 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spiers Memorial Lecture
Introductory lecture: the impact of structure on photoinduced processes in nucleic acids and proteins
Tatiana
Domratcheva
and
Ilme
Schlichting
 *
*
Max Planck Institute for Medical Research, Jahnstr. 29, 69120 Heidelberg, Germany. E-mail: ilme.schlichting@mpimf-heidelberg.mpg.de
First published on 12th March 2018
Abstract
Light is an important environmental variable and most organisms have evolved means to sense, exploit or avoid it and to repair detrimental effects on their genome. In general, light absorption is the task of specific chromophores, however other biomolecules such as oligonucleotides also do so which can result in undesired outcomes such as mutations and cancer. Given the biological importance of light-induced processes and applications for imaging, optogenetics, photodynamic therapy or photovoltaics, there is a great interest in understanding the detailed molecular mechanisms of photoinduced processes in proteins and nucleic acids. The processes are typically characterized by time-resolved spectroscopic approaches or computation, inferring structural information on transient species from stable ground state structures. Recently, however, structure determination of excited states or other short-lived species has become possible with the advent of X-ray free-electron lasers. This review gives an overview of the impact of structure on the understanding of photoinduced processes in macromolecules, focusing on systems presented at this Faraday Discussion meeting.
1. Introduction
Light is an important environmental variable. High energy UV light can damage for example DNA while visible light provides energy for photosynthesis, producing biomass, enabling and sustaining oxygenic life on earth. Organisms have therefore evolved photosensory proteins that sense most of the spectral region, enabling phototaxis or photoavoidance, allowing us to see, and providing clues to e.g. synchronize daily rhythms. Understanding these processes not only has an impact on human health (melanoma, jet lag, photodynamic cancer therapy…) but also on sustainable energy sources by inspiring photovoltaic systems that use solar energy to produce electrical current. Moreover, the use of light-sensitive proteins has transformed cell- and neurobiology. High resolution imaging of live cells is made possible by fluorescent proteins, providing unprecedented insight into various processes. Optogenetics allows manipulation of diverse processes in vivo ranging from vectorial ion transport and signalling by second messengers (Ca2+, cyclic nucleotides) to changing oligomerization states of proteins with high temporal and spatial resolution.For all these reasons, there is great interest in understanding photoinduced processes in nucleic acids and proteins. Recent developments in ultrafast spectroscopy, computational chemistry and in particular time-resolved crystallography have provided unprecedented and novel insights into the very early processes induced by photon absorption, which so far had been difficult to investigate. Taken together, this information provides a detailed understanding of how the early excited-state relaxation couples to the protein matrix and how protein dynamics shapes early photodynamics and the subsequent photochemical events. This Faraday Discussion meeting brings together experts who use different methods to provide detailed insights in the early events in the various photoinduced processes that can take place in nucleic acids and proteins. It will be interesting to see common themes and variations emerge.
2. Photoinduced DNA damage and repair by photoreactivation
Nucleobases strongly absorb UV photons. In single bases the absorbed energy is transformed into heat on a sub-picosecond time scale by nonradiative decay.1 In contrast, the decay of excess electronic energy is significantly slower in DNA oligonucleotides2,3 which are multichromic molecules with numerous overlapping transitions in the UV. These transitions populate different types of excited states including excitons, excimers, exciplexes, and charge transfer (CT) states delocalized on several nucleobases, in addition to the locally excited states of single nucleobases. Stacking and pairing of nucleobases in DNA duplexes favours population of CT states that correspond to intra-strand or inter-strand electron transfer between nucleobases thus resulting in the transient formation of a radical pair. The CT states have been identified as gateways to oxidative damage and photodamage (Banyasz et al., DOI: 10.1039/c7fd00179g; Giussani et al., DOI: 10.1039/c7fd00202e; Jian et al.4), but at the same time they mediate energy dissipation increasing DNA photostability (Martinez-Fernandez and Improta, DOI: 10.1039/c7fd00195a). The processes are typically characterized by time-resolved spectroscopic approaches2,3,5–9 or computation (reviewed in ref. 10), inferring structural information on transient species from stable ground state structures. Known structures of DNA duplexes provide a basis for the development of highly accurate computational-chemistry models in order to study the reactivity of single nucleobases, dimers and higher-order assemblies in a complex DNA environment (Chakraborty et al., DOI: 10.1039/c7fd00188f; Giussani et al., DOI: 10.1039/c7fd00202e; Martinez-Fernandez and Improta, DOI: 10.1039/c7fd00195a). Ultimately, the excitation and relaxation processes of nucleic acids depend not only on oligomeric state and sequence, but also on conformational dynamics (Segarra-Martí et al., DOI: 10.1039/c7fd00201g; Brown et al., DOI: 10.1039/c7fd00186j) and solvent effects (Zhang et al., DOI: 10.1039/c7fd00205j).Although base stacking and pairing in single and double stranded DNA result in significantly longer lived excited states, they rarely cause photochemical damage. In fact, the energy of 99.9% of the absorbed photons is transformed into heat via fast nonradiative decay. Nevertheless, in strong sunlight the far UV region of the solar spectrum can induce 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lesions in DNA per exposed cell per hour. So-called direct DNA damage can occur for example when two adjacent pyrimidine bases react inside DNA upon UV-irradiation to form covalent pyrimidine dimers: either a cyclobutane–pyrimidine-dimer (CPD) (80%) or less frequently (20%) a (6-4) pyrimidine–pyrimidone photoproduct (6-4PP) (Scheme 1a). Both lesions are mutagenic and can eventually lead to cancer. Organisms have thus evolved means to revert this damage. In placental mammals lesion repair occurs via different processes including nucleotide excision. Most other organisms use photolyases, a distinct and ubiquitous class of flavoenzymes which are believed to be ancient DNA repair proteins as evidenced for example by their occurrence in giant DNA viruses.11 Photolyases can reverse the UV-induced DNA damage using the energy of light, a process known as photoreactivation.12 Notably, photoreactivation is also mediated by CT states formed between the enzyme flavin cofactor and the DNA photolesion, and thus involves transient formation of a radical pair that undergoes recombination after cleavage of the interbase covalent bonds. Such a complex repair mechanism critically depends on orchestration of the electron-transfer and bond-cleavage steps by coupling the protein dynamics with the photo-induced dynamics of the charge-transfer radical pair. Structure determination of photolyases complexed with lesion-containing DNA duplexes13–15 greatly contributed to the understanding of the photoreactivation mechanism.
000 lesions in DNA per exposed cell per hour. So-called direct DNA damage can occur for example when two adjacent pyrimidine bases react inside DNA upon UV-irradiation to form covalent pyrimidine dimers: either a cyclobutane–pyrimidine-dimer (CPD) (80%) or less frequently (20%) a (6-4) pyrimidine–pyrimidone photoproduct (6-4PP) (Scheme 1a). Both lesions are mutagenic and can eventually lead to cancer. Organisms have thus evolved means to revert this damage. In placental mammals lesion repair occurs via different processes including nucleotide excision. Most other organisms use photolyases, a distinct and ubiquitous class of flavoenzymes which are believed to be ancient DNA repair proteins as evidenced for example by their occurrence in giant DNA viruses.11 Photolyases can reverse the UV-induced DNA damage using the energy of light, a process known as photoreactivation.12 Notably, photoreactivation is also mediated by CT states formed between the enzyme flavin cofactor and the DNA photolesion, and thus involves transient formation of a radical pair that undergoes recombination after cleavage of the interbase covalent bonds. Such a complex repair mechanism critically depends on orchestration of the electron-transfer and bond-cleavage steps by coupling the protein dynamics with the photo-induced dynamics of the charge-transfer radical pair. Structure determination of photolyases complexed with lesion-containing DNA duplexes13–15 greatly contributed to the understanding of the photoreactivation mechanism.
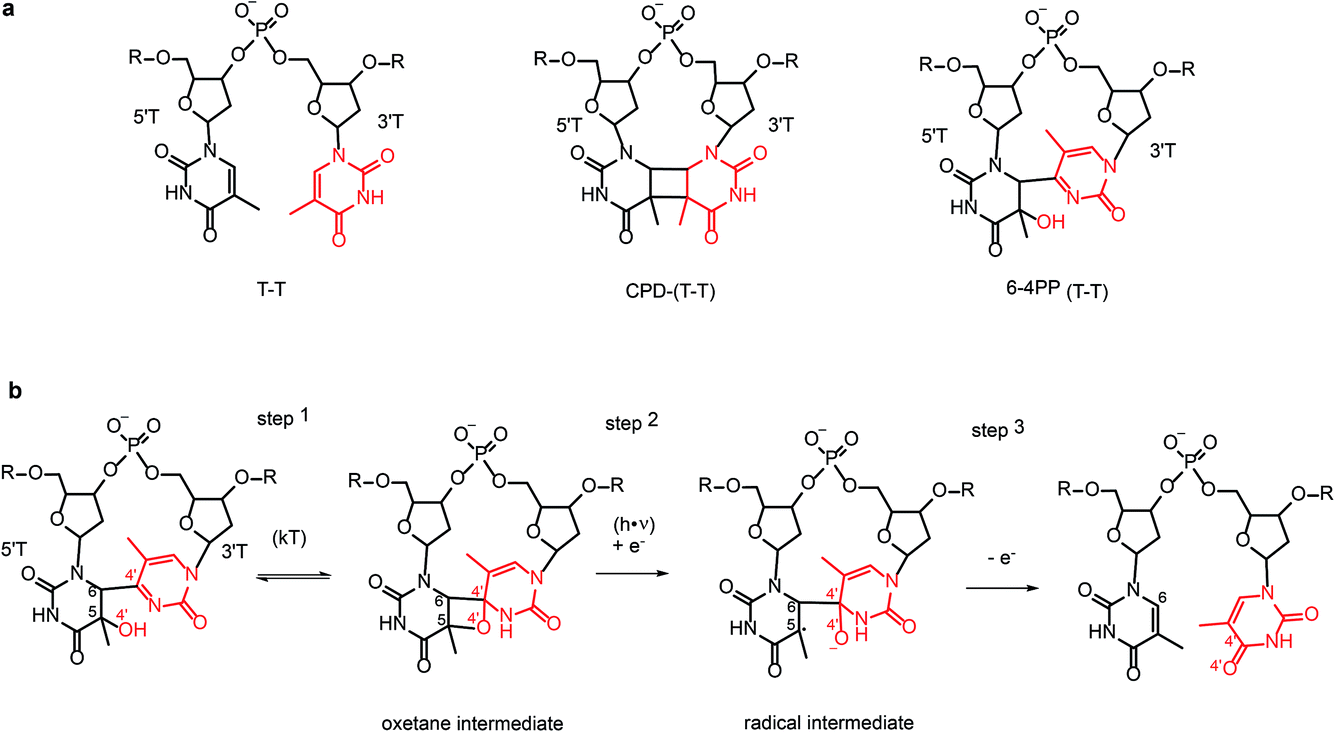 | ||
| Scheme 1 (a) Chemical structure of the T–T dinucleotide and its CPD and 6-4 PP photolesions; (b) sequence of steps involved in the hypothesised repair of 6-4 PP photolesions by 6-4 photolyase via formation and photoreactivation of the oxetane intermediate. The atoms of the 3′ base (3′T) are shown in red. | ||
2.1 DNA repair by photolyase
CPD and 6-4PP lesions are repaired by specific photolyases which are therefore classified as CPD photolyases and 6-4 photolyases, respectively. They share similar primary sequences and structures but can only repair one kind of photoproduct, which they recognize with high specificity. Photolyases absorb blue light by the catalytically active flavin adenine dinucleotide (FAD) cofactor or through energy transfer from an excited antenna chromophore, either 5,10-methenyltetrahydrofolate (MTHF) or 8-hydroxy-5-deazariboflavin (F0), which have absorption maxima of ∼380 and ∼440 nm, respectively. In CPD and 6-4 photolyases the catalytically active cofactor is a fully reduced FADH−. It binds to the enzyme’s active site in an unusual U-shaped configuration, with its isoalloxazine and adenine rings stacked in close distance to the lesion, which is flipped out from the double stranded DNA, pointing into the active site.13–15 Upon photoactivation, the excited FADH− donates an electron to the lesion, initiating the repair reaction. Ultimately back transfer of the electron to the flavin completes the reaction cycle. Repair of the CPD lesion involves splitting of the cyclobutane ring and formation of the two canonical bases. Ultrafast spectroscopy was used to characterize the light-induced electron-transfer steps in CPD photolyase in great detail.16,17 Some controversies remain18,19 concerning the experimental details and the rate of the electron-back transfer.Repair of the 6-4PP lesions to the undamaged T–T or T–C sequence is more complicated than the CPD repair which only involves bond cleavage. To recover the initial two-base sequence, 6-4 photolyase must control the timing of the repair steps which involve not only bond cleavage but also bond formation. The 6-4 PP lesion undergoes transfer of O and H atoms (shown in red in Scheme 1b) from the 5′ base (5′T) to the 3′ base (3′T) base in addition to the cleavage of the C6–C4′ covalent bond.
Before the crystal structure of the 6-4 photolyase complexed to a DNA duplex containing 6-4PP lesion was available20,21 it was believed that 6-4PP rearranges to an oxetane intermediate upon binding to the enzyme (step 1 in Scheme 1b). The excited FADH− cofactor donates an electron to the oxetane intermediate (step 2 in Scheme 1b). Since the oxetane ring is not stable in the radical-anion form it undergoes rearrangement via cleavage of the C5–O4′ bond. Backward electron transfer (step 3) finalizes the enzymatic cycle by recovering reduced FADH− before the repaired DNA is released from the enzyme.
This reaction mechanism was shown to be incorrect, as the crystal structure of 6-4 photolyase complexed with a lesion containing DNA showed a 6-4 pyrimidine-pyrimidone photoproduct lesion instead of the proposed oxetane intermediate. The lack of observation of the oxetane intermediate was not due to a crystallization artefact15 since the crystalline 6-4 photolyase was enzymatically active, repairing the lesion in situ upon illumination to yield the undamaged pyrimidine–pyrimidine bases. The absence of the thermally populated intermediate indicates that all repair steps are coordinated by the protein in the transiently populated radical-pair state with a lifetime of few nanoseconds,22 making 6-4 photolyase an important example of functional coupling between protein dynamics and substrate photodynamics. The availability of a structure of the 6-4 photolyase–lesion containing DNA complex not only falsified the accepted mechanism of the 6-4PP rearrangement, but also spurred a number of new suggestions for the repair mechanism. Most of the mechanisms proposed on the basis of spectroscopic characterization and computational studies include a proton transfer step coupled to the initial photoinduced electron transfer.15,22 According to a two photon mechanism, an oxetane intermediate formed after an initial photoinduced electron-transfer transfer cycle was proposed to undergo rearrangement into undamaged bases via a second photoinduced electron transfer cycle.23,24 An alternative mechanism that avoids proton transfer steps25,26 suggests a direct transfer of the hydroxyl group (O4′H−) from the 5′ base to the 3′ base followed by the cleavage of the C6–C4′ bond.
To determine which one of the currently proposed reaction mechanisms correctly describes the function of 6-4 photolyase, structural information is required of the early intermediates that precede backward electron transfer. Several kinetic intermediates featuring the radical of the photoproduct with the lifetimes ranging from tens of picoseconds to nanoseconds were identified by transient spectroscopy.22 The structure determination will be challenging as spectroscopic studies22 indicate that the population of the nanosecond lifetime intermediate is low, as the photoreaction competes against the futile backward electron transfer from the 6-4 photoproduct anion-radical to the FADH radical, resulting in 10% photoreactivation yield. Despite this complication, new powerful X-ray sources delivering femtosecond pulses enable high resolution insight into the early events following photon absorption by time-resolved crystallography. This opens completely uncharted territory for structural biology that was previously only accessible by ultra-fast spectroscopy.
3. Time-resolved crystallography
Crystals are highly ordered ensembles of molecules. In particular, crystals of macromolecules are often catalytically active due to their high solvent content (typically 30–80%) and relatively few weak interactions between the molecules, providing crystal contacts which allow for reaction-induced conformational changes. In order to determine structures of reaction intermediates by time-resolved crystallography, the reaction needs to be initiated efficiently, gently and faster than the reaction – or step thereof – investigated, in order to resolve the structural changes “in step” and to not wash out the structural changes by averaging different states. Photoinduced reactions can be synchronized easily over macroscopic ensembles of molecules, facilitating the study of various processes at high temporal resolution by an optical pump and X-ray probe technique. However, due to the high molecular concentration in crystals (typically 10–60 mM in protein crystals), optical densities are often very high, complicating if not preventing efficient reaction triggering by light. It is therefore advantageous to use crystals that are as small as possible but still yield high resolution diffraction. Collecting diffraction data of very small crystals and other weakly scattering objects is made possible by X-ray free-electron lasers (XFELs), linear accelerator based X-ray sources that deliver femtosecond pulses with a peak brilliance exceeding that of synchrotron sources by nine orders of magnitude. An additional advantage over synchrotron sources is that the short pulse length of XFELs not only extends the time-resolution from 100 ps (limited by the electron bunch length at synchrotrons) to the chemical timescale of femtoseconds, but also allows outrunning most radiation damage effects.27 Each microcrystal yields a single diffraction pattern upon XFEL pulse exposure before it is destroyed by subsequent damage effects (diffraction before destruction), necessitating high throughput serial data collection, an approach called serial femtosecond crystallography (SFX). Depending on crystallization conditions and the amount of material available, the microcrystals are delivered into the XFEL beam by jetting in thin liquid microstreams, streaming of viscous materials with embedded crystals or rastering of chips (fixed targets) containing the microcrystals.28 All these approaches are compatible with a pump probe data collection scheme and allow data acquisition at ambient temperature, enabling time-resolved experiments on light-sensitive systems.4. Double bond isomerization in photoactive proteins
Light-induced double bond isomerizations occur in the chromophores of various photoreceptors, including tetrapyrrole in phytochromes, p-coumaric acid in photoactive yellow protein, retinal in rhodopsins and 4-(p-hydroxybenzylidene)imidazolidin-5-one in fluorescent proteins (Scheme 2). In photoreceptor proteins, photoinduced change of the chromophore configuration facilitates rearrangement of the hydrogen-bonding network surrounding the chromophore and often causes protonation or deprotonation events that together trigger larger structural changes of the protein resulting in formation of the biologically active signalling state. In fluorescent proteins, change of protein–chromophore interactions induced by isomerization drastically changes the emission properties. Since many of these proteins have important applications in imaging or optogenetics, it is of great practical interest to understand the molecular origins of their colour tuning, specificity, photoactivation and fluorescence efficiency or nature of side-reactions in order to modify these properties in a rational way. To this end, the photoreaction itself needs to be characterized in depth (photophysics, photochemistry) as well as the interactions of the chromophore with the protein matrix. These interactions play a central role in excited-state evolution by introducing steric constraints as well as dynamic effects, controlling the timing of elementary steps and defining coupling between the cofactor photoreaction and protein motions.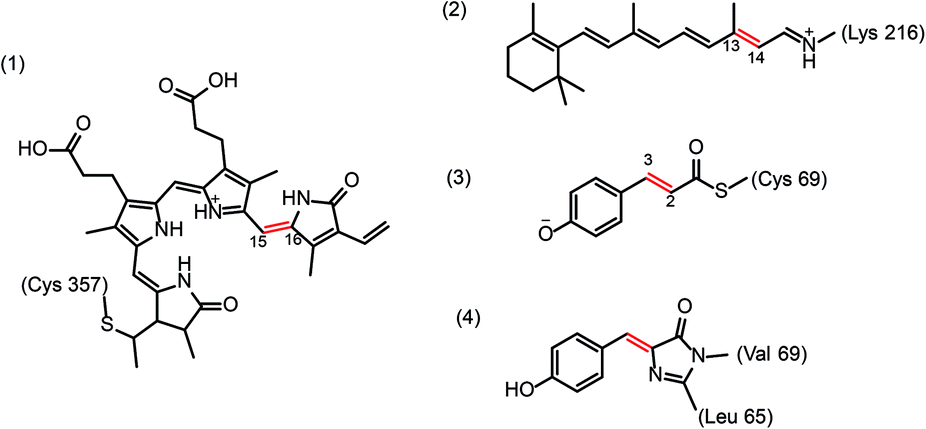 | ||
| Scheme 2 Chemical structures of biological chromophores undergoing photoisomerization around a double bond (shown in red): (1) protonated tetrapyrrole in plant phytochrome, (2) protonated retinal Schiff-base in bacteriorhodopsins, (3) p-coumaric acid (anion) in photoactive yellow protein, and (4) 4-(p-hydroxybenzylidene)imidazolidin-5-one in fluorescent proteins. | ||
4.1 Rhodopsins
Rhodopsins are α-helical membrane proteins that contain a retinal cofactor linked to the protein via a Schiff base with a lysine side chain. The most prominent member of the family is rhodopsin, a G-protein coupled photoreceptor involved in visual phototransduction in animals. Microbial and algal rhodopsins are involved in directed ion transport and have prominent applications in optogenetics. While photoexcited retinal in solution isomerizes at several double bond positions, the protein matrix constrains this process. In visual rhodopsins, 11-cis-retinal is converted to all-trans-retinal whereas microbial rhodopsins convert all-trans retinal to 13-cis. The best characterized microbial rhodopsin is bacteriorhodopsin, a light-driven proton pump. Its photocycle is described by a sequence of distinct spectroscopic states: upon photon absorption a short-lived excited intermediate I decays within ∼0.5 ps to the J intermediate. A hierarchical series of intermediates (J → K → L → M → N → O) with ps (J), μs (K, L) and ms (M, N, O) lifetimes follows before the resting state is reached. The crucial steps enabling proton transport take place at the active site, which comprises Wat402 forming hydrogen bonds with the protonated Schiff base and the anionic side chains of Asp85 and Asp212. Deprotonation of the Schiff base of the photoisomerized retinal and protonation of Asp85 during the L → M transition ultimately results in proton translocation to the extracellular side. Recently, time-resolved SFX was used to visualize the steps from the K-intermediates onward. The K-intermediate, captured 16 ns after photon absorption29 shows a strained 13-cis isomer of retinal and disorder of Wat402 which is close to the Schiff base and part of a water-mediated hydrogen bonded network involved in proton translocation. Most likely, changes in Wat402 relate to the very early events in the photoisomerization reaction. Water-mediated hydrogen bonded networks are not only important for ion conductance in bR but also for stabilizing different conformations in the related retinal containing protein rhodopsin, a G protein coupled receptor (Lesca et al., DOI: 10.1039/c7fd00207f).Insight into the early events following photon absorption has been obtained by ultrafast spectroscopy (e.g.ref. 30–34 and Agathangelou et al., DOI: 10.1039/c7fd00200a) and computation (reviewed in ref. 35 and see e.g. Borin et al., DOI: 10.1039/c7fd00198c). Induced by photoexcitation, charge transfer is followed by bond length alteration (BLA) related to changes of bond character along the retinal polyene chain as well as activation of hydrogen-out-of-plane (HOOP) wagging motions which together facilitate twisting about the isomerizing bond of the chromophore. These molecular motions drive the excited chromophore toward the conical intersection between the S1 and S0 potential energy surfaces.35 The site of retinal isomerization and quantum yield differ markedly between retinal protonated Schiff base bound to a protein and retinal in solution. Similar vibrational signatures of retinal in protein and solution, imply similar starting geometries,36 suggesting that the differences in reactivity likely results from the activation of distinct nuclear motion by energy transfer from the initially populated coordinates.37,38 It is to be expected that these high-frequency retinal modes couple with lower frequency modes of the protein already on an ultrafast timescale.
4.2 Fluorescent proteins
Fluorescent proteins (FP) have revolutionized cell biology by serving as genetically encoded markers that enable visualization of different constituents and processes in vivo. FPs derived from a green fluorescent protein (GFP) consist of a beta barrel that restricts access of water to the chromophore as well as its conformational flexibility. The chromophore is formed by oxidation of amino acids and consists of two π-conjugated phenol and imidazolinone rings linked by a methylene bridge. Variants of GFP can undergo a variety of light-induced transformations:39 FPs can be activated by light from a dark non-fluorescent form to a bright fluorescent form or vice versa. Reversibly photoswitchable FPs can be toggled back and forth between dark and bright forms using light with different wavelength, whereas photoconvertible FPs can be switched between two fluorescent forms of different colours. Photoconvertible and photoswitchable FPs provide the basis for many super-resolution microscopy techniques.Reversibly photoswitchable FPs include Dronpa,40 Padron,41 IrisFP,42 asFP395,43 and rsEGFP2.44 The light-induced interconversion between dark and bright forms involves cis to trans isomerization and a change in protonation state of the chromophore. While the order of events is still under some debate, there is evidence45–47 that isomerization occurs on a picosecond time scale, followed by protonation on a microsecond time scale. The first comprehensive study on the isomerization reaction was performed recently on rsEGFP2 (ref. 44) using a combination of transient absorption spectroscopy, computation and time-resolved crystallography at an XFEL.48 rsEGFP2 can be switched from the fluorescent ON state (cis anionic chromophore) with 488 nm light to the non-fluorescent OFF state (trans protonated chromophore); OFF to ON switching can be done with 400 nm light.44 Since the quantum yield for the ON to OFF reaction is an order of magnitude lower (0.04) than that for the OFF to ON reaction (0.4), only the latter has been investigated in detail48 so far. Analysis of time-resolved absorption spectra showed that within 90 fs after absorption of a 400 nm photon two electronically excited intermediates are populated from the Franck Condon state. They decay with 0.9 ps and 3.7 ps time constants to the ground electronic state, either back to the protonated trans isomer or forward to the still-protonated cis isomer. Unlike GFP49 and Venus (Dhamija et al., DOI: 10.1039/c7fd00187h), rsEGFP2 does not undergo excited state proton transfer as indicated by similar results obtained when the measurements were performed in D2O instead of H2O. Instead, proton transfer occurs on a slower time scale in the ground state similar to that characterized by previous studies on Dronpa45,46 and IrisFP.47
Time-resolved serial femtosecond crystallography measurements were performed to visualize the two short-lived intermediates by collecting diffraction data 1 ps and 3 ps after microcrystals of ON state rsEGFP2 had been excited by an optical pump pulse (see Fig. 1). The 1 ps pump probe delay data showed two low occupancy (∼6–7% total occupancy) intermediates, one with a twisted conformation of the chromophore (T), the other with a planar conformation (P). In the 3 ps delay data the occupancy of the twisted intermediate T was lower, indicating decay of this intermediate. The planar intermediate P could not be identified in 3 ps data, either because it had been fully depleted or because of lower quality of the diffraction data. The 3 ps delay data also indicated population of the ground state cis isomer and repopulation of the OFF state, suggesting that a fraction of the chromophores has isomerized at 3 ps but not yet at 1 ps after photon absorption. In the T intermediate, the phenol and imidazolinone rings of the chromophore are oriented perpendicularly, halfway between the ON- and OFF state conformations. This distorted geometry is in line with the results from QM/MM and classical MD calculations for the S1 excited-state species. The P intermediate was also characterized by calculations and likely corresponds to one of the short-lived intermediates observed by transient absorption spectroscopy.
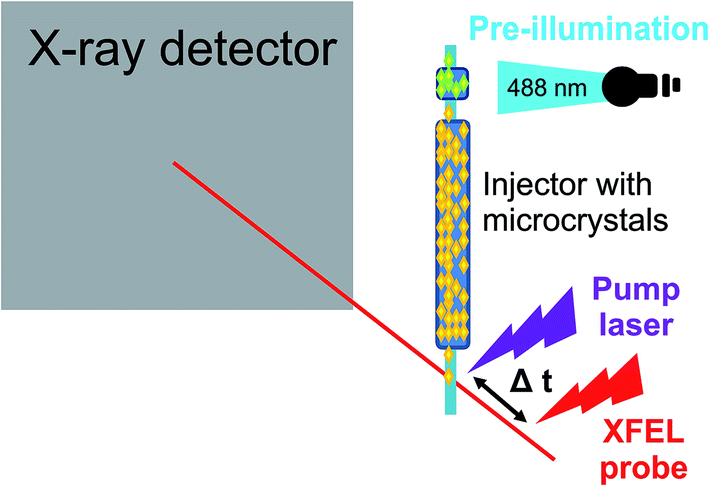 | ||
| Fig. 1 Experimental setup of a time-resolved pump probe experiment to study photoswitching in a fluorescent protein.48 | ||
The conformational changes of the chromophore during trans to cis isomerization are in line with a hula-twist motion.50 Formation of the twisted intermediate is accommodated by changes in the chromophore binding pocket that involve residues Val151 and Thr204 lining chromophore phenol group. Notably, Thr204 has different rotamers in the OFF-, T intermediate-, and ON states, whereas a clash with the side chain of Val151 is avoided by a tilt of the chromophore imidazolinone ring which may cause a translation of the α-helix containing the chromophore. It thus appears as if chromophore twisting was constrained at the phenol side. Accommodation of a twisted chromophore by the protein is believed to be essential for photoswitching in reversibly switching FPs. Since Val151 seems to constrain the twisted conformation (see Fig. 2), it was hypothesized that a smaller amino acid might facilitate photoswitching. Indeed, the Val151Ala variant has a higher photoswitching quantum yield than the wildtype (Val151Ala (wt) OFF-to-ON: 0.77 (0.4); ON-to-OFF 0.064 (0.043)). Thus, detailed knowledge of the structure of the excited state intermediate was used for the first time to rationally engineer a fluorescent protein.48
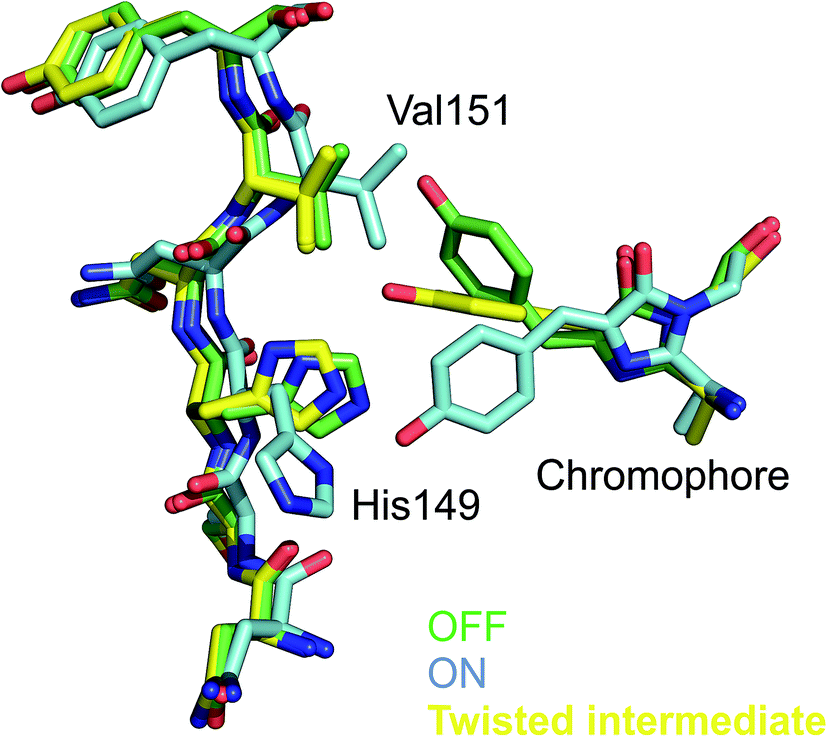 | ||
| Fig. 2 Chromophore conformation in the non-fluorescent OFF (green) and the fluorescent ON (light-blue) state as well in the twisted intermediate of rsEGFP2 observed 1 ps after photoexcitation. The close proximity of the Val151 side chain to the twisted chromophore is noticeable. Indeed, the Val151Ala variant has a higher switching quantum yield.48 | ||
4.3 Photoactive yellow protein
Photoactive yellow protein (PYP) is a blue light photoreceptor in purple photosynthetic bacteria. PYP has a p-coumaric acid (pCA) chromophore attached to a cysteine residue (Cys69) via a thioester bond. In the dark-adapted state, the deprotonated trans chromophore is stabilized by strong hydrogen bonds with Tyr42 and Glu46. Upon exposure to blue light, PYP undergoes a fully reversible branched photocycle from the dark state, through two early spectroscopic intermediates (I0 and I‡0)† that decay on a picosecond and nanosecond timescale to a red-shifted intermediate denoted pR (or I1). On a microsecond timescale pR decays to a blue-shifted state denoted pB (or I2), which then reverts to the dark state. The primary photochemical event activating PYP is the trans to cis isomerization of pCA around the central C3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 double bond.51 The structural changes upon formation of the cis-like configuration (I0) described as volume-saving hula-twist and bicycle bipedal isomerization pathways52 induce rearrangement of hydrogen bonds. The chromophore adopts a planar cis-geometry at the cost of breaking the hydrogen bond between its carbonyl group and the Cys69 backbone. Subsequently, the side-chain configuration of Cys69 changes. Two short hydrogen bonds between the pCA phenolate and Tyr42 and Glu46 break, exposing the pCA phenolate to water and facilitating protonation, which is spectroscopically observed as a blue-shifted intermediate.53 The rearrangement of the hydrogen-bonding network around isomerized pCA propagates the local photochemically-induced conformational changes to the N-terminal region of the protein, resulting in formation of the biologically active signalling state.
C2 double bond.51 The structural changes upon formation of the cis-like configuration (I0) described as volume-saving hula-twist and bicycle bipedal isomerization pathways52 induce rearrangement of hydrogen bonds. The chromophore adopts a planar cis-geometry at the cost of breaking the hydrogen bond between its carbonyl group and the Cys69 backbone. Subsequently, the side-chain configuration of Cys69 changes. Two short hydrogen bonds between the pCA phenolate and Tyr42 and Glu46 break, exposing the pCA phenolate to water and facilitating protonation, which is spectroscopically observed as a blue-shifted intermediate.53 The rearrangement of the hydrogen-bonding network around isomerized pCA propagates the local photochemically-induced conformational changes to the N-terminal region of the protein, resulting in formation of the biologically active signalling state.
The structural changes occurring during PYP photoactivation have been followed by time-resolved crystallography from 100 picoseconds to several seconds52–54 using synchrotron sources. Based on similar diffraction data different reaction schemes were proposed for the nanosecond and microsecond intermediates and the chromophore conformation in the early 100 ps structure was interpreted differently.52,53 In the first intermediate (lifetime ∼600 ps) the pCA chromophore is highly contorted, with its carbonyl group rotated nearly 90° with respect to the plane of the phenolate ring. A hydrogen bond between the pCA carbonyl and the Cys69 backbone constrains the chromophore in this unusual twisted conformation. There were, however, different interpretations52,53,55,56 concerning the geometry of the twisted cofactor, in particular the dihedral angle involving the isomerizing C3C2 double bond (30° (pR0 state53) versus 90° (IT state52)). Recent ultrafast time-resolved crystallography resolved these issues, showing an angle of ∼35° for a structure determined 3 ps after photoexcitation.57 The initial steps along the isomerization were also resolved, indicating a change of the double-bond configuration between 500 fs and 700 fs from a twisted trans conformation of the chromophore to a near cis form with the dihedral angle changing from ∼140° to ∼50°. In the twisted trans conformation the C2C3 double bond is displaced by ∼1 Å from the plane defined by the chromophore structure in the ground state. The hydrogen bonds between the phenolate ring and the Tyr42 and Glu46 side chains (or between the chromophore phenolate ring and the protein) are significantly elongated.
The time delays probed by SFX (0.1 to 3 ps) are within the vibrational dephasing time of the PYP S1 state58 and ground state modes in proteins. However, no oscillatory motions were observed for the protein residues or the cofactor. This could be due to sampling rate or to the inherently low quality of the extrapolated structure factors.
5. Ligand dissociation from myoglobin
Many gaseous ligand-binding heme proteins not only bind their natural ligand but also carbon monoxide (CO). This paves the way for time-resolved studies on ligand dissociation and re-binding exploiting the fact that the iron–CO bond is light-sensitive and dissociates upon absorption of a visible photon. The poster child for such studies is the oxygen storage protein myoglobin (Mb). It was the first protein whose three-dimensional structure was determined almost 60 years ago.59 The structure of the α-helical protein showed that there is no direct path for ligands to bind from the solvent to the heme, implying transient structural changes. Mb has thus become a model system to study protein dynamics, including the vibrational energy flow in proteins.Ligand binding and dissociation in myoglobin have been studied in great detail using numerous approaches. In synchrotron-based time-resolved X-ray crystallography,53 the time-resolution was limited by the 100 ps duration of the X-ray pulse and the structural changes emanating from the breaking ligand bond had already spread throughout the protein, resulting in large scale helix movements.53 It was therefore not possible to identify how and in which order these changes are induced by the breaking of the Fe–CO bond. These initial events were followed by sub-ps time-resolved SFX at an XFEL.60 Within 100 fs, CO appears in the primary docking site and the iron moves 0.2 Å out of the heme plane, in line with prior spectroscopic evidence. Structural changes appear throughout the protein within 500 fs in a quake-like fashion61 again in line with spectroscopic evidence, with the C-, F- and H-helices moving away from the heme and the E- and A-helices moving towards it. This is followed by further displacement of the E-helix and of the distal histidine. These collective movements are predicted by quantum mechanics/molecular mechanics (QM/MM) simulations and also observed in time-resolved solution studies.62 Interestingly, also sub-picosecond dynamics of residues close to the heme were observed. The χ1 torsion angles of Phe43, Val68, and Ile107 show an exponential change with time. Their side chains have a high probability of being hit by the dissociating CO63, which would result in a large momentum transfer, causing a movement of the side chain, followed by an exponential relaxation. In contrast, the χ1 and/or χ2 torsion angles of a number of side chains in close proximity to the heme and of a number of hydrogen bond distances seem to oscillate with time at sub-ps time delays. The oscillations share a 500 ± 150 fs period, which is reminiscent of the doming motion of the heme photoproduct of 430 fs (75 cm−1).64 Based on fs coherence spectroscopy, it has been suggested that electronic rearrangements in the heme iron upon ligand photodissociation can simultaneously excite associated low-frequency vibrational modes.65 The vibrational modes of the heme couple to collective global modes of the protein as suggested earlier,66,67 resulting not only in an immediate collective response of the protein but also the later, large scale motions. This notion of a network of coupled modes is important for the understanding of the nature and time-scales of enzyme catalysis68 and protein dynamics.69,70 Excitation and coupling of collective motions results in anisotropic energy flow through the protein structure. This is likely a general allosteric mechanism for the transport of local binding energy to remote sites in proteins.70
6. Conclusions and challenges
The advent of femtosecond time-resolved crystallography at XFELs and multidimensional spectroscopies allows us to obtain detailed insights into ultrafast protein dynamics, including structures of early intermediates, concerted motions of the excited chromophore and the surrounding protein. This not only provides structural information on vibronic coupling in photosensitive biomolecules, but generally enables correlating information obtained from various time-resolved spectroscopy methods with direct structural information.So far, all time-resolved crystallographic studies using femtosecond laser excitation used significantly higher pump energies than typically used for spectroscopic measurements. Comparing information derived from data that were obtained using very different excitation energies can, however, result in complications.71,72 Goodno et al.71 suggested that at high power excitation conditions, a larger fraction of the excess thermal energy will be transferred via the protein to the solvent via collective modes rather than through the slower diffusive channel. The reason would be a stronger anharmonic coupling between the collective modes of the protein and an increased spatial dispersion of the excess energy.71
Despite this challenge, it is enticing to correlate spectroscopic and structural data by comparing vibrational spectra obtained experimentally and derived computationally using structural models of short-lived species obtained by time-resolved crystallography. This will also facilitate development of computational methods towards an accurate description of interactions between the photoactive chromophore and its molecular environment (protein, DNA, solvent etc.) in particular in the excited state. An accurate description of intermolecular interactions at the quantum-mechanics level73 is still challenging, but necessary since the vibrational frequencies, especially of low-frequency modes, are sensitive to these interactions. Another challenge will be to account for anharmonic coupling enabling energy transfer from the high-frequency modes directly activated by photoexcitation to the low-frequency modes driving the system towards a conical intersection. These modes are observed as vibrational coherences in photoactive proteins, e.g. in rhodopsin and bacteriorhodopsin,37,74–76 but assignment of these coherences to specific vibrations of the chromophore coupled to the vibrations of the protein is still an open question. For a complex many-atom system, it is often unclear how to relate the computed frequencies to the experimental ones without additional experimental data, for instance the frequency shifts caused by site-directed isotope labeling.77 Hence, visualizing the coupled chromophore and protein vibrations by means of femtosecond time-resolved crystallography at XFELs may indeed result in a breakthrough in the identification of collective motions of biomolecules, which would advance our knowledge on the photochemical and functional dynamics of DNA and proteins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Open Access funding provided by the Max Planck Society.References
- C. E. Crespo-Hernandez, B. Cohen, P. M. Hare and B. Kohler, Ultrafast excited-state dynamics in nucleic acids, Chem. Rev., 2004, 104(4), 1977–2019 CrossRef CAS PubMed.
- C. E. Crespo-Hernandez, B. Cohen and B. Kohler, Base stacking controls excited-state dynamics in A.T DNA, Nature, 2005, 436(7054), 1141–1144 CrossRef CAS PubMed.
- C. E. Crespo-Hernandez, K. de la Harpe and B. Kohler, Ground-state recovery following UV excitation is much slower in G x C-DNA duplexes and hairpins than in mononucleotides, J. Am. Chem. Soc., 2008, 130(33), 10844–10845 CrossRef CAS PubMed.
- Y. Jian, E. Maximowitsch, D. Liu, S. Adhikari, L. Li and T. Domratcheva, Indications of 5′ to 3′ Interbase Electron Transfer as the First Step of Pyrimidine Dimer Formation Probed by a Dinucleotide Analog, Chem.–Eur. J., 2017, 23(31), 7526–7537 CrossRef CAS PubMed.
- D. Markovitsi, UV-induced DNA Damage: The Role of Electronic Excited States, Photochem. Photobiol., 2016, 92(1), 45–51 CrossRef CAS PubMed.
- G. W. Doorley, M. Wojdyla, G. W. Watson, M. Towrie, A. W. Parker, J. M. Kelly and S. J. Quinn, Tracking DNA Excited States by Picosecond-Time-Resolved Infrared Spectroscopy: Signature Band for a Charge-Transfer Excited State in Stacked Adenine–Thymine Systems, J. Phys. Chem. Lett., 2013, 4, 2739–2744 CrossRef CAS.
- D. B. Bucher, B. M. Pilles, T. Carell and W. Zinth, Charge separation and charge delocalization identified in long-living states of photoexcited DNA, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(12), 4369–4374 CrossRef CAS PubMed.
- Y. Zhang, J. Dood, A. A. Beckstead, X. B. Li, K. V. Nguyen and C. J. Burrows, et al., Efficient UV-induced charge separation and recombination in an 8-oxoguanine-containing dinucleotide, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(32), 11612–11617 CrossRef CAS PubMed.
- Y. Y. Zhang, K. de La Harpe, A. A. Beckstead, R. Improta and B. Kohler, UV-Induced Proton Transfer between DNA Strands, J. Am. Chem. Soc., 2015, 137(22), 7059–7062 CrossRef CAS PubMed.
- R. Improta, F. Santoro and L. Blancafort, Quantum Mechanical Studies on the Photophysics and the Photochemistry of Nucleic Acids and Nucleobases, Chem. Rev., 2016, 116(6), 3540–3593 CrossRef CAS PubMed.
- M. G. Fischer, I. Kelly, L. J. Foster and C. A. Suttle, The virion of Cafeteria roenbergensis virus (CroV) contains a complex suite of proteins for transcription and DNA repair, Virology, 2014, 466–467, 82–94 CrossRef CAS PubMed.
- R. Dulbecco, Reactivation of ultra-violet-inactivated bacteriophage by visible light, Nature, 1949, 163(4155), 949 CrossRef CAS PubMed.
- A. Mees, T. Klar, P. Gnau, U. Hennecke, A. P. Eker and T. Carell, et al., Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair, Science, 2004, 306(5702), 1789–1793 CrossRef CAS PubMed.
- S. Kiontke, Y. Geisselbrecht, R. Pokorny, T. Carell, A. Batschauer and L. O. Essen, Crystal structures of an archaeal class II DNA photolyase and its complex with UV-damaged duplex DNA, EMBO J., 2011, 30(21), 4437–4449 CrossRef CAS PubMed.
- M. J. Maul, T. R. Barends, A. F. Glas, M. J. Cryle, T. Domratcheva and S. Schneider, et al., Crystal structure and mechanism of a DNA (6-4) photolyase, Angew. Chem., Int. Ed. Engl., 2008, 47(52), 10076–10080 CrossRef CAS PubMed.
- V. Thiagarajan, M. Byrdin, A. P. Eker, P. Muller and K. Brettel, Kinetics of cyclobutane thymine dimer splitting by DNA photolyase directly monitored in the UV, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(23), 9402–9407 CrossRef CAS PubMed.
- Z. Liu, C. Tan, X. Guo, Y. T. Kao, J. Li and L. Wang, et al., Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(36), 14831–14836 CrossRef CAS PubMed.
- K. Brettel and M. Byrdin, DNA photolyase: is the nonproductive back electron transfer really much slower than forward transfer?, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(23), E1462 CrossRef CAS PubMed.
- D. Zhong, A. Sancar and A. Stuchebrukhov, Reply to Brettel and Byrdin: On the efficiency of DNA repair by photolyase, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1463 CrossRef CAS.
- X. Zhao, J. Liu, D. S. Hsu, S. Zhao, J. S. Taylor and A. Sancar, Reaction mechanism of (6-4) photolyase, J. Biol. Chem., 1997, 272(51), 32580–32590 CrossRef CAS PubMed.
- K. Hitomi, H. Nakamura, S. T. Kim, T. Mizukoshi, T. Ishikawa and S. Iwai, et al., Role of two histidines in the (6-4) photolyase reaction, J. Biol. Chem., 2001, 276(13), 10103–10109 CrossRef CAS PubMed.
- J. Li, Z. Liu, C. Tan, X. Guo, L. Wang and A. Sancar, et al., Dynamics and mechanism of repair of ultraviolet-induced (6-4) photoproduct by photolyase, Nature, 2010, 466(7308), 887–890 CrossRef CAS PubMed.
- J. Yamamoto, R. Martin, S. Iwai, P. Plaza and K. Brettel, Repair of the (6-4) photoproduct by DNA photolyase requires two photons, Angew. Chem., Int. Ed. Engl., 2013, 52(29), 7432–7436 CrossRef CAS PubMed.
- K. Sadeghian, M. Bocola, T. Merz and M. Schutz, Theoretical study on the repair mechanism of the (6-4) photolesion by the (6-4) photolyase, J. Am. Chem. Soc., 2010, 132(45), 16285–16295 CrossRef CAS PubMed.
- T. Domratcheva and I. Schlichting, Electronic structure of (6-4) DNA photoproduct repair involving a non-oxetane pathway, J. Am. Chem. Soc., 2009, 131(49), 17793–17799 CrossRef CAS PubMed.
- T. Domratcheva, Neutral histidine and photoinduced electron transfer in DNA photolyases, J. Am. Chem. Soc., 2011, 133(45), 18172–18182 CrossRef CAS PubMed.
- H. N. Chapman, C. Caleman and N. Timneanu, Diffraction before destruction, Philos. Trans. R. Soc., B, 2014, 369, 20130313 CrossRef PubMed.
- I. Schlichting, Serial femtosecond crystallography: the first five years, IUCrJ, 2015, 2(Pt 2), 246–255 CrossRef CAS PubMed.
- E. Nango, A. Royant, M. Kubo, T. Nakane, C. Wickstrand and T. Kimura, et al., A three-dimensional movie of structural changes in bacteriorhodopsin, Science, 2016, 354(6319), 1552–1557 CrossRef CAS PubMed.
- R. A. Mathies, C. H. Brito Cruz, W. T. Pollard and C. V. Shank, Direct observation of the femtosecond excited-state cis–trans isomerization in bacteriorhodopsin, Science, 1988, 240(4853), 777–779 CAS.
- G. R. Loppnow and R. A. Mathies, Excited-state structure and isomerization dynamics of the retinal chromophore in rhodopsin from resonance Raman intensities, Biophys. J., 1988, 54(1), 35–43 CrossRef CAS PubMed.
- T. Kobayashi, T. Saito and H. Ohtani, Real-time spectroscopy of transition states in bacteriorhodopsin during retinal isomerization, Nature, 2001, 414(6863), 531–534 CrossRef CAS PubMed.
- J. Herbst, K. Heyne and R. Diller, Femtosecond infrared spectroscopy of bacteriorhodopsin chromophore isomerization, Science, 2002, 297(5582), 822–825 CrossRef CAS PubMed.
- V. I. Prokhorenko, A. M. Nagy, S. A. Waschuk, L. S. Brown, R. R. Birge and R. J. Miller, Coherent control of retinal isomerization in bacteriorhodopsin, Science, 2006, 313(5791), 1257–1261 CrossRef CAS PubMed.
- S. Gozem, H. L. Luk, I. Schapiro and M. Olivucci, Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores, Chem. Rev., 2017, 117(22), 13502–13565 CrossRef CAS PubMed.
- S. O. Smith, M. S. Braiman, A. B. Myers, J. A. Pardoen, J. M. L. Courtin, C. Winkel, J. Lugtenburg and R. A. Mathies, Vibrational analysis of the all-trans-Retinal Chromophore in light-adapted bacteriorhodopsin, J. Am. Chem. Soc., 1987, 109, 3108–3125 CrossRef CAS.
- M. Liebel, C. Schnedermann, G. Bassolino, G. Taylor, A. Watts and P. Kukura, Direct observation of the coherent nuclear response after the absorption of a photon, Phys. Rev. Lett., 2014, 112(23), 238301 CrossRef CAS PubMed.
- P. J. Johnson, A. Halpin, T. Morizumi, L. S. Brown, V. I. Prokhorenko and O. P. Ernst, et al., The photocycle and ultrafast vibrational dynamics of bacteriorhodopsin in lipid nanodiscs, Phys. Chem. Chem. Phys., 2014, 16(39), 21310–21320 RSC.
- A. Acharya, A. M. Bogdanov, B. L. Grigorenko, K. B. Bravaya, A. V. Nemukhin and K. A. Lukyanov, et al., Photoinduced Chemistry in Fluorescent Proteins: Curse or Blessing?, Chem. Rev., 2017, 117(2), 758–795 CrossRef CAS PubMed.
- M. Andresen, A. C. Stiel, S. Trowitzsch, G. Weber, C. Eggeling and M. C. Wahl, et al., Structural basis for reversible photoswitching in Dronpa, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(32), 13005–13009 CrossRef CAS PubMed.
- M. Andresen, A. C. Stiel, J. Folling, D. Wenzel, A. Schonle and A. Egner, et al., Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy, Nat. Biotechnol., 2008, 26(9), 1035–1040 CrossRef CAS PubMed.
- V. Adam, M. Lelimousin, S. Boehme, G. Desfonds, K. Nienhaus and M. J. Field, et al., Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(47), 18343–18348 CrossRef CAS PubMed.
- M. Andresen, M. C. Wahl, A. C. Stiel, F. Grater, L. V. Schafer and S. Trowitzsch, et al., Structure and mechanism of the reversible photoswitch of a fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(37), 13070–13074 CrossRef CAS PubMed.
- T. Grotjohann, I. Testa, M. Reuss, T. Brakemann, C. Eggeling and S. W. Hell, et al., rsEGFP2 enables fast RESOLFT nanoscopy of living cells, eLife, 2012, 1, e00248 Search PubMed.
- M. M. Warren, M. Kaucikas, A. Fitzpatrick, P. Champion, J. T. Sage and J. J. van Thor, Ground-state proton transfer in the photoswitching reactions of the fluorescent protein Dronpa, Nat. Commun., 2013, 4, 1461 CrossRef PubMed.
- D. Yadav, F. Lacombat, N. Dozova, F. Rappaport, P. Plaza and A. Espagne, Real-time monitoring of chromophore isomerization and deprotonation during the photoactivation of the fluorescent protein Dronpa, J. Phys. Chem. B, 2015, 119(6), 2404–2414 CrossRef CAS PubMed.
- J. P. Colletier, M. Sliwa, F. X. Gallat, M. Sugahara, V. Guillon and G. Schiro, et al., Serial Femtosecond Crystallography and Ultrafast Absorption Spectroscopy of the Photoswitchable Fluorescent Protein IrisFP, J. Phys. Chem. Lett., 2016, 7(5), 882–887 CrossRef CAS PubMed.
- N. Coquelle, M. Sliwa, J. Woodhouse, G. Schiro, V. Adam, A. Aquila, T. R. M. Barends, S. Boutet, M. Byrdin, S. Carbajo, E. De la Mora, R. B. Doak, M. Feliks, F. Fieschi, L. Foucar, V. Guillon, M. Hilpert, M. S. Hunter, S. Jakobs, J. E. Koglin, G. Kovacsova, T. J. Lane, B. Levy, M. Liang, K. Nass, J. Ridard, J. S. Robinson, C. M. Roome, C. Rueckbusch, M. Seaberg, M. Thepaut, M. Cammarata, I. Demachy, M. Field, R. L. Shoeman, D. Bourgeois, J.-P. Colletier, I. Schlichting and M. Weik, Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography, Nat. Chem., 2018, 10(1), 31–37 CrossRef CAS PubMed.
- D. Stoner-Ma, A. A. Jaye, P. Matousek, M. Towrie, S. R. Meech and P. J. Tonge, Observation of excited-state proton transfer in green fluorescent protein using ultrafast vibrational spectroscopy, J. Am. Chem. Soc., 2005, 127(9), 2864–2865 CrossRef CAS PubMed.
- R. S. H. Liu, Photoisomerization by hula-twist: A fundamental supramolecular photochemical reaction, Acc. Chem. Res., 2001, 34(7), 555–562 CrossRef CAS PubMed.
- D. S. Larsen and R. van Grondelle, Initial photoinduced dynamics of the photoactive yellow protein, ChemPhysChem, 2005, 6(5), 828–837 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat and V. Srajer, et al., Volume-conserving trans–cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography, Nat. Chem., 2013, 5(3), 212–220 CrossRef CAS PubMed.
- F. Schotte, H. S. Cho, V. R. Kaila, H. Kamikubo, N. Dashdorj and E. R. Henry, et al., Watching a signaling protein function in real time via 100-ps time-resolved Laue crystallography, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(47), 19256–19261 CrossRef CAS PubMed.
- H. Ihee, S. Rajagopal, V. Srajer, R. Pahl, S. Anderson and M. Schmidt, et al., Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(20), 7145–7150 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat and V. Srajer, et al., Reply to ‘contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein’, Nat. Chem., 2014, 6(4), 259–260 CrossRef CAS PubMed.
- V. R. Kaila, F. Schotte, H. S. Cho, G. Hummer and P. A. Anfinrud, Contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein, Nat. Chem., 2014, 6(4), 258–259 CrossRef CAS PubMed.
- K. Pande, C. D. Hutchison, G. Groenhof, A. Aquila, J. S. Robinson and J. Tenboer, et al., Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein, Science, 2016, 352(6286), 725–729 CrossRef CAS PubMed.
- R. Nakamura, N. Hamada, H. Ichida, F. Tokunaga and Y. Kanematsu, Coherent oscillations in ultrafast fluorescence of photoactive yellow protein, J. Chem. Phys., 2007, 127(21), 215102 CrossRef PubMed.
- J. C. Kendrew, R. E. Dickerson, B. E. Strandberg, R. G. Hart, D. R. Davies and D. C. Phillips, et al., Structure of myoglobin: A three-dimensional Fourier synthesis at 2 A. resolution, Nature, 1960, 185(4711), 422–427 CrossRef CAS PubMed.
- T. R. Barends, L. Foucar, A. Ardevol, K. Nass, A. Aquila and S. Botha, et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation, Science, 2015, 350(6259), 445–450 CrossRef CAS PubMed.
- A. Ansari, J. Berendzen, S. F. Bowne, H. Frauenfelder, I. E. Iben and T. B. Sauke, et al., Protein states and proteinquakes, Proc. Natl. Acad. Sci. U. S. A., 1985, 82(15), 5000–5004 CrossRef CAS.
- M. Levantino, G. Schiro, H. T. Lemke, G. Cottone, J. M. Glownia and D. Zhu, et al., Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser, Nat. Commun., 2015, 6, 6772 CrossRef CAS PubMed.
- R. Elber and M. Karplus, Enhanced Sampling in Molecular Dynamics: Use of the Time-Dependent Hartree Approximation for a Simulation of Carbon Monoxide Diffusion through Myoglobin, J. Am. Chem. Soc., 1990, 112, 9161–9175 CrossRef CAS.
- L. Zhu, J. T. Sage and P. M. Champion, Observation of coherent reaction dynamics in heme proteins, Science, 1994, 266(5185), 629–632 CAS.
- P. M. Champion, F. Rosca, D. Ionascu, W. Cao and X. Ye, Rapid timescale processes and the role of electronic surface coupling in the photolysis of diatomic ligands from heme proteins, Faraday Discuss., 2004, 127, 123–135 RSC.
- R. J. D. Miller, Energetics and Dynamics of Deterministic Protein Motion, Acc. Chem. Res., 1994, 27, 145–150 CrossRef CAS.
- J. T. Sage, S. M. Durbin, W. Sturhahn, D. C. Wharton, P. M. Champion and P. Hession, et al., Long-range reactive dynamics in myoglobin, Phys. Rev. Lett., 2001, 86(21), 4966–4969 CrossRef CAS PubMed.
- V. L. Schramm, Transition States and transition state analogue interactions with enzymes, Acc. Chem. Res., 2015, 48(4), 1032–1039 CrossRef CAS PubMed.
- H. Fujisaki and J. E. Straub, Vibrational energy relaxation in proteins, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(19), 6726–6731 CrossRef CAS PubMed.
- G. Li, D. Magana and R. B. Dyer, Anisotropic energy flow and allosteric ligand binding in albumin, Nat. Commun., 2014, 5, 3100 Search PubMed.
- G. D. Goodno, V. Astinov and R. J. D. Miller, Femtosecond heterodyne-detected four-wave-mixing studies of deterministic protein motions. 2. Protein response, J. Phys. Chem. A, 1999, 103, 10630–10643 CrossRef CAS.
- V. I. Prokhorenko, A. Halpin, P. J. Johnson, R. J. Miller and L. S. Brown, Coherent control of the isomerization of retinal in bacteriorhodopsin in the high intensity regime, J. Chem. Phys., 2011, 134(8), 085105 CrossRef PubMed.
- J. Y. Hasegawa, K. Yanai and K. Ishimura, Quantum mechanical molecular interactions for calculating the excitation energy in molecular environments: a first-order interacting space approach, ChemPhysChem, 2015, 16(2), 305–311 CrossRef CAS PubMed.
- P. J. Johnson, A. Halpin, T. Morizumi, V. I. Prokhorenko, O. P. Ernst and R. J. Miller, Local vibrational coherences drive the primary photochemistry of vision, Nat. Chem., 2015, 7(12), 980–986 CrossRef CAS PubMed.
- J. P. Kraack, T. Buckup, N. Hampp and M. Motzkus, Ground- and excited-state vibrational coherence dynamics in Bacteriorhodopsin probed with degenerate four-wave-mixing experiments, ChemPhysChem, 2011, 12(10), 1851–1859 CrossRef CAS PubMed.
- D. P. Hoffman and R. A. Mathies, Femtosecond Stimulated Raman Exposes the Role of Vibrational Coherence in Condensed-Phase Photoreactivity, Acc. Chem. Res., 2016, 49(4), 616–625 CrossRef CAS PubMed.
- T. Domratcheva, E. Hartmann, I. Schlichting and T. Kottke, Evidence for Tautomerisation of Glutamine in BLUF Blue Light Receptors by Vibrational Spectroscopy and Computational Chemistry, Sci. Rep., 2016, 6, 22669 CrossRef CAS PubMed.
Footnote |
| † There is no commonly agreed-on nomenclature for the various intermediates. |
| This journal is © The Royal Society of Chemistry 2018 |