 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein–protein interactions in AQP regulation – biophysical characterization of AQP0–CaM and AQP2–LIP5 complex formation†
Stefan
Kreida‡
,
Jennifer Virginia
Roche‡
,
Caroline
Olsson
,
Sara
Linse
 and
Susanna
Törnroth-Horsefield
*
and
Susanna
Törnroth-Horsefield
*
Department of Biochemistry and Structural Biology, Lund University, P.O. Box 124, 221 00 Lund, Sweden. E-mail: susanna.horsefield@biochemistry.lu.se
First published on 25th April 2018
Abstract
Protein–protein interactions play important roles in regulating human aquaporins (AQP) by gating as well as trafficking. While structural and functional studies have provided detailed knowledge of AQP transport mechanisms, selectivity as well as gating by conformational changes of loops or termini, the mechanism behind how protein–protein interactions control AQP-mediated water transport through cellular membranes remains poorly characterized. Here we explore the interaction between two human AQPs and regulatory proteins: the interaction between AQP0 and calmodulin, which mediates AQP0 gating, as well as the interaction between AQP2 and LIP5, which is involved in trafficking. Using microscale thermophoresis (MST) and fluorescence anisotropy, two methods that have the advantage of low sample consumption and detergent compatibility, we show that the interactions can be studied using both full-length AQPs and AQP peptides corresponding to the regulatory protein binding sites. However, full-length AQPs gave better reproducibility between methods and for the first time revealed that AQP0 binds CaM in a cooperative manner, which was not seen in experiments using peptides. Our study highlights that, while peptides are great tools for locating binding sites and pinpointing interacting residues, full-length proteins may give additional insights, such as binding mechanism, allostery and cooperativity, important parameters for understanding protein–protein mediated regulation in the cellular context. Our work provides a platform for further studies of AQP regulation that may be of interest for designing drugs that target AQP complexes as well as the development of artificial bio-mimetic water channels for water-purification purposes.
Introduction
In higher eukaryotes, tissue-specific control of trans-membrane water transport is fundamental for organism physiology. This is achieved by regulating aquaporins (AQPs), membrane-bound water channels that facilitate water transport across cellular membranes along osmotic gradients. Eukaryotic AQPs may be regulated by gating, where a conformational change of the protein occludes the water-conducting pore, or by controlling AQP membrane abundance through trafficking between the plasma membrane and intracellular storage sites.1 Regulation of human AQPs is crucial for many physiological processes, including urine concentration, maintaining lens transparency and secretion of isotonic fluids.2 Of the thirteen human AQP isoforms that have been identified, the majority are regulated by trafficking in response to hormones or other cellular signals.3 This is by far best characterized for AQP2 in the kidney collecting duct for which vasopressin-dependent trafficking between storage vesicles and the apical membrane is essential for urine volume regulation.4 In contrast, there is only one conclusively established example of human AQP regulation by gating: the calmodulin-mediated gating of AQP0 in the eye lens fiber cells, which plays an important role in lens water homeostasis.5,6 Both regulatory mechanisms are controlled by protein–protein interactions; trafficking of AQP2 and other AQPs rely on interactions with proteins within the cellular vesicle trafficking machinery that govern the movement of AQPs between cellular membranes7,8 whereas AQP0 gating is achieved by direct binding of calmodulin (CaM) to the cytoplasmic AQP0 surface.5 Regulatory AQP protein–protein interactions can be modulated by post-translational modifications of the AQP C-terminus, allowing for additional control of the regulatory process.9Structural and functional studies of a number of AQPs from several different organisms have shown that all AQPs share a common fold and mechanism for transport and selectivity (Fig. 1A).1 Furthermore, detailed mechanisms for how plant and yeast AQPs may be gated by conformational changes of a cytoplasmic loop and N-terminus respectively are well established.10 In contrast, the mechanism behind how protein–protein interactions control human AQP trafficking and gating remain poorly characterized. Most studies have been done at the cellular level, for example using co-immuno-precipitation or yeast two-hybrid assays.11 Direct interaction between AQPs and a regulatory protein has mainly been probed using AQP peptides corresponding to putative binding sites,5,6,12 which often gives valuable information concerning location of binding sites and specific interacting residues but does not provide the complete biological context. Peptide-based interaction studies may therefore miss additional factors that affect binding affinity, such as allosteric sites and/or the accessibility of the interaction motif. To fully understand human AQP regulation, detailed knowledge of how they interact with regulatory proteins in a full-length context and how this may be controlled by AQP post-translational modifications is essential. Such knowledge could be used to design drugs that target AQP complexes in diseases where AQP regulation is disturbed. Furthermore, an increased understanding of the specific molecular interactions that regulate AQP function in vivo may have implications for the development of artificial water channels for water purification purposes13–15 with novel regulatory properties.
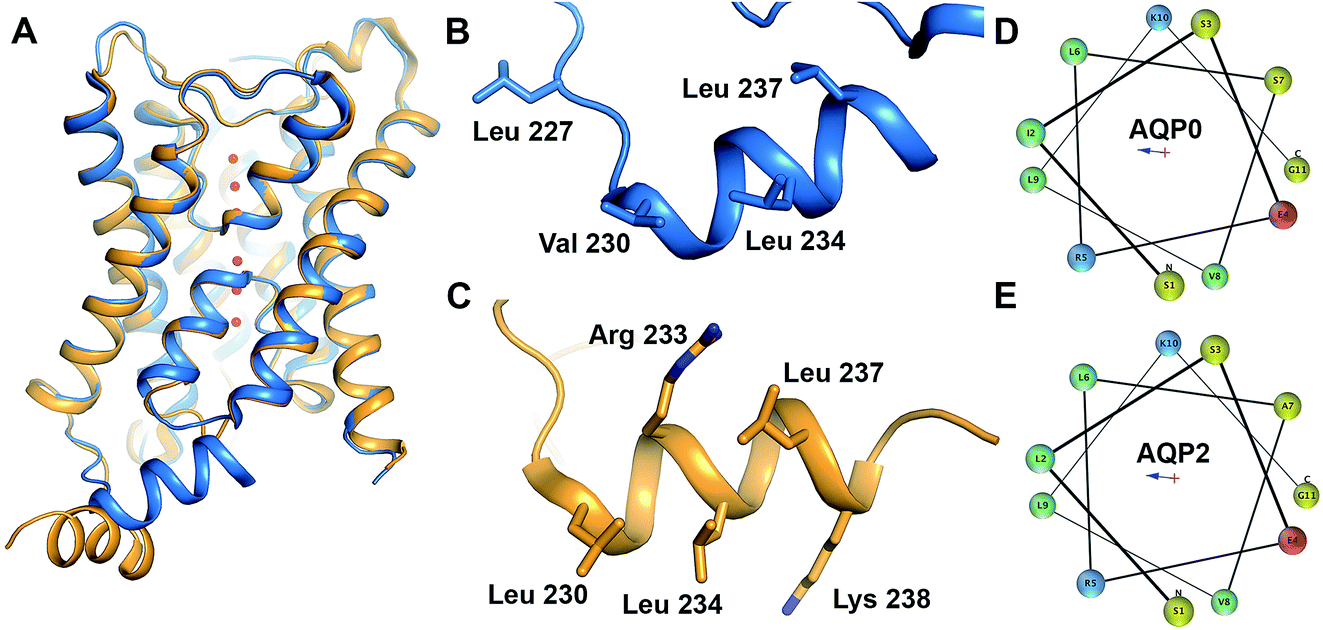 | ||
| Fig. 1 Structure of AQP0 and AQP2. (A) Overlay of crystal structures of human AQP2 (yellow, PDB code 4NEF18) and bovine AQP0 (blue, PDB code 1YMG32). The two structures are very similar except for the position of the C-terminal helix. (B) Proposed CaM-binding site within the C-terminal helix of bovine AQP0. Residues suggested to be involved in the interaction are highlighted in stick representation. (C) Proposed LIP5-binding site within the C-terminal helix of human AQP2. Residues suggested to be involved in the interaction are highlighted in stick representation. Helical wheel representation of the C-terminal helix of (D) human AQP0 and (E) human AQP2 showing its amphipathic character. Residues are colour-coded as follows: aliphatic-green, basic-blue, acidic-red, polar-yellow. | ||
In this study we explore the interaction between AQP0 and CaM as well as AQP2 and the lysosomal trafficking regulatory protein LIP5 (LYST-interacting protein 5), thereby exemplifying protein–protein interactions involved in AQP gating and trafficking respectively.5,16 For AQP0, Ca2+-dependent binding of CaM to a cytoplasmic helix in the AQP0 C-terminus (Fig. 1B) causes a conformational change that mediates pore closure.5 The corresponding helix is also responsible for the interaction between AQP2 and LIP5 (Fig. 1C), which is involved in targeting AQP2 to multivesicular bodies for subsequent degradation or release as exosomes.9 The cytoplasmic C-terminal helix is a common structural feature amongst mammalian AQPs, although its relative location in respect to the tetramer differs between human AQP2 and other mammalian AQPs (Fig. 1A). Comparison of the AQP0 and AQP2 C-terminal helices reveal that they are very similar in sequence and display a clear amphipathic character, with the hydrophobic side proposed to mediate the interaction with CaM and LIP5, respectively (Fig. 1D and E). This may represent a common site for other regulatory protein–protein interactions involving mammalian AQPs.
We have recently characterized the interaction between human AQP2 and mouse LIP5, showing that this interaction is allosterically controlled by C-terminal phosphorylation of AQP2 at sites distal to the proposed LIP5 binding site.9 To our knowledge this is the only study that has investigated AQP protein–protein interactions using isolated full-length proteins in a quantitative manner. The finding that AQP2 phosphorylation at sites outside the LIP5 binding site affects the interaction highlights the importance of using full-length proteins to fully understand AQP protein–protein interactions and its role in AQP regulation. In the case of AQP0, biophysical characterization of its interaction with CaM has only been done using C-terminal AQP0 peptides.5,6 ITC studies have suggested that one CaM binds two copies of the AQP0 C-terminal helix using a two-step binding process where a high-affinity binding event (Kd = 71 nM) is followed by a low-affinity event (Ka = 13 μM).5 In contrast, fluorescence-based studies using dansyl–CaM propose a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding event with a Kd of 0.5 μM and that a phosphorylated AQP0 peptide has significantly lower affinity.17 Interaction studies between full-length AQP0 and CaM are likely to help sort out these discrepancies and may provide additional details regarding the interaction in the biological context.
:1 binding event with a Kd of 0.5 μM and that a phosphorylated AQP0 peptide has significantly lower affinity.17 Interaction studies between full-length AQP0 and CaM are likely to help sort out these discrepancies and may provide additional details regarding the interaction in the biological context.
Here we use full-length human AQP0 and AQP2, as well as peptides corresponding to their C-termini, to characterize their respective interaction with human CaM and LIP5 by microscale thermophoresis (MST) and fluorescence anisotropy. The two methods have been chosen for their low sample consumption, compatibility with detergent-containing buffers and the ability to measure interaction in solution. The interaction between full-length AQP2 as well as the AQP2 peptide and LIP5 could be explored using both MST and fluorescence anisotropy. While both methods gave similar affinity estimates for full-length AQP2, the two methods gave very different results for the AQP2 peptide. MST-studies of the interaction between full-length AQP0 and CaM showed that CaM binds AQP0 in a strongly cooperative manner whereas the same interaction could not be measured by fluorescence anisotropy. For the AQP0 peptide, both methods suggested a non-cooperative interaction of similar affinity as seen for full-length AQP0. In summary, our studies highlight that while peptides can be successfully used to probe protein–protein interactions in many cases, the use of full-length proteins provides more reliable estimates of affinity and is capable of giving important insights about the cooperativity and regulation of the interaction that otherwise may be missed. Furthermore, our finding that AQP0 binds CaM in a cooperative manner provides new insights into the mechanism behind CaM-mediated gating of AQP0.
Experimental methods
Expression and purification of human AQP0 and AQP2
Full-length human AQP0 and AQP2 were expressed in Pichia pastoris as described previously.18,19 The AQP0-construct contained a C-terminal 6 × His-tag whereas the AQP2-construct contained an N-terminal 8 × His-tag. Cells were grown in a fermenter (Belach Bioteknik) and protein expression was induced using methanol for 24–36 hours for AQP2 and 120 hours for AQP0, resulting in a typical yield of 300 g wet cells per litre of culture.50–100 g cells were resuspended in 50 mM phosphate buffer pH 7.5, 5% glycerol and broken using a bead beater (12 × 30 cycles with 30 s waiting in between). The broken cells were centrifuged at 10000 × g (30 min) to remove broken cells and debris after which crude membranes were isolated by centrifugation at 100000 × g (1 h). The membranes were homogenized and washed twice, first using wash buffer (5 mM Tris–HCl pH 9.5, 4 M urea, 2 mM EDTA) followed by membrane buffer (20 mM Tris–HCl pH 8, 20 mM NaCl, 10% glycerol) supplemented with 1 mM PMSF, 2 mM EDTA, with centrifugation at 100000 × g for 2 h in between each washing step. The pellet was finally resuspended in 20 mM Tris pH 8, 20 mM NaCl, 10% glycerol at a concentration of 0.5 mg ml−1 and stored at −80 °C until further use.
Membranes were diluted 1:1 with solubilisation buffer 20 mM Tris, pH 8, 300 mM NaCl, 10% glycerol, 2% DDM (Anatrace) for AQP0, 20 mM Tris–HCl pH 8, 300 mM NaCl, 4% OGNG (Anatrace) for AQP2 supplemented with one cOmplete™ EDTA-free protease inhibitor cocktail tablet (Roche). Final solubilization volume was 50 ml and the final detergent concentration was 1% DDM and 2% OGNG for AQP0 and AQP2 respectively. After solubilisation with continuous stirring at 4 °C for 1–2 h, unsolubilized material was spun down at 100000 × g (30 min). The supernatant was supplemented with 10 mM imidazole and loaded onto a HisTrap column (GE Healthcare) equilibrated with buffer A (20 mM Tris, pH 8, 300 mM NaCl, 10% glycerol, 0.05% DDM for AQP0, 20 mM Tris–HCl pH 8, 300 mM NaCl, 0.2% OGNG for AQP2). The column was washed with 10 column volumes (CV) of buffer A + 75 mM imidazole and eluted with buffer A + 300 mM imidazole. For AQP0, an additional wash step with 5 CV buffer A + 100 mM imidazole was added prior to elution. The AQP-containing fractions were concentrated with a 50 MWCO Vivaspin (GE Healthcare) concentrator and the concentrated sample was loaded on a Superdex 200 10/300 GL (GE Healthcare) gel filtration column equilibrated with 20 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol, 0.05% DDM (AQP0) or 20 mM Tris–HCl pH 8, 300 mM NaCl, 0.2% OGNG (AQP2). Finally, AQP-containing fractions were pooled and concentrated as above and kept on ice for immediate use or flash-frozen in liquid nitrogen and stored at −80 °C.
Dephosphorylation of AQP2
Since AQP2 is shown to be phosphorylated in Pichia pastoris,9 we dephosphorylated it for our experiments by treating with Alkaline Phosphatase (AP) in 5 mM Tris–HCl, pH 7.9, 10 mM NaCl, 1 mM MgCl2 and 0.1 mM DTT and incubated at 30 °C for 2 hours. The detailed procedure of phosphorylation analysis and dephosphorylation is described in Kinoshita et al.20 with slight modification as mentioned in Ampah-Korsah et al.21Cloning, expression and purification of human LIP5
For expression of human LIP5 in E. coli, a codon-optimized gene was purchased from GenScript. The gene was amplified using 5′-GGGTTC GCGGCGCTGGCGCCGCTGCCG-3 (sense) and 5-CTGCTGACCACCGGCCGTGAGGATTACGATATCCCAACTACCGAAAACTTGTATTTTCAGGGTCACCATCACCATCATCATCACCATTAA
GCGGCGCTGGCGCCGCTGCCG-3 (sense) and 5-CTGCTGACCACCGGCCGTGAGGATTACGATATCCCAACTACCGAAAACTTGTATTTTCAGGGTCACCATCACCATCATCATCACCATTAA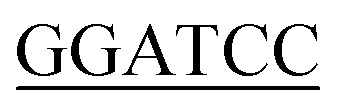 AAG-3′ (antisense) as forward and reverse primers respectively. NdeI and BamHI restriction sites (underlined) were used for cloning into the pET3a vector (Novagene). The final LIP5 construct contains a C-terminal 6 × His-tag preceded by a Tobacco Etch Virus (TEV) protease cleavage site (ENLYFQG). A seven amino acid spacer (DYDIPTT) is included between the last residue of LIP5 and the TEV-site to ensure efficient proteolytic cleavage.
AAG-3′ (antisense) as forward and reverse primers respectively. NdeI and BamHI restriction sites (underlined) were used for cloning into the pET3a vector (Novagene). The final LIP5 construct contains a C-terminal 6 × His-tag preceded by a Tobacco Etch Virus (TEV) protease cleavage site (ENLYFQG). A seven amino acid spacer (DYDIPTT) is included between the last residue of LIP5 and the TEV-site to ensure efficient proteolytic cleavage.
LIP5 was expressed in BL21* (DE3) E. coli (Invitrogen) grown in LB media containing 50 μg ml−1 ampicillin. At an OD600 of 0.8, LIP5 expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3.5–4 h at 30 °C after which the cells were harvested at 6000 × g for 15 minutes. 4 g of cells were resuspended in 50 ml lysis buffer (20 mM Tris pH 8, 250 mM NaCl, 5% glycerol, 10 mM imidazole) supplemented with one cOmplete™ EDTA-free protease inhibitor cocktail tablet (Roche). Cells were broken using 10 × 1 min sonication with one minute interval while kept on ice. Unbroken material was pelleted at 18000 × g at 4 °C for 30 minutes and the cell lysate was filtered before loading onto a 5 ml Ni2+–nitrilotriacetic acid affinity column equilibrated with lysis buffer. After two wash steps with lysis buffer containing 30 mM and 100 mM imidazole respectively, LIP5 was eluted with the same buffer containing 250 mM imidazole. The samples were analyzed on SDS-PAGE and the fractions corresponding to LIP5 were pooled and concentrated using a 10 kDa molecular weight cut off Vivaspin concentration tube (GE Healthcare). The concentrated sample was loaded on a Superdex 200 10/300 GL column (GE Healthcare) with buffer B (20 mM Tris pH 8, 150 mM NaCl, 1 mM DTT). Fractions containing LIP5 were pooled and concentrated as above. The sample was either used directly or flash-frozen in liquid nitrogen and stored at −80 °C after addition of 5% glycerol for storage.
Expression and purification of human CaM
A mutant of human CaM where Ser 17 has been replaced with cysteine (to allow for labelling with cysteine-reactive dyes) was expressed in E. coli and purified as previously described.22Cloning, expression and purification of AQP2 C-terminal peptide
DNA coding for AQP2 residues 227–271 was amplified by PCR from pPICZB encoding full-length human AQP2 (ref. 9) using 5′-GGC ATGCTGGTGCCACGCGGTTCGGCCAAGAGCCTGTCGGAG-3′ (sense) and 5′-GCCTT
ATGCTGGTGCCACGCGGTTCGGCCAAGAGCCTGTCGGAG-3′ (sense) and 5′-GCCTT ACCCTGAAAATACAAGTTTTCGGTAGTTGGGATATCGTAATCGGCCTTGGTACCCCGTGG-3′ as forward and reverse primers respectively. SpeI and XhoI (underlined) were used for cloning into the GST-expressing vector pET-41 Ek/LIC (Novagen). The final construct consists of the AQP2 C-terminus, C-terminally fused to GST with a thrombin cleavage site in between. The construct also contains a 6× His-tag at the C-terminal end of the AQP2 peptide, preceded by a TEV site (ENLYFQG) and a linker region (DYDIPTT) in between the last residue of the AQP2 peptide and the TEV site.
ACCCTGAAAATACAAGTTTTCGGTAGTTGGGATATCGTAATCGGCCTTGGTACCCCGTGG-3′ as forward and reverse primers respectively. SpeI and XhoI (underlined) were used for cloning into the GST-expressing vector pET-41 Ek/LIC (Novagen). The final construct consists of the AQP2 C-terminus, C-terminally fused to GST with a thrombin cleavage site in between. The construct also contains a 6× His-tag at the C-terminal end of the AQP2 peptide, preceded by a TEV site (ENLYFQG) and a linker region (DYDIPTT) in between the last residue of the AQP2 peptide and the TEV site.
GST-AQP2 peptide was expressed in E. coli BL21* grown in LB-media supplemented with 30 μg ml−1 kanamycin. At OD600 = 0.4–0.6, IPTG was added to a total concentration of 0.5 mM and the cultures were induced for 3 hours. Cells were harvested by centrifugation at 10000 × g for 15 min and resuspended in lysis buffer (20 mM Tris pH 7.4, 250 mM NaCl, 5% glycerol, 1 mM DTT) supplemented with 1 cOmplete™ EDTA-free protease cocktail tablet. After breaking the cells using sonication while kept on ice (5 × 1 min with 1 min waiting in between), cell debris was spun down at 15000 × g for 30 min. The supernatant was loaded on a 1 ml GSTrap column (GE Healthcare) equilibrated with lysis buffer and eluted using GST elution buffer (50 mM Tris pH 8, 20 mM glutathione, 1 mM DTT). Fractions containing GST-AQP2 were pooled and concentrated in a Vivaspin concentrator (cutoff 10 kDa) to 1–2 ml. The buffer was changed to thrombin cleavage buffer using a PD10 column (20 mM Tris pH 8, 150 mM NaCl, 2.5 mM CaCl2) and the sample was mixed with 1 U thrombin per mg protein and incubated at 4 °C overnight. The cleaved off AQP2 peptide was isolated by collecting the flow through after ultrafiltration using a Vivaspin concentrator with a molecular weight cutoff of 10 kDa. After changing the buffer to the respective binding assay buffer (see MST and fluorescence anisotropy sections below) using a PD10 column, the AQP2 peptide sample was concentrated using a 3 kDa molecular weight cutoff Vivaspin concentrator and kept at 20 °C until further use.
Protein labelling
AQP2, LIP5 and CaM were labelled with the cysteine-reactive dye C5 Maleimide-Alexa 488 (Thermo Fisher) according to the manufacturer’s instructions. AQP2 was incubated overnight with 20-fold molar excess of dye in 20 mM Tris–HCl pH 7.5, 300 mM NaCl and 0.2% OGNG, CaM was incubated with 3-fold molar excess of dye for 3 h in 20 mM phosphate buffer pH 8 and LIP5 was incubated overnight with 3-fold molar excess of dye in 20 mM Tris, pH 7.5, 100 mM NaCl, 1 mM CaCl2, 5% glycerol. After labelling, excess dye was removed using a PD-10 desalting column.CD spectroscopy
LIP5 and CaM were diluted to 0.2 mg ml−1 in 50 mM NaCl, 20 mM phosphate buffer, pH 8 and 50 mM, 20 mM phosphate buffer, pH 7.5 respectively. For experiments with detergent, DDM, OGNG or OG were added to the buffer to a final concentration of 0.05%, 0.2% and 1% respectively. Far UV CD spectra between 190 and 260 nM were recorded at 25–95 °C with 5 °C intervals for a temperature interval on a Jasco J-815 CD spectrometer. We performed temperature scans in the far UV CD spectrum between 190 and 260 nm, from 20 °C to 95 °C with a 5 °C interval. The sample temperature was controlled by a built-in Peltier controller. Each scan was an average of three individual scans measured with a data pitch of 1 nm and 8 s response time. Measurements were done in a 0.1 cm quartz cuvette.Mean residue ellipticity (MRE) ([θ] × 10−3 deg cm2 dmol−1) was calculated by using the following equation
| MRE = M × θ/(10 × l × c × n) |
where y is the MRE, x is the temperature and x0 is the temperature at which MRE is halfway between the native and denatured state which corresponds to Tm. For the double Boltzmann equation, the data is fitted to

where y is the MRE, x is the temperature and x01 and x02 are the melting temperatures for the two transitions.
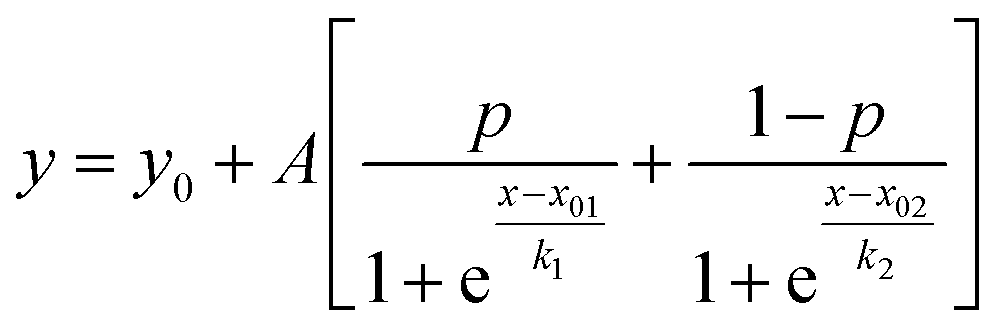
Microscale thermophoresis
For MST experiments characterizing the interaction between full-length AQP2 and LIP5, a 1:1 dilution series of LIP5 (4.9 μM) was made in AQP2 binding assay buffer (20 mM Tris pH 8, 300 mM NaCl), supplemented with 0.2% OGNG and mixed 1:1 with 0.2 μM labelled AQP2. Similarly, for the AQP2 peptide (3.4 μM) a 1:1 dilution series was made in AQP2 binding assay buffer without detergent and mixed 1:1 with 0.18 μM labelled LIP5. Characterization of the interaction between AQP0 and CaM was done using a 2:1 dilution series of full-length AQP0 (50 μM) and a 1:1 dilution series of AQP0 peptide (11 μM), both of which were mixed 1:1 with 50 nM labelled CaM. In the case of the AQP0 peptide, labelled CaM was mixed with unlabelled CaM to a final concentration of 500 nM to avoid sticking to the capillaries. For AQP0, the binding assay buffer was 20 mM Tris, pH 7.5, 100 mM NaCl, 1 mM CaCl2, 5% glycerol which in the case of studies of full-length AQP0 was supplemented with 0.05% DDM. The samples were transferred to Monolith™ NT.115 MST Premium Coated Capillaries (NanoTemper Technologies) and MST traces were recorded at room temperature in a Monolith NT.115 (NanoTemper Technologies) using the MO.Control software. The LED/excitation power setting was 20 for full-length AQP2 and AQP0, and 80 for AQP2 and AQP0 peptides. The MST power setting for FL-AQP2, AQP2 peptide, AQP0 and AQP0 peptide were 40, 80, 40 and 80 respectively. MST data was obtained from three individually prepared dilution series. Data was analyzed using Origin.
Denaturation test
For full-length AQP2 with LIP5 and AQP0 peptide with CaM a variation in the initial fluorescence signal of the capillary scan was observed. In order to ensure that the variation in fluorescence was associated with binding, we performed a standard denaturation test (SD test) as described previously.23 For the SD test of full-length AQP2 with LIP5, four samples were prepared in the same manner as for the MST experiment above, corresponding to high, medium and low concentration of LIP5 (2450 nM, 322 nM and 28 nM) as well as labelled full-length AQP2 alone. For the AQP0 peptide, three samples were prepared corresponding to high and low concentrations of AQP0 peptide (5.375 nM and 0.67 nM) and labelled CaM alone.The samples were subjected to ten minutes centrifugation at 13000 × g to remove any protein precipitate, then mixed with 2× solution containing 4% SDS and 40 mM DTT, heated at 95 °C for 5 minutes to denature the sample. After heating, the sample was centrifuged briefly and loaded into the capillaries. The fluorescence was measured in the Monolith NT.115 using the same settings as for the corresponding MST measurements.
Fluorescence anisotropy
Anisotropy experiments were performed on a Perkin Elmer LS 50B fluorometer. The anisotropy signal was measured for the labelled protein before the addition of the ligand and the grating factor was determined. This grating factor was used to correct all values obtained on the fluorimeter as result of the anisotropy signal. All measurements were done in triplicates.The Ivv and Ivh values were noted, as too low Ivv values would result in higher signal to noise variation. Based on the Ivv (Ivv > 30) value the excitation and emission slit was decided. For FL-AQP2 with LIP5 and AQP2 peptide with LIP5, slit sizes of 7 mm and 10 mm were used. For FL-AQP0 with CaM and AQP0 peptide with CaM the slit size was 5 mm.
For the interaction with FL-AQP2, a 1:1 dilution series of the LIP5 was prepared in 20 mM Tris–HCl, pH 8, 300 mM NaCl, 0.2% OGNG. 0.53 μM of labelled FL-AQP2 was put in the cuvette and sequentially titrated with 2 μl from each sample in the dilution series starting with the lowest concentration of 61.5 nM to a final concentration of 126 μM. The final concentration of the ligand in the cuvette corresponded to 5.9 μM.
For the AQP2 peptide with LIP5, a 1:1 dilution series of the ligand was prepared in 20 mM Tris–HCl, pH 8, 300 mM NaCl. 120 nM of LIP5 labelled with Alexa 488 was put in the cuvette and sequentially titrated with 7 μl of the AQP2 peptide dilution series starting with the lowest concentration of 2.07 nM to a final concentration of 69 μM. The final concentration of the ligand in the cuvette corresponded to 5.5 μM.
A 1:1 dilution series of the ligand was also prepared for the AQP0 peptide in 20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 5% glycerol, 1 mM CaCl2. 1 μM of CaM labelled with Alexa 488 was put in the cuvette and sequentially titrated with 2 μl of AQP0 peptide starting with the lowest stock concentration of 104 nM to a final stock concentration of 340 μM. The final concentration of the ligand in the cuvette corresponded to 127 μM. For full-length AQP0, 0.05% DDM was added to the buffer used for the AQP0 peptide and FL-AQP0 was titrated to the cuvette containing 1 μM of labelled CaM by sequential addition of 2 μl from a stock solution to a final concentration of 18 μM and 90 nM respectively.
Data fitting
The following equations were used for fitting data to a 1:1 binding model:
S1 is the signal from the unbound state, S2 the signal from the complex. Lfree and Ltot are the free and total LIP5 or AQP2 peptide concentration respectively, Ptot the total concentration of fluorescently labelled LIP5 and Kd the dissociation constant. In MST-experiments, Ptot was held constant during the experiment and fitting; but in experiments where, as in fluorescence anisotropy experiments, Ptot varied over the titration, in this case the actual Ptot at each titration point was used in the fitting procedure. Fitting was performed in Origin or using the CaLigator program.24
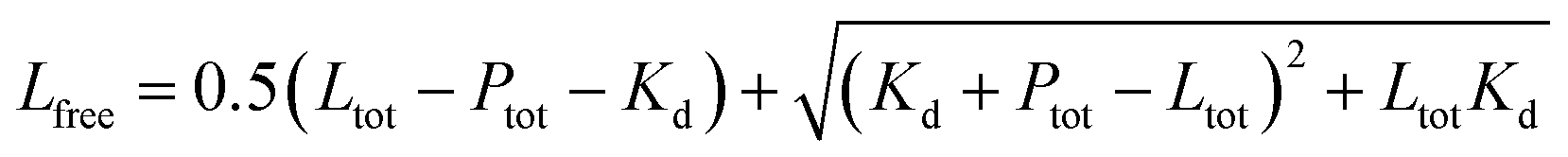
Data were also fitted using a 2:1 binding model, which in some cases produced significantly better fits than the 1:1 binding model, for example in the case of the MST data for AQP0 and CaM. These fits were performed using the CaLigator program24 with the following equation:
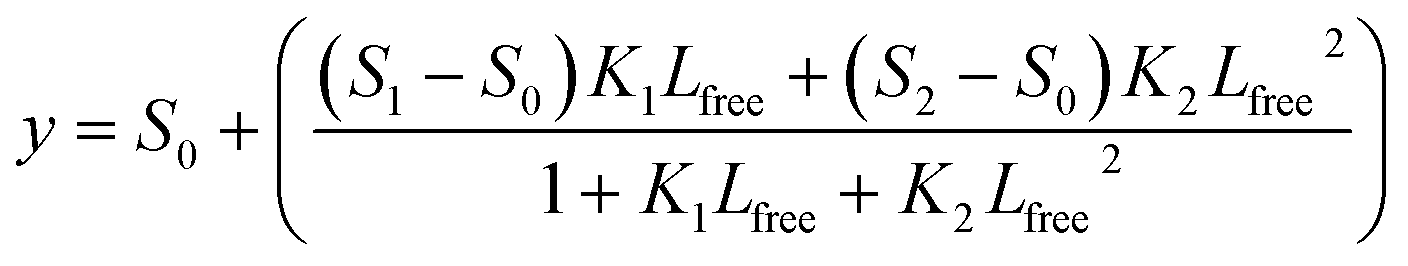
where Ptot is the total protein concentration and Ltot is the total ligand concentration.
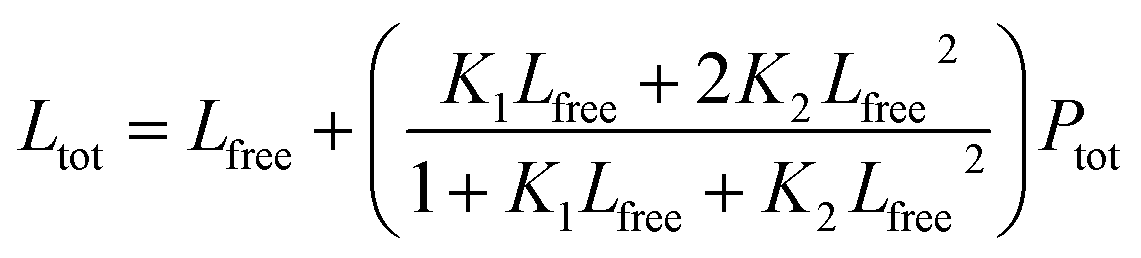
Results and discussion
Stability of CaM and LIP5 in detergent
Since studies of full-length AQP0 and AQP2 require the presence of detergent in the assay buffer, we first explored the effect of the detergent on the stability of the soluble interaction partners CaM and LIP5 using circular dichroism (CD) spectroscopy. Three different detergents that are commonly used for structural and functional studies of membrane proteins were chosen, differing in the critical micelle concentration (CMC) and therefore in their buffer working concentration: n-dodecyl-β-D-maltoside (DDM, CMC ∼0.01%), octyl glucose neopentyl glycol (OGNG, CMC ∼0.058%) and n-octyl-α-D-glucopyranoside (OG, CMC ∼0.3–0.6%). Far UV spectra between 190 and 260 nm were recorded at 25 °C in buffer containing no detergent, 0.05% DDM, 0.2% OGNG and 1% OG respectively. For both CaM and LIP5, the CD spectra were very similar in all four buffers, displaying local minima at 210 and 222 nm that are characteristic for protein containing mainly α-helices (Fig. 2A and C). This indicates that, at room temperature, none of these three detergents at the chosen concentration affects the overall fold of CaM and LIP5.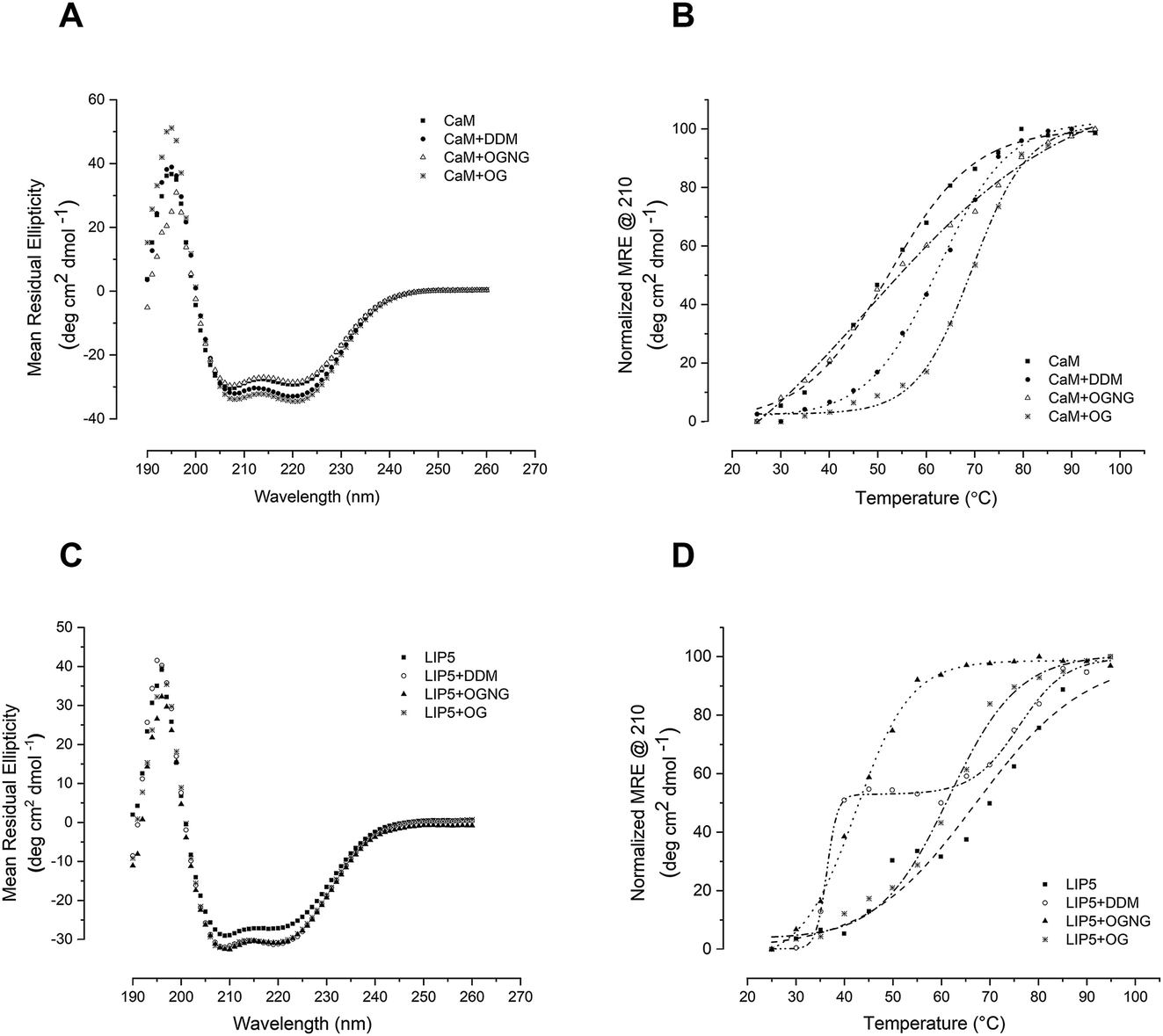 | ||
| Fig. 2 Stability of CaM and LIP5 in detergent. CD spectrum for (A) CaM and (C) LIP5 without detergent and in the presence of 0.05% DDM, 0.2% OGNG or 1% OG. The spectra were recorded at 25 °C. Melting curves for (B) CaM and (D) LIP5 obtained by plotting the Mean Residual Ellipticity (MRE) at 210 nm from CD-spectra obtained at different temperatures. | ||
To investigate whether detergents affect the thermal stability of CaM and LIP5, CD spectra were recorded over a temperature range of 20–95 °C, at 5 °C intervals. Protein melting curves were obtained by plotting the Mean Residual Ellipticity (MRE) at 210 nm against the temperature and fitting the data to a Boltzmann sigmoidal equation. From these curves, the melting temperature, Tm, was determined as the mid-point of the transition between folded and unfolded states (Table 1). For CaM, DDM and OG had a stabilizing effect, causing an increase in Tm by 12 and 19 °C respectively (Tm = 62 ± 0.39 °C in DDM and 69 ± 0.75 °C in OG). As compared to when no detergent was present (Tm = 50 ± 0.75 °C) (Fig. 2B). This fits well with the exposure of a hydrophobic surface on CaM in its Ca2+-bound state,25 which may be further stabilized by hydrophobic interactions with the detergent molecules. In the presence of OGNG, the CaM melting curve displayed a less sharp transition between folded and unfolded states, suggesting a different melting behaviour and with a Tm that was not significantly different from CaM without detergent (48 ± 3.9 °C in OGNG compared to 50 ± 0.75 °C without). In contrast, LIP5 was most stable in the absence of detergent (Tm = 67 ± 1.2 °C), followed by OGNG for which a 5 °C reduction in Tm could be seen (Tm = 62 ± 0.80 °C). In the presence of OG, the stability of LIP5 was significantly lower, giving a Tm of 42 ± 0.39 °C. In DDM, LIP5 showed a clear biphasic melting behaviour with two major transitions with melting temperatures of 36 ± 0.49 °C (Tm1) and 76 ± 0.97 °C (Tm2) respectively. A similar transition, although a lot less pronounced could also be distinguished in the melting curve in the absence of detergent. Since LIP5 has been suggested to be composed of two major domains, an N-terminal domain consisting of tandem MIT alpha-helical motifs and a C-terminal domain for which the structure is not known,26,27 the biphasic melting curves may represent differences in melting behaviour between these two domains.
| Condition | CaM (°C) | LIP5 (°C) |
|---|---|---|
| No detergent | 50 ± 0.8 | 67 ± 1.2 |
| DDM | 62 ± 0.39 | 36 ± 0.49 |
| 76.0 ± 0.97 | ||
| OGNG | 48 ± 3.9 | 62 ± 0.8 |
| OG | 69 ± 0.75 | 42 ± 0.39 |
For the interaction studies below, the detergent used for solubilisation and purification of AQP0 and AQP2 is also used in the binding assay: DDM in the case of AQP0, and OGNG in the case of AQP2. Our CD-data suggest that in both cases, the respective binding partner is stable in the chosen detergent (CaM in DDM and LIP5 in OGNG), supporting the use of these detergents in interaction studies where detergent-solubilized full-length AQP0 and AQP2 are used.
Full-length AQP2 and LIP5 interaction studies
The interaction between full-length AQP2 and LIP5 was investigated using MST. In this technique, the interaction between two molecules is monitored through a change of movement in a thermal gradient.28 A two-fold dilution series of LIP5 was made, resulting in 16 samples. Each sample was mixed 1:1 with a constant concentration of AQP2 that had been fluorescently labelled with the cysteine-reactive dye Alexa-488. After recording MST-traces (Fig. S1A†), the normalized fluorescence, Fnorm, defined as the ratio between the fluorescence after (Fhot) and before (Fcold) heating with an infrared laser, was plotted against the concentration of LIP5. As seen in Fig. 3A, a binding curve could be obtained, describing the interaction between full-length AQP2 and LIP5. However, in order to obtain a reliable estimate of the affinity of the interaction from the thermophoresis data, the initial fluorescence must be constant in all samples. In our experiment, we observed a clear LIP5 concentration-dependence in the initial fluorescence of AQP2 (Fig. 3B). If this is a result of AQP2-binding, the initial fluorescence data can be used to determine the dissociation constant, Kd, instead of the thermophoresis data. To verify that our variation in initial fluorescence is indeed a result of LIP5 binding, we performed a denaturation test where samples with AQP2 concentrations corresponding to the “bound” and “unbound” states were denatured by heating to 95 °C for 5 min in a solution containing 4% SDS and 40 mM DTT.23 After denaturation, the fluorescence levels of the “bound” and “unbound” samples were identical, suggesting that the difference in fluorescence seen in our dilution series is induced by binding rather than loss of sample through ligand-induced surface adsorption or aggregation (Fig. S2†). Hence, we used the initial fluorescence data to quantify the interaction between full-length AQP2 and LIP5, resulting in an estimated Kd of 0.61 ± 0.01 μM (Fig. 3B).
 | ||
| Fig. 3 Interaction between full-length AQP2 and LIP5. (A) Binding curve obtained from plotting the MST signal (normalized fluorescence, Fnorm) against LIP5 concentration. Due to variation in initial fluorescence between capillaries, the curve could not be used to calculate Kd. (B) Plotting the initial fluorescence against the LIP5 concentration resulted in a binding curve that could be fitted to a one-site binding model. Kd for the interaction was estimated to be 0.61 ± 0.01 μM. (C) Binding curve obtained from fluorescence anisotropy measurements. The data was fitted to a 1:1 binding model, resulting in a Kd of 0.32 ± 0.10 μM. | ||
Next, we studied the interaction between full-length AQP2 and LIP5 using fluorescence anisotropy. By determining the difference between vertically and horizontally polarized emissions of a fluorophore and relating this to the rotational correlation time, fluorescence anisotropy can give information about the local and global motion of the fluorophore and thereby also the size of the fluorescently labelled molecule. The technique is therefore useful to study protein–protein interactions.29 For fluorescence anisotropy measurements, a two-fold dilution series of LIP5 was made from which 2 μl of each sample was sequentially added to a cuvette containing fluorescently labelled AQP2. After each addition, the mixture was allowed to incubate for 1 min after which the fluorescence anisotropy was taken as the average from 60 s. Plotting the anisotropy against the LIP5 concentration resulted in a binding curve from which Kd was determined to be 0.32 ± 0.10 μM (Fig. 3C). This is lower than estimated from the MST-experiment above but still in reasonably good agreement.
In a previous study, we studied the interaction between human AQP2 and mouse LIP5 by MST, resulting in a Kd of 0.19 ± 0.04 μM.9 The affinity observed here from fluorescence anisotropy measurements, where we use human LIP5, is thus within error limits the same as that for mouse LIP5. In the earlier study, we used mouse LIP5 that had been fluorescently labelled with an amine reactive dye (NT-647-NHS). Although mouse and human LIP5 are very similar, the sequence identity is 91%, small differences in amino acid sequence or the use of a different fluorescently labelled counterpart with a different dye may explain the observed minor differences in affinity estimated from the MST-experiment.
AQP2 C-terminal peptide and LIP5 interaction studies
In order to study the interaction between LIP5 and a peptide corresponding the human AQP2 C-terminus, we made a construct in which the residues 227–271 of AQP2 were fused to GST and expressed in Escherichia coli. After purification, GST was cleaved off and the AQP2 peptide was isolated. For MST-studies, the AQP2 peptide was diluted in a two-fold dilution series and mixed 1:1 with LIP5 that had been fluorescently labelled with Alexa 488. MST-traces were recorded (Fig. S1B†), and a binding curve was constructed by plotting Fnorm against the LIP5 concentration, from which the Kd was determined (Fig. 4A). The estimated Kd for the interaction between the AQP2 peptide and LIP5 is 0.18 ± 0.07 μM, suggesting a slightly higher affinity than seen for full-length AQP2 by MST but within error margin of the affinity observed by fluorescence anisotropy (Fig. 3).
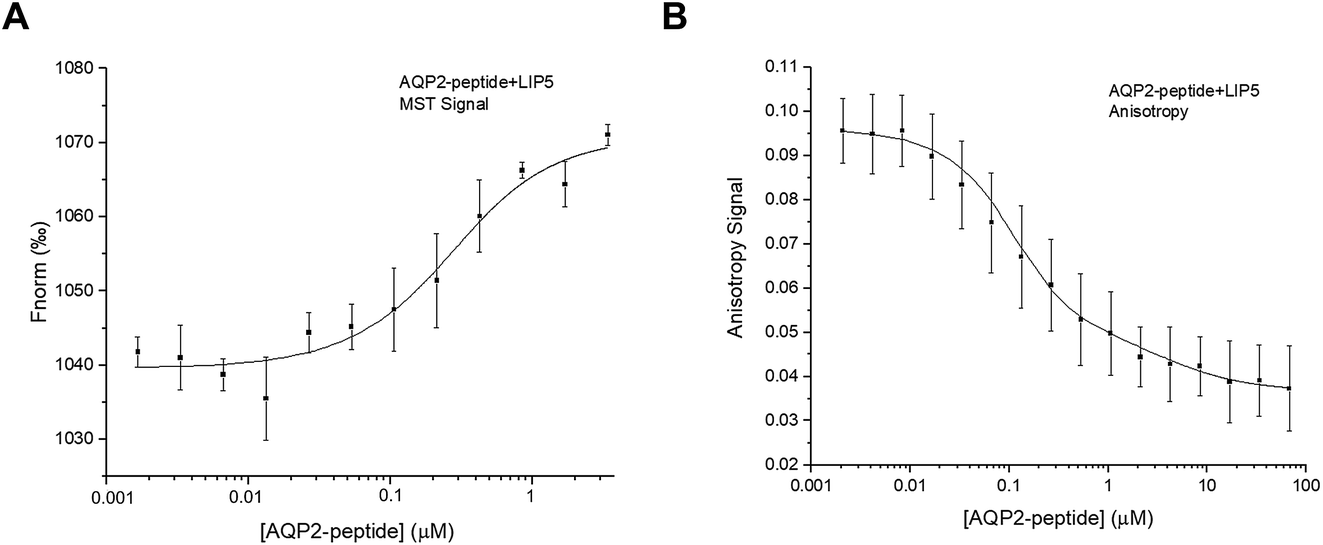 | ||
| Fig. 4 Interaction between AQP2 peptide and LIP5. Binding curves for the interaction between LIP5 and the AQP2 peptide obtained from (A) MST and (B) fluorescence anisotropy experiments. The data in (A) could be fitted to a one-site binding model with an estimated Kd of 0.18 ± 0.07 μM. For (B) the best fit was obtained using a two-site binding model, resulting in Kd1 and Kd2 of 0.05 ± 0.02 and 4.0 ± 1.0 μM respectively. | ||
Fluorescence anisotropy measurements were done similarly as for full-length AQP2, with the exception that 7 μl was sequentially added to the cuvette from the dilution series and each value was an average of five individual measurements. In contrast to what was observed for full-length AQP2, the fluorescence anisotropy decreased with increasing LIP5 concentration (Fig. 4B). This suggests that binding causes the fluorophore to be more mobile, possibly because of an interaction between Alexa 488 and LIP5 that is only present in the unbound state. The data could be fitted by a one-site binding model with Kd = 0.10 ± 0.05 μM. This is similar to the MST-experiment using the same peptide as well as the MST and anisotropy data for full-length AQP2, which all suggested a one-site binding model with a Kd between 0.2 and 0.6 μM. The anisotropy data with the AQP2 peptide could, however, also be fitted by a two-site binding model with a Kd1 of 0.05 ± 0.02 μM for the high affinity site and a Kd2 of 4.0 ± 1.0 μM for the low affinity site. The error square sum decreased by 44% relative to a fit with a single site, suggesting that the two-site binding model more accurately describes the interaction between the AQP2 peptide and LIP5.
Full-length AQP0 and CaM interaction studies
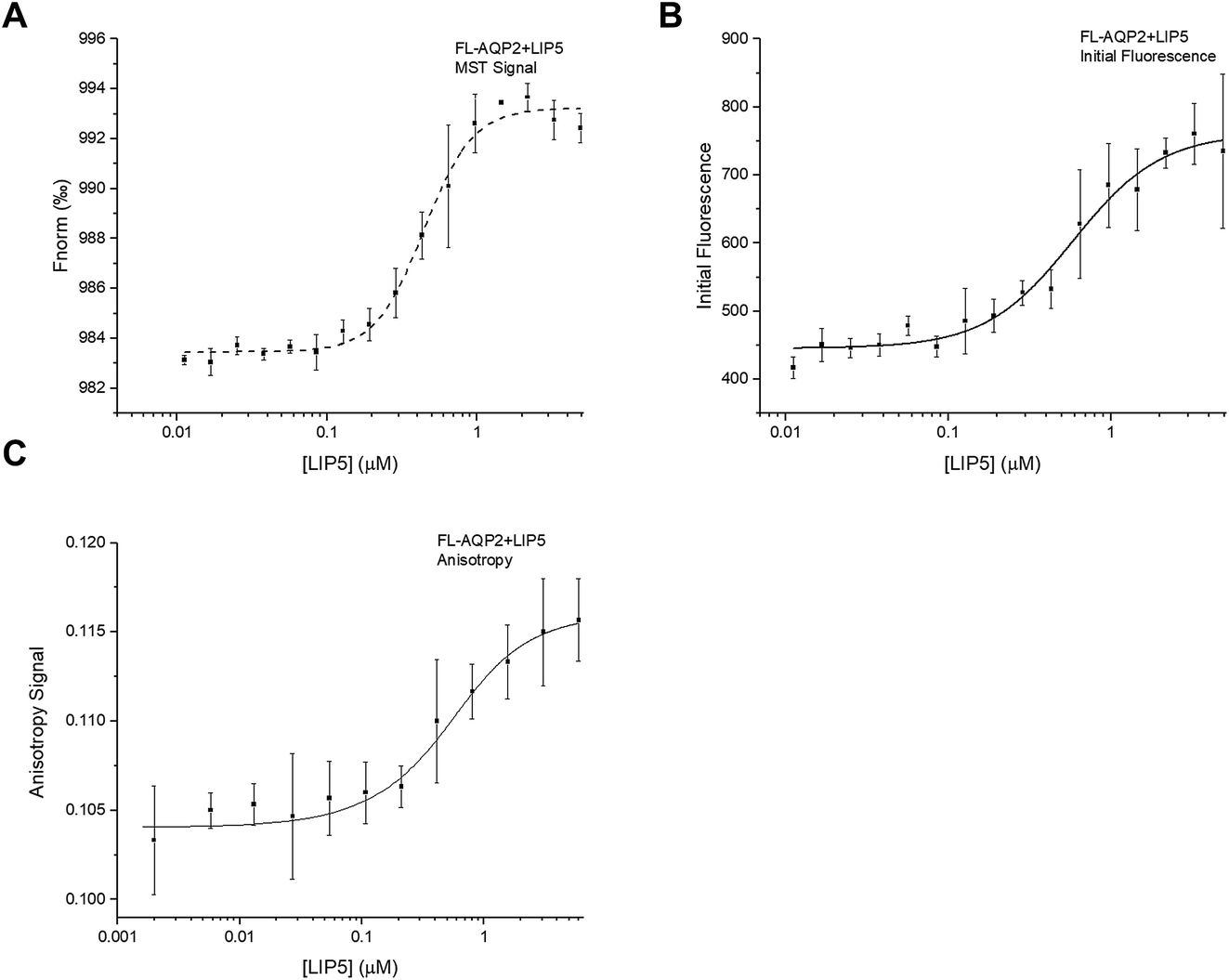
Interaction studies of AQP0 and CaM, were done using a CaM-mutant where Ser 17 is replaced with cysteine in order to allow for labelling using Alexa 488.22 For MST-experiments of full-length AQP0, a 1.5-fold dilution series of AQP0 was made and mixed 1:1 with fluorescently labelled CaM, after which MST-traces were recorded (Fig. S1C†). In contrast to what was seen for the interaction between AQP2 and LIP5, the resulting binding curve could not be fitted to the standard one-site binding model. Instead the data could only be fitted to a two-site model where there is positive cooperativity between the two sites (Fig. 5A). The two macroscopic dissociation constants were determined to be Kd1 = 40 μM and Kd2 = 2.5 μM. An upper limit for the free energy coupling between the two sites, ΔΔG, which is valid for two identical binding sites, was calculated as ΔΔG = −RTln(4Kd1/Kd2) = −10 kJ mol−1, suggesting a very strong positive cooperativity where the ligand affinity for one site is significantly increased when the other site is occupied.30,31 As a consequence of the strong positive cooperativity, the errors in the individual binding parameters are not symmetric; in the direction of increasing positive cooperativity any Kd1 value above 40 μM, any Kd2 value below 2.5 μM, and any ΔΔG value below −10 kJ mol−1 increase the error square sum by maximum 5%, while in the opposite direction the error square sum increases more sharply (Fig. S3†). Because of the positive cooperativity, the product Kd1Kd2 is much more well determined than the individual macroscopic constants.
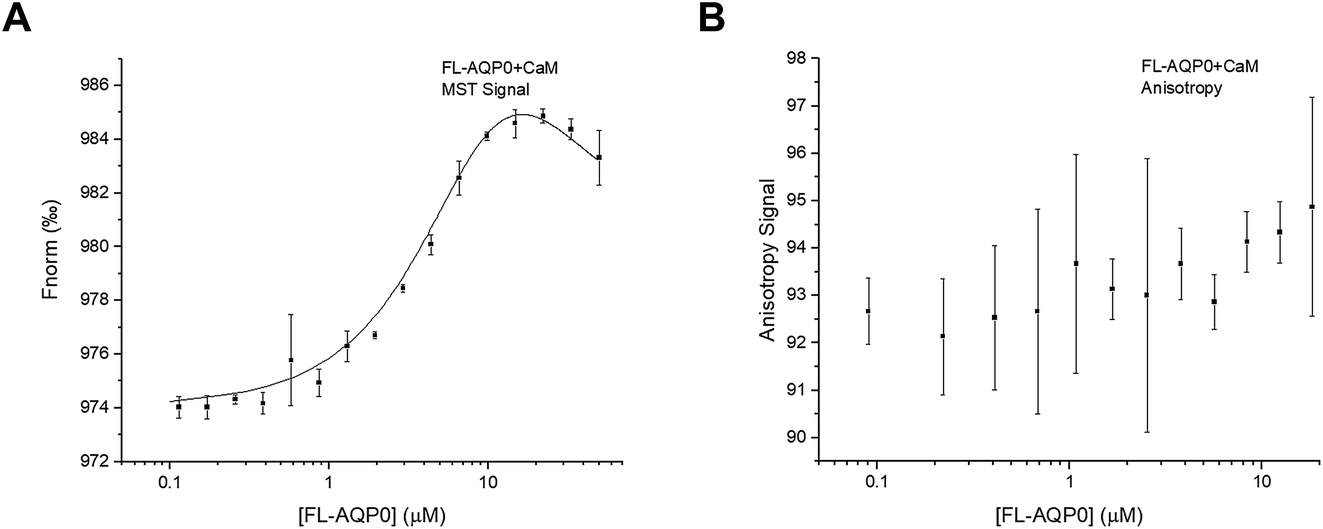 | ||
| Fig. 5 Interaction between full-length AQP0 and CaM. (A) Binding curve obtained from MST by fitting the data to a two-site model with cooperativity between the two sites. The free energy coupling, ΔΔG, between the two sites was <−10.3 kJ mol−1, suggesting strong positive cooperativity. Kd1 and Kd2 were determined to be 40 and 2.5 μM respectively. (B) Fluorescence anisotropy measurements failed to show an interaction between full-length AQP0 and CaM. | ||
Previous ITC-studies of the interaction between an AQP0 C-terminal peptide and CaM, combined with a 30 Å-resolution structural model based on single particle electron microscopy have suggested that one CaM binds two copies of the AQP0 C-terminal helix in an antiparallel arrangement.5 It was proposed in an earlier study that this occurs via a two-step “bind-and-capture” mechanism where CaM first binds one C-terminal helix via a high affinity event (Ka = 1.4 × 107 M−1 corresponding to a Kd of 71 nM) followed by a lower affinity event (Ka = 7.8 × 104 M−1 corresponding to a Kd of 12.8 μM) where the second C-terminal helix is captured. Our data for the interaction between full-length AQP0 and CaM also suggests two binding sites, however, of lower affinity, and that binding at these two sites is a cooperative process where binding of the first C-terminal helix favours binding of the second helix.
Surprisingly, fluorescence anisotropy studies of full-length AQP0 and CaM did not result in a binding curve (Fig. 5B). This may be because the mutated site for fluorophore incorporation, Ser17→Cys, was chosen to be exposed both in free and bound calmodulin,22 thus the fluorophore may be rotating freely both in free CaM and in the complex. We have noticed that the fluorescence of CaM increases in the presence of detergent, and it may be that this enhances the anisotropy signal from Alexa 488 itself.
AQP0 C-terminal peptide and CaM interaction studies
A synthetic peptide corresponding to the proposed CaM-binding site within the human AQP0 C-terminus (residues 223–242) was purchased from GenScript and used for interaction studies with CaM. For MST, a two-fold dilution series of the AQP0 peptide was mixed 1:1 with CaM that had been fluorescently labelled with Alexa 488 as described above. Similarly as for full-length AQP2, the initial fluorescence was shown to be dependent on the CaM-concentration, wherefore the binding curve obtained from plotting Fnorm against the CaM-concentration (Fig. 6A) could not be reliably used for determining Kd. Instead, after using the denaturation test to verify that the variation in initial fluorescence is a result of binding, we estimated the Kd from a binding curve obtained by plotting initial fluorescence against AQP0 peptide concentration (Fig. 6B). The data could be fitted by a one site model (Kd = 5.0 ± 2.0 μM) and there was no improvement in the error if a more complicated model was used.
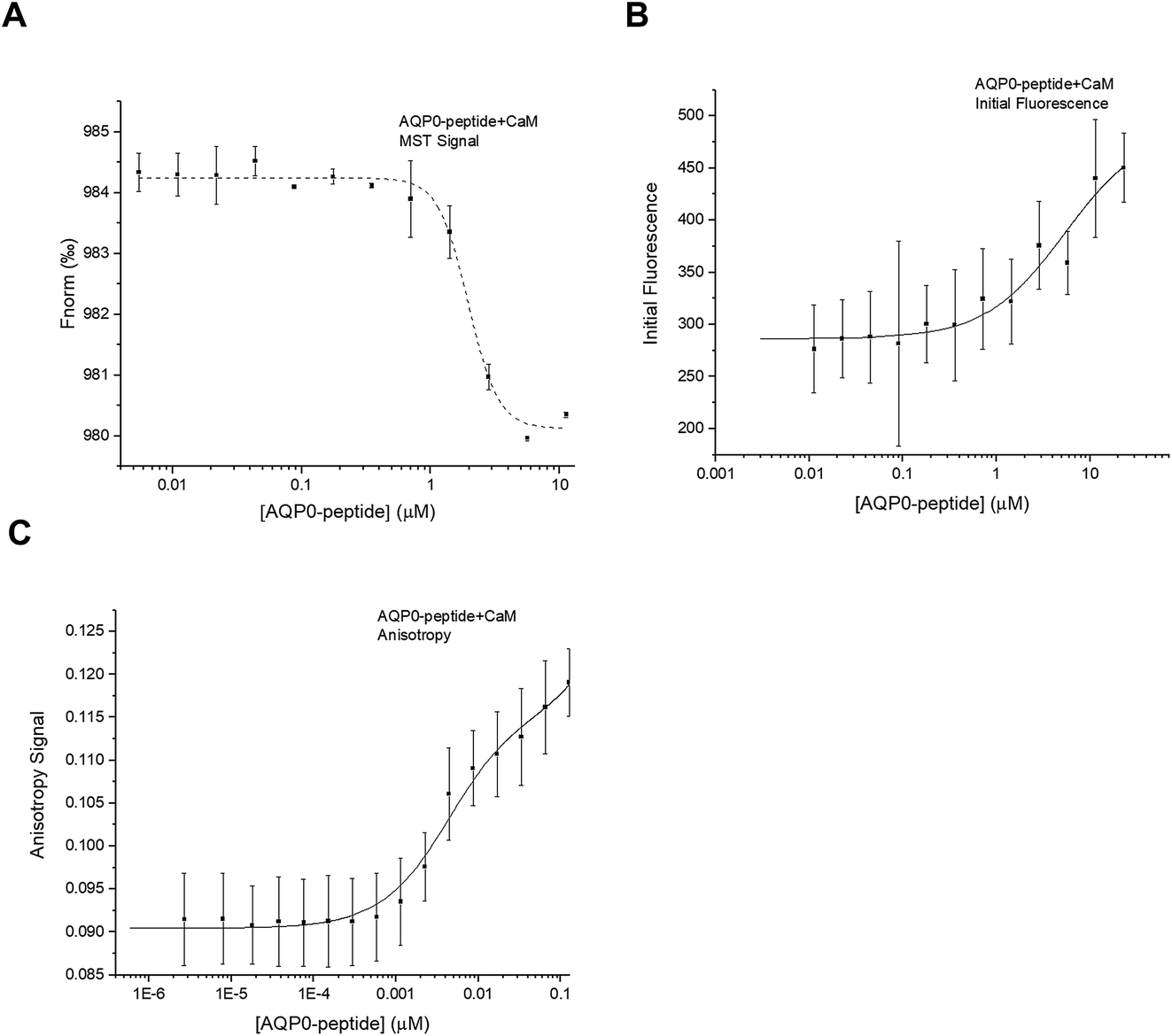 | ||
| Fig. 6 Interaction between AQP0 peptide and CaM. (A) Binding curve obtained from the MST-experiment by plotting Fnorm against the AQP0 peptide concentration. As for the interaction between full-length AQP2 and LIP5, Kd could not be estimated due to a concentration-dependent variation in initial fluorescence. (B) Plotting initial fluorescence against AQP0 peptide concentration resulted in a binding curve that could be fitted to a one-site binding model with a Kd of 5.0 ± 2.0 μM. (C) Binding curve obtained from fluorescence anisotropy measurements. The best fit was obtained by fitting the data to a two-site binding model with Kd1 = 4 ± 1 mM and Kd2 = 1.0 ± 2.0 μM. | ||
Fluorescence anisotropy studies of the interaction between the AQP0 peptide and CaM confirmed these results: a binding curve was obtained that could be fitted to a two-site binding equation, but without cooperativity (Fig. 6C). The Kd-values estimated from fluorescence anisotropy were similar as seen in the MST-experiment: 4.0 ± 1.0 μM and 1.0 ± 2.0 mM, but significantly different than the affinities proposed from the previously published ITC-study using a very similar peptide (Kd1 = 71 nM, Kd2 = 13 μM).5 The fact that the interaction between the AQP0 peptide and CaM but not full-length AQP0 and CaM can be seen using fluorescence anisotropy is surprising, in particular since we use the same fluorescently labelled molecule, CaM labelled with Alexa 488. The discrepancy between the anisotropy studies between full-length AQP0 and AQP0 peptide could indicate that the peptide binds CaM in a different manner, thereby representing a binding mode that is not reproduced in the full-length protein.
Conclusion
Direct quantification of protein–protein interactions involving membrane proteins is often technically challenging because of low sample availability and the need for detergents. To overcome this, peptides corresponding to the membrane protein interaction site are often used. While peptides are very useful for verifying an interaction and locating binding sites and interacting residues, they may miss important parameters that are only present in the context of full-length proteins, for example allosteric regulation or cooperativity between binding sites. In this study we show that MST and fluorescence anisotropy can be successfully used to study the interaction between full-length AQP0 and CaM and full-length AQP2 and LIP5 in the presence of detergent and using low amounts of sample. These studies were then compared with interaction studies using peptides corresponding to the binding sites within the C-terminus of the respective AQP. The estimated Kd-values from all experiments are summarized in Table 2. In both cases, using the full-length AQP was proven to be advantageous. For AQP2, only the full-length protein gave similar results in terms of binding model and affinity from both methods whereas interaction studies using the AQP2 peptide gave very different results when studied by MST and fluorescence anisotropy. Moreover, full-length AQP0 was shown to bind CaM in a cooperative manner while cooperativity could not be seen in the experiments using AQP0 peptide. Our study highlights that using full-length proteins can provide more reliable estimations of affinities and give additional insights into the binding events that are of importance for understanding the interaction from a mechanistic point of view. We are confident that by using both peptides and full-length proteins to study protein–protein interactions involving AQPs, as well as other membrane proteins, we will be able to significantly deepen our understanding of a number of regulatory membrane protein–protein interactions and their role in the cell.In addition to increasing our knowledge of the biological aspects of AQP regulation and how this may be exploited from a pharmaceutical point of view, detailed knowledge of the specific interactions that regulate AQP transport may have implications for the development of artificial water channels. Based on the solution found in nature, artificial water channels seek to mimic the highly efficient and selective water transport mechanism of AQPs, thereby generating a novel strategy for advanced water purification filters.13–15 We envisage that detailed knowledge of the specific molecular interactions that are able to control water transport through AQPs in the cell could enable the development of water filters with completely new regulatory properties, allowing artificial water channels to be controlled using biomimetic solutions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a grant from Vetenskapsrådet (2012-2849) to ST-H.References
- S. Kreida and S. Tornroth-Horsefield, Structural insights into aquaporin selectivity and regulation, Curr. Opin. Struct. Biol., 2015, 33, 126–134, DOI:10.1016/j.sbi.2015.08.004.
- L. S. King, D. Kozono and P. Agre, From structure to disease: the evolving tale of aquaporin biology, Nat. Rev. Mol. Cell Biol., 2004, 5, 687–698 CrossRef PubMed.
- A. C. Conner, R. M. Bill and M. T. Conner, An emerging consensus on aquaporin translocation as a regulatory mechanism, Mol. Membr. Biol., 2013, 30, 1–12, DOI:10.3109/09687688.2012.743194.
- D. Pearce, et al., Collecting duct principal cell transport processes and their regulation, Clin. J. Am. Soc. Nephrol., 2015, 10, 135–146, DOI:10.2215/cjn.05760513.
- S. L. Reichow, et al., Allosteric mechanism of water-channel gating by Ca2+–calmodulin, Nat. Struct. Mol. Biol., 2013, 20, 1085–1092, DOI:10.1038/nsmb.2630.
- S. L. Reichow and T. Gonen, Noncanonical binding of calmodulin to aquaporin-0: implications for channel regulation, Structure, 2008, 16, 1389–1398, DOI:10.1016/j.str.2008.06.011.
- R. Offringa and F. Huang, Phosphorylation-dependent trafficking of plasma membrane proteins in animal and plant cells, J. Integr. Plant Biol., 2013, 55, 789–808, DOI:10.1111/jipb.12096.
- L. M. Traub, Tickets to ride: selecting cargo for clathrin-regulated internalization, Nat. Rev. Mol. Cell Biol., 2009, 10, 583–596, DOI:10.1038/nrm2751.
- J. V. Roche, et al., Phosphorylation of human aquaporin 2 (AQP2) allosterically controls its interaction with the lysosomal trafficking protein LIP5, J. Biol. Chem., 2017, 292, 14636–14648, DOI:10.1074/jbc.M117.788364.
- G. Fischer, et al., Crystal structure of a yeast aquaporin at 1.15 angstrom reveals a novel gating mechanism, PLoS Biol., 2009, 7, e1000130, DOI:10.1371/journal.pbio.1000130.
- J. Sjohamn and K. Hedfalk, Unraveling aquaporin interaction partners, Biochim. Biophys. Acta, 2014, 1840, 1614–1623, DOI:10.1016/j.bbagen.2013.11.012.
- H. B. Moeller, et al., Regulation of the Water Channel Aquaporin-2 via 14-3-3-theta and -zeta, J. Biol. Chem., 2016, 291, 2469–2484, DOI:10.1074/jbc.M115.691121.
- M. Barboiu and A. Gilles, From natural to bioassisted and biomimetic artificial water channel systems, Acc. Chem. Res., 2013, 46, 2814–2823, DOI:10.1021/ar400025e.
- I. Kocsis, et al., Oriented chiral water wires in artificial transmembrane channels, Sci. Adv., 2018, 4, eaao5603, DOI:10.1126/sciadv.aao5603.
- C. S. Lee, et al., Facilitated Water Transport through Graphene Oxide Membranes Functionalized with Aquaporin-Mimicking Peptides, Adv. Mater., 2018, 30, e1705944, DOI:10.1002/adma.201705944.
- B. W. van Balkom, et al., LIP5 interacts with aquaporin 2 and facilitates its lysosomal degradation, J. Am. Soc. Nephrol., 2009, 20, 990–1001 CrossRef PubMed.
- K. M. Rose, et al., Aquaporin 0-calmodulin interaction and the effect of aquaporin 0 phosphorylation, Biochemistry, 2008, 47, 339–347, DOI:10.1021/bi701980t.
- A. Frick, et al., X-ray structure of human aquaporin 2 and its implications for nephrogenic diabetes insipidus and trafficking, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6305–6310, DOI:10.1073/pnas.1321406111.
- F. Oberg, et al., Insight into factors directing high production of eukaryotic membrane proteins; production of 13 human AQPs in Pichia pastoris, Mol. Membr. Biol., 2009, 26, 215–227, DOI:10.1080/09687680902862085.
- E. Kinoshita, et al., Highly sensitive detection of protein phosphorylation by using improved Phos-tag Biotin, Proteomics, 2012, 12, 932–937, DOI:10.1002/pmic.201100639.
- H. Ampah-Korsah, et al., The Aquaporin Splice Variant NbXIP1;1alpha Is Permeable to Boric Acid and Is Phosphorylated in the N-terminal Domain, Front. Plant Sci., 2016, 7, 862, DOI:10.3389/fpls.2016.00862.
- D. J. O’Connell, et al., Integrated protein array screening and high throughput validation of 70 novel neural calmodulin-binding proteins, Mol. Cell. Proteomics, 2010, 9, 1118–1132, DOI:10.1074/mcp.M900324-MCP200.
- P. Linke, et al., An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery, J. Biomol. Screening, 2016, 21, 414–421, DOI:10.1177/1087057115618347.
- I. Andre and S. Linse, Measurement of Ca2+-binding constants of proteins and presentation of the CaLigator software, Anal. Biochem., 2002, 305, 195–205, DOI:10.1006/abio.2002.5661.
- D. C. LaPorte, B. M. Wierman and D. R. Storm, Calcium-induced exposure of a hydrophobic surface on calmodulin, Biochemistry, 1980, 19, 3814–3819 CrossRef PubMed.
- J. J. Skalicky, et al., Interactions of the human LIP5 regulatory protein with endosomal sorting complexes required for transport, J. Biol. Chem., 2012, 287, 43910–43926, DOI:10.1074/jbc.M112.417899.
- C. J. Vild, Y. Li, E. Z. Guo, Y. Liu and Z. Xu, A novel mechanism of regulating the ATPase VPS4 by its cofactor LIP5 and the endosomal sorting complex required for transport (ESCRT)-III protein CHMP5, J. Biol. Chem., 2015, 290, 7291–7303, DOI:10.1074/jbc.M114.616730.
- M. Jerabek-Willemsen, C. J. Wienken, D. Braun, P. Baaske and S. Duhr, Molecular interaction studies using microscale thermophoresis, Assay Drug Dev. Technol., 2011, 9, 342–353, DOI:10.1089/adt.2011.0380.
- C. A. Royer and S. F. Scarlata, Fluorescence approaches to quantifying biomolecular interactions, Methods Enzymol., 2008, 450, 79–106, DOI:10.1016/S0076-6879(08)03405-8.
- S. Forsen and S. Linse, Cooperativity: over the Hill, Trends Biochem. Sci., 1995, 20, 495–497 CrossRef PubMed.
- S. Linse, et al., The role of protein surface charges in ion binding, Nature, 1988, 335, 651–652, DOI:10.1038/335651a0.
- W. E. Harries, D. Akhavan, L. J. Miercke, S. Khademi and R. M. Stroud, The channel architecture of aquaporin 0 at a 2.2-A resolution, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14045–14050, DOI:10.1073/pnas.0405274101.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fd00065d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |