 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Flexible Unit Structure Engine (FUSE) for probe structure-based composition prediction
C.
Collins
 ,
G. R.
Darling
and
M. J.
Rosseinsky
*
,
G. R.
Darling
and
M. J.
Rosseinsky
*
Department of Chemistry, University of Liverpool, Liverpool, UK. E-mail: rossein@liverpool.ac.uk
First published on 11th June 2018
Abstract
Computationally led materials discovery requires efficient methods to generate either exact or approximate crystal structures that span the composition range of a chosen phase space. Here we present a new tool, the Flexible Unit Structure Engine (FUSE), for the generation of approximate ‘probe structures’ to predict regions of composition space where compounds can be experimentally realised. We then test FUSE by applying it to 42 compositions in the Y3+–Sr2+–Ti4+–O2− phase field. FUSE correctly identifies all of the target compounds in the regions of stability and identifies the exact crystal structure for 8 out of the 10 compositions.
1 Introduction
The computational search for new materials at its simplest involves a method for choosing an initial structure to test with a particular choice of elements, and a methodology for evolving this structure1–3 or choosing an alternative from a suitable database.4,5 There have been notable successes from such approaches6,7 to identifying missing elemental compositions or new high-pressure phases of materials. Much effort in inorganic materials chemistry has focused on the discovery of new materials with complex compositions or on the prediction of new compositions that can adopt previously known crystal structures – in either case, the problem becomes very complex once we consider systems containing more than three elements due to the number of possible phase fields and the size of the compositional phase space.In materials discovery our aim is to experimentally discover materials, guided by computational composition prediction. We do not seek to predict the properties of the materials computationally a priori, instead constructing phase fields from combinations of elements or compounds based on an understanding of the likely physical and chemical properties of the constituents, for example combining elements which are known to produce high levels of ionic conductivity. We then use well-established substitution and materials processing techniques to alter or enhance the desired properties of materials as they are experimentally realised.
While compositional complexity has the prospect of yielding multi-functional materials and offers more possibilities to tune functional behaviour, it has the drawback of greatly increasing the computational effort required to explore the range of structures and compositions. The initial choice of elements to investigate is of course determined by chemical knowledge and understanding. For example, Ga-containing oxides have been known to produce high oxide ion conductivity;8,9 it would therefore be desirable to seek new Ga-containing oxides.
Recently we developed a methodology for addressing the complex materials discovery problem, which has been successfully applied to realise two new materials of previously unknown composition and structure. To make the search through complex structure space tractable, we build crystal unit cells from fragments (we refer to these as modules) found in materials of simpler composition containing the chosen elements. These modules are then combined in a chemically sensible fashion (e.g. with the stacking rules of the cubic perovskite structure type) to construct a unit cell containing ions with plausible coordination environments. Relaxation to the local potential energy minimum determines the energy ranking of a structure. The modules are then permuted to generate new structures to be energy ranked. This Extended Module Materials Assembly (EMMA) method10 was employed to aid in the determination of the structure of a large layered perovskite, Y2.24Ba2.28Ca3.48Fe7.44Cu0.56O21, where the modular approach worked well to impose chemically sensible co-ordination chemistry on all of the atoms.
We extended EMMA with the addition of a Monte Carlo basin hopping algorithm to the permutation of modules (MC-EMMA) to obtain a structure searching tool that is sufficiently flexible and fast to permit exploration of the composition space in addition to the structure at given compositions.11 Building crystals by EMMA imposes restrictions on the possible outcomes. In particular, the modules are of fixed composition and they cannot be updated on-the-fly. They also have the same fixed periodicity perpendicular to the stacking direction, although during relaxation the unit cell shape and volume and all of the ionic positions can vary, resulting in crystal structures very different to the initial MC-EMMA constructions.
The best structures generated by MC-EMMA at any given composition in a completely unexplored phase space are typically approximations to the true global energy minimum structure, which we refer to as probe structures. If the probe structures contain physically realistic local environments for the constituent ions, they should then be close enough in energy to a local minimum in comparison to the energies of all the known competing phases (i.e. the convex hull – the energy surface derived from stable compositions) to indicate the possibility of obtaining a new compound with that composition. We can then initiate an experimental synthesis programme targeting the indicated composition, with a successful outcome being the experimental identification of a previously unknown phase.
We chose to explore combinations of Y3+, Sr2+, Ca2+, Ga3+ and O2− to search for possible new materials. These elements have different charge states, ionic radii and bonding characteristics, giving a range of coordination geometries. Although there are related gallate materials with functional optical and transport properties, there were no known materials in this chosen phase-field. With MC-EMMA, we generated probe structures based upon large layered cubic perovskite or melilite structure types. Coarse energy ranking with force-fields and refinement with Density Functional Theory indicated a target region of composition space, and detailed experimental synthesis and structural characterisation led to the identification of two new crystal structures of previously unknown composition. One material is related to the melilite structure and the other is a 4 × 4 × 4 supercell of perovskite. We subsequently modified the perovskite structure to generate a new down conversion phosphor host.
In the present study we introduce a new approach to generating/representing candidate structures that operates below the module level of MC-EMMA, choosing building blocks and the unit cell size and shape within the modules before assembling the full structure with chemically sensible ionic environments. We illustrate this method, the Flexible Unit Structure Engine (FUSE), by applying it to the generation of probe structures to identify the compositions of the stable phases in the experimentally explored Y3+–Sr2+–Ti4+–O2− phase field.12–21 Although we have used FUSE to generate probe structures and implemented structure search by a generalization of the MC basin hopping steps similar to those used in MC-EMMA, we anticipate that the basic construction approach could also be implemented in other structure evolution approaches, such as genetic algorithms or particle swarm optimisation, as used in other inorganic crystal structure prediction methods.22–25
2 Building the unit cell with the Flexible Unit Structure Engine (Fig. 1)
FUSE has been implemented in Python (2.7/3.6 or higher) and is dependent on the atomic simulation environment (ASE)26 and its subsequent dependencies. We generate an initial random seed structure for a given composition in the phase space under consideration in a series of steps outlined in detail below. First we choose randomly from five approximate lattice types, e.g. cubic or trigonal (Table 1), but without imposing internal symmetry, there is no triclinic approximation since we restrict two of the three unit cell angles to 90°. This step determines the number of units (sub-modules) and the number of layers (modules) that these should be spread across. A particular sub-module set is then generated by assigning the in-plane coordination of the cations. This set is split into equal-sized groups and spread across the modules that are used to assemble the 3D crystal structure, with stacking rules appropriate to the chosen lattice type, as implemented previously in EMMA. Before moving to geometry optimisation, we carry out a final error check to reduce the number of cations that are in physically unreasonable environments. Should a structure fail at this point, a fresh structure is generated. The geometry optimisation can be performed with an external Density Functional Theory (DFT) or force-field code.| Approximate lattice | Dimensions (x, y, z = # sub-modules along a, b, c) | γ (°) |
|---|---|---|
| Cubic | x = y, z = 2x | 90 |
| Tetragonal | x = y, z = even | 90 |
| Trigonal | x = y, z | 120 |
| Orthorhombic | x, y, z (z = even) | 90 |
| Monoclinic | x, y, z | 120 |
2.1 The generation of sub-modules (Fig. 1a)
The sub-modules are composed of one cation position accompanied by 0–3 nearest-neighbour anions, as shown in Fig. 1a. This defines eight basic motifs for the sub-modules based upon a single unit of AX3−δ, where A is a cation species, X is an anion species and δ = 0, 1, 2 or 3. For δ = 1 or 2, there are multiple non-equivalent ways in which anions can be arranged, giving one motif containing three anions, three containing two anions, three containing one anion and one motif with zero anions (Fig. 1a). We select randomly from this set, subject to the constraint that the total numbers of each ion type give the correct stoichiometry. Within the computer code, each sub-module is stored as three descriptors: the first labels the cation species, the second stores the motif label, and the final descriptor labels the anion species accompanying the cation in the sub-module. When all of the sub-modules have been generated they are stored in three lists, each containing all the descriptors of one type (e.g. one list contains all of the cation labels).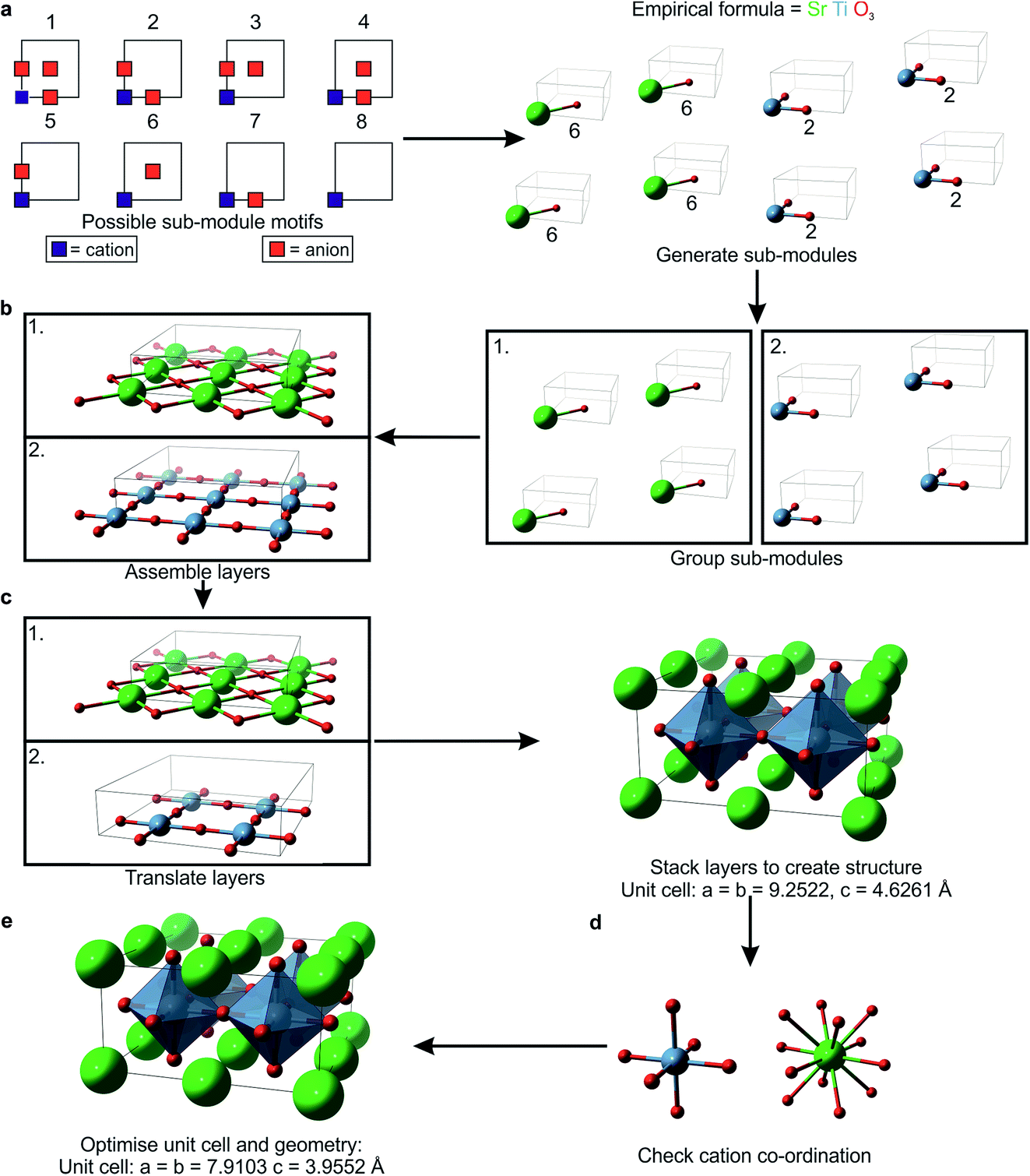 | ||
| Fig. 1 Example of how FUSE generates a structure, with an example cell containing 4 formula units of SrTiO3. (a) The motifs used to generate sub-modules labelled 1–8. FUSE chooses to target a tetragonal unit cell where a = b = 2 sub-modules and c = 2 sub-modules; the eight required sub-modules are then selected, with four of type 6 each containing SrO and four of type 2 each containing TiO2. (b) The unit cell indicates 2 modules; therefore the sub-modules are organised into two groups, which are then assembled into 2D modules. (c) The required translations are applied and the two modules are stacked to produce a full 3D structure. (d) All of the cation co-ordination environments are checked against tabulated data. (e) An external program is called to optimise the geometry and obtain the total energy. | ||
We describe the integer number of sub-modules in a given crystallographic direction using the symbols x, y and z, referring to the a, b and c crystallographic axes, respectively. The chosen lattice type also affects the total number of sub-modules, e.g. for a cubic lattice if we choose to have 2 sub-modules in the x-plane (Table 1), then we should have 2x3 = 16 sub-modules in total. During optimisation, the size of the unit cell, i.e. the number of modules, can grow, so it is not necessary for the initial structures to be very large. In this study we have imposed an arbitrary upper limit of 50 atoms in total in the unit cells of the initial structures. In these small unit cells it might prove impossible to fit the correct numbers of modules to satisfy stoichiometric requirements into the cubic lattice choice, for example. If FUSE is unsuccessful in populating a lattice of a particular type, it will try a different lattice type until a unit cell can be generated. The monoclinic lattice type is the default which can always be populated – in this lattice it will always be possible to at least assemble the sub-modules into a 1D chain.
2.2 The formation of modules from sub-modules (Fig. 1b)
The order in which the sub-modules are stored is randomised and the set is divided into z groups, where z is the number of modules determined by the choice of lattice. x × y sub-modules are selected and stored in separate descriptor arrays for each module. The positions are converted from fractional coordinates to Å scaling by:| Ip = 2(Rcat + Ran) |
2.3 Stacking the modules to produce the 3D structure (Fig. 1c)
To avoid stacking modules with like-charged ions packed on top of each other, each module is assigned a translation to be applied to all of its constituent ions. The translations are selected such that each adjacent module has a different translation instruction. If the selected lattice has γ = 90° the translations are based upon cubic packing, such that they alternate between [0lp, 0lp, 0lp] and [0.5lp, 0.5lp, 0.0lp]. When γ = 120° the translations are based on a close packed structure, with possible translations being [0lp, 0lp, 0lp], [1/3lp, 2/3lp, 0lp] or [2/3lp, 1/3lp, 0lp]. Applying the translations to the module list completes the generation of an initial structure.2.4 Error checking the co-ordination chemistry (Fig. 1d)
With a fully assembled structure, it is possible to determine the number of nearest-neighbour anions for each cation, giving an initial co-ordination number. As each sub-module is of the same size, nearest-neighbour cations and anions are separated by less than lp. To determine the initial co-ordination of a cation, we need to count the number of anions within 0.72lp. If the calculated co-ordination number of a cation differs by more than one from the value listed in the Shannon radii tables,27,28 it is counted as an ‘error’. If the fraction of these co-ordination errors exceeds the threshold btol, then this structure is rejected and a new one is generated until the fraction of errors is acceptable. For the systems investigated in this study, we optimised btol to maintain a high percentage of structures converging successfully in the geometry optimisation, while minimising the time taken to generate a structure with the number of errors below the threshold. We found that a btol value of 0.25 resulted in typically 95% of structures converging, with minimal impact on the time taken to generate structures.2.5 Geometry optimisation
Before relaxing the assembled structure it is important to apply a random displacement to each ion to prevent the optimisation routine failing to relax ions out of artificially high symmetry sites. We use the ASE ‘rattle’ function, which selects random displacements from a normal distribution of a specified standard deviation (in this study we have arbitrarily used 0.025 Å). Having completed the initial structure generation, we generate input files for the chosen geometry optimisation. We have implemented this for the DFT code VASP29 and the force-field based GULP30 code.When the external code has completed its geometry optimisation calculation(s), the co-ordinates and energy of the relaxed structure are recorded by FUSE. During structure evolution, we generate lattices of different sizes containing different numbers of ions; therefore we need to use the energy per atom for ranking structures. If geometry optimisation fails (e.g. due to a physically unreasonable starting geometry, or because the structure cannot relax within the allotted run time), then the energy per atom is artificially set to a very high value to prevent any subsequent search routines from selecting it as a structure to evolve.
3 Using FUSE to search for a probe structure at a given composition
Structure modifications are performed on the unassembled module representation, e.g. we swap ions between modules by simply swapping the appropriate labels within the arrays describing the modules, and then repeat the module stacking and error checking steps above. In principle, the basic FUSE construction could be paired with many existing search algorithms. In this study we implement a Monte Carlo basin-hopping algorithm, building on our earlier study with MC-EMMA11 (Fig. 2).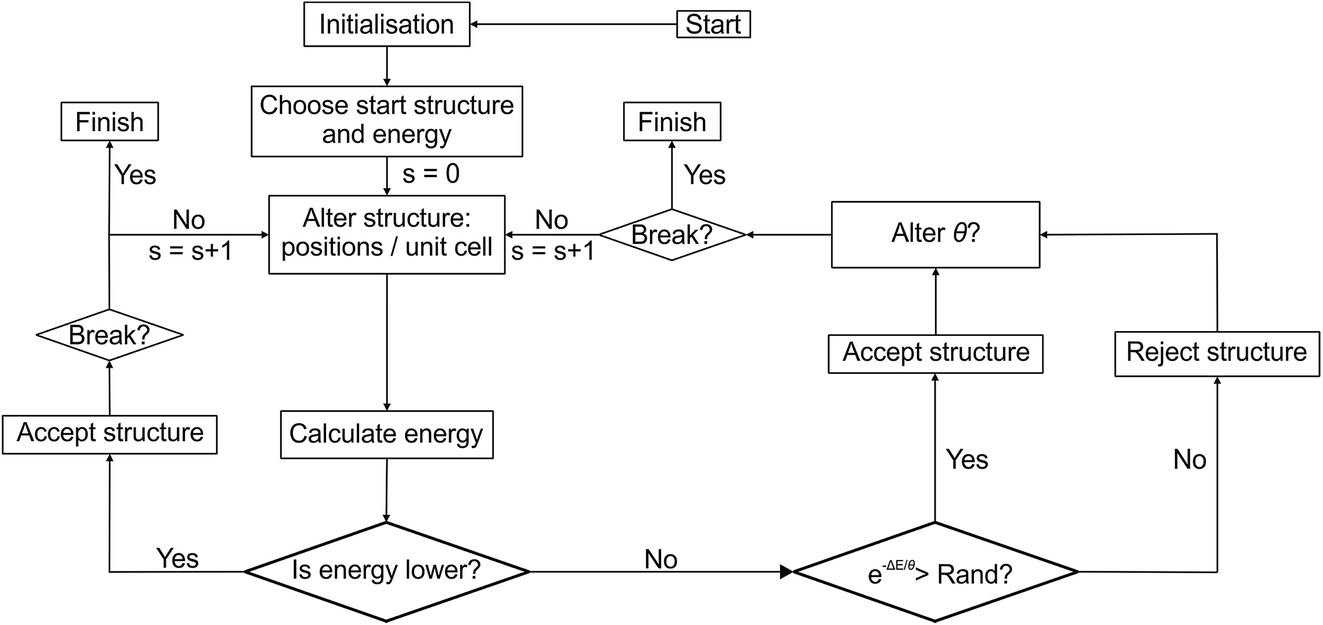 | ||
| Fig. 2 Flow diagram of the Monte Carlo search used by FUSE. Rand indicates a randomly generated number between 0 and 1. | ||
We begin the search for an optimum structure with an initialisation stage; we build structures until we are able to successfully perform geometry optimisation on 1000 structures. The lowest energy structure is then selected for evolution. The initialisation step ensures that the search starts from a physically reasonable structure. Since the number of atoms in the initial structures is limited (to a maximum of 50 in this study), the initialisation has a small computational cost compared to the geometry optimisation of the larger evolving structures. For the systems studied here, we limited the size of structures in the Monte Carlo search to no more than 150 atoms.
In each step of basin-hopping the current evolving structure (CES) can be altered by six different permutations:
3.1 Swap atoms within the structure
We first select whether to swap cations or anions. If there is only one anion or one cation species, FUSE selects to swap the other, i.e. if the only anion in a system is O2−, FUSE will only swap cations. When swapping cations, for example, we simply select two cations of different types from the cation arrays and swap their locations before the modules are assembled into a crystal. Both intra-module and inter-module swapping are allowed. This is a key improvement on our original MC-EMMA basin-hopping, in which the module compositions are fixed. For anions we currently have to keep both the anion type and the motif type together in order to preserve the overall composition of the unit cell. Note that if only one cation and one anion species are present (e.g. for SrO) then this permutation type cannot be used as no swaps will be possible.3.2 Alter the stacking sequence
Two modules within the sequence are switched before the structure is assembled.3.3 Grow the lattice by doubling the structure
If the current number of atoms in the unit cell is less than or equal to half of the permitted maximum, FUSE doubles the structure along a randomly chosen crystallographic axis.3.4 Alter the lattice by changing γ
The lattice angle, γ, is swapped from 90 to 120° or vice versa. This is performed prior to assembling the modules into a structure since the lattice angle determines which translation vectors are used in stacking.3.5 Generate a new random structure of similar volume
A new random structure is generated from the sub-module motifs, as detailed above, but with the maximum number of permitted atoms equal to that for the current structure.3.6 Generate a new random structure
A new random structure is generated with a random number of sub-modules. This differs from the generation of the initial structures in that we do not constrain the unit cell to small values, but allow a choice up to the maximum of number of atoms permitted in structure evolution.During the MC search, we use the simpler permutations more frequently. For the results presented here we use permutation 1 53% of the time. Permutations 2 and 3 each occur 21% of the time and the remaining permutations occur much less frequently, only ∼1% of the time (permutations are rounded to the nearest whole percent).
Once a new trial structure has been generated, geometry optimisation is performed to obtain its energy. The new trial structure replaces the CES if its energy is lower than that of the CES, or randomly if the usual Monte Carlo condition is fulfilled, i.e. if e−ΔE/θ > x, where x is a random number between 0 and 1, ΔE is the energy difference between the CES and the trial structure, and θ is the MC temperature parameter. Higher values of θ give higher probabilities of accepting an uphill energy step. In this study we use a value of θ = 0.02 eV unless the basin-hopping gets trapped in a particular minimum.
The MC loop contains a break condition, at which point the search is considered converged. This occurs when rmax structures have been rejected since finding the structure with the lowest known energy. If the number of structures rejected since the last structure acceptance, r, is greater than 0.1 × rmax the MC temperature parameter is gradually increased. θ is increased by a random number (<0.001) for every ten steps that r is above 0.1 × rmax. While the system is ‘heating up’, FUSE only uses permutation types 5 and 6 (in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio), i.e. the entire crystal structure is replaced. For the example presented in this study, these values were found to be effective in helping the MC search escape into a new basin (as can be seen in Fig. 3a). When the system escapes into a new basin, θ and the permutation frequencies return to the original values.
:1 ratio), i.e. the entire crystal structure is replaced. For the example presented in this study, these values were found to be effective in helping the MC search escape into a new basin (as can be seen in Fig. 3a). When the system escapes into a new basin, θ and the permutation frequencies return to the original values.
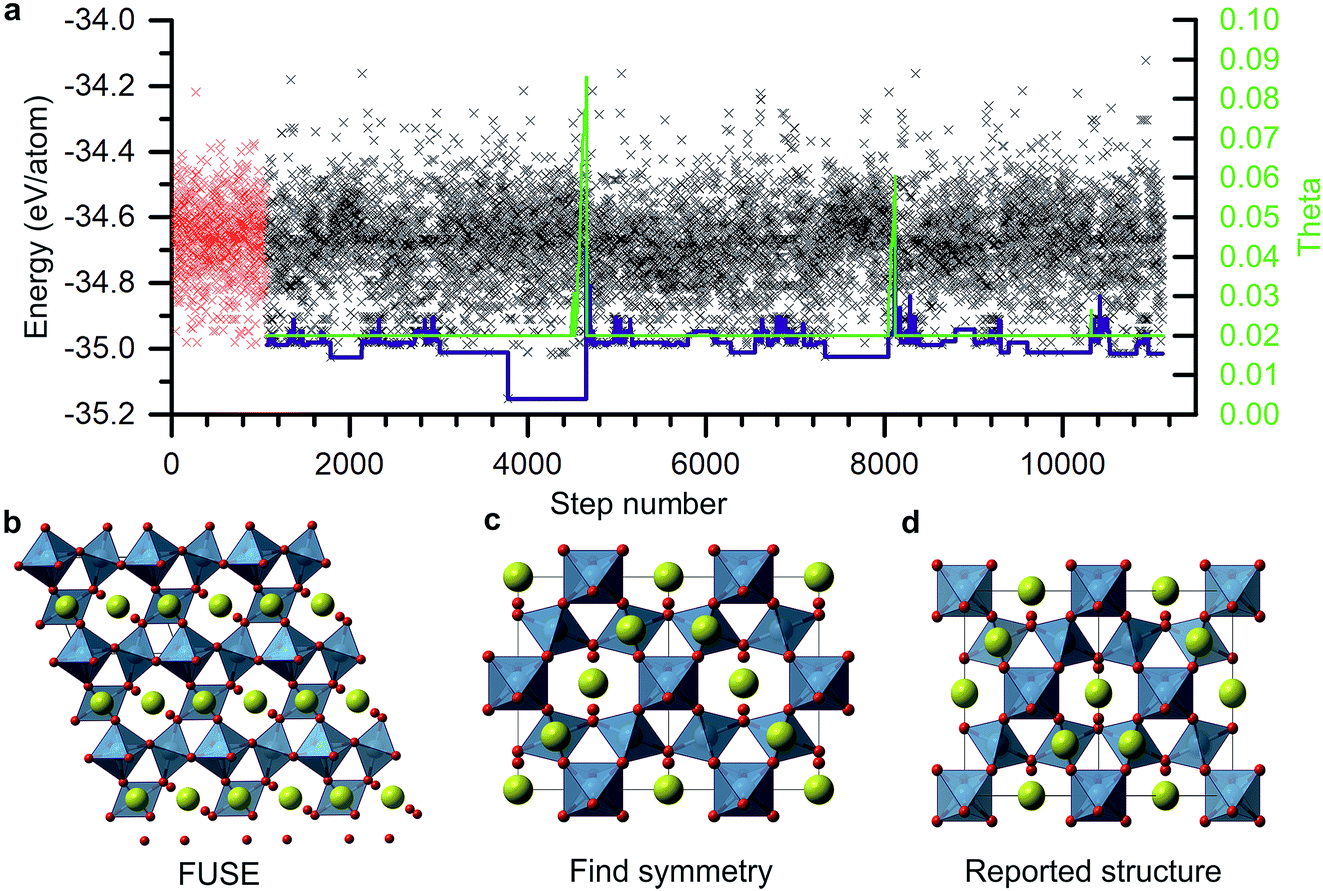 | ||
| Fig. 3 (a) Example FUSE run for Y2Ti2O7. The red markers indicate the 1062 structures sampled in the initialisation loop; black markers indicate structures sampled in the MC search routine; the blue line indicates the energy of the current evolving structure that FUSE is carrying forward in the MC search; the green line indicates the value of the MC temperature parameter (right axis) during the MC search. (b) The raw structure obtained in MC step 2715. (c) The structure obtained after passing the structure in (b) through FINDSYM. (d) The experimental crystal structure for Y2Ti2O7; the FUSE structure differs from the ICSD structure by an origin shift. Atoms are coloured as follows: Y (yellow), Ti (blue) and O (red). | ||
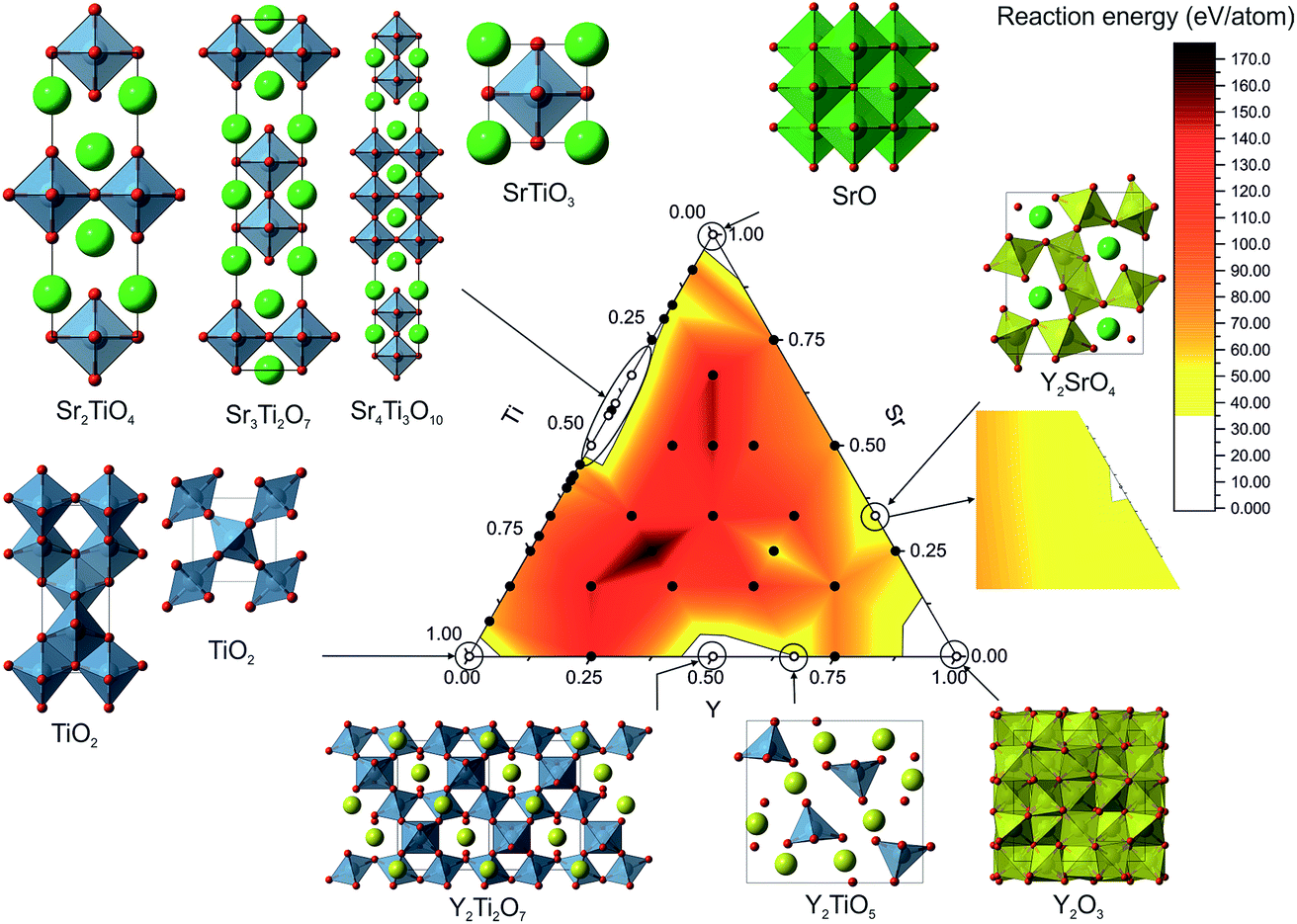 | ||
| Fig. 4 FUSE-based probe structure calculations on the Y–Sr–Ti–O phase field. Points on the phase field indicate where we generated probe structures; open circles indicate compositions at which compounds are reported in the ISCD, with their crystal structures shown outside the phase field; regions highlighted in white indicate regions where the energy is less than 35 meV per atom above the convex hull; inset on the right shows shallow minima found around Y2SrO4. Atoms are coloured as follows: Y (yellow), Ti (blue), Sr (green) and O (red). | ||
During probe structure generation, we performed three independent searches at each composition, taking the lowest energy structure from the three runs as the probe structure for that composition.
4 Testing the methodology: the Y3+–Sr2+–Ti4+–O2− phase field
We used FUSE to explore the Y3+–Sr2+–Ti4+–O2 phase field. Using the Inorganic Crystal Structure Database (ICSD)31 we identified 10 compounds with well-defined structures including rutile, rock-salt, perovskite, pyrochlore and Ruddlesden–Popper phases. This diverse range provided a strong test of our ability to identify energy minima corresponding to different ionic environments using the simple construction encoded in FUSE to generate approximate probe structures.4.1 Geometry optimisation
For the calculations presented within this study, we used the General Utility Lattice Program (GULP)30 for geometry optimisation. Buckingham potentials were used for the short-range component of the potential, with a cut-off radius of 12 Å. For all structures, the unit cell parameters and atomic positions were optimised until the norm of the gradient was lower than 0.001 (a.u.).Parameters for the O2−–O2− and Sr2+–O2− interactions were obtained from the literature.32,33 The parameters for Ti4+–O2− and Y3+–O2− were based upon literature values,22,34 but the A parameters were adjusted so that the lowest energy experimental structures were obtained as the energetic ground states for TiO2 and Y2O3 in comparison to other polymorphs (for TiO2) and random structures generated using FUSE. The final potential parameters are presented in Table 2.
| Interaction | A (eV) | ρ (Å) | C (eV Å−6) |
|---|---|---|---|
| O2−–O2− | 1388.77 | 0.36262 | 175 |
| Y3+–O2− | 23000 |
0.24203 | 0 |
| Sr2+–O2− | 1952.39 | 0.33685 | 19.22 |
| Ti4+–O2− | 4590.7279 | 0.261 | 0 |
5 Results
5.1 The ground-state structure for a single composition: Y2Ti2O7
As an example of a FUSE calculation from within the Y–Sr–Ti–O phase field, we focus on the composition Y2Ti2O7. Experimentally, Y2Ti2O7 crystallises in the pyrochlore structure,16 containing a mixture of octahedrally co-ordinated Ti4+ and eight co-ordinate Y3+ ions in the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The full unit cell contains 88 ions, with 22 in the primitive cell, with four unique sites (Fig. 3d). Obtaining the ground-state structure in the absence of symmetry therefore presents a substantial challenge.
m. The full unit cell contains 88 ions, with 22 in the primitive cell, with four unique sites (Fig. 3d). Obtaining the ground-state structure in the absence of symmetry therefore presents a substantial challenge.
Fig. 3a shows the energies of all the structures generated and tested for this composition. The energy of the CES is shown by the blue line. After the initiation stage the CES cycled up and down in energy before dropping into a deep minimum at MC step 2715. This minimum actually corresponds to the experimental crystal structure and is the lowest energy structure obtained for this composition. Since the energy could not go lower, the MC routine was stuck in this basin until the MC temperature (also shown) was ramped up and the full structure replacement permutations, 5 and 6, were employed. Several other basins were explored, with temperature jumps required to exit two of them. The structure evolution ended after ∼10000 MC steps.
The structure in the lowest energy basin was found to be a small, low symmetry unit cell (Fig. 2b) containing 22 atoms. This was then passed through the FINDSYM program,35 which returned the correct space group and structure, with four unique sites (Fig. 3c), although with an origin shift relative to the reported crystal structure (Fig. 3d).
5.2 Exploring the composition space (Fig. 4)
Across the phase field we sampled 42 different compositions from the binary tie lines and within the ternary space. For each composition we performed three runs, taking the lowest energy structure as the probe structure. The control parameters for FUSE were determined by finding a set for which the correct ground-state structure could be reliably obtained for the Y2Ti2O7 composition. The initialisation loop was run until 1000 structures converged during geometry optimisation or until 1500 structures had been tried. The error tolerance parameter was set to 0.25, the default θ to 0.02, and the run termination parameter rmax was set to 7000 for each composition. The weightings given to the MC moves were not optimised and therefore we hypothesise that given optimisation, the MC search routine would converge on the lowest energy structure more quickly.Using the results from the FUSE calculations, we assembled the convex hull using pymatgen.36 Accounting for the approximate nature of the probe structures, energies lying slightly above the convex hull could still identify islands of stability. In the previous phase diagrams explored using probe structures, we found experimentally realisable materials lying ∼35 meV per atom (ref. 11) above the convex hull and so we used this as a cutoff value to identify stable compositions. All of the known stable compositions were found to lie within this cutoff, i.e. the probe structures correctly identified the compositions of all the known phases. The highest energy above the convex hull was found to be 34 meV per atom for Y2TiO5.15 Plotting the compositional phase space, we observed that only these known compositions were found under the 35 meV per atom threshold. Of these, FUSE obtained the exact ICSD crystal structures for: TiO2 (anatase18 and rutile17), SrO,20 Sr2TiO4,21 Sr3Ti2O7,12 Sr4Ti3O10,12 SrTiO3,19 Y2Ti2O7 (ref. 16) and Y2TiO5.15
6 Comparing probe structures with ICSD structures (Fig. 5)
For SrO, TiO2, Sr2TiO4, SrTiO3 and Y2Ti2O7, the lowest energy structures obtained in FUSE matched the crystal structures in the ICSD database (Fig. 5a–e) when we found symmetry within the probe structure using FINDSYM, although some structures contained an origin shift versus the published structure. For TiO2 FUSE identified the exact crystal structure of both the anatase and rutile polymorphs, with rutile calculated to be the ground-state structure.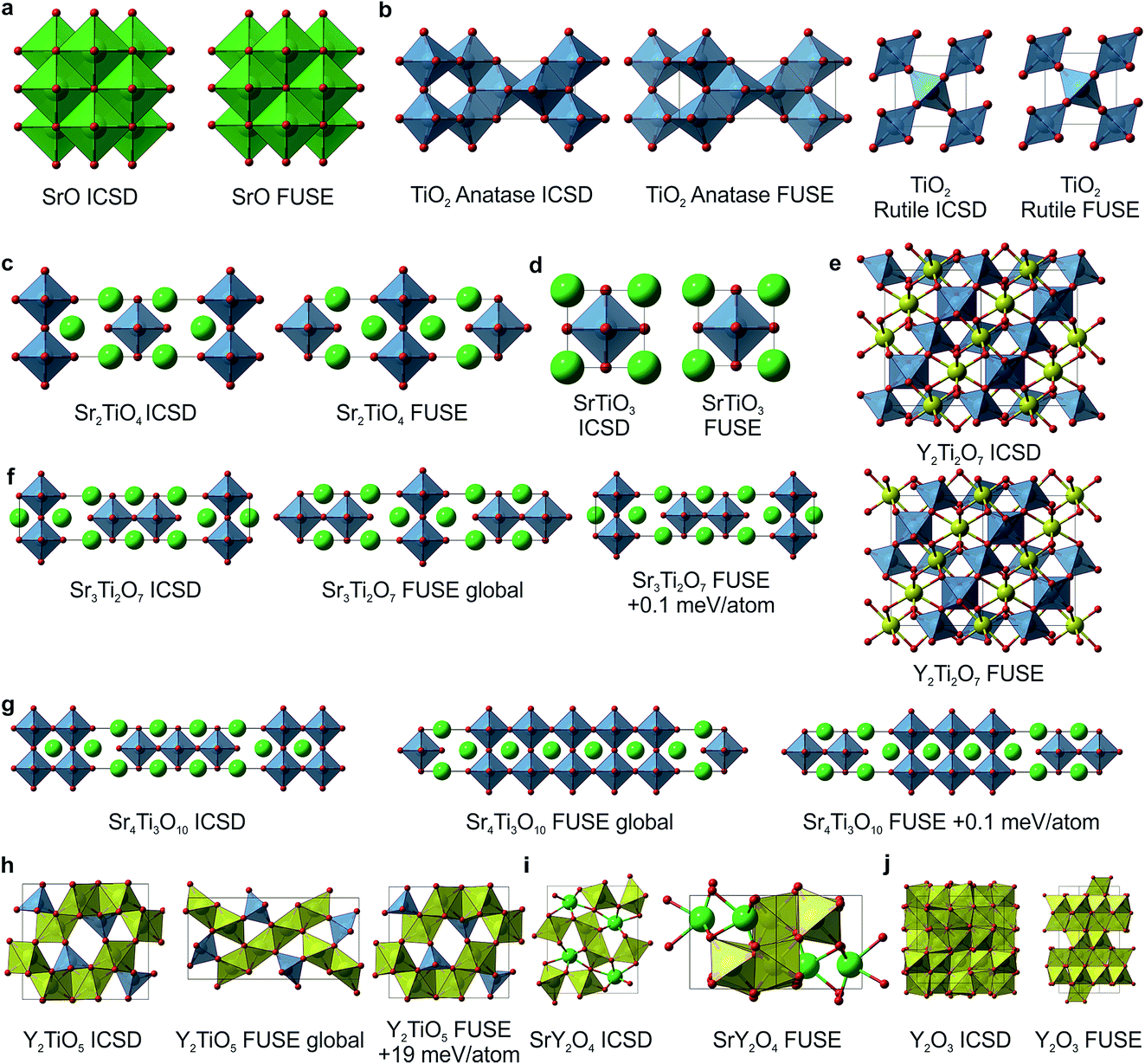 | ||
| Fig. 5 Probe structures for each of the 10 ICSD compounds studied with FUSE in the Y–Sr–Ti–O phase field. (a) SrO in the rock-salt structure. (b) For TiO2 FUSE correctly identifies the rutile polymorph as the ground-state structure (right) and is also able to locate the anatase polymorph (left). (c) The Sr2TiO4 Ruddlesden–Popper n = 1 phase. (d) SrTiO3 perovskite. (e) Y2Ti2O7 pyrochlore structure; FUSE identifies the correct structure although with the origin shifted such that a Y ion is at the origin rather than Ti. (f) The Sr3Ti2O7 Ruddlesden–Popper n = 2 phase; for this composition, there are several similar stacking sequences within 0.2 meV per atom around the minimum energy; the experimental structure is identified by FUSE at +0.1 meV per atom from the minimum energy. (g) The Sr4Ti3O10 Ruddlesden–Popper n = 3 phase; as with (f), there exist several stacking sequences with similar energies, and the experimental structure is within 0.1 meV per atom of the minimum. (h) The Y2TiO5 structure; by force-field calculations, FUSE found the experimental structure at +19 meV per atom above the minimum. (i–j) For SrY2O4 (ref. 14) and Y2O3 (ref. 13) FUSE was unable to locate the experimental structure, although the cation co-ordinations in the probe structures created are similar to those in the experimental structures. Atoms are coloured as follows: Y (yellow), Ti (blue), Sr (green) and O (red). | ||
For the compositions Sr3Ti2O7 and Sr4Ti3O10 (Fig. 5f and g), which both form Ruddlesden–Popper phases, there are multiple stacking sequences which differ by less than 0.2 meV per atom. With the force-field used within this study, the experimental crystal structures were found to be 0.1 meV per atom above the lowest energy structure obtained in FUSE.
For Y2TiO5, the probe structure obtained was found to be 19 meV per atom lower in energy than the experimental structure (Fig. 5h), although the experimental structure was obtained during the FUSE runs, but not identified as the global minimum. This highlights the need to recalculate the energies of the probe structures with more accurate methods (DFT), as we have done previously.11
The compositions SrY2O4 and Y2O3 were correctly identified by the probe structures as lying in regions of stability, however the present MC search was unable to obtain the exact ICSD crystal structures (Fig. 5i and j). The FUSE probe structures for SrY2O4 and Y2O3 were found to be +9 and +40 meV per atom above the ICSD structures. Although we do not set out to fully predict structures, but rather the regions of stable composition, we believe that FUSE will correctly obtain the structures of these two outliers once adjustments are made to the search/construction routine. To demonstrate that FUSE can assemble a model that will relax to the correct structure for Y2O3, we used the FUSE rules to build it by hand. Building a cell for Y2O3 from the mixed anion–cation sub-modules used in the present version of FUSE requires stacking along the [110] axis of the experimental cell. This yields a unit cell containing 160 ions, a greater number than was allowed in the MC search in this study. However, assembling this structure by hand, geometry optimisation (Fig. 6a) followed by symmetry searching did yield the ICSD structure (Fig. 6b and c).
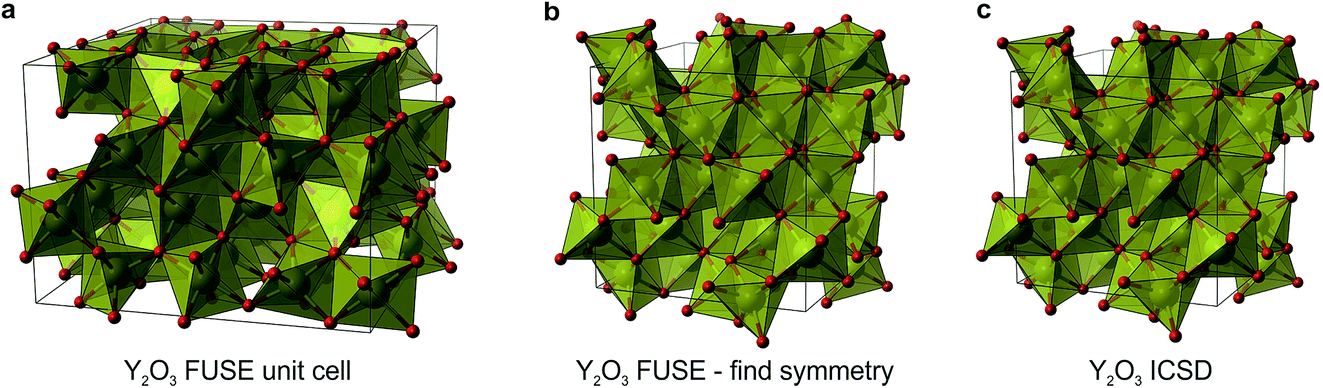 | ||
| Fig. 6 (a) The unit cell of Y2O3 assembled by hand within the FUSE description (after geometry relaxation). (b) The FUSE unit cell of Y2O3 after being run through FINDSYM, reducing the cell to the experimental Ia cell. (c) The ICSD unit cell for Y2O3. Atoms are coloured as follows: Y (yellow) and O (red). | ||
7 Conclusions
In this study, we have presented a new method, FUSE, for probe structure-based inorganic materials discovery, based upon the construction of unit cells from randomly generated modules of varying shape, size and composition. FUSE can generate a wide range of physically plausible structures, with only modest restrictions applied to the type of crystal structure being created. The method was tested by exploring the known Y3+–Sr2+–Ti4+–O2− phase field, and successfully identified all of the compositions where structures are known to form as lying in low energy regions of the phase diagram. Additionally, in the search we found the exact crystal structure of 8 out of the 10 compositions. FUSE provides a new tool specifically designed for the efficient generation of probe structures across the compositional phase space. It has the potential to accelerate the computational prediction of regions of the phase space that can be profitably explored to experimentally realise new materials.8 Data availability
The data used within this study is available from the authors on request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EPSRC for funding under EP/N004884 and the University of Liverpool for access to local high-performance computing resources. We would like to thank Dr F. McBride (University of Liverpool) for useful discussions when developing the FUSE concept and Dr J. B. Claridge (University of Liverpool) for useful discussions when deciding upon the test system for FUSE. All structural images were created using VESTA.37References
- I. V. Pentin, J. C. Schon and M. Jansen, Phys. Chem. Chem. Phys., 2010, 12, 8491–8499 RSC.
- J. C. Schon, K. Doll and M. Jansen, Phys. Status Solidi B, 2010, 247, 23–39 CrossRef.
- S. M. Woodley and R. Catlow, Nat. Mater., 2008, 7, 937–946 CrossRef PubMed.
- G. Hautier, C. Fischer, V. Ehrlacher, A. Jain and G. Ceder, Inorg. Chem., 2011, 50, 656–663 CrossRef PubMed.
- J. C. Schön, Z. Anorg. Allg. Chem., 2014, 640, 2717–2726 CrossRef PubMed.
- H. Chen, G. Hautier and G. Ceder, J. Am. Chem. Soc., 2012, 134, 19619–19627 CrossRef PubMed.
- R. Gautier, X. Zhang, L. Hu, L. Yu, Y. Lin, T. O. L. Sunde, D. Chon, K. R. Poeppelmeier and A. Zunger, Nat. Chem., 2015, 7, 308–316 CrossRef PubMed.
- T. Ishihara, H. Matsuda and Y. Takita, J. Am. Chem. Soc., 1994, 116, 3801–3803 CrossRef.
- X. Kuang, M. A. Green, H. Niu, P. Zajdel, C. Dickinson, J. B. Claridge, L. Jantsky and M. J. Rosseinsky, Nat. Mater., 2008, 7, 498–504 CrossRef PubMed.
- M. S. Dyer, C. Collins, D. Hodgeman, P. A. Chater, A. Demont, S. Romani, R. Sayers, M. F. Thomas, J. B. Claridge, G. R. Darling and M. J. Rosseinsky, Science, 2013, 340, 847–852 CrossRef PubMed.
- C. Collins, M. S. Dyer, M. J. Pitcher, G. F. S. Whitehead, M. Zanella, P. Mandal, J. B. Claridge, G. R. Darling and M. J. Rosseinsky, Nature, 2017, 546, 280–284 CrossRef PubMed.
- S. N. Ruddlesden and P. Popper, Acta Crystallogr., 1958, 11, 54–55 CrossRef.
- M. G. Paton and E. N. Maslen, Acta Crystallogr., 1965, 19, 307–310 CrossRef.
- H. Müller-Buschbaum, Z. Anorg. Allg. Chem., 1968, 358, 138–146 CrossRef.
- W. G. Mumme and A. D. Wadsley, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 1327–1333 CrossRef.
- W. J. Becker and G. Will, Z. Kristallogr. - New Cryst. Struct., 1970, 131, 278–288 CrossRef.
- W. H. Baur and A. A. Khan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2133–2139 CrossRef.
- M. Horn, C. F. Schwerdtfeger and E. P. Meagher, Z. Kristallogr. - New Cryst. Struct., 1972, 136, 273–281 CrossRef.
- R. J. Nelmes, G. M. Meyer and J. Hutton, Ferroelectrics, 1978, 21, 461–462 CrossRef.
- J. Bashir, R. T. A. Khan, N. M. Butt and G. Heger, Powder Diffr., 2002, 17, 222–224 CrossRef.
- K. Kawamura, M. Yashima, K. Fujii, K. Omoto, K. Hibino, S. Yamada, J. R. Hester, M. Avdeev, P. Miao, S. Torii and T. Kamiyama, Inorg. Chem., 2015, 54, 3896–3904 CrossRef PubMed.
- S. M. Woodley, P. D. Battle, J. D. Gale and C. R. A. Catlow, Phys. Chem. Chem. Phys., 1999, 1, 2535–2542 RSC.
- Y. Wang, J. Lv, L. Zhu, S. Lu, K. Yin, Q. Li, H. Wang, L. Zhang and Y. Ma, J. Phys.: Condens. Matter, 2015, 27, 203203 CrossRef PubMed.
- A. O. Lyakhov, A. R. Oganov, H. T. Stokes and Q. Zhu, Comput. Phys. Commun., 2013, 184, 1172–1182 CrossRef.
- J. P. Chris and R. J. Needs, J. Phys.: Condens. Matter, 2011, 23, 053201 CrossRef PubMed.
- L. Ask Hjorth, M. Jens Jørgen, B. Jakob, E. C. Ivano, C. Rune, D. Marcin, F. Jesper, N. G. Michael, H. Bjørk, H. Cory, D. H. Eric, C. J. Paul, J. Peter Bjerre, K. James, R. K. John, K. Esben Leonhard, K. Joseph, K. Kristen, L. Steen, M. Jón Bergmann, M. Tristan, O. Thomas, P. Lars, P. Andrew, R. Carsten, S. Jakob, S. Ole, S. Mikkel, S. T. Kristian, V. Tejs, V. Lasse, W. Michael, Z. Zhenhua and W. J. Karsten, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- Database of Ionic Radii, http://abulafia.mt.ic.ac.uk/shannon/ptable.php, accessed October, 2016.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef.
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291–341 CrossRef.
- The Inorganic Crystal Structure Database, FIZ Karlsruhe, http://www2.fiz-karlsruhe.de/icsd_home.html, accessed February 2017.
- B. W. H. Vanbeest, G. J. Kramer and R. A. Vansanten, Phys. Rev. Lett., 1990, 64, 1955–1958 CrossRef PubMed.
- S. M. Woodley, http://www.ucl.ac.uk/klmc/Potentials/, accessed August 2016.
- R. C. Baetzold, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 11304–11312 CrossRef.
- H. T. Stokes and D. M. Hatch, J. Appl. Crystallogr., 2005, 38, 237–238 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |