmicutes population (P/F, g). (B) Effects on the gut microbiota LPS production. Test agents (CON), vehicle alone; PC, 50 mg kg−1 of silymarin; 27L, 5 × 108 CFU per mouse of LC27; 27H, 2 × 109 CFU per mouse of LC27; 67L, 5 × 108 CFU per mouse of LC27; 67H, 2 × 109 CFU per mouse of LC27; LM, the (1:1 mixture of LC27 and LC67 [2 × 109 CFU per mouse]) were orally administered once a day for 5 days. The mice were sacrificed 15 h after the final administration of test agents. The normal control group (NOR) was treated with saline instead of ethanol and 1% dextrose instead of test agents. All data are represented as mean ± SD (n = 7). Means with the same letters are not significantly different (p < 0.05). | ||
3.4. Effects of LC27 and LC67 on tight junction protein expression and NF-κB activation in ethanol-induced Caco-2 cells
To understand whether LC27 or LC67 could affect the gut membrane permeability, we investigated their effects on the expression of tight junction proteins in Caco-2 cells (Fig. 7). Ethanol treatment significantly suppressed the expression of occludin and claudin-1. However, LC27 or LC67 treatment increased the ethanol-suppressed occludin and claudin-1 expression. These also suppressed ethanol-induced NF-κB and IκB activation. Furthermore, LPS also significantly suppressed the expression of occludin and claudin-1 and increased NF-κB activation, while LC27 or LC67 treatment increased the LPS-induced occludin and claudin-1 expression and suppressed the LPS-induced NF-κB activation.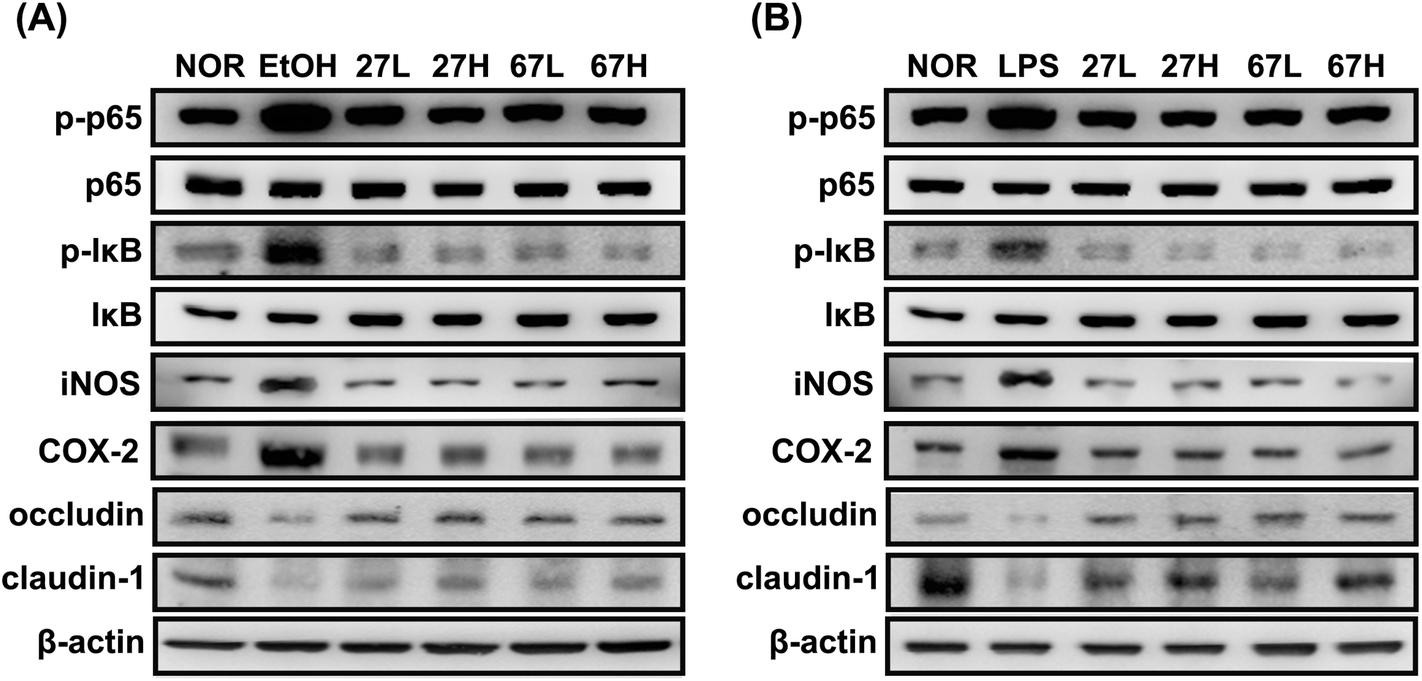 | ||
| Fig. 7 Effects of Lactobacillus plantarum LC27 and Bifidobacterium longum LC67 on the iNOS, COX-2, occludin, and claudin-1 expression and NF-κB activation in ethanol- (A) or LPS-stimulated (B) Caco-2 cells. Cells (2 × 105 cells per mL−1) were incubated with ethanol (50 μL mL−1) or LPS (100 mg mL−1) in the absence or presence of LC27 or LC67 (L, 1 × 103 CFU mL−1; or H, 1 × 105 CFU mL−1). | ||
4. Discussion
Excessive ethanol consumption causes mucosal damage, induction of leukocyte infiltration, chemokine secretion, oxidative stress, and apoptosis in the GI tract including the stomach, leading to GI inflammation and ulcers.32 Moreover, ethanol intake significantly suppressed the number of lactobacilli and bifidobacteria in the feces of mice and increased the number of enterobacteria and enterococci, resulting in the LPS production of gut microbiota.10,11 These accelerate the absorption of macromolecules such as LPS from the GI tract into the blood and liver due to GI inflammation and ulcers, leading to systemic diseases such as hepatitis, hepatic steatosis, and hepatocirrhosis.10,33,34 Therefore, restoring the disturbance of gut microbiota by ethanol intake can be beneficial for hepatic diseases such as hepatic steatosis.In the present study, we observed that the intake of ethanol caused GI inflammation in the stomach, small intestine, and colon. Ethanol intake also disturbed the gut microbiota: it increased the population of Proteobacteria, which potently produce LPS, and reduced the population of Actinobacteria. Furthermore, ethanol treatment increased the gut microbiota LPS production, as well blood LPS levels. Jang et al. reported that LPS-induced GI inflammation increases the membrane permeability of macromolecules, such as LPS, in the GI tract.35 These results suggest that ethanol intake can increase the absorption of gut microbiota macromolecules from the GI tract into the blood due to the overexpression of LPS by the gut microbiota disturbance. However, the oral administration of LC27 or/and LC67 significantly inhibited the ethanol-induced NF-κB activation, TNF-α, and myeloperoxidase activity in the GI tract and reduced the blood LPS level and the P/A ratio. These results suggest that the amelioration of ethanol-induced GI inflammation by LC27 or/and LC67 treatment can reduce the blood LPS level. Furthermore, they inhibited the ethanol-induced expression of iNOS and the production of malondialdehyde and NO. These findings suggest that LC27 and LC67 may alleviate the GI inflammation by inhibiting the activation of neutrophils and macrophages and scavenging radical oxygen species. The activation of neutrophils and macrophages is associated with the activation of NF-κB and up-regulation of proinflammatory cytokines such as TNF-α.36,37 Furthermore, LC27 or/and LC67 decreased ethanol-induced fecal and blood LPS levels and increased ethanol-suppressed tight junction protein expression. These findings suggest that ethanol intake can cause GI inflammation by the disturbance of the gut microbiota composition and the over-production of gut microbiota LPS, resulting in the increase in the gut membrane permeability of gut bacterial LPS into the blood. We also found that the intake of ethanol could cause liver damage and steatosis in mice: it increased TNF-α, iNOS, and COX-2 expression, NF-κB activation, malondialdehyde, NO, and fat accumulation and decreased the tight junction protein expression and AMPK activation in the liver. Ethanol intake caused fat accumulation and increased the α-SMA expression in the liver. All these factors act in concert to exacerbate the liver damage such as hepatitis, hepatic steatosis, and hepatocirrhosis.38,39 LC27 or/and LC67 significantly reduced the ethanol-induced ALT and AST levels in the blood, which are one of the most widely used means for assessing the extent of hepatic damage,40 as well as the TG and TC levels in the liver and blood. LC67 more potently reduced the ALT and AST levels. Its effect is comparable to that of silymarin. Moreover, even if LC27 and LC67 at 1:1, 3:1, and 9:1 ratios were combined, their effects were not different. Overall, LC27 and LC67 additively reduced the ethanol-induced ALT and AST levels.
In addition, Mutlu et al. reported that ethanol intake caused alcohol-induced intestinal dysbiosis, endotoxemia, and steatohepatitis in rats.10 These results suggest that the intake of ethanol can cause alcoholic hepatic steatosis and hepatocirrhosis through GI and liver inflammation and LC27 or/and LC67 can inhibit ethanol-induced liver injury by inhibiting the inflammatory signaling pathway. Moreover, we found that LC27 or LC67 treatment might inhibit the promotion of hepatic fibrosis by suppressing the α-smooth muscle actin expression. These results suggest that LC27 or/and LC67 may attenuate the progression of hepatic fibrosis. Additionally, Lactobacillus fermentum, Lactobacillus rhamnosus CCFM1107, Lactobacillus rhamnosus GG, and Lactobacillus casei MYL01 attenuated alcoholic gastritis and hepatic injury.20,21,41–43Lactobacillus rhamnosus GG downregulated the ethanol-induced expressions of lipogenic and pro-inflammatory genes in the liver, improved intestinal tight junction (TJ) proteins, and reversed the alcohol-disturbed populations of Th17 and regulatory T cells’ population.18,26Lactobacillus rhamnosus CCFM1107 prevented alcoholic liver injury by reducing oxidative stress and restoring the intestinal flora.20 VSL#3, a mixture of eight lactic acid bacteria, also attenuated ethanol-induced liver injury in mice by regulating the expression of tight junction proteins and increasing epithelial permeability.44 These results suggest that lactobacilli and bifidobacteria can alleviate alcohol-induced hepatic steatosis.
5. Conclusion
The oral administration of LC27 or/and LC67 in mice suppressed the ethanol-induced myeloperoxidase activity, TNF-α expression, and NF-κB activation in the GI tract. These also restored the ethanol-induced gut microbiota disturbance and suppressed the fecal and blood LPS levels. Treatment with LC27 or/and LC67 increased the ethanol-suppressed expression of gut tight junction proteins, as well as reduced ethanol-induced blood ALT, AST, and TNF-α levels. Treatment with LC27 or/and LC67 alleviated ethanol-induced TG, TC, LDL-cholesterol, and TNF-α levels in the blood and liver, as well as suppressed the NF-κB activation and malondialdehyde and NO levels in the liver. Conclusively, LC27 and LC67 may simultaneously alleviate ethanol-induced GI inflammation, liver injury, and steatosis by inhibiting LPS-mediated NF-κB activation through restoration of the gut microbiota disturbance.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
This research is supported by “Rediscovery of the Past R&D Result” through the Ministry of Trade, Industry and Energy (MOTIE) and the Korea Institute for Advancement of Technology (KIAT) (Grant No.: N0002485).References
- B. Gao and R. Bataller, Gastroenterology, 2011, 141, 1572–1585 CrossRef PubMed.
- R. S. O'Shea and A. J. McCullough, Clin. Liver Dis., 2005, 9, 103–134 CrossRef PubMed.
- G. Malaguarnera, M. Giordano, G. Nunnari, G. Bertino and M. Malaguarnera, World J. Gastroenterol., 2014, 20, 16639–16648 CrossRef PubMed.
- G. Szabo, Gastroenterology, 2015, 148, 30–36 CrossRef PubMed.
- K. J. Atkinson and R. K. Rao, Am. J. Physiol.: Gastrointest. Liver Physiol., 2001, 280, G1280–G1288 CrossRef PubMed.
- H. K. Na and J. Y. Lee, Int. J. Mol. Sci., 2017, 18, E1116 CrossRef PubMed.
- L. R. Hoyt, M. J. Randall, J. L. Ather, D. P. DePuccio, C. C. Landry, X. Qian, Y. M. Janssen-Heininger, A. van der Vliet, A. E. Dixon, E. Amiel and M. E. Poynter, Redox Biol., 2017, 12, 883–896 CrossRef PubMed.
- W. C. Chiu, Y. L. Huang, Y. L. Chen, H. C. Peng, W. H. Liao, H. L. Chuang, J. R. Chen and S. C. Yang, Food Funct., 2015, 6, 1692–1700 RSC.
- P. Chen and B. Schnabl, Gut Liver, 2014, 8, 237–241 CrossRef PubMed.
- E. Mutlu, A. Keshavarzian, P. Engen, C. B. Forsyth, M. Sikaroodi and P. Gillevet, Alcohol.: Clin. Exp. Res., 2009, 33, 1836–1846 CrossRef PubMed.
- G. Capurso and E. Lahner, Best Pract. Res., Clin. Gastroenterol., 2017, 31, 579–588 CrossRef PubMed.
- N. Matsushita, T. Osaka, I. Haruta, H. Ueshiba, N. Yanagisawa, M. Omori-Miyake, E. Hashimoto, N. Shibata, K. Tokushige, K. Saito, S. Tsuneda and J. Yagi, Scand. J. Immunol., 2016, 83, 109–118 CrossRef PubMed.
- E. S. Orman, G. Odena and R. Bataller, J. Gastroenterol. Hepatol., 2013, 28(Suppl 1), 77–84 CrossRef PubMed.
- J. Behnsen, E. Deriu, M. Sassone-Corsi and M. Raffatellu, Cold Spring Harbor Perspect. Med., 2013, 3, a010074 Search PubMed.
- H. Kumar, S. Salminen, H. Verhagen, I. Rowland, J. Heimbach, S. Bañares, T. Young, K. Nomoto and M. Lalonde, Curr. Opin. Biotechnol., 2015, 32, 99–103 CrossRef PubMed.
- E. O. Petrof, Antiinflamm. Antiallergy Agents Med. Chem., 2009, 8, 260–269 CrossRef PubMed.
- G. D. Kang and D. H. Kim, Int. Immunopharmacol., 2016, 43, 179–186 CrossRef PubMed.
- B. Kim, K. Y. Park, Y. Ji, S. Park, W. Holzapfel and C. K. Hyun, Biochem. Biophys. Res. Commun., 2016, 473, 530–536 CrossRef PubMed.
- K. A. Kim, J. J. Jeong and D. H. Kim, J. Funct. Foods, 2015, 13, 183–191 CrossRef.
- F. Tian, F. Chi, G. Wang, X. Liu, Q. Zhang, Y. Chen, H. Zhang and W. Chen, J. Microbiol., 2015, 53, 856–863 CrossRef PubMed.
- E. K. Kwon, G. D. Kang, W. K. Kim, M. J. Han and D. H. Kim, J. Funct. Foods, 2017, 38, 389–398 CrossRef.
- H. Zhao, C. Zhao, Y. Dong, M. Zhang, Y. Wang, F. Li, X. Li, C. McClain, S. Yang and W. Feng, Toxicol. Lett., 2015, 234, 194–200 CrossRef PubMed.
- S. Segawa, Y. Wakita, H. Hirata and J. Watari, Int. J. Food Microbiol., 2008, 128, 371–377 CrossRef PubMed.
- Y. Wang, I. Kirpich, Y. Liu, Z. Ma, S. Barve, C. J. McClain and W. Feng, Am. J. Pathol., 2011, 179, 2866–2875 CrossRef PubMed.
- H. J. Kang and S. H. Im, J. Nutr. Sci. Vitaminol., 2016, 61(Suppl), S103–S105 Search PubMed.
- R. C. Chen, L. M. Xu, S. J. Du, S. S. Huang, H. Wu, J. J. Dong, J. R. Huang, X. D. Wang, W. K. Feng and Y. P. Chen, Toxicol. Lett., 2016, 241, 103–110 CrossRef PubMed.
- I. A. Lee, Y. J. Park, H. K. Yeo, M. J. Han and D. H. Kim, J. Agric. Food Chem., 2010, 58, 10929–10934 CrossRef PubMed.
- K. A. Kim, W. Gu, I. A. Lee, E. H. Joh and D. H. Kim, PLoS One, 2012, 7, e47713 CrossRef PubMed.
- S. M. Lim, H. S. Choi and D. H. Kim, Am. J. Chin. Med., 2017, 45, 1033–1046 CrossRef PubMed.
- N. A. SHirwany and M. H. Zou, Front Biosci., 2014, 19, 447–474 CrossRef.
- B. Hinz, G. Celetta, J. J. Tomasek, G. Gabbiani and C. Chaponnier, Mol. Biol. Cell, 2001, 12, 2730–2741 CrossRef PubMed.
- C. Bode and J. C. Bode, Alcohol Health Res. World, 1997, 21, 76–83 Search PubMed.
- Y. Tang, C. B. Forsyth, A. Farhadi, J. Rangan, S. Jakate, M. Shaikh, A. Banan, J. Z. Fields and A. Keshavarzian, Alcohol.: Clin. Exp. Res., 2009, 33, 1220–1230 CrossRef PubMed.
- H. Sung, S. W. Kim, M. Hong and K. H. Suk, World J. Gastroenterol., 2016, 22, 6673–6682 CrossRef PubMed.
- S. E. Jang, S. M. Lim, J. J. Jeong, H. M. Jang, H. J. Lee, M. J. Han and D. H. Kim, Mucosal Immunol., 2018, 11, 369–379 CrossRef PubMed.
- S. Ghosh and M. S. Hayden, Nat. Rev. Immunol., 2008, 8, 837–848 CrossRef PubMed.
- R. L. Szabady and B. A. McCormick, Front. Immunol., 2013, 4, 220 Search PubMed.
- A. D. Dhanda, R. W. Lee, P. L. Collins and C. A. McCune, World J. Gastroenterol., 2012, 18, 5504–5513 CrossRef PubMed.
- H. Kawaratani, T. Tsujimoto, A. Douhara, H. Takaya, K. Moriya, T. Namisaki, R. Noguchi, H. Yoshiji, M. Fujimoto and H. Fukui, Mediators Inflammation, 2013, 2013, 495156 Search PubMed.
- S. E. Woods, M. Hitchcock and A. Meyer, Am. Fam. Physician, 1993, 47, 1171–1178 Search PubMed.
- Y. H. Chiu, J. J. Tsai, S. L. Lin and M. Y. Lin, J. Dairy Sci., 2014, 97, 2009–2016 CrossRef PubMed.
- H. Suo, X. Zhao, Y. Qian, P. Sun, K. Zhu, J. Li and B. Sun, Nutrients, 2016, 8, 155 CrossRef PubMed.
- Y. Wang, I. Kirpich, Y. Liu, Z. Ma, S. Barve, C. J. McClain and W. Feng, Am. J. Pathol., 2011, 179, 2866–2875 CrossRef PubMed.
- C. Loguercio, A. Federico, C. Tuccillo, F. Terracciano, M. V. D'Auria, C. De Simone and C. Del Vecchio Blanco, J. Clin. Gastroenterol., 2005, 39, 540–543 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |