 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-file transport of water through membrane channels
Andreas
Horner
 and
Peter
Pohl
*
and
Peter
Pohl
*
Johannes Kepler University Linz, Institute of Biophysics, Gruberstr. 40, 4020 Linz, Austria. E-mail: peter.pohl@jku.at
First published on 18th June 2018
Abstract
Water at interfaces governs many processes on the molecular scale from electrochemical and enzymatic reactions to protein folding. Here we focus on water transport through proteinaceous pores that are so narrow that the water molecules cannot overtake each other in the pore. After a short introduction into the single-file transport theory, we analyze experiments in which the unitary water permeability, pf, of water channel proteins (aquaporins, AQPs), potassium channels (KcsA), and antibiotics (gramicidin-A derivatives) has been obtained. A short outline of the underlying methods (scanning electrochemical microscopy, fluorescence correlation spectroscopy, measurements of vesicle light scattering) is also provided. We conclude that pf increases exponentially with a decreasing number NH of hydrogen bond donating or accepting residues in the channel wall. The variance in NH is responsible for a more than hundredfold change in pf. The dehydration penalty at the channel mouth has a smaller effect on pf. The intricate link between pf and the Gibbs activation energy barrier, ΔG‡t, for water flow suggests that conformational transitions of water channels act as a third determinant of pf.
1 Water at interfaces
Interfacial water is of crucial importance in many technological, environmental and biological processes. For example, it is ubiquitously present as a thin film at hydrophilic surfaces in humid air, it governs electrochemical reactions on liquid–solid interfaces or enzymatic reactions in the active centers of proteins, it is an important determinant for protein folding and it regulates the uptake and release of gases at the liquid–vapor interface of oceans and clouds.1,2 Substrates influence the structure and properties of adjacent water molecules. Examples are provided by one-dimensional clusters formed at metal surfaces such as Cu(110)3 and oscillatory density profiles within a distance of 10 Å near solid–aqueous interfaces.4One of the manifold functional consequences of the altered physicochemical properties of water is illustrated by the decrease in the dielectric constant from 78 to roughly four on top of a solid surface (mica).5 A similar observation has been made in the immediate vicinity of biological membranes.6 Yet another example is provided by the water assisted movement of protons at solid surfaces7,8 or along membranes.9–11 In contrast to hydrophilic substances, water reduces the number of hydrogen bonds adjacent to extended hydrophobic substrates.12 As a result, the first layer of water molecules retracts from the surface – adopting a vapor-like structure.13 A non-hydrogen bonded OH group (dangling OH) emerges that points toward the hydrophobic surface.14 In neuroscience such liquid–vapor oscillations were hypothesized to occur in hydrophobic segments of membrane channels.15 This so-called hydrophobic gating may be responsible for keeping the channel functionally closed in the absence of the neurotransmitter. Otherwise, the vapor-filled constriction zone of the channel would be wide enough to allow the passage of both water molecules and ions.
Water structuring on the interface may be expected to have implications on water mobility. Assessment by NMR spectroscopy revealed that water reorientation and translation are 3–5 times slower than in the bulk.16,17 The level of mobility reduction is highly heterogeneous at the surface of lipid bilayers18 and proteinaceous structures. That is, the dynamics of water reproduce the chemical features of amino acids.1 The differences in water motion usually decay within five water layers from the surface.19 Protein crowding may prevent the decay from happening. It was reported to leave the number of H-bonds practically unchanged while affecting the H-bond dynamics.20 Very little is known about their effect on fluid flow through narrow channels.
2 Hydrodynamics
It is clear that the macroscopic laws of hydrodynamics (e.g. Hagen–Poiseuille’s law) do not apply to narrow biological channels.1 That is, the channel length and diameter can no longer act as major determinants of fluid flow. Moreover, it is impossible to imagine that the outermost water layer adheres to the channel wall, since otherwise there would be no flow across pores that are only one water molecule wide. Already tens of nanometers wide, carbon nanotubes show an enhanced permeability that can only be described by Poiseuille flow with slippage inside the nanotube.21 In the extreme case of biological channels specialized in water transport (aquaporins, AQPs), slippage may be perfect, i.e. the water molecules may retain a mobility that is close to the mobility of bulk water.22Contributions aiming at a theoretical description of the hydrodynamics in very narrow channels are scarce. Existing ones represent the channels as cylindrical pipes and ignore the interaction of the permeating water molecules with the channel wall. Commonly, partial water dehydration at the channel entrance is assumed to be the major energetic barrier for facilitated water transport.23 In contrast, water movement through the single-file region of the channel is treated as if it was not offering any resistance.24 In consequence, the predictive power of such models for water flow through biological channels is limited. For carbon nanotubes the situation is different. Here, the dehydration penalty may be rate determining. Existing models envision water flow as (i) a collective motion of the entire single-file water column in a random walk fashion25 or as (ii) the movement of a vacancy in the direction opposite to that of water movement.26 An overview of single-file water in artificial nanopores has been given previously,27 allowing this review to focus on biological channels.
Here we reproduce a short outline of the most influential model of the past decades that has been proposed by Alan Finkelstein28 long before water selective channel proteins, i.e. AQPs, were discovered. The model envisions the work W required to move N water molecules through the channel to be equal to:
| W = vwNΔΠ | (1) |
| W = FΠL | (2) |
It is illuminating to follow Finkelstein’s derivation of unitary water channel permeability, pf, till the end, even though we disagree with eqn (2) on the grounds that there is no experimental evidence for W to be proportional to L. We should also keep in mind that eqn (2) implies the dehydration penalty be much smaller than W.
Expressing FΠ from eqn (1) and (2) and setting it equal to the frictional force Fγ:
| Fγ = Nγv | (3) |
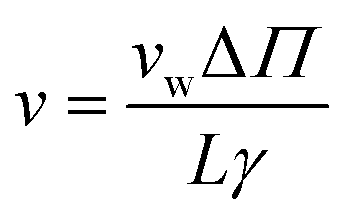 | (4) |
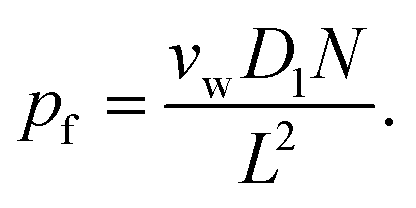 | (5) |
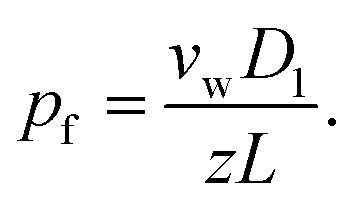 | (6) |
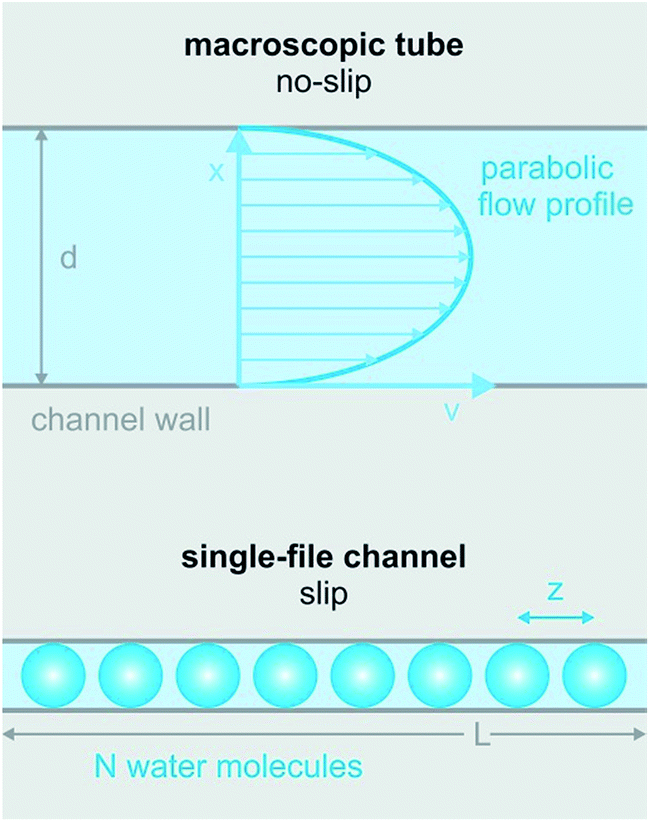 | ||
| Fig. 1 The difference between macroscopic and sub-nanometer hydrodynamics. Hagen–Poiseuille’s law envisions a parabolic streaming profile through macroscopic tubes due to a no-slip condition at the channel wall. In contrast, there cannot be a parabolic streaming profile in single-file flow (eight water molecules are drawn in blue – not to scale). Thus, the no-slip condition is not obeyed in narrow biological channels. L denotes the length of the channel, N denotes the number of single-file water molecules and z denotes the distance between adjacent water molecules. | ||
Eqn (6) states that pf is inversely proportional to L. This important result is a direct consequence of the assumption W ∼ L made by eqn (2). The problem with eqn (6) is that D1 is a purely theoretical parameter. There is no isolated single-water molecule in the pore. Water moves in a column of N water molecules. This is thought to reduce its mobility N times,31i.e. the diffusion coefficient Dw of a water molecule in the pore is equal to  . Such an observation has been made for hard spheres in a cylindrical pipe. The presence of a constriction zone as well as the availability of hydrogen bond donors and acceptors in the wall of a membrane channel may confound the analysis. That is, the same reservations that have been raised against eqn (2) apply here.
. Such an observation has been made for hard spheres in a cylindrical pipe. The presence of a constriction zone as well as the availability of hydrogen bond donors and acceptors in the wall of a membrane channel may confound the analysis. That is, the same reservations that have been raised against eqn (2) apply here.
Most interestingly, the two questionable length dependencies (for Dw and W) cancel each other out when substituting D1 for Dw. Inserting N × z for L transforms eqn (6) into:
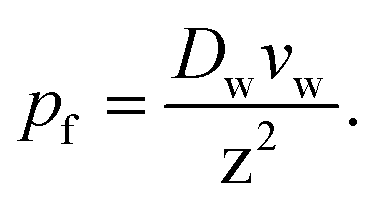 | (7) |
Eqn (7) has independently been derived to extract pf values from molecular dynamics simulations.25,32 It shows that pf and Dw are interchangeable parameters for fully occupied water channels. The drawback of eqn (7) is that it does not allow for making any predictions about how fast a given channel will conduct water. That information is hidden behind Dw and can only be unveiled by an analysis of the interaction between the pore lining residues and the permeating water molecules.
3 Water mobilities inside narrow membrane channels vary widely
Eqn (6) predicts very limited variability of pf for membrane channels. In the case of homogeneous interactions with the channel wall, i.e. for an invariant D1, pf may vary no more than two times between different channels. The simple reason is that known single-file regions are at least four water molecules long (potassium channels) and accommodate no more than eight water molecules (AQPs).The attempt to verify the prediction (eqn (6)) experimentally faced large obstacles. Progress in determining the pf value of water channeling proteins has been slow due to large experimental difficulties. In contrast to ion channel abundance, which can simply be measured by observing conductivity steps when a constant voltage is applied across a membrane patch, water channel density in the membrane patch is difficult to assess. The background conductivity of lipid bilayers to water is much too large to enable single channel recordings of AQPs. Thus, AQP counting often relies on immunohistochemistry or other biochemical assays with limited accuracy. For example, determining the water channel concentration via the bicinchoninic acid assay (BCA assay) depends on a standard curve from a known, standard protein. There is a good chance that the membrane protein does not interact with the dye in a similar fashion to the standard water soluble protein (commonly bovine serum albumin, BSA) because the relative amount of hydrophobic amino acids insoluble in membrane proteins must be different. Since the BCA reaction is influenced by cysteine, tyrosine, and tryptophan residues, the membrane protein concentration may be overestimated by a large margin.
In addition to counting problems, unstirred layers, i.e. stagnant water layers, in the immediate membrane vicinity, may hamper the results. The simple reason is that upon entering the hyperosmotic solution water dilutes the osmolyte within these unstirred layers and thus diminishes the driving force for the transmembrane water flux.33,34 Finally, other methodological problems that arise when (i) extracting water efflux from time dependent changes of the vesicle or cell volume and (ii) using light scattering to continuously measure the volume of a shrinking particle, have contributed to the great variability of reported pf values.
The difference in data quality is reflected in the color code of Fig. 2. The pf values of single-file channels that have been obtained (i) by a direct count of the reconstituted channels and (ii) by accounting for unstirred layer effects are indicated in red. Data that (i) are based on a biochemical or immunohistochemical estimate of the actual protein abundance or (ii) may have suffered from unstirred layers or other methodological problems enter Fig. 2 in pink.
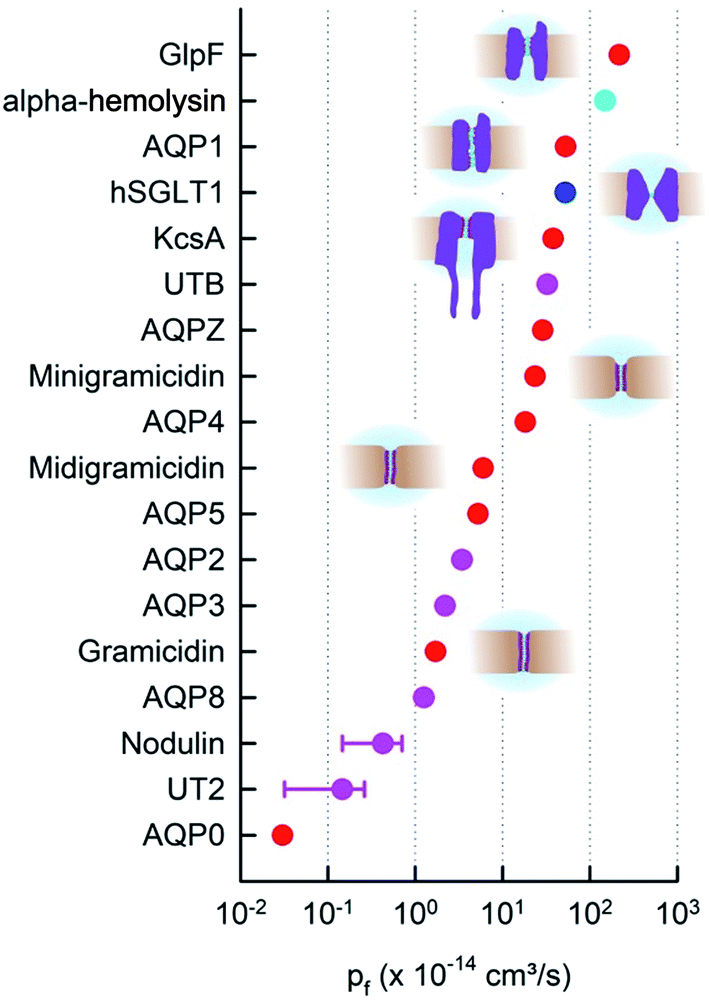 | ||
| Fig. 2 The pf values of membrane channels and transporters. The red and pink dots indicate pores with a known single-file region. Whether the sodium–glucose cotransporter SGLT1 (blue) has a single-file region is not entirely clear. The beta-barrel α-hemolysin (cyan) accommodates multiple water molecules in its cross-section.35 Moreover, red and blue dots indicate quantitative pf measurements in terms of protein counting. hSGLT1,36 KcsA, GlpF, AQP1, AQPZ,22 AQP4,37 and AQP538 were counted with fluorescence correlation spectroscopy (FCS). The bacterial cation selective gramicidin channels39 were measured electrically and AQP0 (ref. 40) with electron microscopy (EM). AQP8 (ref. 41) is also permeable to ammonia and urea. Vice versa, urea transporters Ut-B and UT-2 are permeable to water.42 | ||
In contrast to its expected small variability (eqn (6)), the experimental pf values of single-file channels, and thus the intraluminal water mobilities, vary over four orders of magnitude (Fig. 2). The most efficient water channel seems to be the aqua-glycerol facilitator GlpF from E. coli.22 Water retains bulk-like mobility within the channel. Dw amounts to about 5 × 10−5 cm2 s−1 according to eqn (6). The slowest water movement has been observed for AQP0 (ref. 40), also called the major intrinsic protein of the lens. The water mobility drops four orders of magnitude below the bulk mobility, as indicated by Dw 4 × 10−9 cm2 s−1. We conclude that the variation in L cannot explain the variability in pf (and Dw).
Most interestingly, water traverses both AQP0 and GlpF in single-file.43,44 That is, mechanisms must exist that regulate Dw to adopt such different values, while keeping L nearly constant. As outlined below, the availability of hydrogen bond donating or receiving residues in the channel wall is one of the major determinants of water mobility,22 the presence of charged residues at the channel mouth is another.37 These factors seem to be more important than even an increased channel diameter. For example, the lumen of α-hemolysin is much wider than the diameter of a single water molecule, i.e. water molecules and ions may overtake each other in the channel, and yet, its pf value is smaller than that of GlpF (Fig. 2). In addition to interactions between permeating water molecules and water pathway lining residues, there may be other factors – like channel gating – that add to the variability in pf.
4 Length dependence of pf
Based on Finkelstein’s theory, pf is widely assumed to linearly decrease with L (eqn (6)). However, experimental evidence in support of the postulate is scarce. As outlined above, linearity may not be expected in the case of an inhomogeneous channel that by virtue of a constriction zone may impose a larger barrier to water permeation in one spot than in the remainder of the channel. However, it is also not clear whether the postulate works for a homogeneous channel: the water molecules in the channel are likely to be hydrogen-bonded to residues in the channel wall. These hydrogen bonds should all break at once in order to allow the advancement of the water chain as a whole. The probability of such an event cannot generally be expected to depend linearly on the total number of water molecules in the chain.To test this prediction, we designed homogeneous peptide channels of different lengths.39 Gramicidin A served as a template. The wild-type channel is a dimer that consists of two penta-deca peptides which each span only one membrane leaflet. We shortened either one or both monomers in such a dimer and covalently linked the two halves to form midigramicidin and minigramicidin (Fig. 3B). To this end we had channels that accommodated seven,45 six39 and five39 water molecules. They spontaneously inserted into planar lipid bilayers as was indicated by their ion conducting activity, which also served to count the channels. That is, we first measured single channel conductance g in separate experiments and assumed that the total electrical conductivity G of reconstituted bilayers with many thousands of such channels was equal to g multiplied by the number n of reconstituted channels: G = n × g.
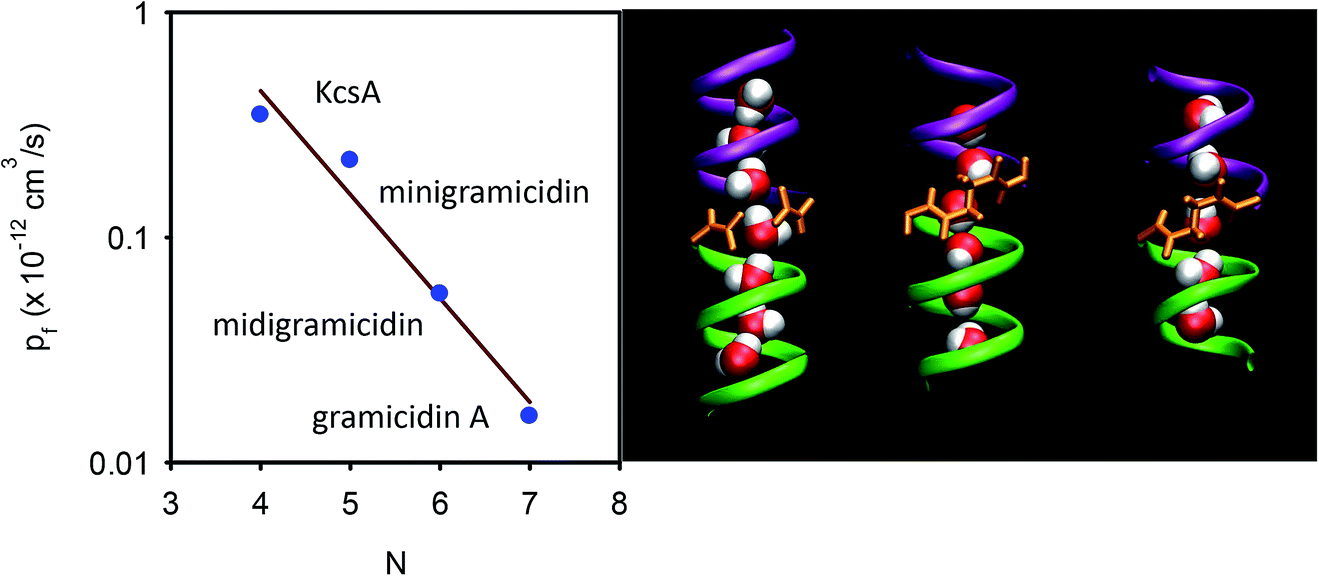 | ||
| Fig. 3 Dependence of pf on the length of the single-file. (A) The number N of water molecules that line up in a single-file configuration is a crucial determinant of pf. For the KcsA experiments, see section 6. (B) The length dependence is derived by exploiting three different gramicidin channels (from left to right): wild-type gramicidin A, midigramicidin, and minigramicidin, in addition to the bacterial potassium channel KcsA. The two gramicidin monomers are covalently linked together in the shorter channel versions. The right panel is taken from Saparov et al., Phys. Rev. Lett., 2006.39 | ||
Both G and the integral water permeability Pf of these membranes were assessed simultaneously. The Pf measurements were performed by exploiting scanning electrochemical microscopy (SEM). The method itself is outlined in more detail in the next section. In brief, a volume flux is induced by imposing an osmotic gradient across the planar bilayer and the steady-state solute concentration change in the immediate membrane vicinity is recorded by an ion-selective microelectrode in a spatially resolved manner.34pf can be derived by plotting Pf as a function of G:
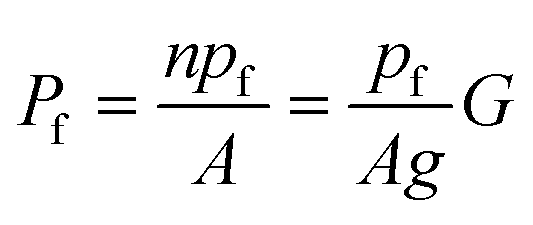 | (8) |
In contrast to theoretical predictions (eqn (6)), pf decreases exponentially with N (or L since L = Nz) (Fig. 3A).39 This is a very important result because it (i) allows mechanistic insight and (ii) has important methodological implications:
(i) The interactions of water molecules in a water chain with residues in the channel wall cannot be represented as a linear superposition of frictional forces, i.e. it is not reflected by a model in which hard spheres roll through a pipe. Instead, the concerted formation or breakage of multiple hydrogen bonds between the water chain and channel wall could be reconciled with the exponential dependence of pf on N.
(ii) Measurements of the ratio of pf and the diffusional permeability pd are unlikely to reveal N, since both parameters have been derived assuming that the energy for moving a water molecule across the channel linearly depends on L (eqn (2)).
p d is measured in the absence of an osmotic gradient. That is, a tracer (e.g. D2O or THO) is added to one compartment and its appearance is monitored on the other side of the membrane. The integral diffusive permeability Pd of a membrane is equal to Pf in the absence of membrane channels.28 As soon as water channeling proteins are present, Pf is always larger than Pd.28 The reason is that pf > pd. Finkelstein derived the dependence of pd ∼ 1/N2 in a fashion similar to pf ∼ 1/N (eqn (2)–(6)). We do not reproduce the derivation here, because it rests on the incorrect assumption that eqn (2) is valid. Accordingly, the predicted ratio pf/pd = N was never confirmed experimentally. Finkelstein himself reports pf/pd ≈ 5 for gramicidin A,46 while molecular dynamics simulations show seven molecules in the pore39 (Fig. 3B). Mathai et al. report pf/pd ≈ 13 for AQP1,47 instead of about eight molecules that according to AQP1’s structure may be placed into the single-file region.29
5 Experimental approaches for determining pf
Two principally different approaches for membrane water permeability measurements can be distinguished: (a) tracer experiments that are carried out in osmotic equilibrium, and (b) experiments in which an osmotic gradient is established across the membrane. Tracer experiments using NMR spectroscopy48 or monitoring the efflux of a radioactive isotope49 are very convenient means to determine Pd in the absence of membrane channels. However they are seldom used for the characterization of water channel permeability – the reason being that there are more convenient and accurate ways of finding N than via the pf/pd ratio. Consequently, we will not dwell on equilibrium methods, but focus instead on some selected methods for determining pf.These methods rely on the presence of a transmembrane osmotic gradient. The assessment of the resulting osmotic flux can be carried out in two principally different ways (Fig. 4): the osmotic flux is monitored either (α) in steady-state or quasi steady-state, or (β) in a kinetic approach. In (α) the volumes of both the donating and receiving compartments are large as compared to the volume of the transmembrane flux per minute, so that the osmotic gradient does not significantly change with time (Fig. 5), whereas in (β) vesicles or cells that enclose a rather limited internal volume are deflated or enlarged (Fig. 7).
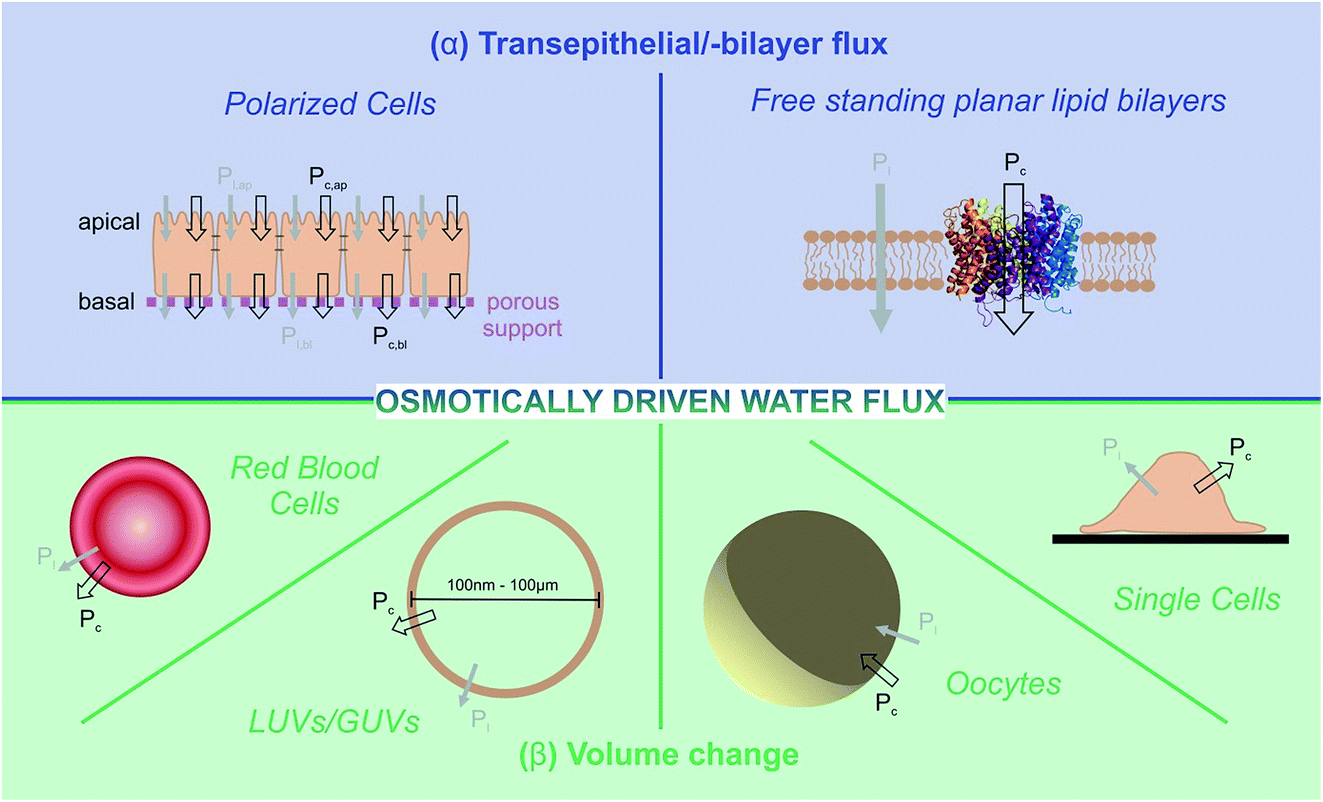 | ||
| Fig. 4 Two different approaches allow estimations of integral osmotic water permeability Pf: (α) continuous flux experiments through a confluent cell monolayer or a free standing planar lipid bilayer (blue background), or (β) volume adaptation of cells or vesicles after an osmotic challenge (green background). In both cases the overall permeability Pf is the sum of the permeabilities Pf,l and Pf,c of the lipid matrix and water conducting channels, respectively. The measurement systems include artificial free standing planar lipid bilayers as well as large or giant unilamellar vesicles (LUVs/GUVs) on the one hand and erythrocytes, oocytes, confluent cell monolayers or single cells on the other hand. | ||
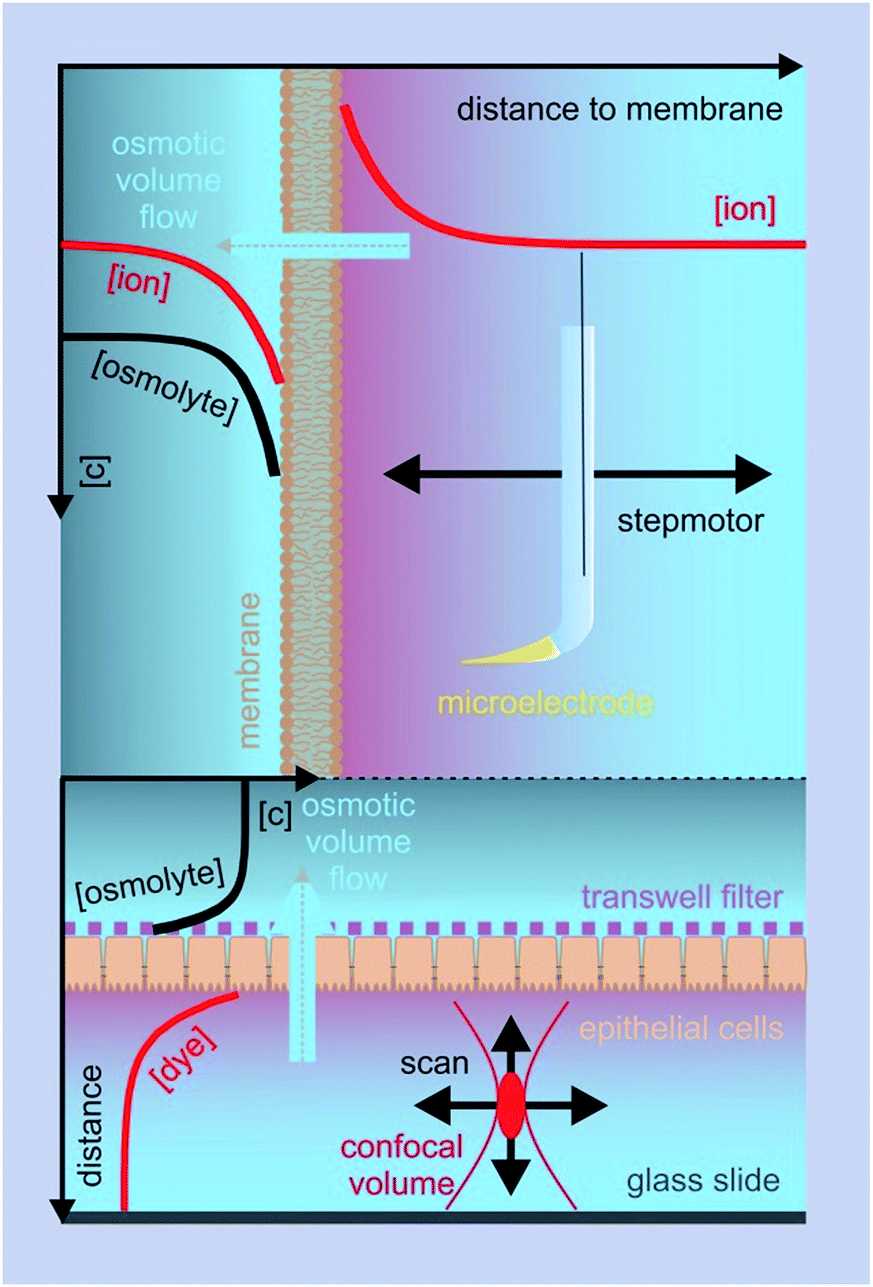 | ||
| Fig. 5 Steady-state or quasi steady-state water flux monitored by means of SEM or FCS, respectively. Top: a hydraulic step motor moves the ion selective microelectrode perpendicular to a free standing planar lipid bilayer. The electrode records the osmotically induced change in ion concentration within the unstirred layer as a function of the distance to the lipid bilayer. The lipid bilayer is folded across an aperture (∼100–400 μm diameter) in a Teflon septum that separates the two compartments. Bottom: the focal volume is moved in lateral and/or axial directions through the lower compartment formed by the glass slide and the cell monolayers on a porous filter support. The number of fluorescent dye molecules in the focal volume is derived from fluctuations in the fluorescence intensity. | ||
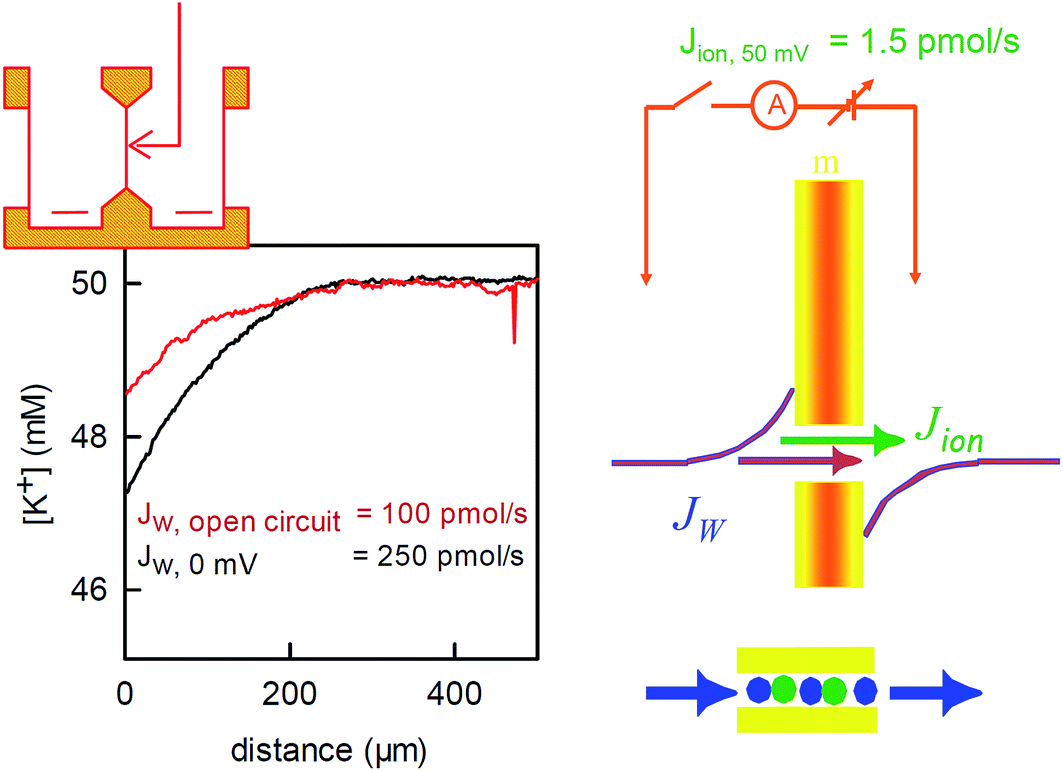 | ||
| Fig. 6 Water flux through the reconstituted bacterial potassium channel KcsA. The potassium channels were reconstituted into planar bilayer lipid membranes. An osmotic gradient (1 M urea) induced a water flux across the membrane (from left to right in the scheme on the top of the left panel). A scanning potassium selective microelectrode in the hyperosmotic compartment recorded the resulting solute dilution (scheme in the middle of the right panel) as a function of the distance to the membrane (graph on the left, the graph has been modified from Saparov et al., PNAS, 2004 (ref. 58) Copyright (2004) National Academy of Sciences). The concentration profile was used to calculate the water flux Jw. Disrupting the electrical connection between the reference electrodes on both sides of the membrane (scheme on the right) decreased Jw. The effect is due to the streaming potential that develops under open circuited conditions. The potential acts to inhibit the ion flow. In turn, the water flow across the channel also stops, since the water molecules cannot overtake the ions in the single-file region (scheme on the right, bottom). The remaining water flow (red concentration profile) passes across the lipid matrix. Water flux across the potassium channels is responsible for the difference between the red and the black profiles. It amounts to 150 pmol s−1. The simultaneously measured ion flow is 100 times smaller. | ||
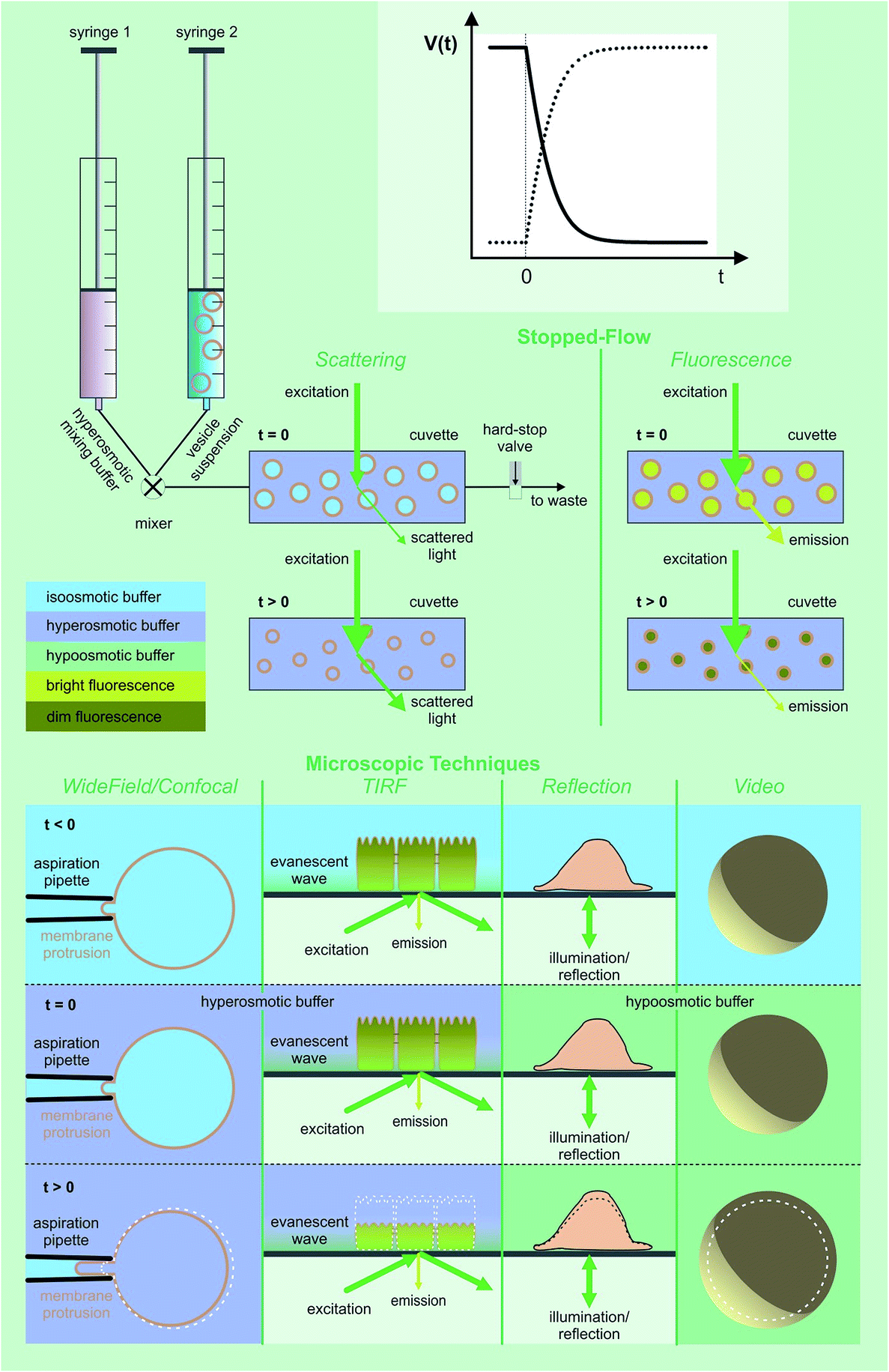 | ||
| Fig. 7 Assessment of volume changes inflicted by an osmotic challenge. The volume V(t) of LUVs and erythrocytes can be derived from the light scattering intensity or the fluorescence of a dye that is encapsulated in self-quenching concentrations. The rapid change in the external osmolyte concentration is achieved in a stopped-flow device (see main text for details). The Pf value of giant unilamellar vesicles (GUVs) can be obtained by using the microaspiration technique.72,73 GUV deflation lengthens the lipid projection length inside the aspiration pipette. The method is sensitive to small osmotic gradients. Alternatively, water efflux can be monitored by measuring the contact area of the surface adhered GUVs during deswelling.74V(t) of adherent cells can either be studied via (i) laser scanning reflection microscopy,75,76 (ii) the dilution of an encapsulated dye in total internal reflection microscopy (TIRFM) mode,77 or (iii) laser scanning confocal microscopy.78 Oocyte swelling rates can be tracked by video microscopy.79,80 However, extensive internal unstirred layers81 as well as the resistance of the oolemma to unfolding36 may lead to an underestimation of Pf. | ||
α Transepithelial/-bilayer flux
Osmotic transepithelial or transbilayer water fluxes dilute the solution they enter and concentrate the solution they leave.50 Consequently, reporter molecules in one or both compartments can be used to assess Pf.34 As such, metal ions are most convenient, since they are usually present in the surrounding solutions. Scanning ion selective microelectrodes are well suited to record the corresponding concentration profiles in the membrane vicinity. In contrast, FCS requires the addition of aqueous fluorescent dyes (Fig. 5). The sensitivity of both methods greatly depends on the size of the unstirred layer, the osmotic gradient, Pf, and the diffusion constant of the reporter molecule.34,51 Hence, bigger molecules, which have a smaller diffusion constant, are characterized by a more pronounced concentration shift at the interface. Thus, a clever selection of fluorescently labeled reporter molecules, like dextrans, antibodies or lipid vesicles, may result in a higher sensitivity of FCS measurements as compared to SEM. Sensitivity can be further enhanced by extending the unstirred layers. For example, a small distance between the epithelial cell monolayer and the glass slide (Fig. 5 (bottom)), ensures that transport through the resulting cleft occurs only by diffusion. Both SEM and FCS allow visualization of the effect of inhibitors or other pharmacologically interesting substances on the water flux in one experiment, i.e. on one and the same sample.36,38,41,52 In contrast, demonstrating the effect of an inhibitor on the rate of water efflux from vesicles or cells (see below) requires performing two subsequent experiments (i.e. using two different samples): one in the presence and one in the absence of the inhibitor.SEM was used to measure the water flux and solvent drag through gramicidin derivatives,39,45,53 to functionally characterize several AQPs,41,52,54–56 the translocation complex SecYEG,57 and the potassium channel KcsA,58 and to investigate different routes of epithelial water flow.59 Magnetic stirrers in both compartments ensure that steady-state is reached within a reasonable amount of time.60 Because the volume of the two compartments is large as compared to the flow volume, solute bulk concentrations can be assumed to be constant during the experiment. The steady-state solute concentration distribution in the unstirred layer can be used to determine the velocity vt of water flow across the barrier:
C(x) = Cs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e(−vtx/D)+(ax3/3D) e(−vtx/D)+(ax3/3D) | (9) |
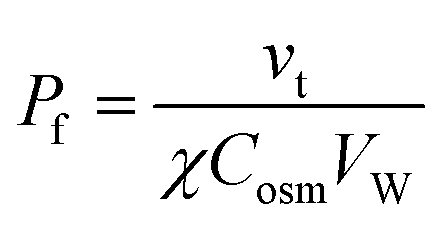 | (10) |
In a similar fashion, fluorophore dilution or up concentration adjacent to the cell monolayer was used to calculate the pf value of AQP5 (ref. 38) and the sodium–glucose cotransporter hSGLT1.36
6 Water flux through K+ channels
To corroborate the notion that pf exponentially depends on length, we substituted gramicidin for another pore: the bacterial potassium channel KcsA. A further shortened gramicidin channel would otherwise have required a matching decrease of bilayer thickness, which would have resulted in rather unstable planar bilayers. The problem does not arise with KcsA since this membrane channel has a “normal” hydrophobic thickness. Its single-file region is much shorter. It is limited to the selectivity filter, which is only four water molecules long. The remaining space is occupied by a water filled cavity that is wide enough to cause negligible resistance to water flow.The structure of the selectivity filter shows water molecules separated by ions.62,63 Commonly it is believed that the water molecules act as spacers in order to reduce the electrostatic repulsion between the ions.64 An alternative model that envisioned a water free selectivity filter65 has not been confirmed experimentally.66 In contrast, a selectivity filter that contains a reduced amount of ions or is completely free of ions was suggested by streaming potential measurements67,68 or by scanning electrochemical microscopy58 (Fig. 6), respectively.
The application of an osmotic pressure across a planar bilayer that is reconstituted with potassium channels results in water flow. The flux of potassium ions that are dragged by the water across the channels can be estimated in terms of the short circuit current (Fig. 6). The ion flux was at least a hundred times smaller than the water flow that was simultaneously measured across the channels by scanning potassium selective microelectrodes.58 The true ion to water flux ratio is most probably larger, since (i) Jw is underestimated, because its calculation assumes an impermeable solute, and (ii) the number of dragged ions is overestimated because the Nernst potential for potassium (compare Fig. 6) drives additional ions through the channel in the direction of the water flow. However, even the lower limit of 100 water molecules per one ion indicates that most of the time, the selectivity filter does not contain ions. That is, KcsA behaves like a water channel and can be used to extend the plot of pf(N) beyond the gramicidin data (Fig. 3).
Counting the channels based on their electrical activity resulted in an unreasonably high pf value. It corresponded to water molecules that were much more mobile than bulk water molecules.58 Channel inactivation provided a possible explanation for the conundrum. That is, 90% of the channels were electrically silent and yet they provided a water pathway.69
Evidence was obtained by counting the channels that were reconstituted into LUVs via their fluorescence.69 The label was attached to an artificially introduced cysteine in the channel. Since LUVs are smaller than the focal volume, we introduced a two-step procedure: first the number of reconstituted vesicles was counted by FCS. Subsequently, the vesicles were dissolved by detergent and the number of channel-containing micelles was counted (see below for a more detailed description). The ratio of the two counts indicated the number of channels per vesicle. The water conducting ability of the reconstituted channels was derived by (i) exposing the proteoliposomes to an osmotic gradient and (ii) continuously monitoring vesicle deflation in terms of the intensity I(t) of the scattered light. In a subsequent publication we derived (i) an analytical equation that links Pf to vesicle volume and (ii) an expression that allows calculation of the vesicle volume from I(t)22 (see methodological details in the next section). Together with several point mutations in the channel that prevented both gating70 and inactivation,71 these technological advancements resulted in the pf value that is depicted in Fig. 3. It confirms the hypothesis that shortening the length of the single-file region augments pf.
β Cell or vesicle deflation as a means to obtain pf
Exposure of cells or vesicles at time t = 0 to a hyperosmotic solution that contains membrane impermeable solutes of concentration cout leads to a Pf dependent volume shrinkage: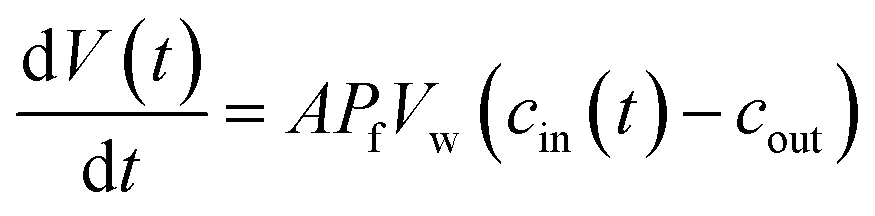 | (11) |
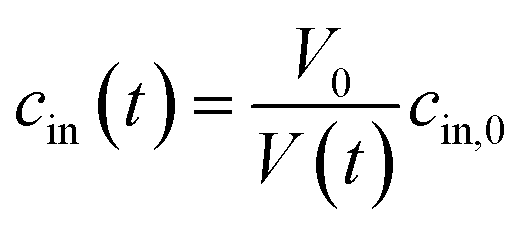 | (12) |
Here we focus on the use of proteoliposomes (PLs), i.e. reconstituted large unilamellar vesicles (LUVs), for Pf measurements (Fig. 7 (top)). PLs are osmotically challenged and the resulting decrease in vesicular volume is derived by monitoring the intensity of the scattered light82,83 or by measuring the fluorescence of an encapsulated dye in self-quenching concentrations.84,85
A variety of approaches exist to extract Pf from V(t):
(α) First, numerically the differential–algebraic equations eqn (11) and (12) are solved for numerous Pf values. In a second step, the theoretical curves are fitted by a monoexponential function to find the characteristic time constants τc for vesicle deflation. Third, the recorded V(t) data are subjected to the same fitting procedure and the experimental constant τ for vesicle deflation is determined. Matching τ to the nearest τc value serves to identify Pf in a fourth step.85
(β) Pf is directly calculated from τ
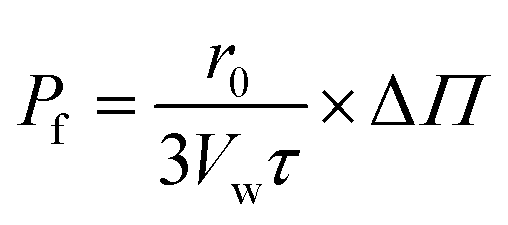 | (13) |
(γ) The analytical solution of eqn (11) is used:22
 | (14) |
The approximate solutions (β) may lead to a large error in Pf – more than an order of magnitude in size.89 Moreover, Pf erroneously seems to depend on the osmotic gradient. In contrast, scanning electrochemical microscopy58 and fluorescence self-quenching experiments (analyzed using a numerical solution)90 show that Pf does not depend on the osmotic gradient. Eqn (14) has been used to calculate the pf values of several AQPs,22,37 hSGLT1,36 and KcsA.91 It may be substituted by the following approximation that ensures acceptable accuracy:89
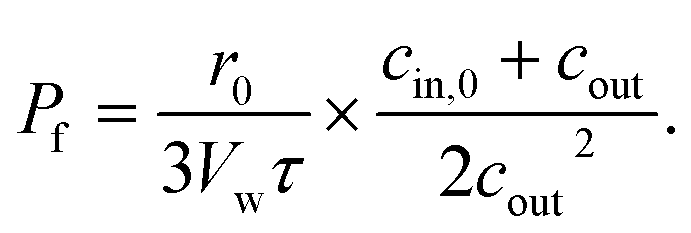 | (15) |
Protein reconstitution is likely to result in two different vesicle populations: bare lipid vesicles and PLs.92 Consequently, two Pf values and two V(t) distributions have to be considered.22,89 Adapting the Rayleigh–Gans–Debye relation and comprising the change in size and refractive index of the lipid vesicles revealed a second order dependency with coefficients a, b, d:22
| I(t) = a + b × V(t) + d × V2(t). | (16) |
In contrast, eqn (15) assumes a linear dependence of V(t) on I(t), which, in the relevant range of r0 (∼30 to 100 nm), leads to an acceptable accuracy. r0 has to be known with great accuracy as the error in Pf linearly depends on the ratio of assumed to real vesicle radii.89
γ Protein counting
Accurate methods of transmembrane protein density estimations are based on direct visualization of single proteins in electrophysiology,39,53,57,58 EM,40 atomic force microscopy (AFM),22 or FCS.22,36,37,69,93 Even though the latter method relies on fluorescence labeling of the channel, FCS is a very convenient, reliable and time efficient method which can be used in vivo as well as in vitro. The protein concentration in the plasma membrane of polarized or non-polarized cells, GUVs or free standing planar lipid bilayers can be directly assessed. LUVs are too small to directly count the number of reconstituted proteins. Instead, two-step procedures are required. First the number of PLs is determined in the protein channel and second the vesicles are dissolved by detergent and the number of fluorescent micelles is counted in the focal volume.22,36,37,69,93 Depending on the protein, the addition of a mild detergent will lead to the formation of protein oligomers containing micelles and further addition of a harsh detergent leads to the formation of protein monomers containing micelles. In the ideal case, the ratio of fluorescent particles in the protein channel leads to an average number n of protein oligomers per proteoliposome, the number of protomers per oligomer, and the ratio of bare lipid vesicles to PLs. For the procedure to work, every protomer must bear a label. If the labeling efficiency is insufficient, the number of protomers per oligomer cannot be accurately determined. The reliability of this protein counting assay was verified by counting the channel density with AFM after spreading AQP-containing PLs on a solid support94 or on a cushion-supported membrane.95 Comparing the channel densities from AFM and FCS measurements revealed satisfactory agreement between the two complementary methods.227 Water flow through AQPs
Extending the dependence of pf(N) (Fig. 3) to longer channels required us to include a totally different class of proteins: AQPs. We first focused on human AQP1 (ref. 52 and 76) that is present in blood cells and in kidneys, on AQPZ,96 and on the aqua-glycerol facilitator GlpF,56 both from E. coli. In contrast to the water selective AQPZ, GlpF facilitates glycerol transport in addition to water.97 Channel reconstitution into liposomes at different protein to lipid ratios, followed by exact measurements of protein abundance using both FCS and high-speed-AFM, and the subsequent assessment of water efflux from osmotically challenged LUVs, allowed the total vesicle membrane permeability Pf to be plotted as a function of the membrane protein concentration.22 The slopes of these dependencies are indicative for pf.Although the pores of both AQPZ and AQP1 accommodate a long single-file that consists of eight water molecules, the pf value of AQP1 exceeds the one of the much shorter KcsA. This observation clearly shows that N cannot generally serve as a determinant of pf. Obviously, the exponential dependence of pf on N is not due to hopping of the water chain along N well-separated binding sites. The simple reason is that the number of water binding sites in both gramicidin or KcsA exceeds N. There are so many hydrogen bond forming residues in the walls of the single-file regions in both types of channels that the distance between two sets of different hydrogen bonding patterns is smaller than the diameter of a water molecule. In contrast, long stretches of the AQP pore contain only a few residues that are able to act as hydrogen bond donors or acceptors.22 That is, the total density of these residues is very different in the three types of channels. Plotting pf as a function of the number NH of potential hydrogen bond donors and acceptors in the walls of the single-file region revealed a logarithmic dependence (Fig. 8). The plot also includes GlpF, which has a shorter single-file region than AQP1 and thus its NH value is also smaller. Accordingly, GlpF turned out to be the most efficient water facilitator (Fig. 2). Water molecules retain their bulk-like mobility within it. The pf value of GlpF is only topped by nanotubes25,98 – possibly because carbon nanotubes are unable to form any hydrogen bonds within the pore. It is important to note that the pf value for carbon nanotubes has been obtained in silico and is not yet confirmed experimentally.99
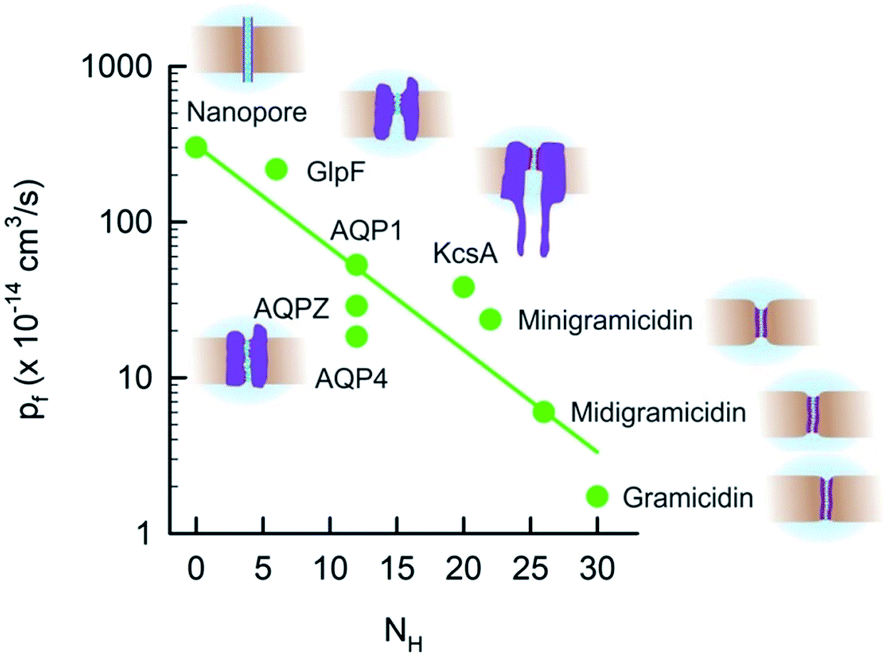 | ||
| Fig. 8 The unitary water channel conductance pf depends exponentially on the number NH of available hydrogen bond donors and acceptors. The data for AQP4 have been taken from ref. 37 and all other data are from ref. 22. | ||
The exponential dependence of pf on NH (Fig. 8) predicts that the pf value of carbon nanotubes does not depend on the tube length because NH = 0 remains invariant. Indeed, the pf value of single-file carbon nanotubes was found in silico to depend very weakly on L.100 For example, tubes that accommodate 17 instead of 3 water molecules have only a 20% lower pf value.
Strikingly, the same invariance of pf on L was obtained in molecular dynamics simulations that used a set of D, L polyalanine peptides with a typical β-helix gramicidin-A folding motif.23 Instead of being governed by hydrogen bond dynamics, water movement was limited by the dehydration penalty at the channel mouth. The reason for the discrepancy between the in vitro and in silico experiments is not entirely clear. A subsequent study identified transient channel blockages by the head groups of adjacent lipids as being responsible for the large quantitative differences in the pf values of the in silico and in vitro studies.101
Fig. 8 clearly shows that NH cannot be the only determinant of water flow through single-file channels. The three water channels – AQP4, AQP1, and AQPZ – offer the same number of hydrogen bonding residues within the water pathway – and yet their pf values significantly differ from each other. An accompanying paper in this volume of Faraday Discussions proposes that the difference in pf is due to the dehydration penalty that the water molecules face upon entering the single-file region:37 if only the hydrogen bonds to the preceding and following water molecules remain intact, the water molecules lose two of their four hydrogen bonds that they form in bulk. The energetic penalty of this process depends on the local environment at the channel mouth. Since positively charged residues are weaker hydrated than negatively charged amino acid side chains,102,103 they may contribute to a pf increase. This hypothesis has been confirmed experimentally: the slowest AQP in Fig. 8, AQP4, offers the smallest number of positive charges. AQP1 has more than twice as many positively charged residues placed at the channel mouths and, accordingly, facilitates water transport three times faster.
We conclude that the network of hydrogen bonds at the channel mouth is important for the fine-tuning of pf (Fig. 9). That is, hydrogen bonds in the single-file region are responsible for a variability in pf that extends over 2 to 3 orders of magnitude, whereas the hydrogen bonded network in the channel mouth alters pf two to three fold.
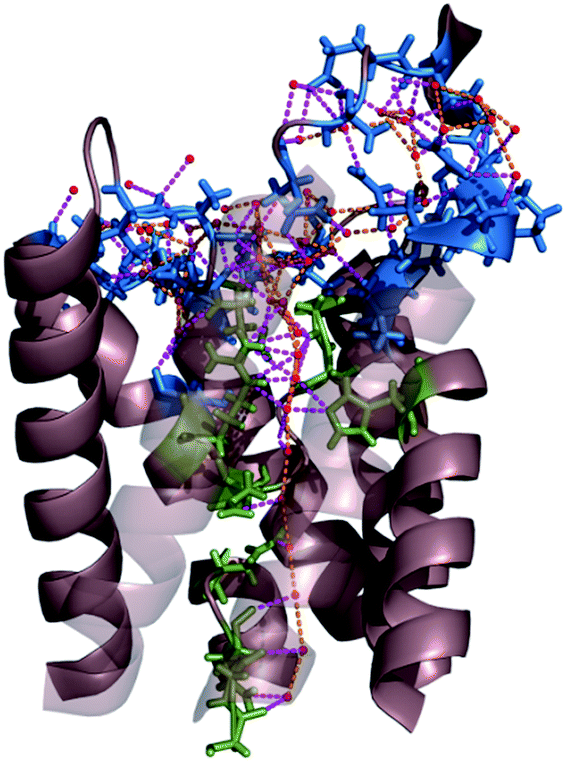 | ||
| Fig. 9 Network of hydrogen bonds within the single-file region of AQP and at the channel mouth. The residues that interact with single-file water molecules and with water molecules in the entrance region are depicted in green and blue, respectively. The H-bonds between water molecules and H-bonds between permeating water molecules and channel residues are shown in orange and pink, respectively. For clarity, the two front helices of yeast AQP1 [Protein Data Bank (PDB) #3Z0J] appear transparent. | ||
8 Energetics of water transport
H2O crosses lipid bilayer membranes by a solubility diffusion mechanism.104 Accordingly, a Gibbs activation energy barrier, ΔG‡l, of 10.5 to 12.4 kcal mol−1 can be estimated for lipid membranes from two parameters of bulk hydrocarbons: 7.8 to 8.9 kcal mol−1 for the enthalpy of the partition coefficient and 2.6 to 3.4 kcal mol−1 for the activation of water diffusion.105 Some fine-tuning was achieved 40 years later by quantifying the impact of two more determinants of water diffusion across cholesterol-free bilayers:82 the area per lipid and the hydrocarbon thickness.84,106The link between ΔG‡l and membrane permeability appears to be very robust – it also works for cholesterol-containing membranes. To obtain ΔG‡t, Pf is measured at different temperatures and ln(Pf) is plotted over 1/T. Cholesterol insertion into pure diphytanoyl phosphatidyl membranes increased ΔG‡l from 12 to 17–18 kcal mol−1 and reduced Pf to less than half of its initial value (at 30 °C).107 Molecular dynamics simulations exploit the link between permeability and ΔG‡l to accurately predict the membrane permeability of water and other small solutes from (i) local partition coefficients and (ii) local diffusion coefficients.108
Similarly, the Gibbs activation energy barrier ΔG‡t for facilitated water transport is intricately linked to pf. Two scenarios can be distinguished:105
(α) Water inside the pores has essentially bulk properties. In this case, the effective energy barrier for permeation is the activation energy for the self-diffusion of water, about 4.6 kcal mol−1.109
(β) The value of 4.6 kcal mol−1 can be taken as a lower limit if water does not retain bulk properties inside the pore. The upper limit has been estimated to be about 15 kcal mol−1, assuming that in addition to the transport activation energy (4.6 kcal mol−1), the energy necessary for a water molecule to pass out of the solution and into the pore has to be considered. It is equal to 10.5 kcal mol−1 – the enthalpy of vaporization.105
Transition state theory allows a closer view on the link between pf and ΔG‡t. We first introduce the “hopping rate” r with which the water chain moves forward or backward:25
| r = pfvw. | (17) |
Second we link r to ΔG‡t:
| r = ν0exp(−ΔG‡t/kBT) | (18) |
| pf = ν0vwexp(−ΔG‡t/kBT). | (19) |
Plotting the experimentally obtained pf values that have been obtained by accurately counting the number of reconstituted channels for which the corresponding ΔG‡t values are known (Fig. 10), we find a satisfactory agreement between the prediction made by eqn (19) and the experiments.
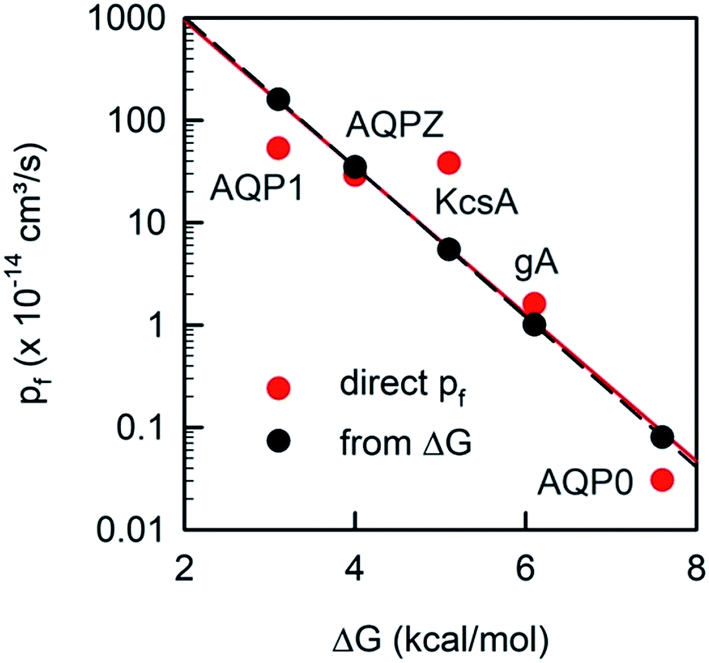 | ||
| Fig. 10 The unitary water permeability pf and the Gibbs activation energy barrier ΔG‡t are intricately linked. The red dots represent experimental results: ΔG‡t has been reported for AQP1,85 AQPZ,96 KcsA,58 gA,110 and AQP0.111pf has also been obtained for AQP1,22 AQPZ,22 KcsA,22 gA,45 and AQP0.40 The black dots are calculated according to eqn (19). | ||
Artificial water channels that could mimic the selectivity and extraordinarily fast flow possible in aquaporins would have a wide range of potential applications.112 Molecular dynamics simulations suggest that this goal is achievable: 8.1 Å wide nanotubes conduct water in single-file, while hydrated sodium and chloride ions are too large to pass in silico.98,100
In contrast, a recently published experimental study reports not only water, but also K+ and C− transport across 10 nm long nanotubes with a pore diameter of 0.68 nm.113 The single nanotube conductivity at 100 mM K+ amounted to ∼30 pS, which is comparable to that of potassium selective channels like Shaker114 or Kir2.1.115 How the nanotube achieves such efficiency without electrostatically attracting K+ to internal binding sites is unclear. Since surrogates for the water molecules of hydration of K+ are missing, the fully hydrated ion should be conducted. If this was the case, water molecules should be able to overtake each other in the channel, and yet H2O is believed to cross the tube in single-file.
At pH = 7.8, ΔG‡t for water transport across these tubes is reported to be equal to 24.1 kcal mol−1.113Eqn (19) predicts an immeasurable small pf value in the order of 10−28 cm3 s−1 and yet the authors claim pf = 6.8 10−13 cm3 s−1. Such a disentanglement between the rate of a process and its activation energy is impossible because it violates fundamental thermodynamic laws.99 Moreover, the measured ΔG‡t value exceeds the upper limit of water transport through a pore of ∼15 kcal mol−1 (see the preceding section). In their rebuttal in Science116 as well as in their contribution in the current Faraday Discussions volume,117 the authors do not provide evidence that water or ions pass the nanotubes. On the contrary, the inhibitory effect of calcium on both the ion and water fluxes is compatible with the view that calcium binding to the lipid alters lipid packing, which may partly seal the nanotube induced leak for water and ions in the bilayer.
9 Additional factors that affect pf
The example of AQP0 shows that factors besides the number of hydrogen bonds and surface charges may have an effect on pf: AQP0 offers 16 residues that accept or donate hydrogen bonds in its pore forming wall,22 and yet the pf value is orders of magnitude smaller than that suggested by Fig. 8. Conformational transitions of the protein may explain the discrepancy. AQP0 was found in a closed118 as well as an open state.119 In addition, AQP0 was reported to be modulated by pH120 and calmodulin binding.121 Hence, the low pf value of AQP0 (Fig. 2) may be due to a considerable amount of time the channel resides in its closed state.The crystal structure of AqpZ also revealed two distinct conformations – different positions of Arg-189,122 yet its pf value corresponds very well to the prediction that can be made from NH. The explanation is provided by recent NMR spectroscopy experiments. They revealed no indication of R189 gating in reconstituted AQPZ.123
Geometry effects may also play a minor role in fluid flow across biological channels and are believed to be important determinants for flow across nanotubes.124 A theoretical analysis of water transport through AQPs suggested that the form of the vestibule may boost AQP’s pf value by several-fold. The pf value was lowest for the channels with a cylindrical geometry, and highest for the channels with an hourglass shape.24 However, the geometry effects were derived from a continuum description that was unable to capture the water movement inside the central constriction zone. Contrary to the intuitive assumption that resistance is highest in the narrowest part of the channel, permeation through this one-water-molecule-wide part of the AQP pore was thought to occur free of any resistance.24 Experimental studies that would confirm this in silico prediction are still missing.
Conclusions
We conclude that the major determinant of water flow across biological channels is the number of hydrogen bonds that water molecules may form on their way through the channel. Additional, but much less important factors, are: positive surface charges at the channel mouth, the presence of a closed conformational state of the channel, and possibly also the geometry of the vestibule at the channel mouth.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grant P23466 of the Austrian Science Fund (FWF) to P. P.References
- M.-C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673–7697 CrossRef PubMed.
- O. Björneholm, M. H. Hansen, A. Hodgson, L.-M. Liu, D. T. Limmer, A. Michaelides, P. Pedevilla, J. Rossmeisl, H. Shen, G. Tocci, E. Tyrode, M.-M. Walz, J. Werner and H. Bluhm, Chem. Rev., 2016, 116, 7698–7726 CrossRef PubMed.
- J. Carrasco, A. Michaelides, M. Forster, S. Haq, R. Raval and A. Hodgson, Nat. Mater., 2009, 8, 427–431 CrossRef PubMed.
- P. Fenter and N. C. Sturchio, Prog. Surf. Sci., 2004, 77, 171–258 CrossRef.
- O. Teschke, G. Ceotto and E. F. de Souza, Chem. Phys. Lett., 2000, 326, 328–334 CrossRef.
- A. L. Lomize, I. D. Pogozheva and H. I. Mosberg, J. Chem. Inf. Model., 2011, 51, 930–946 CrossRef PubMed.
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 CrossRef PubMed.
- L. R. Merte, G. Peng, R. Bechstein, F. Rieboldt, C. A. Farberow, L. C. Grabow, W. Kudernatsch, S. Wendt, E. Lægsgaard, M. Mavrikakis and F. Besenbacher, Science, 2012, 336, 889–893 CrossRef PubMed.
- A. Springer, V. Hagen, D. A. Cherepanov, Y. N. Antonenko and P. Pohl, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 14461–14466 CrossRef PubMed.
- E. Weichselbaum, M. Osterbauer, D. G. Knyazev, O. V. Batishchev, S. A. Akimov, T. H. Nguyen, C. Zhang, G. Knor, N. Agmon, P. Carloni and P. Pohl, Sci. Rep., 2017, 7, 4553 CrossRef PubMed.
- N. Agmon, H. J. Bakker, R. K. Campen, R. H. Henchman, P. Pohl, S. Roke, M. Thämer and A. Hassanali, Chem. Rev., 2016, 116, 7642–7672 CrossRef PubMed.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef PubMed.
- J. Mittal and G. Hummer, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 20130–20135 CrossRef PubMed.
- L. F. Scatena, M. G. Brown and G. L. Richmond, Science, 2001, 292, 908–912 CrossRef PubMed.
- O. Beckstein and M. S. Sansom, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 7063–7068 CrossRef PubMed.
- D. S. Grebenkov, Y. A. Goddard, G. Diakova, J. P. Korb and R. G. Bryant, J. Phys. Chem. B, 2009, 113, 13347–13356 CrossRef PubMed.
- B. Halle, Philos. Trans. R. Soc. Lond. B Biol. Sci., 2004, 359, 1207–1223 CrossRef PubMed ; discussion 1223–1204, 1323–1208.
- O. B. Tarun, C. Hannesschlaeger, P. Pohl and S. Roke, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 4081–4086 CrossRef PubMed.
- D. Laage, T. Elsaesser and J. T. Hynes, Chem. Rev., 2017, 117, 10694–10725 CrossRef PubMed.
- J. T. King, E. J. Arthur, C. L. Brooks 3rd and K. J. Kubarych, J. Am. Chem. Soc., 2014, 136, 188–194 CrossRef PubMed.
- E. Secchi, S. Marbach, A. Nigues, D. Stein, A. Siria and L. Bocquet, Nature, 2016, 537, 210–213 CrossRef PubMed.
- A. Horner, F. Zocher, J. Preiner, N. Ollinger, C. Siligan, S. A. Akimov and P. Pohl, Sci. Adv., 2015, 1, e1400083 Search PubMed.
- G. Portella, P. Pohl and B. L. de Groot, Biophys. J., 2007, 92, 3930–3937 CrossRef PubMed.
- S. Gravelle, L. Joly, F. Detcheverry, C. Ybert, C. Cottin-Bizonne and L. Bocquet, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 16367–16372 CrossRef PubMed.
- A. Berezhkovskii and G. Hummer, Phys. Rev. Lett., 2002, 89, 064503 CrossRef PubMed.
- T. Chou, Phys. Rev. Lett., 1998, 80, 85–88 CrossRef.
- J. Kofinger, G. Hummer and C. Dellago, Phys. Chem. Chem. Phys., 2011, 13, 15403–15417 RSC.
- A. Finkelstein, Water movement through lipid bilayers, pores, and plasma membranes, Wiley & Sons, New York, 1987 Search PubMed.
- B. L. de Groot and H. Grubmuller, Science, 2001, 294, 2353–2357 CrossRef PubMed.
- U. Kosinska Eriksson, G. Fischer, R. Friemann, G. Enkavi, E. Tajkhorshid and R. Neutze, Science, 2013, 340, 1346–1349 CrossRef PubMed.
- D. G. Levitt and G. Subramanian, Biochim. Biophys. Acta, 1974, 373, 132–140 CrossRef.
- F. Zhu, E. Tajkhorshid and K. Schulten, Phys. Rev. Lett., 2004, 93, 224501 CrossRef PubMed.
- P. H. Barry and J. M. Diamond, Physiol. Rev., 1984, 64, 763–872 CrossRef PubMed.
- P. Pohl, S. M. Saparov and Y. N. Antonenko, Biophys. J., 1997, 72, 1711–1718 CrossRef PubMed.
- S. Paula, M. Akeson and D. Deamer, Biochim. Biophys. Acta, 1999, 1418, 117–126 CrossRef.
- L. Erokhova, A. Horner, N. Ollinger, C. Siligan and P. Pohl, J. Biol. Chem., 2016, 291, 9712–9720 CrossRef PubMed.
- A. Horner, C. Siligan, A. Cornean and P. Pohl, Faraday Discuss., 2018 10.1039/c8fd00050f.
- L. Erokhova, A. Horner, P. Kugler and P. Pohl, J. Biol. Chem., 2011, 286, 39926–39932 CrossRef PubMed.
- S. M. Saparov, J. R. Pfeifer, L. Al-Momani, G. Portella, B. L. de Groot, U. Koert and P. Pohl, Phys. Rev. Lett., 2006, 96, 148101 CrossRef PubMed.
- G. A. Zampighi, M. Kreman, K. J. Boorer, D. D. Loo, F. Bezanilla, G. Chandy, J. E. Hall and E. M. Wright, J. Membr. Biol., 1995, 148, 65–78 CrossRef PubMed.
- S. M. Saparov, K. Liu, P. Agre and P. Pohl, J. Biol. Chem., 2007, 282, 5296–5301 CrossRef PubMed.
- B. Yang and A. S. Verkman, J. Biol. Chem., 1998, 273, 9369–9372 CrossRef PubMed.
- T. Gonen, Y. Cheng, P. Sliz, Y. Hiroaki, Y. Fujiyoshi, S. C. Harrison and T. Walz, Nature, 2005, 438, 633–638 CrossRef PubMed.
- E. Tajkhorshid, P. Nollert, M. O. Jensen, L. J. Miercke, J. O’Connell, R. M. Stroud and K. Schulten, Science, 2002, 296, 525–530 CrossRef PubMed.
- P. Pohl and S. M. Saparov, Biophys. J., 2000, 78, 2426–2434 CrossRef PubMed.
- P. A. Rosenberg and A. Finkelstein, J. Gen. Physiol., 1978, 72, 341–350 CrossRef PubMed.
- J. C. Mathai, S. Mori, B. L. Smith, G. M. Preston, N. Mohandas, M. Collins, P. C. van-Zijl, M. L. Zeidel and P. Agre, J. Biol. Chem., 1996, 271, 1309–1313 CrossRef PubMed.
- D. Huster, A. J. Jin, K. Arnold and K. Gawrisch, Biophys. J., 1997, 73, 855–864 CrossRef PubMed.
- J. Brahm, J. Gen. Physiol., 1982, 79, 791–819 CrossRef PubMed.
- R. Fettiplace and D. A. Haydon, Physiol. Rev., 1980, 60, 510–550 CrossRef PubMed.
- P. Pohl, S. M. Saparov and Y. N. Antonenko, Biophys. J., 1998, 75, 1403–1409 CrossRef PubMed.
- S. M. Saparov, D. Kozono, U. Rothe, P. Agre and P. Pohl, J. Biol. Chem., 2001, 276, 31515–31520 CrossRef PubMed.
- S. M. Saparov, Y. N. Antonenko, R. E. Koeppe and P. Pohl, Biophys. J., 2000, 79, 2526–2534 CrossRef PubMed.
- S. M. Saparov, M. J. Borgnia, P. Agre and P. Pohl, Biophys. J., 2000, 78, 389A Search PubMed.
- S. M. Saparov, U. Rothe, M. J. Borgnia, P. Agre and P. Pohl, in Molecular biology and physiology of water and solute transport, ed. S. Hohmann and S. Nielsen, Kluwer Academic/Plenum Publishers, New York, 2000, ch. 1.6, pp. 41–48 Search PubMed.
- S. M. Saparov, S. P. Tsunoda and P. Pohl, Biol. Cell, 2005, 97, 545–550 CrossRef PubMed.
- S. M. Saparov, K. Erlandson, K. Cannon, J. Schaletzky, S. Schulman, T. A. Rapoport and P. Pohl, Mol. Cell, 2007, 26, 501–509 CrossRef PubMed.
- S. M. Saparov and P. Pohl, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 4805–4809 CrossRef PubMed.
- R. Mollajew, F. Zocher, A. Horner, B. Wiesner, E. Klussmann and P. Pohl, Biophys. J., 2010, 99, 3647–3656 CrossRef PubMed.
- P. Pohl, S. M. Saparov and Y. N. Antonenko, Prog. Biophys. Mol. Biol., 1996, 65, 97 Search PubMed.
- A. V. Krylov, P. Pohl, M. L. Zeidel and W. G. Hill, J. Gen. Physiol., 2001, 118, 333–340 CrossRef PubMed.
- J. H. Morais-Cabral, Y. Zhou and R. MacKinnon, Nature, 2001, 414, 37–42 CrossRef PubMed.
- D. A. Doyle, C. J. Morais, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef PubMed.
- S. Berneche and B. Roux, Nature, 2001, 414, 73–77 CrossRef PubMed.
- D. A. Kopfer, C. Song, T. Gruene, G. M. Sheldrick, U. Zachariae and B. L. de Groot, Science, 2014, 346, 352–355 CrossRef PubMed.
- H. T. Kratochvil, J. K. Carr, K. Matulef, A. W. Annen, H. Li, M. Maj, J. Ostmeyer, A. L. Serrano, H. Raghuraman, S. D. Moran, J. L. Skinner, E. Perozo, B. Roux, F. I. Valiyaveetil and M. T. Zanni, Science, 2016, 353, 1040–1044 CrossRef PubMed.
- C. Miller, Biophys. J., 1982, 38, 227–230 CrossRef PubMed.
- C. Alcayaga, X. Cecchi, O. Alvarez and R. Latorre, Biophys. J., 1989, 55, 367–371 CrossRef PubMed.
- T. Hoomann, N. Jahnke, A. Horner, S. Keller and P. Pohl, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 10842–10847 CrossRef PubMed.
- A. N. Thompson, D. J. Posson, P. V. Parsa and C. M. Nimigean, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 6900–6905 CrossRef PubMed.
- L. G. Cuello, V. Jogini, D. M. Cortes and E. Perozo, Nature, 2010, 466, 203–208 CrossRef PubMed.
- K. Olbrich, W. Rawicz, D. Needham and E. Evans, Biophys. J., 2000, 79, 321–327 CrossRef PubMed.
- W. Rawicz, B. A. Smith, T. J. McIntosh, S. A. Simon and E. Evans, Biophys. J., 2008, 94, 4725–4736 CrossRef PubMed.
- A. Berthaud, F. Quemeneur, M. Deforet, P. Bassereau, F. Brochard-Wyart and S. Mangenot, Soft Matter, 2016, 12, 1601–1609 RSC.
- K. Maric, B. Wiesner, D. Lorenz, E. Klussmann, T. Betz and W. Rosenthal, Biophys. J., 2001, 80, 1783–1790 CrossRef PubMed.
- S. P. Tsunoda, B. Wiesner, D. Lorenz, W. Rosenthal and P. Pohl, J. Biol. Chem., 2004, 279, 11364–11367 CrossRef PubMed.
- J. Farinas, V. Simanek and A. S. Verkman, Biophys. J., 1995, 68, 1613–1620 CrossRef PubMed.
- M. Zelenina and H. Brismar, Eur. Biophys. J., 2000, 29, 165–171 CrossRef PubMed.
- J. Fischbarg, K. Y. Kuang, J. C. Vera, S. Arant, S. C. Silverstein, J. Loike and O. M. Rosen, Proc. Natl. Acad. Sci. U.S.A., 1990, 87, 3244–3247 CrossRef.
- G. M. Preston, T. P. Carroll, W. B. Guggino and P. Agre, Science, 1992, 256, 385–387 CrossRef PubMed.
- P. P. Duquette, P. Bissonnette and J. Y. Lapointe, Proc. Natl. Acad. Sci. U.S.A., 2001, 98, 3796–3801 CrossRef PubMed.
- D. F. Savage and R. M. Stroud, J. Mol. Biol., 2007, 368, 607–617 CrossRef PubMed.
- D. Kozono, X. Ding, I. Iwasaki, X. Meng, Y. Kamagata, P. Agre and Y. Kitagawa, J. Biol. Chem., 2003, 278, 10649–10656 CrossRef PubMed.
- J. C. Mathai, S. Tristram-Nagle, J. F. Nagle and M. L. Zeidel, J. Gen. Physiol., 2008, 131, 69–76 CrossRef PubMed.
- M. L. Zeidel, S. V. Ambudkar, B. L. Smith and P. Agre, Biochemistry, 1992, 31, 7436–7440 CrossRef PubMed.
- M. P. van Heeswijk and C. H. van Os, J. Membr. Biol., 1986, 92, 183–193 CrossRef PubMed.
- M. J. Borgnia, D. Kozono, G. Calamita, P. C. Maloney and P. Agre, J. Mol. Biol., 1999, 291, 1169–1179 CrossRef PubMed.
- R. E. Farmer and R. I. Macey, Biochim. Biophys. Acta, 1970, 196, 53–65 CrossRef.
- C. Hannesschläger, T. Barta, C. Siligan and A. Horner, Sci. Rep., 2018, 8, 8516 CrossRef PubMed.
- P. Y. Chen, D. Pearce and A. S. Verkman, Biochemistry, 1988, 27, 5713–5718 CrossRef PubMed.
- A. Horner, F. Zocher, J. Preiner, N. Ollinger, C. Siligan, S. A. Akimov and P. Pohl, Biophys. J., 2015, 108, 182A CrossRef.
- N. Jahnke, O. O. Krylova, T. Hoomann, C. Vargas, S. Fiedler, P. Pohl and S. Keller, Anal. Chem., 2014, 86, 920–927 CrossRef PubMed.
- D. G. Knyazev, A. Lents, E. Krause, N. Ollinger, C. Siligan, D. Papinski, L. Winter, A. Horner and P. Pohl, J. Biol. Chem., 2013, 288, 17941–17946 CrossRef PubMed.
- J. Preiner, A. Horner, A. Karner, N. Ollinger, C. Siligan, P. Pohl and P. Hinterdorfer, Nano Lett., 2015, 15, 759–763 CrossRef PubMed.
- A. Karner, B. Nimmervoll, B. Plochberger, E. Klotzsch, A. Horner, D. G. Knyazev, R. Kuttner, K. Winkler, L. Winter, C. Siligan, N. Ollinger, P. Pohl and J. Preiner, Nat. Nanotechnol., 2017, 12, 260–266 CrossRef PubMed.
- P. Pohl, S. M. Saparov, M. J. Borgnia and P. Agre, Proc. Natl. Acad. Sci. U.S.A., 2001, 98, 9624–9629 CrossRef PubMed.
- M. J. Borgnia and P. Agre, Proc. Natl. Acad. Sci. U.S.A., 2001, 98, 2888–2893 CrossRef PubMed.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef PubMed.
- A. Horner and P. Pohl, Science, 2018, 359, eaap9173 CrossRef PubMed.
- A. Kalra, S. Garde and G. Hummer, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 10175–10180 CrossRef PubMed.
- G. Portella, T. Polupanow, F. Zocher, D. A. Boytsov, P. Pohl, U. Diederichsen and B. L. de Groot, Biophys. J., 2012, 103, 1698–1705 CrossRef PubMed.
- K. D. Collins, Biophys. J., 1997, 72, 65–76 CrossRef PubMed.
- M. Y. Kiriukhin and K. D. Collins, Biophys. Chem., 2002, 99, 155–168 CrossRef PubMed.
- A. Finkelstein, J. Gen. Physiol., 1976, 68, 127–135 CrossRef PubMed.
- H. D. Price and T. E. Thompson, J. Mol. Biol., 1969, 41, 443–457 CrossRef PubMed.
- J. F. Nagle, J. C. Mathai, M. L. Zeidel and S. Tristram-Nagle, J. Gen. Physiol., 2008, 131, 77–85 CrossRef PubMed.
- P. J. Milianta, M. Muzzio, J. Denver, G. Cawley and S. Lee, Langmuir, 2015, 31, 12187–12196 CrossRef PubMed.
- F. Zocher, D. van der Spoel, P. Pohl and J. S. Hub, Biophys. J., 2013, 105, 2760–2770 CrossRef PubMed.
- J. H. Wang, C. V. Robinson and I. S. Edelman, J. Am. Chem. Soc., 1953, 75, 466–470 CrossRef.
- B. A. Boehler, J. De Gier and L. L. M. Van Deenen, Biochim. Biophys. Acta, 1978, 512, 480–488 CrossRef.
- M. Kumar, J. E. Habel, Y. X. Shen, W. P. Meier and T. Walz, J. Am. Chem. Soc., 2012, 134, 18631–18637 CrossRef PubMed.
- J. K. Holt, H. G. Park, Y. Wang, M. Stadermann, A. B. Artyukhin, C. P. Grigoropoulos, A. Noy and O. Bakajin, Science, 2006, 312, 1034–1037 CrossRef PubMed.
- R. H. Tunuguntla, R. Y. Henley, Y.-C. Yao, T. A. Pham, M. Wanunu and A. Noy, Science, 2017, 357, 792–796 CrossRef PubMed.
- L. Heginbotham and R. MacKinnon, Biophys. J., 1993, 65, 2089–2096 CrossRef PubMed.
- H. Choe, H. Sackin and L. G. Palmer, J. Gen. Physiol., 2000, 115, 391–404 CrossRef PubMed.
- R. H. Tunuguntla, Y. Zhang, R. Y. Henley, Y.-C. Yao, T. A. Pham, M. Wanunu and A. Noy, Science, 2018, 359, eaaq1241 CrossRef PubMed.
- R. Tunuguntla, A. Y. Hu, Y. Zhang and A. Noy, Faraday Discuss., 2018 10.1039/c8fd00068a.
- T. Gonen, P. Sliz, J. Kistler, Y. F. Cheng and T. Walz, Nature, 2004, 429, 193–197 CrossRef PubMed.
- W. E. Harries, D. Akhavan, L. J. Miercke, S. Khademi and R. M. Stroud, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 14045–14050 CrossRef PubMed.
- K. L. Nemeth-Cahalan and J. E. Hall, J. Biol. Chem., 2000, 275, 6777–6782 CrossRef PubMed.
- S. L. Reichow, D. M. Clemens, J. A. Freites, K. L. Nemeth-Cahalan, M. Heyden, D. J. Tobias, J. E. Hall and T. Gonen, Nat. Struct. Mol. Biol., 2013, 20, 1085–1092 CrossRef PubMed.
- J. Jiang, B. V. Daniels and D. Fu, J. Biol. Chem., 2006, 281, 454–460 CrossRef PubMed.
- Y. Zhao, H. Xie, L. Wang, Y. Shen, W. Chen, B. Song, Z. Zhang, A. Zheng, Q. Lin, R. Fu, J. Wang and J. Yang, J. Am. Chem. Soc., 2018, 140, 7885–7895 CrossRef PubMed.
- S. Gravelle, L. Joly, C. Ybert and L. Bocquet, J. Chem. Phys., 2014, 141, 18C526 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |